

Abstract
The dynein axonemal assembly factor (Dnaaf) protein family is involved in preassembly and stability of dynein arms before they are transported into the cilia. In humans, mutations in DNAAF genes lead to several diseases related to cilia defects such as primary ciliary dyskinesia (PCD; OMIM: 612518). Patients with PCD experience malfunctions in cilia motility, which can result in inflammation and infection of the respiratory tract among other defects. Previous studies have identified that a mutation in DNAAF2 results in PCD and that 40% of these patients also experience laterality defects. In an outbred genetic background, Dnaaf2 homozygotes die after birth and have left/right defects among other phenotypes. Here we characterize a novel null allele of Dnaaf2 obtained from the International Mouse Phenotyping Consortium. Our data indicate that on a defined C57bl/6NJ genetic background, homozygous Dnaaf2 mouse embryos fail to progress beyond organogenesis stages with many abnormalities including left–right patterning defects. These findings support studies indicating that hypomorphic mutations of human DNAAF2 can result in ciliary dyskinesia and identify Dnaaf2 as an essential component of cilia function in vivo.
Introduction
Cilia are evolutionarily conserved structures that are present in most eukaryotes (1). Although initially thought to be vestigial, it is now accepted that the majority of mammalian cell types retain a primary cilium, and many investigations have shown that cilia play important and essential roles in various signaling pathways and developmental processes (2–4). Furthermore, defects in cilia assembly greatly influence the function of cilia. There are two types of cilia; motile and primary (non-motile) cilia. Motile cilia exert mechanical force and are usually found in epithelial cells, whereas primary cilia predominantly function as a sensory organelle and are essential for initiating cellular signaling in adult tissues such as the kidney and in embryonic structures including the node (5,6).
Embryonic cells contain both motile and primary cilia (7). Primary cilia play a particularly significant role during gastrulation—the dynamic morphogenetic process that results in the organization of the three primary germ layers. At the onset of gastrulation an epithelial-to-mesenchymal transition occurs on the posterior of the embryo forming a transient structure known as the primitive streak, which extends through the midline of the embryo and contributes to the definition of the anterior–posterior axis (8). The streak extends from the proximal/posterior to the distal tip of the embryo, and the most distal/anterior tip of the primitive streak forms a unique flattened horseshoe-shaped structure, which contains organizer activity. Elegant studies have shown that this transient structure (the node) contains cilia that generate a leftward flow resulting in the first molecular break in left–right symmetry of the developing embryo. This cilia-induced nodal flow is essential for left–right axis specification (7), and although the mechanism remains controversial, it is clear that defects in this leftward flow can result in situs inversus or situs ambiguous (9,10).
Bilateral asymmetry requires specific spatiotemporal gene expression (11). In the mouse embryo, the leftward flow generated at the node results in asymmetric Nodal expression, which produces asymmetric gene expression in the lateral plate mesoderm (12,13). Nodal promotes expression of Pitx2, a gene that is normally expressed only on the left-side lateral plate mesoderm (in response to the presence of Nodal on the left side of the node) (14). Sonic hedgehog (Shh) has been shown to be upstream of Nodal, and in Shh null mouse embryos the left–right axis is compromised with a variety of laterality defects including misexpression of Nodal, Pitx2, Lefty1 and other left–right specific expression (14). It has been hypothesized that the asymmetric gene expression is generated by both motile and primary cilia at the node (14). This idea is supported by studies of mutant mice with immotile cilia as well as mutants without any nodal cilia (15,16). In both cases, genes that are typically expressed on either the left or right are found to be bilaterally or abnormally expressed in the mutant embryos.
The dynein axonemal assembly factor (Dnaaf) protein family is involved in the preassembly and stability of dynein arms before they are transported into the cilia (17–21). In humans, mutations in DNAAF genes lead to several diseases related to cilia defects such as primary ciliary dyskinesia (PCD) (19,21–23). Patients with PCD experience malfunctions in cilia motility, which can result in inflammation and infection in the respiratory tract among other defects (24). Previous studies have identified that a mutation in DNAAF2 results in PCD and that 40% of these patients also experience laterality defects (17,21,25). Mammalian cells lacking Dnaaf2 have defects in both the outer and inner dynein arms, resulting in a loss of cilia motility (17,18). Malfunction of cilia during embryonic development can also result in laterality defects and left–right axis abnormalities (7).
As mentioned above, human DNAAF2 can result in ciliary dyskinesia and laterality defects. However, little is known about the role of Dnaaf2 during embryogenesis. The Knockout Mouse Project (KOMP) is part of a global initiative to generate a null mutation in every gene within the mouse genome. The goal of the project is to elucidate the function of every gene and evaluate potential involvement in human health and disease. While the major focus has been on identifying genetic mutations resulting in adult phenotypes related to human disease, a large percentage of knockout (KO) alleles result in homozygous embryonic lethal phenotypes (26,27). Here we report that in the absence of Dnaaf2, embryos fail to develop beyond early organogenesis, with abnormalities including left–right patterning defects. Consistent with finding in other species, our data suggest that Dnaaf2 does not interact directly with left–right signaling pathways but is required for proper cilia function and nodal flow.
Results
Expression of Dnaaf genes
We first determined the expression of Dnaaf2 in various adult mouse tissues. Since Dnaaf2 is associated with proper cilia function, it is not surprising that the gene is expressed in multiciliated tissues including brain, kidney, liver, lung, ovary, oviduct, spleen and testis (Fig. 1A). This result also correlates well with a previous study of Dnaaf2 in mouse tissues (19). We also examined the expression of the other Dnaaf family members in adult mouse tissues. Past work has identified DNAAF1 to be crucial for neural tube development in humans (28), where its main role is to provide stability for the ciliary structure. DNAAF2, DNAAF3 and DNAAF4 (18–20,29) are associated with laterality and cilia motility in humans, and Dnaaf2 was shown to interact with Dnaaf4 in mouse respiratory epithelial cells (20). DNAAF5 is essential for transporting ciliary machinery to the motile axoneme (30). Unlike other DNAAF members, DNAAF5 does not interact with the chaperone proteins and has been found to colocalize with DNAAF2 and other proteins to form the early preassembly complex (29). Consistent with the role of the Dnaaf family, these genes are all expressed in oviduct and testis (Fig. 1A) where cilia play an essential role in tubal transport in vivo and tubule formation and cellular signaling in testicular somatic cells in vitro (31–33). Specifically, Dnaaf1 is expressed at high level in brain, ovary, oviduct and testis but expressed at low level in heart, kidney, liver, lung and spleen; Dnaaf2, Dnaaf4 and Dnaaf5 are expressed in all tissues examined (brain, heart, kidney, liver, lung, ovary, oviduct, spleen and testis); and Dnaaf3 expression is largely restricted to lung, ovary, oviduct and testis (Fig. 1A). Previous studies have shown that DNAAF4 and DNAAF5 physically interact with DNAAF2 (20,29), so it is perhaps not surprising that we observe similar expression patterns for these genes.
Figure 1.
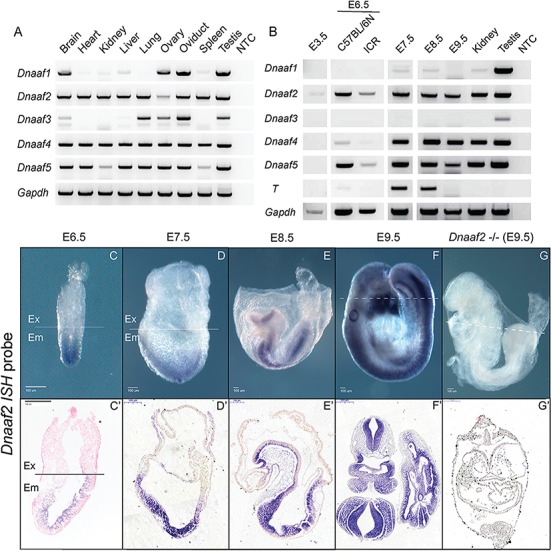
Dnaaf2 is expressed in various adult mouse tissues and across different stages of mouse embryos. (A and B) RNA was freshly extracted from adult mouse tissues, and cDNA was synthesized for reverse transcription polymerase chain reaction (RT-PCR) analysis. T (brachyury) and Gapdh were used for indicating embryonic stages and loading control. (A) Dnaaf2 is expressed in multiple ciliated adult mouse tissues. (C–G) To more closely examine Dnaaf2 expression during development, embryos were collected at the indicated embryonic day (E), and whole-mount ISH was performed using Dnaaf2 antisense probe that is generated from intron-spanning primers that are complementary to the protein-coding regions of exon 1 and exon 3 (Fig. 3C). During embryo development, Dnaaf2 is expressed throughout the embryonic lineage beginning at E6.5 and develops tissue specificity at organogenesis (C′–F′). Sagittal section, C′, D′ and E′; transverse section, F′ and G′; −/− denotes Dnaaf2 mutant embryo; dotted line denotes the transverse section shown in C′, F′ and G′; Ex denotes extraembryonic portion; and Em denotes embryonic portion.
It is currently unknown when or where Dnaaf family members are expressed during embryonic development. Therefore, we assessed Dnaaf family gene expression in wild-type embryos at specific developmental stages in mouse embryos: blastocyst (E3.5), gastrulation (E6.5–8.5) and organogenesis (E9.5) stage embryos (Fig. 1B).
Dnaaf2 is the only family member expressed in the blastocysts during preimplantation. Dnaaf4 and Dnaaf5 have similar temporal expression beginning at E6.5. Conversely, Dnaaf1 and Dnaaf3 are not expressed at appreciable levels during any embryonic stage tested (Fig. 1B). It is worth noting that besides Dnnaf2, Dnaaf4 and Dnaaf5 are expressed robustly beginning at the onset of gastrulation. The expression pattern of Dnaaf2, Dnaaf4 and Dnaaf5 overlaps at the onset of gastrulation suggesting the importance of these Dnaaf members in promoting proper functional cilia at this specific stage of development. To evaluate spatiotemporal and tissue-specific expression, we performed whole-mount in situ hybridization (ISH) with two distinct antisense probes using intron-spanning primers designed against Dnaaf2 (only one shown). Dnaaf2 mRNA is present specifically in the epiblast at E6.5 and in all newly specified embryonic lineages at E7.5 but is conspicuously absent from all extraembryonic lineages, even those that are primitive streak derived including the allantois and yolk sac mesoderm (Fig. 1C and D). Importantly, Dnaaf2 is expressed in the embryonic node at E7.5. In late gastrulation and organogenesis stage embryos, Dnaaf2 expression becomes further restricted to the lateral plate mesoderm, the neural tube, gut tube and first branchial arch (Fig. 1E and F). As expected, Dnaaf2 signal was completely absent in Dnaaf2−/− embryos (Fig. 1G) confirming the specificity of the ISH expression patterns observed.
Dnaaf2 function is essential for normal embryo development
To determine the role of Dnaaf2, we obtained a Dnaaf2 KO allele generated at the Jackson Laboratories as part of the KOMP (26). This mutation was produced by replacing the majority of the endogenous locus with a lacZ reporter gene, resulting in deletion of 8457 bases (Dnaaf2tm1.1(KOMP)Vlcg). A previous large-scale screen of viability in newly generated alleles identified a lack of Dnaaf2 embryos at E15.5 (26). Surprisingly, one previous study reports that a KO of Dnaaf2 is viable with subtle adult phenotypes (19). We believe this discrepancy is most likely explained due to genetic background and functional redundancy (discussed further below). Therefore, in order to identify the Dnaaf2 loss-of-function phenotype in a C57bl/6NJ background, we performed heterozygous intercrosses and analyzed embryos between E6.5 and E13.5 (Fig. 2A). Genotypes of Dnaaf2 homozygous null embryos were verified with Dnaaf2-specific polymerase chain reaction (PCR) primers (Fig. 2D). Comparing the morphology of Dnaaf2−/− embryos (hereafter referred to as `−/−’, `mutants’ or `mutant embryos’) to wild-type and heterozygous littermates (hereafter referred to as `control’, `+/−’ or `+/+’ embryos), mutants show a significant developmental delay as early as gastrulation stages (E6.5–7.5, Fig. 2A). Mutants continue to develop but are grossly retarded by E8.5–9.5 and are significantly malformed and dying at later stages (E11.5 and 13.5). In addition to the developmental delay, the most obvious phenotype observed is that mutants do not initiate embryonic turning, which is essential for continued growth and development of both the placenta and embryo (Fig. 2A). At E9.5, mutant embryos have not established a vascular network in the yolk sac. This difference is most apparent when comparing the yolk sac of E11.5 mutant embryo and its littermate (Fig. 2B and C). Failure to develop the proper cardiovascular network greatly affects embryo development and the viability of the growing embryo (34). We also observe improper closure of the neural tube in the brain of most mutant embryos, most apparent in E9.5 mutant embryos (Fig. 2A). At E11.5, mutant embryos display pericardial edema, which could be a secondary defect of malformation of the cardiovascular network, which in turn may contribute to the delay in heart development (Fig. 2A). The tailbud region of the mutant embryos at E11.5 also displays an abnormal cavity (Fig. 2A). This may further indicate a failure of functional connection between the embryo and the placenta. Since the placenta appears to develop normally in mutant embryos, the improper development of the vascular network in the yolk sac likely disrupts umbilical cord development (35) and hence contributes to the overall delay in development (Fig. 2A).
Figure 2.
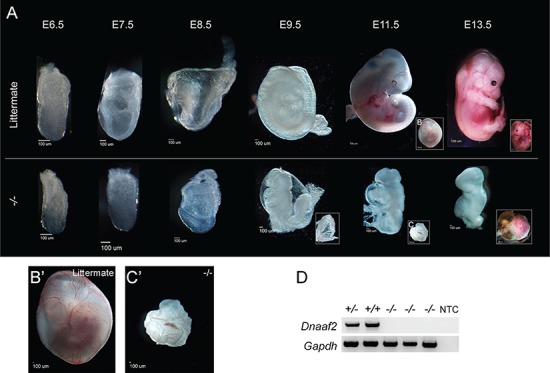
Dnaaf2 −/− mouse embryos are delayed in development when compared to littermate. Embryos were collected at the indicated embryonic day (E). The lower panel denotes the embryo lacking both copies of the Dnaaf2 allele, and the upper panel denotes their respective wild-type or heterozygous littermates (A). Dnaaf2−/− embryos are delayed in development and do not proceed through the turning of the embryos. Particularly, the E11.5 yolk sac vasculature of the Dnaaf2−/− embryo is dramatically different from its littermate (B, B′, C and C′). No Dnaaf2 homozygous mutant embryos are recovered at time of delivery. RT-PCR result verifies that the mutant embryos do not contain the Dnaaf2 alleles (D). B′ and C′ are magnified views of B and C. Gapdh is used as a loading control. NTC denotes no template control; +/+ denotes wild-type embryo; +/− denotes Dnaaf2 heterozygous embryo; −/− denotes Dnaaf2 mutant embryo. Scale bar for each image is included below each embryo.
Dnaaf2 is required for proper left–right patterning
Since there are known human DNAAF2 mutations associated with laterality defects and the embryonic node is known to be essential for establishment of left–right asymmetry, we determined if and when the embryonic node forms in Dnaaf2 mutant mouse embryos. Interestingly, although the formation and extension of the primitive streak occur at the appropriate developmental time (~E6.5) a visible node cannot be observed in E7.5 mutant embryos, a stage at which all littermates have a distinct node at the distal end of the embryo. A morphological node structure does appear in ~E8.5 mutants but is quite small in size and less well defined compared with controls of any stage (Fig. 3, compare A to B).
Figure 3.
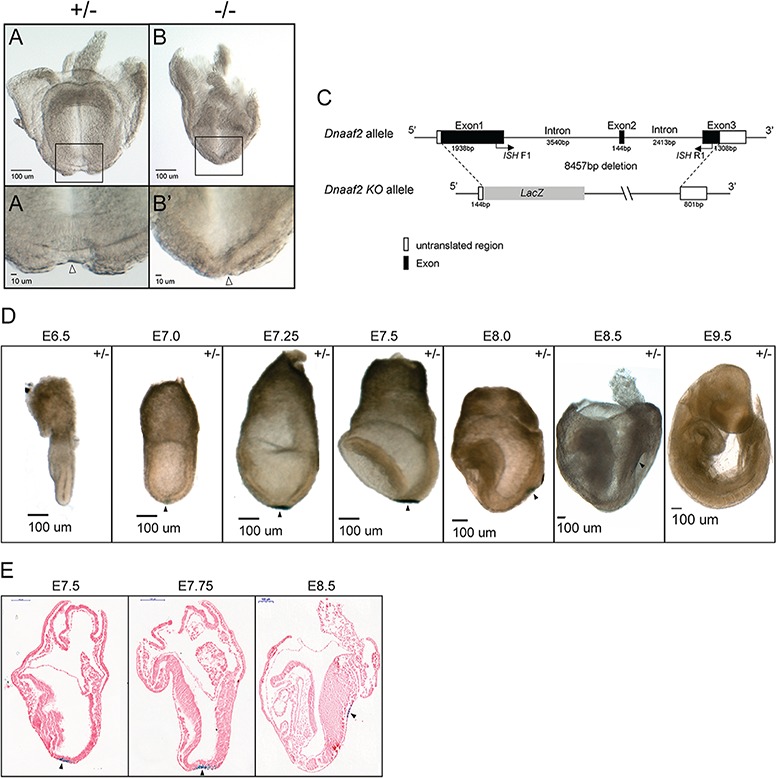
Dnaaf2 −/− embryos have a morphologically distinct node. Embryos were collected at embryonic day (E) 8.5 and examined under a microscope for node assessment (A and B). Dnaaf2−/− embryos do have a morphologically distinct node (black hollow triangle in A′ and B′). (A′ and B′) Magnified view of node region is indicated with a black arrow. The construct of the Dnaaf2 KO allele is generated by replacing a portion of the Dnaaf2 genomic sequence with a cassette containing a LacZ reporter gene (C). ISH F1 and ISH R1 denote the intron-spanning primers used for the generation of Dnaaf2 ISH probe. X-gal staining reveals LacZ expression precisely in the node but not in any other tissues (black triangle in D). ISH F1 and ISH R1 are the forward and reverse primers for Dnaaf2 ISH probe generation. Frontal view, A and B; lateral view, D and E; sagittal section, E. +/− denotes Dnaaf2 heterozygous embryo; −/− denotes Dnaaf2 mutant embryo.
As mentioned above, the Dnaaf2 KO allele is generated by deleting a portion of the Dnaaf2 genomic sequence and replacing it with a lacZ reporter gene (Fig. 3C). We decided to utilize the lacZ reporter gene and perform X-gal staining to examine the lacZ expression in Dnaaf2 heterozygous embryos collected from E6.5 to E9.5 (Fig. 3D). Surprisingly, lacZ expression is localized at the distal tip of the embryo, precisely in the primitive node. Node-specific lacZ is observed at all stages of development at which the node is apparent. LacZ expression is absent in all other embryonic tissues and stages (Fig. 3D and E). Given the function of Dnaaf2 and documented role in dynein arm complex assembly, Dnaaf2 expression in the node is not unexpected. However, the lacZ expression pattern is strikingly different from the Dnaaf2 mRNA expression pattern observed by multiple distinct Dnaaf2 ISH probe, which indicates that the wide-spread embryonic expression of Dnaaf2 mRNA is not confined to the node (Figs 1C–G and 3D and E). Since genomic sequence (both introns and exons) was removed when generating the KO allele, the most likely explanation is that these sequences contain enhancer and/or regulatory elements that normally confer wide-spread expression of Dnaaf2.
To evaluate organizer development in Dnaaf2 mutant embryos, we examined the localization of Cthrc1, which has been shown to be expressed in the node as it is forming (36). Cthrc1 ISH does indeed show expression in mutant embryos in a more diffuse but overall similar pattern as in control littermates (Fig. 4A–C). We further examined mutant embryos by immunofluorescent co-detection of Arl13B and brachyury (T) to determine if the mutant node contains cilia. While we do observe Arl13b/T double-positive cells indicating cilia are present in the mutant node (Fig. 4D, D′, E and E′), there are fewer Arl13b-positive cells in the mutant node, consistent with the apparent reduced node size. This observation is strikingly apparent when compared to a developmental node-stage control (wild-type E7.5 node compared to mutant E8.5 node—compare Fig. 4E, E′, F and F′).
Figure 4.
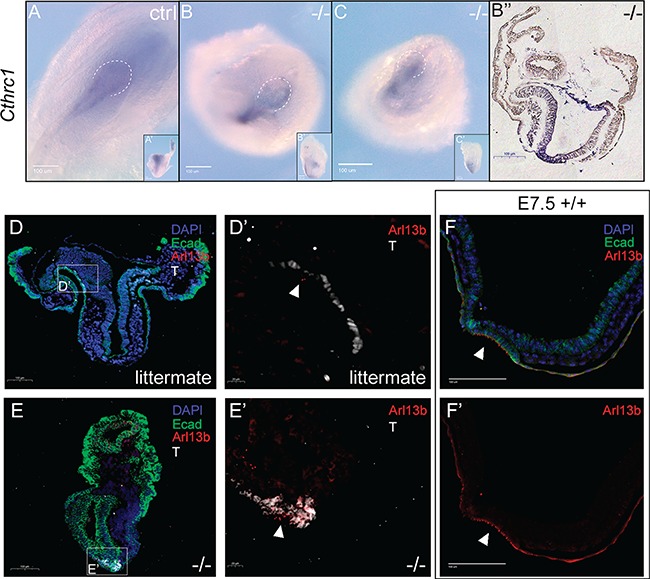
Dnaaf2 −/− embryos contain a structure of a node and a reduction of cilia. Embryos were collected at E8.5. (A–C) Whole-mount ISH was performed with a Cthrc1 antisense probe. (D–F′) Embryos were sagitally sectioned and immunofluorescently labeled with e-cadherin (green), T (white) and Arl13b (red), and the nuclei were counterstained with DAPI (blue). Similar to the littermate, Dnaaf2−/− embryos appear to have a node structure (A, B and C). The white dotted line marked the boundary of the node of the embryos. Though Dnaaf2−/− embryos were delayed in development when compared to the wild-type (E7.5 +/+) embryos (D, D′, E and E′), Dnaaf2−/− embryos appear to have a structurally normal node with cilia (B′) as expected in a E7.5 embryo (F and F′). However, there seems to be a reduction in cilia localization at the node in the Dnaaf2−/− embryos (E′ and F′). The white arrows indicate the localization of the cilia; ctrl denotes stage-matched embryo; +/+ denotes wild-type embryo; −/− denotes Dnaaf2 mutant embryo. Posterior view, A; ventral view, B and C; lateral view, A′, B′ and C′; sagittal section of B, B′′.
The node contains cilia that generate a leftward flow, which gives rise to left–right asymmetry (7). Having established that Dnaaf2 mutants do form a node (albeit delayed and small), we sought to determine if Dnaaf2−/− embryos establish left–right asymmetry. We therefore examined expression of genes with left–right specific patterns. Pitx2 expression is normally confined to the left lateral plate mesoderm. Utilizing whole-mount ISH, we find that Pitx2 is expressed inappropriately on the right side in 25% of mutant embryos (n = 4, Fig. 5A–C). Similarly, we examined Hand1, a gene that is expressed on the outer curvature of the left ventricle (but not the right ventricle) in control embryos. Our results show abnormal expression of Hand1 in all mutants examined and that mutant embryos experience reverse heart looping (Fig. 5D–F). These observations indicate that Dnaaf2 function is required for proper establishment of left–right patterning during embryogenesis possibly because of abnormal node function.
Figure 5.
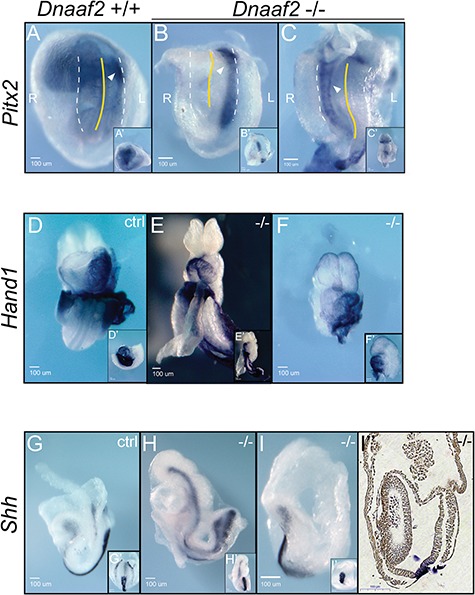
Left–right patterning gene expression in Dnaaf2−/− embryos. Embryos were collected at E8.5 (A, B, G and I) and E9.5 (C–F and H), and whole-mount ISH was performed with Pitx2 (A–C), Hand1 (D–F) or Shh (G–I) antisense probes. (A–C) Pitx2, a gene that is normally expressed in the left lateral plate mesoderm, is mis-expressed in the right lateral plate mesoderm in 25% of Dnaaf2−/− mutants. White triangles indicate Pitx2 expression; white dotted lines indicate the lateral-most sides of each embryo. Yellow dotted line shows midline of each embryo. R denotes the right side of the embryo, and L denotes the left side of the embryo. (D–F) Hand1, a gene that is expressed at the outer curvature of the left ventricle, is found on the opposite side in Dnaaf2−/− mutants. The looping of the heart is also reversed in the Dnaaf2−/− embryos. (G–I) Shh expression is not altered in the Dnaaf2−/− embryos. Both Dnaaf2−/− and wild-type embryos express Shh at the notochord. The white arrows refer to the expression of the indicated gene; ctrl denotes stage-matched embryo; −/−denotes Dnaaf2 mutant embryo. Ventral view, A–C; lateral view, A′, G–I; frontal view, B′–C′ and D–F; lateral view, G′ and H′; ventral view, I′; sagittal sectioned of I, I′′.
Left–right specification, including expression of Pitx2, is downstream of Shh (14). Since we observed laterality defect in Pitx2 expression, we next determined if Shh is appropriately expressed in mutant embryos. Whole-mount ISH indicates that the notochord (a product of the node) displays a normal pattern of Shh in mutant embryos (Fig. 5G–I). These results confirm that there is normal developmental timing of Shh expression. Since Pitx2 and Hand1 are expressed at the proper developmental stage, the inappropriate lateral expression observed is not due to defects in transcriptional regulation of these loci, but rather defects in left–right pattering. This observation also suggests that Dnaaf2 is required for the lateral specific establishment of these genes, most likely due to non- or poorly functional cilia and abrogated nodal flow in the absence of functional Dnaaf2 protein.
As mentioned earlier, we observe an obvious developmental delay (Fig. 2) and shortened posterior axis in the E9.5 Dnaaf2−/− embryos (Fig. 5G–I) when compared to the littermate or stage-matched control embryos. We speculate that the truncated phenotype observed is due to cells undergoing apoptosis at the posterior end of the embryo. As predicted, we observe many apoptotic cells in the posterior of E9.5 Dnaaf2−/− embryos, whereas the stage-matched controls have very few (Fig. 6C and D). We also find a heightened density of the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive cells in the neural tube of the mutant, which correlates well with the expression of Dnaaf2 in mouse embryos (Fig. 1F). At E8.5, many apoptotic cells are already present in the embryonic portion of the mutants (Fig. 6A and B), also consistent with embryonic specific expression of Dnaaf2 (Fig. 1). Appropriate growth and proliferation are essential for the embryo to advance. Turning of the embryo is an important process required for gut tube closure as well as the transition to fetal position for embryogenesis to proceed (37). The accumulation and increase in apoptotic cells may be a major reason for the early lethality of the Dnaaf2−/− embryos.
Figure 6.
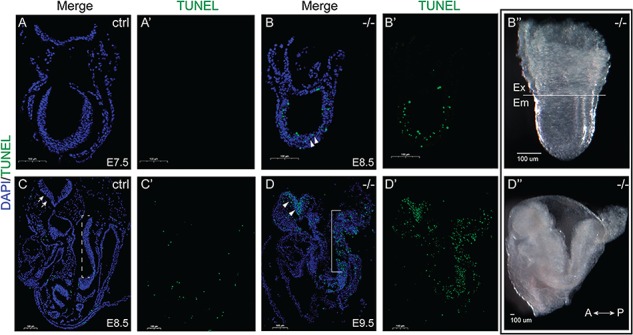
TUNEL assay in Dnaaf2−/− embryos and their stage-matched control embryos (A and B; C and D). Embryos were collected at the indicated embryonic day (A–D) and processed for TUNEL assay. (A–D) Compared to stage-matched control embryos, E8.5 Dnaaf2−/− embryo displays apoptotic cells in the embryonic portion (white triangles in B). At E9.5, TUNEL-positive cells accumulate at the neural tube (white triangles in D) and the tail region of the Dnaaf2−/− embryos (white triangles and bracket in D), whereas the stage-matched control embryo experience minimal apoptosis (white arrows and dotted bracket in C). B′′ and D′′ are whole-mount images of embryos sectioned for B and D. Sagittal sectioned, A–D. Ex denotes extraembryonic portion; em denotes embryonic portion; A denotes anterior axis; P denotes posterior axis; ctrl denotes stage-matched embryo; −/− denotes Dnaaf2 mutant embryo.
Discussion
Cilia are evolutionarily conserved cellular organelles that are found across a wide spectrum of species. Although once believed to be vestigial structures, it is now appreciated that most cells contain cilia and that cilia play critical roles in many signaling pathways and cellular functions (1–4,7). Research from the past decades has supported the essential role of cilia during normal embryogenesis (1,7). In humans, DNAAF2 encodes a protein required for the preassembly of dynein arms, which is essential for cilia function and has been associated with diseases and disorders that are due to cilia defects (17,19,21). Here, for the first time we characterize the role of Dnaaf2 during embryogenesis. Our data show that in the absence of Dnaaf2 function, mouse embryos are delayed beginning at early gastrulation and do not progress past E9.5 stages.
Since cilia are essential for breaking left–right symmetry through of nodal flow, we carefully examined the nodes in mutant embryos. We show that the node is present morphologically and specified molecularly, although it is reduced in size with fewer cilia containing cells. We also show that although Shh expression is normal, left–right axis specification is disrupted in Dnaaf2 mutant embryos.
Combined, these results suggest that the left–right defects present in Dnaaf2 mutant embryos are due to a malfunction of cilia in the node. We speculate that the motility of these cilia is compromised due to dynein arm structural defects or malfunction in the absence of Dnaaf2, as suggested by previous reports in cell culture models (17,19). It is likely that the lack of Dnaaf2 disrupts or weakens the leftward flow of the cilia at the node and which could result in the improper neural tube closure present in E9.5 Dnaaf2 null embryos as cilia have also been implicated to play important roles in brain development (38–40). The embryonic yolk sac is one of the fetal membranes that contributes to the placenta (35). A previous large-scale mouse KO study shows a linkage between malformations in placental development and vascular, heart and brain defects in mid-gestation embryos (41). At E11.5, Dnaaf2 mutant embryos display an obvious defect in yolk sac vasculature. Therefore, it is likely that yolk sac defects are present as early as E8.5, which may certainly contribute to the plethora of embryonic defects described above. It is also interesting that we observe an overall truncation phenotype in E9.5 Dnaaf2−/− embryos as past studies have shown that defects in cilia greatly affect planar cell polarity signaling, which is essential for the convergence and extension movements during gastrulation (42,43).
As mentioned above, the knock-in reporter LacZ expression is narrowly expressed precisely in the node, a pattern that is quite different from Dnaaf2 mRNA expression (compare Fig. 1C–G and Fig. 3D and E). We speculate that there must be enhancer or regulatory elements in the intronic sequence that direct wide-embryonic expression, and hence when a large portion of the Dnaaf2 genomic sequence is removed, the node-specific reporter expression is the result. Further investigation of the promoter and intronic regions of the Dnaaf2 locus may resolve this discrepancy and may elucidate a node-specific regulatory element, which may be of interest for future studies.
Intriguingly, when generated on an outbred ICR background, Dnaaf2/Ktu homozygous KO pups are found at birth, but 90% of these pups die prior to weaning (19). Importantly, similar to the phenotypes presented herein, these Ktu KO newborns exhibit situs inversus totalis and hydrocephaly (19). Importantly, we attempted to recapitulate this later phenotype by breeding the Dnaaf2tm.1.1 onto an ICR background (data not shown). Although we did not recover newborn mutants at generation N5, homozygous mutant embryos displayed better developmental progress and could be recovered at later stages than C57bl/6NJ homozygotes. These data imply ICR-specific functional redundancy that compensates for Dnaaf2 loss.
It is also curious that Dnaaf4 and Dnaaf5 are robustly expressed at slightly later stages than Dnaaf2. This raises the possibility that functional redundancy combined with subtle differences in the timing of expression of these genes may explain the variation observed in Dnaaf2 phenotypes (lethal in C57bl/6NJ versus viable in ICR). Interestingly, DNAAF2 loss of function in humans does not affect survival (17,44)—further indicating that DNAAF family member redundancies can compensate for the loss of the DNAAF2 in humans but not C57bl/6NJ mice. It may therefore be of interest to map subtle differences between ICR and C57bl/6NJ embryonic Dnaaf expression patterns as well as to explore patterns of Dnaaf in other mammalian species.
Materials and Methods
Animals
The Dnaaf2 KO allele was generated by the Jackson Laboratory (stock no. 024570: Dnaaf2tm1.1(KOMP)Vlcg). In brief, 8457 bp of the Dnaaf2 genomic sequence was replaced with the lacZ reporter in C57BL/6NJ mouse embryonic stem cells removing part of exon 1, intron 1 and exon 2, intron 2 and part of exon 3 (Fig. 3C).
Embryo retrieval and genotyping
Dnaaf2 −/− embryos were generated by heterozygote intercosses. Males and females were housed together, and the presence of the vaginal plugs was defined as E0.5. Genotypes of individual embryos were determined by PCR using primers (given 5′ to 3′): wild type (ATCTAAGCCCCGTCGGTTAC and ACCGTGAAGGCTTAAGAGCA) and KO (CGGTCGCTACCATTACCAGT and TAGCTAGGCATTGGGATTGC). Specific genotyping protocol is available at https://www.jax.org/strain/024570 (stock no. 024570). All results shown were repeated on a minimum of three mutant embryos except for E11.5 and E13.5 embryos shown in Figure 2. Use of animals was approved by the University of Massachusetts Institutional Animal Care and Use Committee (IACUC).
RNA extraction and RT-PCR analysis
RNA was extracted using Roche High Pure RNA Isolation Kit (Roche Diagnostics, Indianapolis, Indiana USA, 11828665001). cDNA synthesis was performed using Bio-Rad iScript cDNA Synthesis (Bio-Rad, Hercules, California USA, 1708890). RT-PCR was performed for 35 cycles of 30 s at 60°C, 72°C and 95°C with the following primers with indicated gene names unless otherwise specified (given 5′ to 3′): Dnaaf1 (359 bp): GCCTCTTCCTGCAAGTGAAC and GTGCTTCAGACGGACAGTGA; Dnaaf2 (583 bp): TCTGCCACAAACTCTTCACG and GCCCAGCAGGATCACTACAT; Dnaaf3 (301 bp): GGCACCGGTTATGGTAGTGT and CTTGGAGCCCCATCTTCTC; Dnaaf4 (319 bp): AAACTATTTGGCAGCGGTTG and CCGAATCTTCTCTGCATCGT; Dnaaf5 (348 bp): CCTCAAGAATTCAGGGGACA and AGCTGCTCGCAGTAGGTAGC; T (313 bp): CATGTACTCTTTCTTGCTGG and GGTCTCGGGAAAGCAGTGGC; Gapdh (452 bp): ACCACAGTCCATGCCATCAC and TCCACCACCCTGTTGCTGTA.
RT-PCR analyses for E6.5 C57BL/6NJ and ICR mouse embryos in Figure 2 were performed under the following condition: 33 cycles of 30 s at 60°C, 72°C and 95°C.
Whole-mount ISH
Freshly dissected embryos were fixed in 4% paraformaldehyde for 2–4 h on ice and subjected to modification from Tremblay et al. (2001) (45). Embryos were then dehydrated in methanol and stored up to 1 month. Embryos were rehydrated in a series of methanol in 1× PBT. Subsequently, the embryos were rinsed in 1× PBT for 5 min twice and bleached in 6% hydrogen peroxide for 1 h. Embryos were then rinsed with 1× PBT, proceeded with proteinase K treatment for 6–8 min and incubated with glycine in 1× PBT for 5 min. After rinsing with 1× PBT for 5 min twice, embryos were fixed with 4% paraformaldehyde with 0.02% glutaraldehyde for 20 min. Embryos were rinsed in 1× PBT for 5 min twice and incubated in 1:1 hybridization buffer/PBT for 10 min and subsequently in hybridization buffer for 10 min. The embryos were then incubated with 150 ng RNA probes in hybridization buffer at 70°C overnight. The next day, embryos were washed with solutions containing formamide, 20× SSC, 1 m citric acid, 20% SDS and Tween 20 for 30 min at 70°C for the first 3 washes and at 65°C for the last 3 washes. Following this, the embryos were rinsed in 1× maleic acid buffer containing levamisole for 5 min 3 times. The embryos were then blocked in maleic acid buffer with 2% Boehringer blocking reagent for 1 h at room temperature and then incubated with the antibody solution containing Anti-Digoxigenin-AP (Roche Diagnostics, Indianapolis, Indiana USA, 11093274910), heat activated sheep serum (Sigma-Aldrich, St. Louis, Missouri USA, S2263) and levamisole (Sigma-Aldrich, St. Louis, Missouri USA, L9756) at 4°C overnight. The following day, the embryos were washed with 1× maleic acid buffer containing levamisole for at least 1 h 6 times. The embryos were then rinsed with alkaline phosphatase buffer containing 1 m Tris-HCl pH 9.5, 5 m NaCl, 1 m MgCl2 and Tween 20 for 10 min 3 times and developed in BM purple solution (Roche Diagnostics, Indianapolis, Indiana USA, 11442074001) until the color has developed. The embryos were then washed with 1× PBT containing 0.5 m EDTA, fixed in 4% paraformaldehyde overnight, dehydrated in a series of ethanol, cleared in xylene, embedded with paraffin and sectioned at 7 um thick. Antisense probes were generated using E7.5 embryonic cDNA. RT-PCR was performed for 35 cycles of 30 s at 60°C, 72°C and 95°C with the following primers with indicated genes. The amplicons were then cloned into the TOPO TA Cloning Vectors using the TOPO TA Cloning kit for Sequencing (Invitrogen, Waltham, Massachusetts USA, 450030) according to the manufacturer’s instructions.
Probes used were the following: Cthrc1 (554 bp): TGCTGCTGCTACAGTTGTCC and CCCTTCACAGAGTCCTTCCA; Dnaaf2 (intron-spanning primers designed to generate an amplicon that includes exon 1 to exon 3; 583 bp): TCTGCCACAAACTCTTCACG and GCCCAGCAGGATCACTACAT; Hand1 (574 bp): CCATCATCACCACTCACACC and GCGCCCTTTAATCCTCTTCT; Pitx2 (46); and Shh (47).
X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) staining
Freshly dissected embryos were fixed in X-gal buffer containing 0.2% glutaraldehyde and 1% formaldehyde on ice for 15 min and subjected to modification from Tremblay et al. (2000) (48). In brief, the fixed embryos were washed with X-gal buffer (PBS, 5 mm EGTA, 2 mm MgCl:6H2O, 0.2% NP-40, 0.2 mm deoxycholate) for 10 min 3 times and stained with X-gal stain (X-gal buffer, 5 mm potassium ferricyanide and 5 mm potassium ferrocyanide and 0.5 mg/ml X-gal) overnight at 37°C. Subsequently, embryos were dehydrated in ethanol, cleared in xylene, embedded with paraffin and sectioned at 7 um thick. The sectioned embryos were deparaffinated and rehydrated for subsequent processing. Eosin staining was performed by immersing rehydrated sectioned embryos in eosin Y solution for 15–20 s, followed by 95% ethanol for 2 min and then 100% ethanol for 2 min and lastly cleared in xylene. Slides were then sealed with Cytoseal 60. X-gal stained sections were imaged with a Panoramic MIDI II slide scanner (3DHISTECH, Budapest, Hungary).
Immunofluorescence
Freshly dissected embryos were fixed in 4% paraformaldehyde overnight, dehydrated in ethanol, cleared in xylene, embedded with paraffin and sectioned at 7 um thick. The sectioned embryos were deparaffinated and rehydrated for subsequent procedures. Antigen retrieval was performed by boiling in 0.01 m Tris-Base pH 10 with 0.05% Tween 20 for 4 min and allowed to cool to room temperature. The slides were then rinsed with 1× PBT for 2 min twice. Primary antibodies were incubated at 4°C overnight in a humid chamber. The slides were then rinsed with 1× PBT for 15 min 3 times, followed by incubation with secondary antibodies at room temperature for 1 h in a humid chamber. Subsequently, the slides were rinsed with 1× PBS for 15 min 3 times. Nuclei were counterstained with (DAPI) in PBS (1:10000) for 3 min. Slides were rinsed with 1× PBS and sealed with ProLong Gold. Immunofluorescence slides were imaged with Panoramic MIDI II slide scanner (3DHISTECH).
Primary antibodies were used in the following conditions: T (Santa Cruz Biotechnology, Inc, Dallas, Texas USA; 1:100); Ecad (Abcam, Cambridge, Massachusetts USA; 1:1000); and Arl13b (Proteintech, Rosemont, Illinois USA; 17711-1-AP, 1:250). Secondary antibodies were used in the following conditions: Alexa fluor 488 donkey anti-mouse (Thermo Fisher Scientific, Waltham Massachusetts USA; A-21202, 1:500); 647 donkey anti-goat (Thermo Fisher Scientific, Waltham Massachusetts USA; A-21447, 1:500); and 546 donkey anti-rabbit (Thermo Fisher Scientific, Waltham Massachusetts USA; A10040, 1:500). DAPI concentration (Molecular Probes, Waltham, Massachusetts USA; 1:10000).
Assessment of apoptosis
Apoptosis was assessed using In Situ Cell Death Detection Kit (Roche Diagnostics, Indianapolis, Indiana USA, 11684795910). Freshly dissected embryos were fixed in 4% paraformaldehyde overnight, dehydrated in ethanol, cleared in xylene, embedded with paraffin and sectioned at 7 um thick. The sectioned embryos were deparaffinated and rehydrated for the TUNEL technology according to manufacturer’s protocol.
Imaging
Digital images of whole-mount embryos were captured on a Nikon SMZ-1500 stereomicroscope equipped with a Spot Idea Digital Camera and Spot software (v4.6). Images of sectioned embryos were taken with a Pannoramic MIDI slide scanner or Nikon Eclipse 50i equipped with a Spot Idea Digital Camera and Spot software (v4.6) or Nikon Eclipse Ti inverted microscope with an Andor DR-228C camera and Nikon NIS Elements AR software.
Acknowledgements
We would like to thank members of the Mager and Tremblay laboratories for providing useful input throughout the project.
Conflict of Interest statement. None declared.
Funding
Funded in part by National Institutes of Health (R01HD083311 to J.M.).
References
- 1. Sung C.H. and Leroux M.R. (2013) The roles of evolutionarily conserved functional modules in cilia-related trafficking. Nat. Cell Biol., 15, 1387–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Veland I.R., Awan A., Pedersen L.B., Yoder B.K. and Christensen S.T. (2009) Primary cilia and signaling pathways in mammalian development, health and disease. Nephron Physiol., 111, 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Goetz S.C. and Anderson K.V. (2010) The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet., 11, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Basten S.G. and Giles R.H. (2013) Functional aspects of primary cilia in signaling, cell cycle and tumorigenesis. Cilia, 2, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pazour G.J. and Witman G.B. (2003) The vertebrate primary cilium is a sensory organelle. Curr. Opin. Cell Biol., 15, 105–110. [DOI] [PubMed] [Google Scholar]
- 6. Shah A.S., Ben-Shahar Y., Moninger T.O., Kline J.N. and Welsh M.J. (2009) Motile cilia of human airway epithelia are chemosensory. Science, 325, 1131–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Drummond I.A. (2012) Cilia functions in development. Curr. Opin. Cell Biol., 24, 24–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Downs K.M. (2009) The enigmatic primitive streak: prevailing notions and challenges concerning the body axis of mammals. Bioessays, 31, 892–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Supp D.M., Witte D.P., Potter S.S. and Brueckner M. (1997) Mutation of an axonemal dynein affects left–right asymmetry in inversus viscerum mice. Nature, 389, 963–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pennekamp P., Menchen T., Dworniczak B. and Hamada H. (2015) Situs inversus and ciliary abnormalities: 20 years later, what is the connection? Cilia, 4, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Levin M. (2004) Left–right asymmetry in embryonic development: a comprehensive review. Mech. Dev., 122, 3–25. [DOI] [PubMed] [Google Scholar]
- 12. Shiratori H. and Hamada H. (2006) The left–right axis in the mouse: from origin to morphology. Development, 133, 2095–2104. [DOI] [PubMed] [Google Scholar]
- 13. Brennan J., Norris D.P. and Robertson E.J. (2002) Nodal activity in the node governs left–right asymmetry. Genes Dev., 16, 2339–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tsukui T., Capdevila J., Tamura K., Ruiz-Lozano P., Rodriguez-Esteban C., Yonei-Tamura S., Magallón J., Chandraratna R.A., Chien K., Blumberg B. et al. (1999) Multiple left–right asymmetry defects in Shh(−/−) mutant mice unveil a convergence of the Shh and retinoic acid pathways in the control of Lefty-1. Proc. Natl. Acad. Sci. U. S. A., 96, 11376–11381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nonaka S., Tanaka Y., Okada Y., Takeda S., Harada A., Kanai Y., Kido M. and Hirokawa N. (1998) Randomization of left–right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell, 95, 829–837. [DOI] [PubMed] [Google Scholar]
- 16. Supp D.M., Brueckner M., Kuehn M.R., Witte D.P., Lowe L.A., McGrath J., Corrales J. and Potter S.S. (1999) Targeted deletion of the ATP binding domain of left–right dynein confirms its role in specifying development of left–right asymmetries. Development, 126, 5495–5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Qi Z., Leblond G., O’Toole E., Hara C. et al. (2008) Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature, 456, 611–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mitchison H.M., Schmidts M., Loges N.T., Freshour J., Dritsoula A., Hirst R.A., O'Callaghan C., Blau H., Al M., Olbrich H. et al. (2012) Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet., 44, 381–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Matsuo M., Shimada A., Koshida S., Saga Y. and Takeda H. (2013) The establishment of rotational polarity in the airway and ependymal cilia: analysis with a novel cilium motility mutant mouse. Am. J. Physiol. Lung Cell. Mol. Physiol., 304, L736–L745. [DOI] [PubMed] [Google Scholar]
- 20. Tarkar A., Loges N.T., Slagle C.E., Francis R., Dougherty G.W., Tamayo J.V., Shook B., Cantino M., Schwartz D., Jahnke C. et al. (2013) DYX1C1 is required for axonemal dynein assembly and ciliary motility. Nat. Genet., 45, 995–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Raidt J., Wallmeier J., Hjeij R., Onnebrink J.G., Pennekamp P., Loges N.T., Olbrich H., Häffner K., Dougherty G.W., Omran H. et al. (2014) Ciliary beat pattern and frequency in genetic variants of primary ciliary dyskinesia. Eur. Respir. J., 44, 1579–1588. [DOI] [PubMed] [Google Scholar]
- 22. Afzelius B. (2004) Cilia-related diseases. J. Pathol., 204, 470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Brown J.M. and Witman G.B. (2014) Cilia and diseases. Bioscience, 64, 1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zihlif N., Paraskakis E., Tripoli C., Lex C. and Bush A. (2006) Markers of airway inflammation in primary ciliary dyskinesia studied using exhaled breath condensate. Pediatr. Pulmonol., 41, 509–514. [DOI] [PubMed] [Google Scholar]
- 25. Maimoona A., Zariwala M.R.K. and Omran H. (2007) Genetic defects in ciliary structure and function. Annu. Rev. Physiol., 69, 423–450. [DOI] [PubMed] [Google Scholar]
- 26. Dickinson M.E., Flenniken A.M., Ji X., Teboul L., Wong M.D., White J.K., Meehan T.F., Weninger W.J., Westerberg H., Adissu H. et al. (2016) High-throughput discovery of novel developmental phenotypes. Nature, 537, 508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ayadi A., Birling M.C., Bottomley J., Bussell J., Fuchs H., Fray M., Gailus-Durner V., Greenaway S., Houghton R., Karp N. et al. (2012) Mouse large-scale phenotyping initiatives: overview of the European Mouse Disease Clinic (EUMODIC) and of the Wellcome Trust Sanger Institute Mouse Genetics Project. Mamm. Genome, 23, 600–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Miao C., Jiang Q., Li H., Zhang Q., Bai B., Bao Y. and Zhang T. (2016) Mutations in the motile cilia gene DNAAF1 are associated with neural tube defects in humans. G3 (Bethesda), 6, 3307–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Horani A., Ustione A., Huang T., Firth A.L., Pan J., Gunsten S.P., Haspel J.A., Piston D.W. and Brody S.L. (2018) Establishment of the early cilia preassembly protein complex during motile ciliogenesis. Proc. Natl. Acad. Sci. U. S. A., 115, 1221–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Diggle C.P., Moore D.J., Mali G., zur Lage P., Ait-Lounis A., Schmidts M., Shoemark A., Munoz A.G., Halachev M.R., Gautier P. et al. (2014) HEATR2 plays a conserved role in assembly of the ciliary motile apparatus. PLoS Genet., 10, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lyons R.A., Saridogan E. and Djahanbakhch O. (2006) The reproductive significance of human fallopian tube cilia. Hum. Reprod. Update, 12, 363–372. [DOI] [PubMed] [Google Scholar]
- 32. Ou Y., Dores C., Rodriguez-Sosa J.-R., van der Hoorn F.A. and Dobrinski I. (2014) Primary cilia in the developing pig testis. Cell Tissue Res., 358, 597–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dores C., Alpaugh W., Lin S., Biernaskie J. and Dobrinski I. (2017) Primary cilia on porcine testicular somatic cells and their role in hedgehog signaling and tubular morphogenesis in vitro. Cell Tissue Res., 368, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Garcia M.D. and Larina I.V. (2014) Vascular development and hemodynamic force in the mouse yolk sac. Front. Physiol., 5, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Freyer C. and Renfree M.B. (2009) The mammalian yolk sac placenta. J. Exp. Zool. B Mol. Dev. Evol., 312, 545–554. [DOI] [PubMed] [Google Scholar]
- 36. Yamamoto S., Nishimura O., Misaki K., Nishita M., Minami Y., Yonemura S., Tarui H. and Sasaki H. (2008) Cthrc1 selectively activates the planar cell polarity pathway of Wnt signaling by stabilizing the Wnt-receptor complex. Dev. Cell, 15, 23–36. [DOI] [PubMed] [Google Scholar]
- 37. Spence J.R., Lauf R. and Shroyer N.F. (2011) Vertebrate intestinal endoderm development. Dev. Dyn., 240, 501–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Han Y.G. and Alvarez-Buylla Y. (2010) Role of primary cilia in brain development and cancer. Curr. Opin. Neurobiol., 20, 58–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Faubel R., Westendorf C., Bodenschatz E. and Eichele G. (2016) Cilia-based flow network in the brain ventricles. Science, 353, 176–178. [DOI] [PubMed] [Google Scholar]
- 40. Foerster P., Daclin M., Asm S., Faucourt M., Boletta A., Genovesio A. and Spassky N. (2017) mTORC1 signaling and primary cilia are required for brain ventricle morphogenesis. Development, 144, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Perez-Garcia V., Fineberg E., Wilson R., Murray A., Mazzeo C.I., Tudor C., Sienerth A., White J.K., Tuck E., Ryder E.J. et al. (2018) Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature, 555, 463–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ross A.J., May-Simera H., Eichers E.R., Kai M., Hill J., Jagger D.J., Leitch C.C., Paul Chapple J., Munro P.M., Fisher S. et al. (2005) Disruption of Bardet–Biedl syndrome ciliary proteins perturbs planar cell polarity in vertebrates. Nat. Genet., 37, 1135–1140. [DOI] [PubMed] [Google Scholar]
- 43. Wallingford J.B. (2006) Planar cell polarity, ciliogenesis and neural tube defects. Hum. Mol. Genet., 15, R227–R234. [DOI] [PubMed] [Google Scholar]
- 44. Lek M., Karczewski K.J., Minikel E.V., Samocha K.E., Banks E., Fennell T., O’Donnell-Luria A.H., Ware J.S., Hill A.J., Cummings B.B. et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tremblay K.D., Dunn N.R. and Robertson E.J. (2001) Mouse embryos lacking Smad1 signals display defects in extra-embryonic tissues and germ cell formation. Development, 1283609–3621. [DOI] [PubMed] [Google Scholar]
- 46. Ryan A.K., Blumberg B., Rodriguez-Esteban C., Yonei-Tamura S., Tamura K., Tsukui T., de la Peña J., Sabbagh W., Greenwald J., Choe S. et al. (1998) Pitx2 determines left–right asymmetry of internal organs in vertebrates. Nature, 394, 545–551. [DOI] [PubMed] [Google Scholar]
- 47. Echelard Y., Epstein D.J., St-Jacques B., Shen L., Mohler J., McMahon J.A. and McMahon A.P. (1993) Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell, 75, 1417–1430. [DOI] [PubMed] [Google Scholar]
- 48. Tremblay K.D., Hoodless P.A., Bikoff E.K. and Robertson E.J. (2000) Formation of the definitive endoderm in mouse is a Smad2-dependent process. Development, 127, 3079–3090. [DOI] [PubMed] [Google Scholar]