

Abstract
Lipid-modifying enzymes serve crucial roles in cellular processes such as signal transduction (producing lipid-derived second messengers), intracellular membrane transport (facilitating membrane remodeling needed for membrane fusion/fission), and protein clustering (organizing lipid domains as anchoring platforms). The lipid products crucial in these processes can derive from different metabolic pathways, thus it is essential to know the localization, substrate specificity, deriving products (and their function) of all lipid-modifying enzymes. Here we discuss an emerging family of these enzymes, the lysophosphatidic acid acyltransferases (LPAATs), also known as acylglycerophosphate acyltransferases (AGPATs), that produce phosphatidic acid (PA) having as substrates lysophosphatidic acid (LPA) and acyl-CoA. Eleven LPAAT/AGPAT enzymes have been identified in mice and humans based on sequence homologies, and their localization, specific substrates and functions explored. We focus on one member of the family, LPAATδ, a protein expressed mainly in brain and in muscle (though to a lesser extent in other tissues); while at the cellular level it is localized at the trans-Golgi network membranes and at the mitochondrial outer membranes. LPAATδ is a physiologically essential enzyme since mice knocked-out for Lpaatδ show severe dysfunctions including cognitive impairment, impaired force contractility and altered white adipose tissue. The LPAATδ physiological roles are related to the formation of its product PA. PA is a multifunctional lipid involved in cell signaling as well as in membrane remodeling. In particular, the LPAATδ-catalyzed conversion of LPA (inverted-cone-shaped lipid) to PA (cone-shaped lipid) is considered a mechanism of deformation of the bilayer that favors membrane fission. Indeed, LPAATδ is an essential component of the fission-inducing machinery driven by the protein BARS. In this process, a protein-tripartite complex (BARS/14-3-3γ/phosphoinositide kinase PI4KIIIβ) is recruited at the trans-Golgi network, at the sites where membrane fission is to occur; there, LPAATδ directly interacts with BARS and is activated by BARS. The resulting formation of PA is essential for membrane fission occurring at those spots. Also in mitochondria PA formation has been related to fusion/fission events. Since PA is formed by various enzymatic pathways in different cell compartments, the BARS-LPAATδ interaction indicates the relevance of lipid-modifying enzymes acting exactly where their products are needed (i.e., PA at the Golgi membranes).
Keywords: acyltransferase, AGPAT, LPAAT, phosphatidic acid, lysophosphatidic acid, Golgi complex, membrane fission, BARS
Introduction
Lysophosphatidic acid acyltransferases are an emerging family of enzymes that catalyze the production of PA using LPA and acyl-CoA (Korbes et al., 2016). The product of this enzymatic reaction, PA is involved in several essential cellular functions based on its unique properties as: (i) precursor for the biosynthesis of all glycerophospholipids and triacylglycerol (TAG); (ii) important membrane remodeling metabolite involved in the intracellular transport; and (iii) precursor of bioactive lipid mediators implicated in cell survival, proliferation, and tumor progression (Kume and Shimizu, 1997; Lu et al., 2005; Eto et al., 2014). The LPA is first synthetized, in the de novo-Kennedy pathway, via acylation of glycerol 3-phosphate (G3P) by the glycerol-3-phosphate acyltransferase (GPAT) enzymes that use acyl-CoAs as donors. Then, another fatty acid moiety (often an unsaturated one in eukaryotes) is incorporated at the sn-2 position on the LPA glycerol backbone to form PA by the 1-acylglycerol-3-phosphate acyltransferase enzyme (AGPAT, also known as LPA acyltransferase: LPAAT).
Eleven AGPAT enzymes, named AGPAT 1-11, have been identified in mice and humans based on the homology of their primary sequences (Kume and Shimizu, 1997). All AGPATs display highly conserved structural motif (Coleman and Mashek, 2011) and exhibit acyltransferase activity, using acyl-CoA and lysophospholipid as acyl-donor and acyl-acceptor, respectively (Kume and Shimizu, 1997). AGPAT enzymes have been named based on their substrate specificities and order in which their cloning have been reported. AGPATs 1, 2, 3, 4, and 5 specifically prefer LPA to form PA and, as such, are also known as LPAATα, β, γ, δ and ε, respectively, while AGPATs 6-11 are classified as lysophospholipid acyltransferases (LPLATs) or GPATs based on their substrate specificities (Yamashita et al., 2014).
Of note, recent studies indicate that each LPAAT enzyme is responsible for the production of a distinct and specific pool of PA required to affect downstream lipid metabolism that mediates specific cellular and organelle membrane lipid composition, which, in turn, controls physiological functions (Bradley and Duncan, 2018). Loss of function of a single LPAAT influences different downstream lipid biosynthetic pathways generating distinct pathophysiological consequences, including embryonic lethality, lipodystrophy, impaired spatial learning and memory (Bradley and Duncan, 2018). Moreover, LPAAT enzymes show differential subcellular localization together with a differential expression profiles within tissues or organs as well as preference toward specific fatty acyl-CoA donor moieties (Shindou et al., 2009). These aspects may explain the existence of different LPAAT isoforms in nature able to specifically modulate downstream glycerolipid pathways in different tissues and organs for maintenance of normal physiology. As a consequence, loss of specific LPAAT cannot be functionally and biochemically replaced by other LPAATs (Bradley and Duncan, 2018). Tissue distribution patterns reveal an ubiquitous expression of LPAATα and LPAATγ, while LPAATβ, δ, and ε display distinct tissue-specific profiles (Takeuchi and Reue, 2009).
Following a brief overview on phylogenetic tree of LPAAT genes and on the unique properties of PA, the product of the reaction catalyzed by these enzymes, we analyze the conserved acyltransferase motifs required for enzymatic activity with the spotlight on LPAATδ member. We first discuss the membrane topology of human LPAATδ based on the recently resolved structure of bacterial LPAAT. We then highlight the role of the specific LPAATδ-produced PA pool in lipid metabolism and tissue functions. Finally, we report on recent advances in our understanding of the membrane fission event involving LPAATδ and occurring at the trans-side of the Golgi complex. Specifically, we discuss how this enzyme assembles with other proteins in a protein complex and how this machinery is regulated and operates in the formation of the basolaterally directed post-Golgi carriers.
LPAAT Family
The LPAAT enzymes are an ancient gene family that were first functionally described in 1956 (Kennedy and Weiss, 1956). It belongs to the subgroup 3 of the membrane bound O-acyltransferase (MBOAT) superfamily (Chang et al., 2011; Korbes et al., 2016). LPAAT genes were found in all three domains of life: Archaea, Bacteria and Eukarya where the LPAAT enzymes play key metabolic roles. At least one LPAAT gene was found in each of the eukaryotic genome examined (Korbes et al., 2016). Some eukaryotic species contain a few LPAAT genes (e.g., thirteen in the soya bean Glycine max), whereas yeast Saccharomyces cerevisiae possesses only one LPAAT gene, SLC1 (Korbes et al., 2016). Five LPAAT enzymes namely LPAATα-LPAATε are present in mammals (Korbes et al., 2016; Bradley and Duncan, 2018).
The phylogenetic tree analyses of LPAAT genes from prokaryotic to eukaryotic species based on phosphate acyltransferase (PlsC) domain identified three distinct clusters designed as cluster I, II, and III (Korbes et al., 2016). Cluster I is the most ancient and contains plant LPAATα and LPAATβ, prokaryotic and fungal LPAATs, as well as animal LPAATα and LPAATβ. Cluster II consists of animal AGPAT6, AGPAT10, AGPAT7, AGPAT9, and AGPAT11. Cluster III is divided into subclusters: IIIa contains animal LPAATγ, LPAATδ and plant LPAATβ, LPAATγ; IIIb is composed of plant LPAATδ, LPAATε, fungal LPAAT and animal AGPAT8; and IIIc includes animal LPAATε. Of note, two different proteins LPAATγ and LPAATδ in animals appeared due to a duplication event (Korbes et al., 2016). The sequence of human LPAATδ is more similar to the sequence of human LPAATγ (ca. 60%) than to that of other three human LPAATs, see Supplementary Tables S1, S3. A few residues are typical of the members of subcluster IIIa: they are highly conserved in LPAATγ and LPAATδ orthologs, but are rare among other animal LPAATs. A good example of such residues is tryptophan (W, at position 106 in human LPAATδ), five residues downstream of aspartic acid (D) belonging to the conserved NHxxxxD sequence, see Supplementary Tables S1–S4.
Lysophosphatidic acid acyltransferase γ, as LPAATα, is ubiquitously expressed with high mRNA levels in adipose tissue, liver and heart (Yuki et al., 2009). LPAATγ, in addition of being localized, like LPAATα and LPAATβ, at the ER (Agarwal et al., 2011; Yamashita et al., 2014), is a Golgi-resident enzyme that controls Golgi structure and retrograde transport from the Golgi complex to the ER. Depletion of LPAATγ causes Golgi membrane fragmentation and severe impairment of COPI-coated vesicle formation (Schmidt and Brown, 2009; Yang et al., 2011). Indeed, inhibition of LPAATγ enzyme impairs the release of COPI buds as vesicles from the Golgi complex. This is due to the role of LPAATγ activity in controlling the membrane fission of these buds from the Golgi membranes (Yang et al., 2011). Of note, LPAATγ prefers C20:4 and C22:6 fatty acyl-CoA donors (Yuki et al., 2009; Koeberle et al., 2010, 2012) and this might explain the original evidence of acyl-CoA’s involvement in COPI-coated vesicle formation (Pfanner et al., 1989).
Here we selectively focus on LPAATδ because of its emerging role in membrane remodeling (fission and fusion) and related diseases caused by defective lipid metabolism and/or altered membrane composition, while for an extensive and accurate description of the other LPAAT enzymes the reader is referred to excellent reviews (Yamashita et al., 2014; Bradley and Duncan, 2018).
Phosphatidic Acid: the Product of the LPAAT-Catalyzed Enzymatic Reaction
Phosphatidic acid is composed of a three-carbon glycerol backbone, to which two fatty acyl chains are ester-linked at positions C-1 and C-2, and a phosphate is ester-linked at position C-3. On the basis of their shapes, lipids can be divided into three classes (Cullis and De Kruijff, 1979; Corda et al., 2002; Chernomordik and Kozlov, 2003; and references therein): cone-shaped lipids, inverted-cone-shaped lipids, and cylindrical lipids. Lipids whose tails are wider than headgroups are cone-shaped, lipids whose headgroups are wider than tails are inverted-cone-shaped, and lipids whose headgroups are approximately as wide as tails are cylindrical. PA is a cone-shaped lipid, whereas LPA possesses an inverted-cone shape (Kooijman et al., 2005b; Figure 1). Thus, the interconversion from LPA to PA mediated by LPAAT enzymes destabilizes the organization of the lipid bilayer causing a distortion of this bilayer, a process that supports membrane fission (Barr and Shorter, 2000; Shemesh et al., 2003; Pagliuso et al., 2016). Moreover, both PA and LPA are charged negatively (Kooijman et al., 2005a). Hence, PA possesses an unique combination of cone shape and negative charge (Tanguy et al., 2019). Such unique property of PA allows it to recruit various proteins to the membrane (Stace and Ktistakis, 2006). According to the electrostatic/hydrogen bond switch mechanism (Kooijman et al., 2007), the PA charge is able to change from -1 to -2 due to the deprotonation of PA headgroup, caused by formation of the hydrogen bond of positively charged amino acid residue (lysine or arginine) with this headgroup. This change can stabilize the protein-lipid interaction (Kooijman et al., 2007). Due to its phosphomonoester headgroup whose pKa value is within physiological range, PA acts as a pH biosensor (Young et al., 2010; Shin and Loewen, 2011). Eventhough the content of PA in the cellular membrane is usually low, ∼1–4% (Zinser et al., 1991; Young et al., 2010; Zegarlinska et al., 2018), this lipid is involved in many important biological processes such as induction of membrane curvature, membrane trafficking, recruitment of proteins to membranes, regulation of catalytic activity of various enzymes, signal transduction, cytoskeletal organization and regulation of gene expression (Kooijman and Burger, 2009; Raben and Barber, 2017; Kameoka et al., 2018; Zegarlinska et al., 2018; Tanguy et al., 2019; and references therein). Various phospholipids, such as PC, PE, PI and CL are synthesized through the common precursor, PA (Yamashita et al., 2014). In the Lands’ cycle, these phospholipids can be deacylated and reacylated at the sn-2 position in a coordinated and concerted control cycle by phospholipases A2 (PLA2s) and LPLATs actions, respectively (Bankaitis, 2009; Ha et al., 2012). This cycle is essential to generate the membrane asymmetry and diversity that support membrane fluidity and curvature required for fundamental biological functions. Thus, PA is involved, directly or indirectly, in the biosynthesis of most phospholipids (Kassas et al., 2017). Hence, also enzymes that catalyze the synthesis of PA play very important role in biological processes.
FIGURE 1.
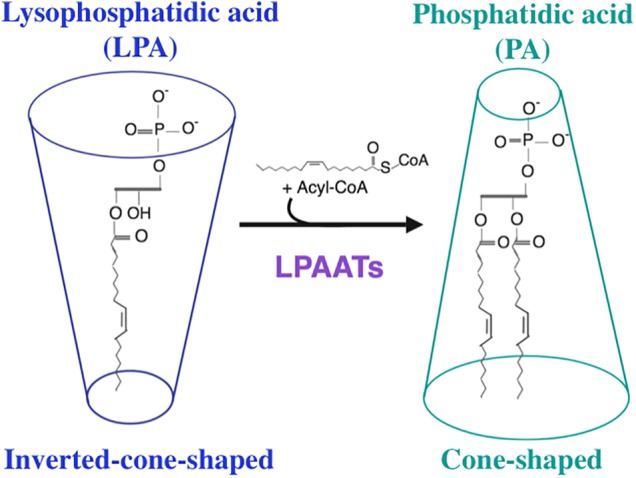
Enzymatic reaction catalyzed by lysophosphatidic acid acyltransferase enzymes. LPAATs promote the addition of an unsaturated fatty acid from Acyl-CoA (as acyl donor) to lysophosphatidic acid (LPA, as acyl acceptor) to form phosphatidic acid (PA). This reaction converts an inverted-cone-shaped lipid (LPA) into a cone-shaped lipid (PA).
In all organisms, PA can be produced by one of three major routes (Foster et al., 2014; Vodicka et al., 2015; Kassas et al., 2017): de novo synthesis in which the final step is acylation of LPA by LPAAT enzymes, phosphorylation of DAG by DAGKs, and hydrolysis of phospholipids by PLD (Figure 2). LPAATs are members of the family of AGPAT enzymes that specifically use LPA as acyl acceptor (Yamashita et al., 2014).
FIGURE 2.
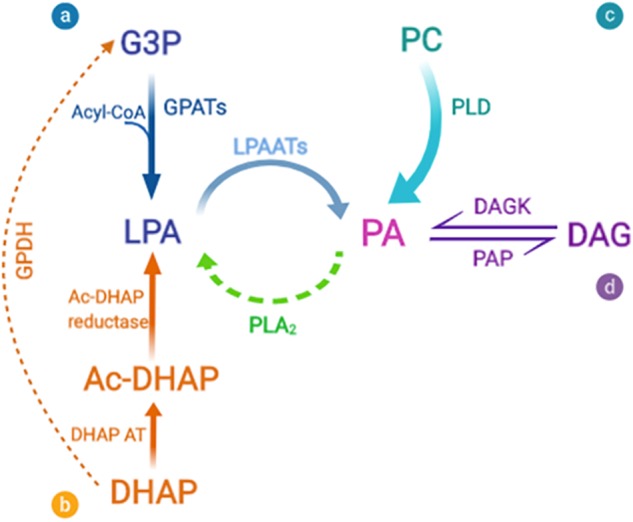
Biosynthetic pathways for phosphatidic acid production. Phosphatidic acid (PA) can be generated by three major routes: (a-b) The de novo synthesis via lysophosphatidic acid (LPA) formation that occurs via two different acylation pathways. (a) The first and main synthesis route is the glycerol 3-phosphate (G3P) pathway. G3P is acylated by a glycerol 3-phosphate acyltransferase (GPAT) to form LPA. (b) The second pathway of LPA formation involves acylation of dihydroxyacetone phosphate (DHAP) by DHAP acyltransferase (DHAP AT) via the 1-acylDHAP (Ac-DHAP) pathway followed by Ac-DHAP reductase-mediated reduction. LPA can then be further acylated by the addition of an unsaturated fatty acid (generally arachidonate) to form PA, via a lysophosphatidic acid acyltransferase (LPAAT). The inverse reaction is mediated by phospholipase A2 (PLA2), which thus converts PA into LPA. Both of these reactions are of particular significance for the geometry of the membrane bilayers, since cone-shaped PA is converted to inverted-cone-shaped LPA (and vice versa), thus facilitating rapid changes in membrane curvature. (c) PA is also formed by the breakdown of other phospholipids, and in particular by the activity of phosphatidylcholine (PC)-specific phospholipase D (PLD). (d) Finally, PA can be dephosphorylated by PA phosphatases (PAPs), to form diacylglycerol (DAG), a strongly conical component of the bilayer, which due to its small and uncharged headgroup, has spontaneous transbilayer movement (flip-flop). The opposite reaction is catalyzed by diacylglycerol kinases (DAGKs). PA and DAG have been shown to be in dynamic equilibrium, and this mechanism can affect the composition and curvature of both leaflets of the bilayer.
Acyltransferase Motifs in LPAATs
The LPAAT enzymes contain four catalytic motifs I-IV (Takeuchi and Reue, 2009; Rottig and Steinbuchel, 2013; Yamashita et al., 2014), and here we report the sequence alignment of LPAATs from sixty species of different biological kingdoms, see Supplementary Tables S1, S3.
Motifs I and III are the most conserved, whereas motif IV is the least conserved. Motifs I and III are suggested to be involved in the interaction with the acyl acceptor, whereas motif IV is suggested to interact with acyl donor (Yamashita et al., 2014; Robertson et al., 2017; and references therein).
Catalytic motif I contains a conserved NHxxxxD sequence. In this sequence, the residue following histidine (H) is usually hydrophilic, whereas the 5th residue upstream this H and the residue preceding aspartic acid (D) are almost always hydrophobic, see Supplementary Tables S1, S3. The H belonging to this sequence acts as a general base to abstract proton from the hydroxyl group which has to be acylated (Pagac et al., 2011; Yamashita et al., 2014; and references therein). The role of D belonging to the NHxxxxD sequence is to maintain the lone pair of electrons on the Nε2 nitrogen of catalytic H so as to abstract a proton (Robertson et al., 2017; and references therein). Among sixty LPAATs from different species presented in the Supplementary Tables S1, S3, the H belonging to the catalytic motif I is absolutely conserved, whereas the D is conserved almost absolutely (present in 59 out of 60 sequences).
Catalytic motif II contains a conserved arginine (R). The residue that precedes this R is always (or almost always) hydrophilic, whereas amino acid residue that precedes this hydrophilic residue is usually hydrophobic. In LPAATs from animals and fungi, this R often belongs to the (F/Y)xxR pair, see Supplementary Tables S1, S3.
Catalytic motif III contains a conserved EGTR sequence. In most LPAAT enzymes, although not in mammalian LPAATδ, amino acid residue that is two residues upstream of glutamic acid (E) belonging to the EGTR sequence is phenylalanine (F), see Supplementary Tables S1, S3. In PlsC, a LPAAT from bacterium Thermotoga maritima, the only LPAAT for which the crystal structure is available, conserved R belonging to this EGTR sequence is ideally positioned to bind the 3′-phosphate of LPA (Robertson et al., 2017). Among sixty LPAATs from different species presented in Supplementary Tables S1, S3, E and glycine (G) of the catalytic motif III are also almost absolutely conserved. They are present, respectively, in 58 and 59 of the 60 sequences shown in Supplementary Tables S1, S3.
Catalytic motif IV contains a conserved proline (P), see (Yamashita et al., 2014; Korbes et al., 2016) and Supplementary Tables S1, S3. In most LPAAT enzymes, two residues preceding this P are hydrophobic, see Supplementary Tables S1, S3. Sometimes the residue following this P also plays a role in catalysis (Yamashita et al., 2014).
In mammalian LPAATα, mutations of conserved residues belonging to the calatylic motifs lead to the strong inhibition of catalytic activity. Among these mutations are the following: mutation of H of the motif I to alanine (A), mutations of D of the motif I to E or asparagine (N), mutation of R of the motif II to A, mutation of E of the motif III to D or glutamine (Q), mutation of G of the motif III to leucine (L), mutation of R of the motif III to A or lysine (K) (Yamashita et al., 2007, 2014).
Naturally occurring mutations in human LPAATβ, the best studied and characterized of all mammalian LPAATs, are associated with type one Berardinelli-Seip CGL (Agarwal et al., 2002; Magre et al., 2003; Agarwal, 2012; Subauste et al., 2012) and Brunzell syndrome (Fu et al., 2004). Some of the mutations causing the CGL affect residues belonging to the catalytic motifs and/or were shown to result in partial or complete inhibition of LPAATβ enzymatic activity. Specifically: (i) mutation of E to K at position 172 (E172K) belonging to the conserved EGTR sequence, within the catalytic motif III (Magre et al., 2003; Haghighi et al., 2012); (ii) deletion mutation 140delF affects F that belongs to the conserved FxxR pair within the catalytic motif II (Haque et al., 2005); and (iii) mutation of serine (S) to N at position 100 (S100N) changes the sequence within the catalytic motif I (Cortes et al., 2009).
Finally, single mutations H96A, D101A, and E176A in mouse LPAATγ (Yuki et al., 2009) and quintuple mutation N95A/H96A/D101A/E176A/G177A in human LPAATγ (Schmidt and Brown, 2009) lead to complete inhibition of LPAAT activity. Residues N95, H96, and D101 belong to the catalytic motif I, whereas residues E176 and G177 belong to the catalytic motif III.
Animal LPAATδ Enzyme
Lysophosphatidic acid acyltransferase δ is a member of the LPAAT family (Lu et al., 2005; and references therein) that exhibits catalytic activity with LPA, but not with most other major lysophospholipids, such as LPC, LPE, LPS, LPI, LPG, MLCL, DLCL or with glycerol 3-phosphate (G3P) (Eto et al., 2014; Bradley et al., 2015). The unsaturated acyl-CoAs C22:6, C20:4, C18:1 followed by C16:0 are the preferred substrates of this enzyme with Vmax = 23.2 ± 2.4 nmol/min/mg and Km = 42.9 ± 2.9 μM for 18:0-LPA (Eto et al., 2014) and Vmax = 38 ± 1 nmol/min/mg, Km = 29 ± 1 μM for 18:1-LPA (Pagliuso et al., 2016). The highest LPAATδ catalytic activity was observed at pH 7.4 using 18:1-LPA and 18:1-acyl-CoA, with a minor decrease when pH rose to 7.6 and a sharp decrease when pH was lowered to 7.2 (Bradley et al., 2015).
The human Lpaatδ gene is on chromosome 6. Both human Lpaatδ and mouse Lpaatδ possess seven introns and five exons while Arabidopsis and rice Lpaatδ possess two introns and three exons with similar size (Korbes et al., 2016). Mammalian LPAATδ (residue numbers are for the human ortholog, UniProt accession number Q9NRZ5) contains a few conserved residues belonging to four catalytic motifs, including N95, H96 and D101 of the motif I, F143 and R146 of the motif II, E176 and G177 of the motif III, P206 of the motif IV. The H96A mutant of mouse LPAATδ shows much lower catalytic activity as compared to the wild type enzyme (Eto et al., 2014), and H96V mutant of human LPAATδ is inactive (Pagliuso et al., 2016), demonstrating that H96 residue is essential for catalysis. Moreover, this H96V mutation in human LPAATδ leads to the inhibition of the fission step during post-Golgi carrier formation indicating that LPAATδ catalytic activity is required for this process (Pagliuso et al., 2016). Based on the published results of the mutagenesis experiments with LPAATα (Yamashita et al., 2007, 2014) and LPAATγ (Yuki et al., 2009), we expect that the following mutations will also inhibit the catalytic activity of LPAATδ: D101A, D101E, D101N, R146A, E176A, E176D, E176Q, G177L, R179A, and R179K.
A crystal structure of PlsC, a LPAAT from bacterium Thermotoga maritima (T. maritima), was reported with the studies of the functional role of various PlsC residues (Robertson et al., 2017). Based on comparison among TmPlsC and other LPAATs, we can derive some conclusions concerning the structure of mammalian LPAATδ.
In TmPlsC, the phosphate group of LPA was suggested to interact with the highly conserved residues R159 (within catalytic motif III) and K105 (Robertson et al., 2017). This K105 is located between the catalytic motif I and a highly conserved P (P112 in TmPlsC). This K is highly conserved across all biological kingdoms except Archaea, see Supplementary Tables S1, S3. In human LPAATδ, K123 followed by K124 correspond to K105 from TmPlsC, see Supplementary Tables S1, S3. Based on this sequence alignment, we can conclude that in LPAATδ, K123 or, perhaps, K124 interact with the phosphate group of LPA. In the orthologs of animal LPAATδ, K corresponding to K123 of human LPAATδ is conserved, see Supplementary Tables S2, S4. We expect that in human LPAATδ, the catalytic activity of the K123A/K124A double mutant will be negligible.
Membrane Topology of Human LPAATδ
The TmPlsC crystal structure is consistent with the organization in two domains: the N-terminal two-helix motif and the αβ-domain that contains all four catalytic motifs (Robertson et al., 2017). This molecule does not contain any TMDs, but its α1 helix (belonging to the N-terminal two-helix motif) enters and exits on the same side of the membrane, due to the presence of the G25G26 kink within this helix.
In Supplementary Tables S1, S3 the sequence alignment of TmPlsC, human LPAATδ and many other LPAATs from different biological kingdoms is indicated. One of TmPlsC aromatic residues suggested to interact with the apolar interior of the lipid bilayer, tryptophan W116 (Robertson et al., 2017) is highly conserved, see Supplementary Tables S1, S3. This W is located between catalytic motifs I and II and, more precisely, between highly conserved P (P112 in TmPlsC) and catalytic motif II. The W in this location is present in the LPAAT enzymes from many species. In human LPAATδ, W134 and W136 are located between highly conserved P (P130 in LPAATδ, part of highly conserved PxxG motif) and catalytic motif II. In LPAATδ, tryptophans at these positions are highly conserved throughout evolution, see Supplementary Tables S2, S4. We hypothesize that W134 and W136 from LPAATδ, like W116 from TmPlsC, interact with the apolar interior of the lipid bilayer.
The membrane topology of a few LPAATs, such as human LPAATα (Yamashita et al., 2007), human LPAATγ (Schmidt et al., 2010), Saccharomyces cerevisiae SLC1 (Pagac et al., 2011) and peanut LPAT4 (Chen et al., 2012), was studied experimentally as well as using bioinformatics. All enzymes reported in these studies are localized to the ER or Golgi membranes and contain a TMD between catalytic motifs I and II, while no TMD is foreseen between catalytic motifs II and III (see Figure 3 of Yamashita et al., 2014). This topological organization is quite unusual (motif I on one side of the membrane, and motifs II and III on the other side) and it can be explained by the need to bring in close proximity catalytic motifs I–IV that may penetrate into the membrane from the cytosolic or luminal side to act in concert (Yamashita et al., 2007, 2014; Chen et al., 2012).
Eto et al. (2014) predicted six TMDs in LPAATδ by HMMTOP transmembrane topology prediction server (Tusnady and Simon, 1998, 2001).
We would like to point out that no long hydrophobic stretches between catalytic motifs II and III and between catalytic motifs III and IV are present in LPAATδ (see Supplementary Tables S2, S4). Accordingly, it is reasonable to propose that no TMD characterizes the organization of this LPAATδ segment, and, hence, the three catalytic motifs II, III, and IV are located on the same side of the membrane. It should be noted, however, that the stretch L126AYVPIIGWMWYF138 between catalytic motifs I and II of human LPAATδ is very hydrophobic and that similar hydrophobic stretches are present in this location in LPAATδ orthologs from other animal species (Supplementary Tables S2, S4). It may be hypothesized that this stretch is a TMD and, hence, in LPAATδ, as in all four eukaryotic LPAATs mentioned above, a TMD is present between catalytic motifs I and II. However, this stretch (13 residues) is unusually short, and the vast majority of TMDs are longer (Singh and Mittal, 2016). As a consequence, if this stretch is indeed a TMD, the location of catalytic motifs I and II of LPAATδ on the different sides of the membrane would not favor catalysis (Yamashita et al., 2007; Schmidt et al., 2010; Pagac et al., 2011; Chen et al., 2012).
In order to bring some clarity, we decided to use CCTOP server1, a web-based application providing prediction of membrane protein topology. This server utilizes ten different state-of-the-art topology prediction methods, including HMMTOP (Dobson et al., 2015). We studied membrane topology of human LPAATδ using CCTOP. We found that, according to this prediction, with reliability 81.1, human LPAATδ contains only three TMDs: one TMD is upstream of the catalytic motif I, whereas two more TMDs are downstream of the catalytic motif IV. Hence, according to this prediction, all four catalytic motifs are located on the same side of the membrane (Figure 3).
FIGURE 3.

Proposed membrane topology of human LPAATδ. The transmembrane domains (TMD; in turquoise) were predicted by CCTOP prediction server (http://cctop.enzim.ttk.mta.hu) (Dobson et al., 2015) and indicated as TMD1 (amino acids 15-38), TMD2 (amino acids 308-327), and TMD3 (amino acids 333-352). The four Catalytic Motifs of LPAATδ are indicated as I-IV in purple, and their amino acid sequences are reported in the sequence alignment as Motif I (amino acids 93-103), Motif II (amino acids 140-146), Motif III (amino acids 173-182), and Motif IV (amino acids 204-209). Highly conserved proline P130 and glycine G133 in the PxxG motif between catalytic motifs I and II are highlighted. Lysines K123 and K124 are highlighted. The N-terminus of the protein is predicted to be located in the lumen while the C-terminus is predicted to be located in the cytoplasm (as indicated).
We hypothesize that human LPAATδ, like TmPlsC, does not contain any TMDs between catalytic motifs I and II, and all four catalytic motifs are located on the same side of the membrane (Figure 3). Moreover, we hypothesize that in human LPAATδ, the L126AYVPIIGWMWYF138 stretch forms an α-helix, and this helix, like the α1 helix in TmPlsC, enters and exits on the same side of the membrane. In the α1 helix in TmPlsC, such topology is facilitated by the presence of the G25G26 kink in the middle of this helix. We believe that in human LPAATδ, the motif P130xxG133 plays a role of such kink. This PxxG motif is highly conserved among various LPAATs, see Supplementary Tables S1, S3, and is conserved across animal LPAATδ orthologs, see Supplementary Tables S2, S4. In proteins, kinks often coincide with the hinges (flexible regions that decouple pre-hinge and post-hinge portions of protein segment) (Cordes et al., 2002; Bright and Sansom, 2003; Hall et al., 2009). Molecular hinges often contain P residues (Sansom and Weinstein, 2000; Cordes et al., 2002; Bright and Sansom, 2003; Hall et al., 2009). The flexibility introduced by a P residue can be increased in the presence of G (Sansom and Weinstein, 2000; Cordes et al., 2002; Bright and Sansom, 2003) close to P, usually not more than four residues between P and G (Bright and Sansom, 2003).
The function of such hypothetical conformation of the L126AYVPIIGWMWYF138 stretch (entering and exiting on the same side of the membrane) might be, as suggested to the two-helix motif of TmPlsC (Robertson et al., 2017), to be a “fishing bobber” that suspends the LPAATδ active site close to the lipid molecules involved in reaction. Similar function of the short hydrophobic stretch between catalytic motifs I and II can be hypothesized also in other LPAATs.
Physiological Functions of LPAATδ Enzyme in Mouse Model
Lysophosphatidic acid acyltransferase δ is most highly expressed in brain (Eto et al., 2014; Bradley et al., 2015, 2016, 2017), but also in muscle (Bradley and Duncan, 2018) and, to a lesser extent, in other tissues, such as lungs, intestines, epidermis, and spleen. At the level of the mouse central nervous system, LPAATδ is abundant at the brain stem, cortex, hippocampus, cerebellum and olfactory bulbs (Bradley et al., 2015).
The characterization of the physiological functions of LPAATδ is based on the analysis of the dysfunctions associated with Lpaatδ gene knockout in mouse model. The absence of this enzyme results in a wide range of alterations that include: cognitive impairment, impaired force contractility and altered visceral white adipose tissue depots.
The level of PI, PE, and PC is significantly lower (by 52, 32, and 38%, respectively) in the brain of Lpaatδ- mice (Bradley et al., 2015). However, the loss of LPAATδ did not influence the level of brain PA (Bradley et al., 2015) and this can be explained by redundancies in the LPAAT family. Indeed, an up-regulation of LPAATε, LPAATα and, even more, of LPAATβ has been found in the brain of LPAATδ- mice indicating a degree of compensation in PA synthesis (Bradley et al., 2015). Of note, this adaptation in PA brain content was not able to compensate the reduced levels of the other phospholipid species (PI, PC, and PE) observed in the brain of Lpaatδ- mice. This implies that the LPAATδ enzyme produces a specific pool of PA that is used as precursor to support the biosynthesis of PI, PC, and PE. The levels of the other major brain phospholipids were not affected indicating an unique function of LPAATδ in regulating the PI, PC and PE as downstream Kennedy pathway derivatives (Bradley et al., 2015).
Lpaatδ- mice have significant impairments in spatial learning and memory (Bradley et al., 2017). This phenomenon can be partly attributed to the drastically lower brain content of the NMDA receptor subunits (namely NR1, NR2A, and NR2B), and of the GluR1 subunit of the AMPA receptor. NMDA receptor and AMPA receptor are two glutamate-gated transmembrane proteins involved in synaptic plasticity and memory (Bradley et al., 2017). Such a significantly reduced neural content of these subunits, in turn, might be explained by the noticeable decrease in PI, PE, and PC in the brain of Lpaatδ- mice. The presence of these glycerophospholipids might be required to preserve the biophysical properties of neuronal membranes in order to generate the conditions for the correct assembly and function of these receptors (Bradley et al., 2017). Specific lipids have been reported to have a modulatory role in the structure and function of many membrane proteins (Opekarova and Tanner, 2003; Lee, 2004; Bogdanov et al., 2008; Zhukovsky et al., 2013; and references therein). We expect that phospholipids whose synthesis depends (directly or indirectly) on LPAATδ are required for the native conformation and normal function of a few other membrane proteins, and the absence of LPAATδ might lead to the disruption of the function of these proteins.
Lysophosphatidic acid acyltransferase δ possesses catalytic activity for DHA-CoA (Eto et al., 2014). DHA is required for neurite outgrowth in hippocampal neurons (Calderon and Kim, 2004), and its reduced levels is associated with the development of cognitive and neurodegenerative disorders. Hence, the high level of LPAATδ expression in the brain is consistent with DHA being abundant among brain phospholipids (Yamashita et al., 2014), suggesting an important role of LPAATδ in maintaining DHA in neural membranes (Eto et al., 2014).
Lysophosphatidic acid acyltransferase δ was found in various muscle types. However, it was detected at highest levels in soleus, a red oxidative fiber-type that is rich in mitochondria (Bradley and Duncan, 2018). This is consistent with the localization of LPAATδ to the outer mitochondrial membrane (Bradley et al., 2015). Lpaatδ- mice showed increased PA and PE contents on fiber-type composition that in turn is suggested to impair the force contractility in soleus. These effects seem not associated to LPAATδ-related mitochondria dysfunction; as such, the Lpaatδ- mice did not exhibit impaired mitochondrial function or reduced mitochondrial content (Bradley et al., 2015). In Bradley et al. (2018), a compensatory mechanism in PA synthesis was also reported. Indeed, LPAATβ and LPAATε are specifically upregulated in soleus of Lpaatδ- mice, but not LPAATγ and LPAATα.
Lysophosphatidic acid acyltransferase δ is also highly expressed in white adipose tissue (WAT), particularly in epididymal and perirenal WAT (Prasad et al., 2011; Mardian et al., 2017). Male mice deficient in Lpaatδ gene have significant (by 40%) increase in the epididymal WAT weight with no effects on perirenal and inguinal WAT, as well as brown adipose tissue. The high PA and TAG levels in the epididymal WAT of Lpaatδ- mice is associated with an increase in adipocyte size rather than in adipocyte number. This is explained by an impaired lipolysis process due to reduced expression levels of adipose triglyceride lipase and phosphorylated hormone-sensitive lipase (Mardian et al., 2017). Here, a compensatory upregulation of LPAATα, LPAATβ, LPAATγ, and LPAATε occurs only in the perirenal WAT and not in the epididymal WAT. This adequate compensation mechanism is associated with normal tissue glycerolipid contents and, in turn, with normal tissue function (Mardian et al., 2017).
Interestingly, the loss of Lpaatδ gene pointed to the functional role of the specific pool of PA generated by LPAATδ enzyme. Indeed, although total PA level may be compensated in Lpaatδ- mice tissue by the induction of other LPAATs, the above studies indicated that the pool of PA generated by adaptive mechanisms is not able to functionally replace the LPAATδ-mediated production of PA and the downstream phospholipid derivatives that support specific cellular and tissue demands (as indeed shown in brain, soleus muscle and epididymal WAT in Lpaatδ- mice) (Bradley and Duncan, 2018; and references therein).
Subcellular Localization of LPAATδ
According to Eto et al. (2014), murine LPAATδ localizes to the ER. Other authors reported that in mouse brain, LPAATδ resides on the MOM, but not on the MIM (Bradley et al., 2015). We recently demonstrated that both human and murine LPAATδ are targeted to both trans-Golgi membranes and mitochondria (Pagliuso et al., 2016).
In accordance with the endosymbiotic hypothesis, mitochondria of eukaryotes evolved from aerobic bacteria (Gray, 2012). As expected from this hypothesis, the MOM has similar composition to the plasma membrane and/or ER that may have surrounded symbiotic bacteria (Kuroda et al., 1998; and references therein), and it is not surprising that same or similar proteins are present in all these membranes (Kuroda et al., 1998; Colombo et al., 2005; Bhatt et al., 2008; Tamir et al., 2013; Marchi et al., 2014). We thus concluded that LPAATδ might localize both to the ER (from which it is transported to Golgi) and to the mitochondria (Pagliuso et al., 2016).
In addition to LPAATδ, there are other multipass transmembrane proteins that are targeted to both mitochondria and ER/Golgi. Mammalian diacylglycerol acyltransferase-2 (DGAT2) is a good example of such protein. Like LPAATδ, this enzyme is an acyltransferase, and reaction catalyzed by this enzyme, like LPAATδ-catalyzed reaction, belongs to the Kennedy pathway (Mcfie et al., 2011). DGAT2 contains two TMDs (Mcfie et al., 2014) and is localized to the ER, to the mitochondrial outer membrane, and to the lipid droplets (Mcfie et al., 2011). Mitochondrial targeting sequence contains few residues in the cytosolic portion of the protein, just upstream of the first TMD (Stone et al., 2009). This is a typical location of mitochondrial targeting signals (Rapaport, 2003; Stone et al., 2009). ER targeting signal resides within the first TMD (Mcfie et al., 2011), whereas lipid dropet targeting sequence is in the C-terminal cytosolic region of DGAT2 (Mcfie et al., 2018).
We carefully hypothesize that LPAATδ, like DGAT2, might contain separate targeting signals for mitochondria and for Golgi apparatus. The balance of LPAATδ amount between MOM and ER/Golgi membranes might be determined by comparative affinity of two signals for their respective organelles (Yogev and Pines, 2011). We expect that mutagenesis experiments will allow to specify the localization of these putative targeting signals. Possibly, as in the case of DGAT2, positively charged residues in the cytosolic portion of LPAATδ, just upstream of the putative second TMD, belong to the mitochondrial targeting sequence.
Role of LPAATδ Enzyme in Membrane Fission of Golgi Membranes
Brefeldin A ADP-ribosylated substrate is the shorter splice isoform of CtBP1 protein, a member of the C-terminal binding protein (CtBP) family, known as CtBP1-S/BARS (Spano et al., 1999; Nardini et al., 2003; Valente et al., 2005; Corda et al., 2006). BARS (from here on) is a dual-function protein that in its dimeric NADH-bound conformation acts as transcriptional regulator in the nucleus, whereas in its p21-activated kinase 1 (PAK1)-phosphorylated monomeric conformation mediates membrane fission in the cystoplasm (Nardini et al., 2003, 2009; Yang et al., 2005; Colanzi et al., 2007, 2013; Liberali et al., 2008; Valente et al., 2013). BARS is a key member of a protein complex that is required for various membrane fission processes including basolaterally-directed post-Golgi carrier formation (Bonazzi et al., 2005; Valente et al., 2012), COPI-coated vesicle formation (Yang et al., 2005, 2008; Valente et al., 2012), macropinocytosis (Liberali et al., 2008; Valente et al., 2012), fluid-phase endocytosis and Golgi partitioning in mitosis (Hidalgo Carcedo et al., 2004; Bonazzi et al., 2005; Colanzi and Corda, 2007; Colanzi et al., 2007).
We and others have previously shown that the BARS-induced fission on isolated Golgi membranes correlates with PA production starting from LPA and acyl-CoA and that this LPAAT catalytic reaction supports membrane fission (Weigert et al., 1999; Kooijman et al., 2003; Shemesh et al., 2003; Pagliuso et al., 2016). This LPAAT activity is associated with, rather than intrinsic to BARS (Gallop et al., 2005) as shown by the fact that: (i) the minimal BARS domain able to support COPI-coated vesicle fission does not incorporate this activity (Yang et al., 2005); and (ii) during purification of recombinant BARS from E. coli, bacterial LPAAT, known as PlsC, specifically binds BARS (Gallop et al., 2005; Pagliuso et al., 2016). These data prompted the search for evolutionary conserved interaction between BARS and the LPAAT enzymes from bacteria to mammals. We have shown that BARS, at the TGN, is incorporated in a well-defined protein complex (Valente et al., 2012), where it binds to and activates the LPAATδ enzyme catalyzing the production of a PA pool required to support membrane fission of the basolaterally-directed post-Golgi carriers (Pagliuso et al., 2016; Figure 4). Specifically, as the cargo protein reaches the TGN membranes, BARS upon PAK1-mediated phosphorylation at serine 147, that induces its monomeric fission-prone conformation, assembles in a complex where it binds to the 14-3-3γ adaptor (Valente et al., 2012, 2013). Through a 14-3-3γ dimer, BARS is in a tripartite core complex with the phosphoinositide kinase PI4KIIIβ and binds proteins implicated in post-Golgi carrier formation, such as ARF, NCS-1 (also known as frequenin) and PKD (Valente et al., 2012, 2013; Figure 4). This complex allows to spatially and temporally couple the budding/tubulation of post-Golgi carriers with their fission. The reversible and regulated formation of this complex enhances the efficiency of the fission machinery that assembles along the tubular carrier precursor emerging out of the TGN on the site where then fission will take place (Valente et al., 2012, 2013; Figure 4).
FIGURE 4.

Putative mechanism of BARS-mediated membrane fission during post-Golgi carrier formation. Schematic representation of tubular carrier precursor that emerged out of specialized TGN-export domain during the budding/tubulation step (I), a process that precedes membrane fission (II). Upon fission event, the free post-Golgi carriers are released and directed toward the basolateral plasma membrane (usually with the employment of microtubular tracks and microtubule-based motors). At the protein level: upon activation ARF recruits PI4KIIIβ and PKD kinases onto the Golgi membranes and activates the PI4KIIIβ kinase leading to PtdInsP4 production. PI4KIIIβ kinase can be also activated by NCS-1, a well-known ARF interactor. ARF and the produced PtdInsP4-membrane pool promote the recruitment of FAPPs, which endorse membrane bending and budding/tubulation. PI4KIIIβ and BARS are then phosphorylated by PKD and PAK1, respectively (see text for details) allowing to the formation of a tripartite core complex where the phosphorylated kinase-active form of PI4KIIIβ is bridged to the phosphorylated fission-active form of BARS through 14-3-3γ dimer. Once incorporated into this complex, BARS binds to and activates LPAATδ enzyme leading to the local membrane conversion of LPA into PA. At the lipid level this enzymatic conversion of LPA into PA is central for the completion of the fission event. This figure is created using the web-based tool BioRender (https://biorender.com/library/).
Recently we showed that, when incorporated into this complex, BARS binds to and activates LPAATδ and that this LPAATδ-mediated production of PA is required for fission of post-Golgi carriers (Pagliuso et al., 2016).
Many membrane fission processes require the presence of specific lipids. These lipids could be named lipid ligands (Gopaldass et al., 2017), or lipid factors (Danne et al., 2017), or lipid cofactors (Ramachandran, 2018). In some cases, fission might be energized via the energy used in the synthesis of lipid cofactors (Gopaldass et al., 2017). We claim that PA produced in the reaction catalyzed by LPAATδ is a lipid cofactor for BARS-mediated fission.
Lysophosphatidic acid acyltransferase δ localizes to the Golgi and to the MOM (Bradley et al., 2015; Pagliuso et al., 2016), see above. PA has been shown to play a role in mitochondrial dynamics by regulating membrane fusion and fission (Frohman, 2015; Kameoka et al., 2018; and references therein). We can hypothesize that in mitochondria, the role of a PA-producing enzyme as LPAATδ is also related, among other things, to membrane fission, similar to the role of LPAATδ at the Golgi complex. Such possible dual role of LPAATδ can be somewhat analogous to the function of hFis1 that is dual targeted to mitochondria and peroxisomes and regulates membrane fission of both organelles (Delille and Schrader, 2008).
Differently from BARS, another member of the CtBP family RIBEYE possesses acyltransferase activity (Schwarz et al., 2011). In mammals, CtBPs are encoded by two genes, CtBP1 and CtBP2. CtBP1 has two splicing variants, CtBP1-L and BARS (CtBP1-S/BARS), whereas CtBP2 has three splicing variants, CtBP2-L, CtBP2-S, and RIBEYE. RIBEYE contains a large N-terminal domain that is unrelated to the CtBPs (Maxeiner et al., 2016; Schwarz and Schmitz, 2017) and a C-terminal domain. This C-terminal domain is very similar to CtBP2 and BARS in the NAD(H)-binding domain and substrate-binding domain (ca. 88.5%; Kumar et al., 2002; Nardini et al., 2003) but with relatively different residues in the C-terminal region (ca. 50.6%; Kumar et al., 2002; Nardini et al., 2003). These differences in the C-terminal region could explain the intrinsic LPAAT activity owned by RIBEYE and not by BARS (Schwarz et al., 2011). Indeed, as reported, this is the region responsible for LPAAT activity and substrate binding (Schwarz et al., 2011). Moreover, this RIBEYE enzymatic activity was not due to contaminating proteins copurifying with RIBEYE (Schwarz et al., 2011). It has been proposed that PA generated by RIBEYE at synaptic ribbons facilitates synaptic vesicle trafficking (Schwarz et al., 2011). We hypothesize that in RIBEYE (UniProt accession number Q9EQH5-2), the N792H793xxxxD798 segment belongs to the LPAAT catalytic motif I, whereas E701GTR704 segment belongs to the LPAAT catalytic motif III and, perhaps, F366xxR369 pair belongs to the LPAAT catalytic motif II. However, we should consider that in LPAATs, the residue preceding D is almost always hydrophobic, see Supplementary Tables S1, S3, whereas in rat RIBEYE, this residue is hydrophilic N797. Moreover, the localization of two catalytic motifs in RIBEYE (motif III upstream of motif I) is very unusual, because in all (or almost all) other LPAATs, including LPAATδ, motif III is downstream of motif I (Korbes et al., 2016). Hence, most probably, LPAAT activity of RIBEYE evolved independently of the other LPAATs. We suppose that in RIBEYE dimer (whose crystal structure is not available yet), N792H793xxxxD798 motif of one protomer might be close to the E701GTR704 motif of another protomer. In the literature (Yamashita et al., 2007, 2014; Yuki et al., 2009; Eto et al., 2014; Pagliuso et al., 2016; and references therein), the catalytic activity-disrupting mutants of residues belonging to the catalytic motifs of LPAATs are reported, see above. Based on these data, we expect that in some of the mutants H793A, H793V, D798A, D798E, D798N, R369A, E701A, E701D, E701Q, G702L, R704A, R704K, the LPAAT activity of RIBEYE will be inhibited.
Following on the above considerations, we hypothesize that an evolutionary ancestor of BARS also possessed acyltransferase activity. BARS residues N232, H233 and D238 might form a vestige of an LPAAT catalytic motif I, whereas the E141GTR144 stretch might be a vestige of an LPAAT catalytic motif III. Possibly, an ancestor of BARS was simultaneously an LPAAT and a fission-inducing protein, it catalyzed production of PA and used this lipid as a cofactor in membrane fission reaction, similar to Agrobacterium tumefaciens PmtA that is an enzyme producing MMPE and a membrane fission-inducing protein that uses this lipid as a cofactor in fission reaction (Danne et al., 2017). Perhaps, later BARS gradually lost its LPAAT activity and simultaneously acquired an ability to bind and activate LPAATδ, and to use PA produced by LPAATδ as a cofactor in membrane fission reaction. Interestingly, the BARS residue R144 belonging to the putative vestigial catalytic motif E141GTR144 also belongs to the Rxx(pS) motif (pS is phosphorylated serine) that is involved in the interaction with 14-3-3γ adaptor protein needed for the formation of fully functional fission-inducing protein complex (Valente et al., 2012). Hypothetically, an ancestor of BARS gradually acquired the ability to bind 14-3-3γ, but in the course of this, the sequence motif E141GTR144, overlapping with the 14-3-3γ-binding site, lost the ability to be involved in LPAAT reaction. That is why, we hypothesize that simultaneously with acquiring the ability to bind 14-3-3γ, an ancestor of BARS gradually lost acyltransferase activity and acquired an ability to bind and activate LPAATδ.
Conclusion
Lipid-modifying enzymes such as those of the LPAAT family discussed above are now recognized as central actors in diverse cell functions. Their central role is also testified by the neuronal and muscle pathologies, among others, that are linked to mutations in their structure. This is rather unexpected since the numerous isoforms of the different LPAATs could suggest that redundancy is there to protect the organism from the lack of a given membrane component, PA in this case.
We can assume that redundancy cures several defects, but still there are very specific functions in cell compartments or tissues that are finely controlled by the activity of a single member of the LPAAT family.
The challenge in this field is to build a complete picture of the lipid-modifying enzyme localization, function and regulation. This knowledge will help designing the approaches and tools that will allow complementing their activity; with these procedures, we may anticipate the treatment of diseases caused by defective lipid metabolism and/or altered membrane composition.
Author Contributions
All authors wrote the manuscript. AF prepared the figures. MZ, CV and AF prepared the tables.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the funding agencies listed above for supporting our research.
Abbreviations
- AGPAT
acylglycerophosphate acyltransferase
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- ARF
ADP ribosylation factor
- BARS
brefeldin A ADP-ribosylated substrate
- C14:0
myristic acid
- C16:0
palmitic acid
- C18:0
stearic acid
- C18:1
oleic acid
- C20:4
arachidonic acid
- CGL
congenital generalized lipodystrophy
- CL
cardiolipin
- CoA
coenzyme A
- DAG
diacylglycerol
- DAGKs
diacylglycerol kinases
- DHA
docosahexaenoyl acid
- DHA-CoA
docosahexaenoyl-CoA
- DHAP
dihydroxyacetone phosphate
- DLCL
dilysocardiolipin
- ER
endoplasmic reticulum
- FAPP
four-phosphate-adaptor protein
- G3P
glycerol 3-phosphate
- GPAT
glycerophosphate acyltransferase
- LPA
lysophosphatidic acid
- LPAAT
lysophosphatidic acid acyltransferase
- LPC
lysophosphatidylcholine
- LPE
lysophosphatidylethanolamine
- LPG
lysophosphatidylglycerol
- LPI
lysophosphatidylinositol
- LPS
lysophosphatidylserine
- MIM
mitochondrial inner membrane
- MLCL
monolysocardiolipin
- MMPE
monomethyl-phosphatidylethanolamine
- MOM
mitochondrial outer membrane
- NCS-1
neuronal calcium sensor-1
- NMDA
N-methyl D-aspartate
- PA
phosphatidic acid
- PAK1
p21-activated kinase
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PI
phosphatidylinositol
- PI4KIIIβ
phosphatidylinositol 4-kinase IIIβ
- PKD
protein kinase D
- PLA2s
phospholipases A2
- PLD
phospholipase D
- TAG
triacylglycerol
- TGN
trans-Golgi network
- TMD
transmembrane domain
- TmPlsC
Thermotoga maritima PlsC
- WAT
white adipose tissue
Funding. Research work in the authors’ laboratories was supported by the Italian Association for Cancer Research (AIRC) (to DC IG10341 and IG18776; to AL IG20786 and IG15767), the AIRC-Fondazione Cariplo TRansforming IDEas in Oncological research project (TRIDEO) (to CV IG17524), the PRONAT project, the PRIN project No 20177XJCHX, the SATIN POR project 2014–2020 and the Italian-MIUR Cluster project Medintech (CNT01_00177_962865).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fcell.2019.00147/full#supplementary-material
References
- Agarwal A. K. (2012). Lysophospholipid acyltransferases: 1-acylglycerol-3-phosphate O-acyltransferases. From discovery to disease. Curr. Opin. Lipidol. 23 290–302. 10.1097/MOL.0b013e328354fcf4 [DOI] [PubMed] [Google Scholar]
- Agarwal A. K., Arioglu E., De Almeida S., Akkoc N., Taylor S. I., Bowcock A. M., et al. (2002). AGPAT2 is mutated in congenital generalized lipodystrophy linked to chromosome 9q34. Nat. Genet. 31 21–23. 10.1038/ng880 [DOI] [PubMed] [Google Scholar]
- Agarwal A. K., Sukumaran S., Cortes V. A., Tunison K., Mizrachi D., Sankella S., et al. (2011). Human 1-acylglycerol-3-phosphate O-acyltransferase isoforms 1 and 2: biochemical characterization and inability to rescue hepatic steatosis in Agpat2(-/-) gene lipodystrophic mice. J. Biol. Chem. 286 37676–37691. 10.1074/jbc.M111.250449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankaitis V. A. (2009). The Cirque du Soleil of Golgi membrane dynamics. J. Cell Biol. 186 169–171. 10.1083/jcb.200907008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr F. A., Shorter J. (2000). Membrane traffic: do cones mark sites of fission? Curr. Biol. 10 R141–R144. [DOI] [PubMed] [Google Scholar]
- Bhatt K., Feng L., Pabla N., Liu K., Smith S., Dong Z. (2008). Effects of targeted Bcl-2 expression in mitochondria or endoplasmic reticulum on renal tubular cell apoptosis. Am. J. Physiol. Renal Physiol. 294 F499–F507. [DOI] [PubMed] [Google Scholar]
- Bogdanov M., Mileykovskaya E., Dowhan W. (2008). Lipids in the assembly of membrane proteins and organization of protein supercomplexes: implications for lipid-linked disorders. Subcell. Biochem. 49 197–239. 10.1007/978-1-4020-8831-5_8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonazzi M., Spano S., Turacchio G., Cericola C., Valente C., Colanzi A., et al. (2005). CtBP3/BARS drives membrane fission in dynamin-independent transport pathways. Nat. Cell Biol. 7 570–580. 10.1038/ncb1260 [DOI] [PubMed] [Google Scholar]
- Bradley R. M., Bloemberg D., Aristizabal Henao J. J., Hashemi A., Mitchell A. S., Fajardo V. A.et al. (2018). Lpaatδ/Agpat4 deficiency impairs maximal force contractility in soleus and alters fibre type in extensor digitorum longus muscle. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1863 700–711. 10.1016/j.bbalip.2018.04.001 [DOI] [PubMed] [Google Scholar]
- Bradley R. M., Duncan R. E. (2018). The lysophosphatidic acid acyltransferases (acylglycerophosphate acyltransferases) family: one reaction, five enzymes, many roles. Curr. Opin. Lipidol. 29 110–115. 10.1097/MOL.0000000000000492 [DOI] [PubMed] [Google Scholar]
- Bradley R. M., Mardian E. B., Bloemberg D., Aristizabal Henao J. J., Mitchell A. S., Marvyn P. M., et al. (2017). Mice Deficient in lysophosphatidic acid acyltransferase delta (Lpaatdelta)/acylglycerophosphate acyltransferase 4 (Agpat4) have impaired learning and memory. Mol. Cell. Biol. 37:e00245-17. 10.1128/MCB.00245-17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley R. M., Mardian E. B., Marvyn P. M., Vasefi M. S., Beazely M. A., Mielke J. G., et al. (2016). Data on acylglycerophosphate acyltransferase 4 (AGPAT4) during murine embryogenesis and in embryo-derived cultured primary neurons and glia. Data Brief 6 28–32. 10.1016/j.dib.2015.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley R. M., Marvyn P. M., Aristizabal Henao J. J., Mardian E. B., George S., Aucoin M. G., et al. (2015). Acylglycerophosphate acyltransferase 4 (AGPAT4) is a mitochondrial lysophosphatidic acid acyltransferase that regulates brain phosphatidylcholine, phosphatidylethanolamine, and phosphatidylinositol levels. Biochim. Biophys. Acta 1851 1566–1576. 10.1016/j.bbalip.2015.09.005 [DOI] [PubMed] [Google Scholar]
- Bright J. N., Sansom M. S. P. (2003). The flexing/twirling helix:? exploring the flexibility about molecular hinges formed by proline and glycine motifs in transmembrane helices. J. Phys. Chem. B 107 627–636. 10.1021/jp026686u [DOI] [Google Scholar]
- Calderon F., Kim H. Y. (2004). Docosahexaenoic acid promotes neurite growth in hippocampal neurons. J. Neurochem. 90 979–988. 10.1111/j.1471-4159.2004.02520.x [DOI] [PubMed] [Google Scholar]
- Chang C. C. Y., Sun J., Chang T.-Y. (2011). Membrane-bound O-acyltransferases (MBOATs). Front. Biol. 6:177 10.1007/s11515-011-1149-z [DOI] [Google Scholar]
- Chen S. L., Huang J. Q., Lei Y., Zhang Y. T., Ren X. P., Chen Y. N., et al. (2012). Identification and characterization of a gene encoding a putative lysophosphatidyl acyltransferase from Arachis hypogaea. J. Biosci. 371029–1039. 10.1007/s12038-012-9277-4 [DOI] [PubMed] [Google Scholar]
- Chernomordik L. V., Kozlov M. M. (2003). Protein-lipid interplay in fusion and fission of biological membranes. Annu. Rev. Biochem. 72 175–207. 10.1146/annurev.biochem.72.121801.161504 [DOI] [PubMed] [Google Scholar]
- Colanzi A., Corda D. (2007). Mitosis controls the Golgi and the Golgi controls mitosis. Curr. Opin. Cell Biol. 19 386–393. 10.1016/j.ceb.2007.06.002 [DOI] [PubMed] [Google Scholar]
- Colanzi A., Grimaldi G., Catara G., Valente C., Cericola C., Liberali P., et al. (2013). Molecular mechanism and functional role of brefeldin A-mediated ADP-ribosylation of CtBP1/BARS. Proc. Natl. Acad. Sci. U.S.A. 110 9794–9799. 10.1073/pnas.1222413110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colanzi A., Hidalgo Carcedo C., Persico A., Cericola C., Turacchio G., Bonazzi M., et al. (2007). The Golgi mitotic checkpoint is controlled by BARS-dependent fission of the Golgi ribbon into separate stacks in G2. EMBO J. 26 2465–2476. 10.1038/sj.emboj.7601686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman R. A., Mashek D. G. (2011). Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem. Rev. 111 6359–6386. 10.1021/cr100404w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo S., Longhi R., Alcaro S., Ortuso F., Sprocati T., Flora A., et al. (2005). N-myristoylation determines dual targeting of mammalian NADH-cytochrome b5 reductase to ER and mitochondrial outer membranes by a mechanism of kinetic partitioning. J. Cell Biol. 168 735–745. 10.1083/jcb.200407082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corda D., Colanzi A., Luini A. (2006). The multiple activities of CtBP/BARS proteins: the Golgi view. Trends Cell Biol. 16 167–173. 10.1016/j.tcb.2006.01.007 [DOI] [PubMed] [Google Scholar]
- Corda D., Iurisci C., Berrie C. P. (2002). Biological activities and metabolism of the lysophosphoinositides and glycerophosphoinositols. Biochim. Biophys. Acta 1582 52–69. 10.1016/s1388-1981(02)00137-3 [DOI] [PubMed] [Google Scholar]
- Cordes F. S., Bright J. N., Sansom M. S. (2002). Proline-induced distortions of transmembrane helices. J. Mol. Biol. 323 951–960. 10.1016/s0022-2836(02)01006-9 [DOI] [PubMed] [Google Scholar]
- Cortes V. A., Curtis D. E., Sukumaran S., Shao X., Parameswara V., Rashid S., et al. (2009). Molecular mechanisms of hepatic steatosis and insulin resistance in the AGPAT2-deficient mouse model of congenital generalized lipodystrophy. Cell Metab. 9 165–176. 10.1016/j.cmet.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullis P. R., De Kruijff B. (1979). Lipid polymorphism and the functional roles of lipids in biological membranes. Biochim. Biophys. Acta 559 399–420. 10.1016/0304-4157(79)90012-1 [DOI] [PubMed] [Google Scholar]
- Danne L., Aktas M., Unger A., Linke W. A., Erdmann R., Narberhaus F. (2017). Membrane remodeling by a bacterial phospholipid-methylating enzyme. mBio 8:e02082-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delille H. K., Schrader M. (2008). Targeting of hFis1 to peroxisomes is mediated by Pex19p. J. Biol. Chem. 283 31107–31115. 10.1074/jbc.M803332200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobson L., Remenyi I., Tusnady G. E. (2015). CCTOP: a Consensus Constrained TOPology prediction web server. Nucleic Acids Res. 43W408–W412. 10.1093/nar/gkv451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto M., Shindou H., Shimizu T. (2014). A novel lysophosphatidic acid acyltransferase enzyme (LPAAT4) with a possible role for incorporating docosahexaenoic acid into brain glycerophospholipids. Biochem. Biophys. Res. Commun. 443 718–724. 10.1016/j.bbrc.2013.12.043 [DOI] [PubMed] [Google Scholar]
- Foster D. A., Salloum D., Menon D., Frias M. A. (2014). Phospholipase D and the maintenance of phosphatidic acid levels for regulation of mammalian target of rapamycin (mTOR). J. Biol. Chem. 289 22583–22588. 10.1074/jbc.R114.566091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frohman M. A. (2015). Role of mitochondrial lipids in guiding fission and fusion. J. Mol. Med. 93 263–269. 10.1007/s00109-014-1237-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M., Kazlauskaite R., Baracho Mde F., Santos M. G., Brandao-Neto J., Villares S., et al. (2004). Mutations in Gng3lg and AGPAT2 in Berardinelli-Seip congenital lipodystrophy and Brunzell syndrome: phenotype variability suggests important modifier effects. J. Clin. Endocrinol. Metab. 89 2916–2922. 10.1210/jc.2003-030485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallop J. L., Butler P. J., Mcmahon H. T. (2005). Endophilin and CtBP/BARS are not acyl transferases in endocytosis or Golgi fission. Nature 438 675–678. 10.1038/nature04136 [DOI] [PubMed] [Google Scholar]
- Gopaldass N., Fauvet B., Lashuel H., Roux A., Mayer A. (2017). Membrane scission driven by the PROPPIN Atg18. EMBO J. 36 3274–3291. 10.15252/embj.201796859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M. W. (2012). Mitochondrial evolution. Cold Spring Harb. Perspect. Biol. 4:a011403. 10.1101/cshperspect.a011403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha K. D., Clarke B. A., Brown W. J. (2012). Regulation of the Golgi complex by phospholipid remodeling enzymes. Biochim. Biophys. Acta 1821 1078–1088. 10.1016/j.bbalip.2012.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi A., Razzaghy-Azar M., Talea A., Sadeghian M., Ellard S., Haghighi A. (2012). Identification of a novel nonsense mutation and a missense substitution in the AGPAT2 gene causing congenital generalized lipodystrophy type 1. Eur. J. Med. Genet. 55 620–624. 10.1016/j.ejmg.2012.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall S. E., Roberts K., Vaidehi N. (2009). Position of helical kinks in membrane protein crystal structures and the accuracy of computational prediction. J. Mol. Graph. Model. 27 944–950. 10.1016/j.jmgm.2009.02.004 [DOI] [PubMed] [Google Scholar]
- Haque W., Garg A., Agarwal A. K. (2005). Enzymatic activity of naturally occurring 1-acylglycerol-3-phosphate-O-acyltransferase 2 mutants associated with congenital generalized lipodystrophy. Biochem. Biophys. Res. Commun. 327 446–453. 10.1016/j.bbrc.2004.12.024 [DOI] [PubMed] [Google Scholar]
- Hidalgo Carcedo C., Bonazzi M., Spano S., Turacchio G., Colanzi A., Luini A., et al. (2004). Mitotic Golgi partitioning is driven by the membrane-fissioning protein CtBP3/BARS. Science 305 93–96. 10.1126/science.1097775 [DOI] [PubMed] [Google Scholar]
- Kameoka S., Adachi Y., Okamoto K., Iijima M., Sesaki H. (2018). Phosphatidic acid and cardiolipin coordinate mitochondrial dynamics. Trends Cell Biol. 28 67–76. 10.1016/j.tcb.2017.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassas N., Tanguy E., Thahouly T., Fouillen L., Heintz D., Chasserot-Golaz S., et al. (2017). Comparative characterization of phosphatidic acid sensors and their localization during frustrated phagocytosis. J. Biol. Chem. 292 4266–4279. 10.1074/jbc.M116.742346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennedy E. P., Weiss S. B. (1956). The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 222 193–214. [PubMed] [Google Scholar]
- Koeberle A., Shindou H., Harayama T., Shimizu T. (2010). Role of lysophosphatidic acid acyltransferase 3 for the supply of highly polyunsaturated fatty acids in TM4 Sertoli cells. FASEB J. 24 4929–4938. 10.1096/fj.10-162818 [DOI] [PubMed] [Google Scholar]
- Koeberle A., Shindou H., Harayama T., Yuki K., Shimizu T. (2012). Polyunsaturated fatty acids are incorporated into maturating male mouse germ cells by lysophosphatidic acid acyltransferase 3. FASEB J. 26 169–180. 10.1096/fj.11-184879 [DOI] [PubMed] [Google Scholar]
- Kooijman E. E., Burger K. N. (2009). Biophysics and function of phosphatidic acid: a molecular perspective. Biochim. Biophys. Acta 1791 881–888. 10.1016/j.bbalip.2009.04.001 [DOI] [PubMed] [Google Scholar]
- Kooijman E. E., Carter K. M., Van Laar E. G., Chupin V., Burger K. N., De Kruijff B. (2005a). What makes the bioactive lipids phosphatidic acid and lysophosphatidic acid so special? Biochemistry 44 17007–17015. 10.1021/bi0518794 [DOI] [PubMed] [Google Scholar]
- Kooijman E. E., Chupin V., Fuller N. L., Kozlov M. M., De Kruijff B., Burger K. N., et al. (2005b). Spontaneous curvature of phosphatidic acid and lysophosphatidic acid. Biochemistry 44 2097–2102. 10.1021/bi0478502 [DOI] [PubMed] [Google Scholar]
- Kooijman E. E., Chupin V., De Kruijff B., Burger K. N. (2003). Modulation of membrane curvature by phosphatidic acid and lysophosphatidic acid. Traffic 4 162–174. 10.1034/j.1600-0854.2003.00086.x [DOI] [PubMed] [Google Scholar]
- Kooijman E. E., Tieleman D. P., Testerink C., Munnik T., Rijkers D. T., Burger K. N., et al. (2007). An electrostatic/hydrogen bond switch as the basis for the specific interaction of phosphatidic acid with proteins. J. Biol. Chem. 282 11356–11364. 10.1074/jbc.m609737200 [DOI] [PubMed] [Google Scholar]
- Korbes A. P., Kulcheski F. R., Margis R., Margis-Pinheiro M., Turchetto-Zolet A. C. (2016). Molecular evolution of the lysophosphatidic acid acyltransferase (LPAAT) gene family. Mol. Phylogenet. Evol. 96 55–69. 10.1016/j.ympev.2015.12.001 [DOI] [PubMed] [Google Scholar]
- Kumar V., Carlson J. E., Ohgi K. A., Edwards T. A., Rose D. W., Escalante C. R., et al. (2002). Transcription corepressor CtBP is an NAD(+)-regulated dehydrogenase. Mol. Cell 10 857–869. 10.1016/s1097-2765(02)00650-0 [DOI] [PubMed] [Google Scholar]
- Kume K., Shimizu T. (1997). cDNA cloning and expression of murine 1-acyl-sn-glycerol-3-phosphate acyltransferase. Biochem. Biophys. Res. Commun. 237 663–666. 10.1006/bbrc.1997.7214 [DOI] [PubMed] [Google Scholar]
- Kuroda R., Ikenoue T., Honsho M., Tsujimoto S., Mitoma J. Y., Ito A. (1998). Charged amino acids at the carboxyl-terminal portions determine the intracellular locations of two isoforms of cytochrome b5. J. Biol. Chem. 273 31097–31102. 10.1074/jbc.273.47.31097 [DOI] [PubMed] [Google Scholar]
- Lee A. G. (2004). How lipids affect the activities of integral membrane proteins. Biochim. Biophys. Acta 1666 62–87. 10.1016/j.bbamem.2004.05.012 [DOI] [PubMed] [Google Scholar]
- Liberali P., Kakkonen E., Turacchio G., Valente C., Spaar A., Perinetti G., et al. (2008). The closure of Pak1-dependent macropinosomes requires the phosphorylation of CtBP1/BARS. EMBO J. 27 970–981. 10.1038/emboj.2008.59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B., Jiang Y. J., Zhou Y., Xu F. Y., Hatch G. M., Choy P. C. (2005). Cloning and characterization of murine 1-acyl-sn-glycerol 3-phosphate acyltransferases and their regulation by PPARalpha in murine heart. Biochem. J. 385 469–477. 10.1042/bj20041348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magre J., Delepine M., Van Maldergem L., Robert J. J., Maassen J. A., Meier M., et al. (2003). Prevalence of mutations in AGPAT2 among human lipodystrophies. Diabetes 52 1573–1578. 10.2337/diabetes.52.6.1573 [DOI] [PubMed] [Google Scholar]
- Marchi S., Patergnani S., Pinton P. (2014). The endoplasmic reticulum-mitochondria connection: one touch, multiple functions. Biochim. Biophys. Acta 1837 461–469. 10.1016/j.bbabio.2013.10.015 [DOI] [PubMed] [Google Scholar]
- Mardian E. B., Bradley R. M., Aristizabal Henao J. J., Marvyn P. M., Moes K. A., Bombardier E., et al. (2017). Agpat4/Lpaatdelta deficiency highlights the molecular heterogeneity of epididymal and perirenal white adipose depots. J. Lipid Res. 58 2037–2050. 10.1194/jlr.M079152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxeiner S., Luo F., Tan A., Schmitz F., Sudhof T. C. (2016). How to make a synaptic ribbon: RIBEYE deletion abolishes ribbons in retinal synapses and disrupts neurotransmitter release. EMBO J. 35 1098–1114. 10.15252/embj.201592701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcfie P. J., Banman S. L., Kary S., Stone S. J. (2011). Murine diacylglycerol acyltransferase-2 (DGAT2) can catalyze triacylglycerol synthesis and promote lipid droplet formation independent of its localization to the endoplasmic reticulum. J. Biol. Chem. 286 28235–28246. 10.1074/jbc.M111.256008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mcfie P. J., Banman S. L., Stone S. J. (2018). Diacylglycerol acyltransferase-2 contains a c-terminal sequence that interacts with lipid droplets. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1863 1068–1081. 10.1016/j.bbalip.2018.06.008 [DOI] [PubMed] [Google Scholar]
- Mcfie P. J., Jin Y., Banman S. L., Beauchamp E., Berthiaume L. G., Stone S. J. (2014). Characterization of the interaction of diacylglycerol acyltransferase-2 with the endoplasmic reticulum and lipid droplets. Biochim. Biophys. Acta 1841 1318–1328. 10.1016/j.bbalip.2014.06.004 [DOI] [PubMed] [Google Scholar]
- Nardini M., Spano S., Cericola C., Pesce A., Massaro A., Millo E., et al. (2003). CtBP/BARS: a dual-function protein involved in transcription co-repression and Golgi membrane fission. EMBO J. 22 3122–3130. 10.1093/emboj/cdg283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardini M., Valente C., Ricagno S., Luini A., Corda D., Bolognesi M. (2009). CtBP1/BARS Gly172–>Glu mutant structure: impairing NAD(H)-binding and dimerization. Biochem. Biophys. Res. Commun. 381 70–74. 10.1016/j.bbrc.2009.02.010 [DOI] [PubMed] [Google Scholar]
- Opekarova M., Tanner W. (2003). Specific lipid requirements of membrane proteins–a putative bottleneck in heterologous expression. Biochim. Biophys. Acta 1610 11–22. 10.1016/s0005-2736(02)00708-3 [DOI] [PubMed] [Google Scholar]
- Pagac M., De La Mora H. V., Duperrex C., Roubaty C., Vionnet C., Conzelmann A. (2011). Topology of 1-acyl-sn-glycerol-3-phosphate acyltransferases SLC1 and ALE1 and related membrane-bound O-acyltransferases (MBOATs) of Saccharomyces cerevisiae. J. Biol. Chem. 286 36438–36447. 10.1074/jbc.M111.256511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuso A., Valente C., Giordano L. L., Filograna A., Li G., Circolo D., et al. (2016). Golgi membrane fission requires the CtBP1-S/BARS-induced activation of lysophosphatidic acid acyltransferase delta. Nat. Commun. 7:12148. 10.1038/ncomms12148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfanner N., Orci L., Glick B. S., Amherdt M., Arden S. R., Malhotra V., et al. (1989). Fatty acyl-coenzyme A is required for budding of transport vesicles from Golgi cisternae. Cell 59 95–102. 10.1016/0092-8674(89)90872-6 [DOI] [PubMed] [Google Scholar]
- Prasad S. S., Garg A., Agarwal A. K. (2011). Enzymatic activities of the human AGPAT isoform 3 and isoform 5: localization of AGPAT5 to mitochondria. J. Lipid Res. 52 451–462. 10.1194/jlr.M007575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raben D. M., Barber C. N. (2017). Phosphatidic acid and neurotransmission. Adv. Biol. Regul. 63 15–21. 10.1016/j.jbior.2016.09.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramachandran R. (2018). Mitochondrial dynamics: the dynamin superfamily and execution by collusion. Semin. Cell Dev. Biol. 76 201–212. 10.1016/j.semcdb.2017.07.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rapaport D. (2003). Finding the right organelle. Targeting signals in mitochondrial outer-membrane proteins. EMBO Rep. 4 948–952. 10.1038/sj.embor.embor937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson R. M., Yao J., Gajewski S., Kumar G., Martin E. W., Rock C. O., et al. (2017). A two-helix motif positions the lysophosphatidic acid acyltransferase active site for catalysis within the membrane bilayer. Nat. Struct. Mol. Biol. 24 666–671. 10.1038/nsmb.3436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottig A., Steinbuchel A. (2013). Acyltransferases in bacteria. Microbiol. Mol. Biol. Rev. 77 277–321. 10.1128/MMBR.00010-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom M. S., Weinstein H. (2000). Hinges, swivels and switches: the role of prolines in signalling via transmembrane alpha-helices. Trends Pharmacol. Sci. 21 445–451. 10.1016/s0165-6147(00)01553-4 [DOI] [PubMed] [Google Scholar]
- Schmidt J. A., Brown W. J. (2009). Lysophosphatidic acid acyltransferase 3 regulates Golgi complex structure and function. J. Cell Biol. 186 211–218. 10.1083/jcb.200904147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt J. A., Yvone G. M., Brown W. J. (2010). Membrane topology of human AGPAT3 (LPAAT3). Biochem. Biophys. Res. Commun. 397 661–667. 10.1016/j.bbrc.2010.05.149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz K., Natarajan S., Kassas N., Vitale N., Schmitz F. (2011). The synaptic ribbon is a site of phosphatidic acid generation in ribbon synapses. J. Neurosci. 31 15996–16011. 10.1523/JNEUROSCI.2965-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz K., Schmitz F. (2017). RIBEYE(B)-domain binds to lipid components of synaptic vesicles in an NAD(H)-dependent, redox-sensitive manner. Biochem. J. 474 1205–1220. 10.1042/BCJ20160886 [DOI] [PubMed] [Google Scholar]
- Shemesh T., Luini A., Malhotra V., Burger K. N., Kozlov M. M. (2003). Prefission constriction of Golgi tubular carriers driven by local lipid metabolism: a theoretical model. Biophys. J. 85 3813–3827. 10.1016/s0006-3495(03)74796-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin J. J., Loewen C. J. (2011). Putting the pH into phosphatidic acid signaling. BMC Biol. 9:85. 10.1186/1741-7007-9-85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindou H., Hishikawa D., Harayama T., Yuki K., Shimizu T. (2009). Recent progress on acyl CoA: lysophospholipid acyltransferase research. J. Lipid Res. 50(Suppl.), S46–S51. 10.1194/jlr.R800035-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sievers F., Wilm A., Dineen D., Gibson T. J., Karplus K., Li W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S., Mittal A. (2016). Transmembrane domain lengths serve as signatures of organismal complexity and viral transport mechanisms. Sci. Rep. 6:22352. 10.1038/srep22352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spano S., Silletta M. G., Colanzi A., Alberti S., Fiucci G., Valente C., et al. (1999). Molecular cloning and functional characterization of brefeldin A-ADP-ribosylated substrate. A novel protein involved in the maintenance of the Golgi structure. J. Biol. Chem. 274 17705–17710. 10.1074/jbc.274.25.17705 [DOI] [PubMed] [Google Scholar]
- Stace C. L., Ktistakis N. T. (2006). Phosphatidic acid- and phosphatidylserine-binding proteins. Biochim. Biophys. Acta 1761 913–926. 10.1016/j.bbalip.2006.03.006 [DOI] [PubMed] [Google Scholar]
- Stone S. J., Levin M. C., Zhou P., Han J., Walther T. C., et al. (2009). The endoplasmic reticulum enzyme DGAT2 is found in mitochondria-associated membranes and has a mitochondrial targeting signal that promotes its association with mitochondria. J. Biol. Chem. 284 5352–5361. 10.1074/jbc.m805768200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subauste A. R., Das A. K., Li X., Elliott B. G., Evans C., El Azzouny M., et al. (2012). Alterations in lipid signaling underlie lipodystrophy secondary to AGPAT2 mutations. Diabetes 61 2922–2931. 10.2337/db12-0004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi K., Reue K. (2009). Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am. J. Physiol. Endocrinol. Metab. 296 E1195–E1209. 10.1152/ajpendo.90958.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamir S., Zuris J. A., Agranat L., Lipper C. H., Conlan A. R., Michaeli D., et al. (2013). Nutrient-deprivation autophagy factor-1 (NAF-1): biochemical properties of a novel cellular target for anti-diabetic drugs. PLoS One 8:e61202. 10.1371/journal.pone.0061202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanguy E., Wang Q., Moine H., Vitale N. (2019). Phosphatidic acid: from pleiotropic functions to neuronal pathology. Front. Cell. Neurosci. 13:2. 10.3389/fncel.2019.00002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tusnady G. E., Simon I. (1998). Principles governing amino acid composition of integral membrane proteins: application to topology prediction. J. Mol. Biol. 283 489–506. 10.1006/jmbi.1998.2107 [DOI] [PubMed] [Google Scholar]
- Tusnady G. E., Simon I. (2001). The HMMTOP transmembrane topology prediction server. Bioinformatics 17 849–850. 10.1093/bioinformatics/17.9.849 [DOI] [PubMed] [Google Scholar]
- Valente C., Luini A., Corda D. (2013). Components of the CtBP1/BARS-dependent fission machinery. Histochem. Cell Biol. 140 407–421. 10.1007/s00418-013-1138-1 [DOI] [PubMed] [Google Scholar]
- Valente C., Spano S., Luini A., Corda D. (2005). Purification and functional properties of the membrane fissioning protein CtBP3/BARS. Methods Enzymol. 404 296–316. 10.1016/s0076-6879(05)04027-9 [DOI] [PubMed] [Google Scholar]
- Valente C., Turacchio G., Mariggio S., Pagliuso A., Gaibisso R., Di Tullio G., et al. (2012). A 14-3-3gamma dimer-based scaffold bridges CtBP1-S/BARS to PI(4)KIIIbeta to regulate post-Golgi carrier formation. Nat. Cell Biol. 14 343–354. 10.1038/ncb2445 [DOI] [PubMed] [Google Scholar]
- Vodicka P., Mo S., Tousley A., Green K. M., Sapp E., Iuliano M., et al. (2015). Mass spectrometry analysis of wild-type and knock-in Q140/Q140 Huntington’s disease mouse brains reveals changes in glycerophospholipids including alterations in phosphatidic acid and lyso-phosphatidic acid. J. Huntingtons Dis. 4 187–201. 10.3233/JHD-150149 [DOI] [PubMed] [Google Scholar]
- Weigert R., Silletta M. G., Spano S., Turacchio G., Cericola C., Colanzi A., et al. (1999). CtBP/BARS induces fission of Golgi membranes by acylating lysophosphatidic acid. Nature 402 429–433. 10.1038/46587 [DOI] [PubMed] [Google Scholar]
- Yamashita A., Hayashi Y., Matsumoto N., Nemoto-Sasaki Y., Oka S., Tanikawa T., et al. (2014). Glycerophosphate/Acylglycerophosphate acyltransferases. Biology 3 801–830. 10.3390/biology3040801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita A., Nakanishi H., Suzuki H., Kamata R., Tanaka K., Waku K., et al. (2007). Topology of acyltransferase motifs and substrate specificity and accessibility in 1-acyl-sn-glycero-3-phosphate acyltransferase 1. Biochim. Biophys. Acta 1771 1202–1215. 10.1016/j.bbalip.2007.07.002 [DOI] [PubMed] [Google Scholar]
- Yang J. S., Gad H., Lee S. Y., Mironov A., Zhang L., Beznoussenko G. V., et al. (2008). A role for phosphatidic acid in COPI vesicle fission yields insights into Golgi maintenance. Nat. Cell Biol. 10 1146–1153. 10.1038/ncb1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. S., Lee S. Y., Spano S., Gad H., Zhang L., Nie Z., et al. (2005). A role for BARS at the fission step of COPI vesicle formation from Golgi membrane. EMBO J. 24 4133–4143. 10.1038/sj.emboj.7600873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. S., Valente C., Polishchuk R. S., Turacchio G., Layre E., Moody D. B., et al. (2011). COPI acts in both vesicular and tubular transport. Nat. Cell Biol. 13 996–1003. 10.1038/ncb2273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yogev O., Pines O. (2011). Dual targeting of mitochondrial proteins: mechanism, regulation and function. Biochim. Biophys. Acta 1808 1012–1020. 10.1016/j.bbamem.2010.07.004 [DOI] [PubMed] [Google Scholar]
- Young B. P., Shin J. J., Orij R., Chao J. T., Li S. C., Guan X. L., et al. (2010). Phosphatidic acid is a pH biosensor that links membrane biogenesis to metabolism. Science 329 1085–1088. 10.1126/science.1191026 [DOI] [PubMed] [Google Scholar]
- Yuki K., Shindou H., Hishikawa D., Shimizu T. (2009). Characterization of mouse lysophosphatidic acid acyltransferase 3: an enzyme with dual functions in the testis. J. Lipid Res. 50 860–869. 10.1194/jlr.M800468-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zegarlinska J., Piascik M., Sikorski A. F., Czogalla A. (2018). Phosphatidic acid - a simple phospholipid with multiple faces. Acta Biochim. Pol. 65 163–171. 10.18388/abp.2018_2592 [DOI] [PubMed] [Google Scholar]
- Zhukovsky M. A., Lee P. H., Ott A., Helms V. (2013). Putative cholesterol-binding sites in human immunodeficiency virus (HIV) coreceptors CXCR4 and CCR5. Proteins 81 555–567. 10.1002/prot.24211 [DOI] [PubMed] [Google Scholar]
- Zinser E., Sperka-Gottlieb C. D., Fasch E. V., Kohlwein S. D., Paltauf F., Daum G. (1991). Phospholipid synthesis and lipid composition of subcellular membranes in the unicellular eukaryote Saccharomyces cerevisiae. J. Bacteriol. 173 2026–2034. 10.1128/jb.173.6.2026-2034.1991 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.