

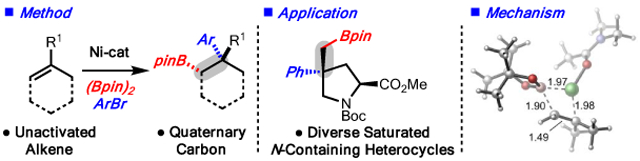
Abstract
A method for the Ni-catalyzed arylboration of unactivated monosubstituted, 1,1-disubstituted and trisubstituted alkenes is disclosed. The reaction is notable in that it converts highly substituted alkenes, arylbromides and diboron reagents to products that contain a quaternary carbon and a synthetically versatile carbon-boron bond with control of stereoselectivity and regioselectivity. In addition, the method is demonstrated to be useful for the synthesis of saturated nitrogen heterocycles, which are important motifs in pharmaceutical compounds. Finally, due to the unusual reactivity demonstrated, the mechanistic details of the reaction were studied with both computational and experimental techniques.
Graphical Abstract
INTRODUCTION:
The use of cross coupling reactions to forge C-C bonds has transformed the way in which molecules are constructed.1 These reactions work particularly well for the synthesis of Csp2-Csp2 bonds, but the translation of these methods towards the preparation of Csp3-Csp2 bonds is considerably more challenging due to rapid β-hydride elimination of alkyl-metal intermediates and slower rates of transmetalation.1 Furthermore, the synthesis of quaternary carbons exacerbates the aforementioned challenges.2,3,4,5
An emerging approach towards Csp3-Csp2 cross-coupling is to utilize alkenes as conjunctive reagents.6 This strategy is attractive since the nucleophilic or electrophilic component does not need to be pregenerated, and widely available alkenes are employed. Our lab has been interested in the development of an approach that involves the cross coupling of aryl halides with Csp3-alkyl metal intermediates that are catalytically generated by boryl metalation of alkenes.7,8 These arylboration reactions are valuable as they allow for carbodifunctionalization by stereospecific transformation of the C-B bond to other functional groups.9 Early work from our group and others focused on Pd/Cu, Ni/Cu and Pd-catalyzed arylboration of activated alkenes (Scheme 1A).7,10,11
Scheme 1.

Arylboration of Alkenes
Our group recently disclosed a Ni-catalyzed arylboration on challenging unactivated 1,2-disubstituted alkenes (Scheme 1B).12,13,14 However, arylboration reactions on monosubstituted, 1,1-disubstituted, and trisubstituted alkenes were generally low yielding and poorly selective. Overcoming these challenges in the arylboration of 1,1-disubstituted and trisubstituted alkenes is particularly desirable as it leads to the construction of quaternary carbons. Carboboration of 1,1-disubstituted alkenes has only been demonstrated with select α-alkyl alkenylarenes, whereas reactions of trisubstituted alkenes are not known.15 Additionally, selective 1,2-arylboration of monosubstituted alkenes has not yet been reported. Thus, the ability to carry out arylboration reactions of unactivated alkenes with various substitution patterns remains an unmet challenge.16
Herein we disclose a new method that overcomes the aforementioned challenges and allows for arylboration of unactivated monosubstituted, 1,1-disubstituted and trisubstituted alkenes (Scheme 1C). In the latter classes of substrates, the reaction involves stereospecific cross coupling of an in situ-generated, stereodefined, tertiary alkyl-[Ni] complex, resulting in vicinal alkene difunctionalization that displays complete stereochemical control. Moreover, the method outlined herein represents a new strategy for the synthesis of a diverse range of saturated nitrogen heterocycles, which are valuable building blocks in pharmaceutical synthesis (Scheme 1D).17 Finally, a detailed analysis of the reaction mechanism is presented, with computational and experimental data that reveals the details of this unique reaction manifold.
RESULTS AND DISCUSSION: SCOPE
A key challenge identified in our initial report was the regioselectivity in the arylboration of terminal alkene substrates. For example, vinylcyclohexane (1) underwent 1,2-arylboration (product 2) in moderate yield and with concomitant formation of 1,1-arylboration adduct 3 (Scheme 2). Based on preliminary mechanistic studies outlined in the initial report,12 the 1,2-arylboration product 2 likely arises via addition of a [Ni]-Bpin complex to the alkene, followed by capture of the subsequent alkyl-[Ni]-intermediate 4 with arylbromide. The formation of the 1,1-arylboration adduct 3 presumably arises by β-hydride elimination of the alkyl-[Ni]-intermediate followed by reinsertion and ArBr capture via 5.18
Scheme 2.

Initial Investigations
To mitigate the formation of the 1,1-arylboration adduct 3, ligands that occupy coordination sites and prevent the β-agostic interaction necessary for β-hydride elimination were investigated. However, traditional ligands such as amines or phosphines inhibited the reaction. Since THF is likely coordinated to Ni during catalysis, other oxygen-derived Lewis basic additives were evaluated (Scheme 3). Early screens identified that amide additives suppressed the formation of the 1,1-arylboration product. Ultimately, 20 equiv of dimethylacetamide (DMA) was found to be optimal, both for suppressing formation of 1,1-arylboration product 3 and enhancing the yield of the desired 1,2-arylboration product 2. Other related amides were inferior to DMA, and amides capable of bidentate coordination inhibited the reaction. Use of less than 20 equiv of DMA was slightly less effective, while increased quantities of DMA provided no additional benefit. A 4:1 mixture of THF:DMA, or approximately 20 equiv of DMA, was identified as optimal for reactivity and simple reaction setup.
Scheme 3.

Evaluation of Lewis Basic Additives
Application of the new conditions to a range of monosubstituted alkenes resulted in formation of the 1,2-arylboration products in good yields and suppressed 1,1-arylboration (Scheme 4).19 Several points are noteworthy: 1) reaction of branched alkenes (products 2, 6-8, 13) resulted in high regioselectivity (>10:1 rr). In particular, sterically demanding t-Bu-ethylene (product 7) can participate in the reaction; however, in this case the 1,1-arylboration isomer formed in 11% yield. The increase in the amount of 1,1-arylboration in this case is likely due to a reduced rate of reaction of the alkyl-[Ni]-complex (analogous to 4) with the arylbromide, which allows for β-hydride elimination to compete. 2) Reaction with non-branched alkenes led to product formation with good yield but modest regioselectivity (products 9-12). 3) Reaction of allylbenzene formed only 1,2-arylboration product 12, despite the potential for facile β-hydride elimination with the benzylic hydrogens.16 4) Ringopening products resulting from reaction of vinylcyclopropane were only formed in 3% yield (product 8). 5) In several cases, 3.0 equiv PhBr (vs. 1.5 equiv PhBr) were necessary to obtain high yields, presumably resulting in faster trapping of the alkyl-Ni-complex and outcompeting β-hydride elimination or other decomposition reactions.
Scheme 4.

Reaction with Monosubstituted Alkenes
Given the remarkable effect of DMA on reactivity and selectivity, we desired to explore the reactivity of other classes of challenging alkenes. Selective 1,2-arylboration of 1,1-disubstituted alkenes was particularly difficult under previously reported conditions12 due to low yield and formation of the 1,1-arylboration regioisomer.20 For these reactions to proceed, a sterically encumbered tertiary alkyl-[Ni]-complex must be generated and then undergo cross coupling with an ArBr faster than β-hydride elimination. Under the new conditions with DMA, alkenes with a variety of substitution patterns functioned well in the arylboration reaction (Scheme 5). Proximal functionalities, such as an ester (product 15), acetal (product 18), and Boc-protected amine (products 19-20) functioned smoothly in the reaction. Sterically demanding substrates underwent the arylboration, albeit in slightly diminished yields (product 16-17). Symmetric trisubstituted alkenes also perform well in the reaction and generate a single regioisomer in every case (products 20, 22-23).
Scheme 5.

Reaction with Various Alkenes
One of the most significant aspects of this study is the reaction of asymmetric trisubstituted alkenes, due to the opportunity for stereocontrol (Scheme 5). Reaction of Z- and E-alkenes led to the formation of different diastereomers resulting from a syn-arylboration in each case (products 24 and 25, respectively). This process has also been extended to reaction of a cycloalkene to generate 26. In all cases the reaction likely proceeds via the formation of stereodefined tertiary alkyl-[Ni]-complexes 27-29. These Ni-complexes do not appear to undergo epimerization or β-hydride elimination, thus highlighting their remarkable stability and reactivity.
A wide range of arylbromides was evaluated with alkene 30 (Scheme 6). In all cases, the reaction afforded a single observable regioisomer, and the undesired 1,1-arylboration product was not detected (with the exception of product 40). Notably, aryl bromides bearing electron-donating (product 33), electron-withdrawing (products 32, 34-37), and sterically demanding substituents (product 40) function smoothly. In addition, functional groups such as an ester (product 35), acetal (product 43), primary alcohol (product 39), tertiary amine, (product 42), amide (products 41, 44), and select heterocycles (products 45-47)21 are tolerated. Finally, reaction with an alkenylbromide demonstrated that alkenylboration is a viable process (product 38).
Scheme 6.

Reaction with Various (Het)Arylbromides
Substrates in which the arylbromide and alkene units are tethered were also examined (Scheme 7). In both cases (substrates 48 and 50), the reaction formed products 49 and 51, respectively, in accordance with formation of the more substituted alkyl-[Ni]-complex from borylnickelation. Furthermore, the formation of 51 in 20:1 dr (consistent with the starting alkene geometry isomeric ratio) represents the synthesis of a N-containing heterocycle.
Scheme 7.

Intramolecular Variants
Saturated nitrogen heterocycles, such as piperdine, pyrazine and pyrrolidine are among the top-ten ring systems in FDA approved drugs.17a,b Given the pharmaceutical industry’s recent push to evaluate molecules with increased saturation, development of methods to access these motifs efficiently is highly relevant.17c,d We sought to apply the present arylboration to the synthesis of a diverse range of saturated nitrogen heterocycles from readily available unsaturated nitrogen heterocycles.
Under the optimized conditions, a variety of unsaturated nitrogen heterocycles underwent arylboration (Scheme 8). In the case of pyrrolidines with disubstituted and trisubstituted, endo- and exocyclic alkenes (52, 54,22 58, 60 and 70) function well. In the case of stereoisomeric alkenes 58 and 60, the products are formed from a syn-arylboration pathway. A similar range of piperidine ring systems was tolerated in the reaction (alkenes 62, 64, 66, and 68). In addition, an azetadine-based substrate was shown to function smoothly (alkene 56).
Scheme 8.

Synthesis of Saturated Nitrogen Heterocycles
The ability of the method to function well with chiral non-racemic substrates was also demonstrated with alkene 72 (prepared from commercially available N-Boc hydroxyproline methyl ester in two steps) (Scheme 9). In this case, reaction with bromobenzene delivered product 73 in 72% yield and 18:1 dr without epimerization of the C1 stereocenter. Based on the stereochemistry of the product, borylnickelation occurred from the least hindered face opposite the ester substituent according to the model (74) illustrated in Scheme 9
Scheme 9.

Diastereoselective Alkene Functionalization
The method is also amenable to gram scale synthesis as illustrated in Scheme 10 (products 26 and 76). As a demonstration of the robustness of the reaction conditions, the synthesis of 26 was achieved without the aid of a glovebox using standard Schlenk techniques.
Scheme 10.

Gram Scale Reactions and Further Functionalization
The Bpin unit can easily be converted to other functional groups through various transformations such as oxidation (product 77) and Matteson homologation (product 78) (Scheme 10). The homologation product could also be elaborated by Pd-catalyzed cross coupling to provide 79.23 Thus, through the sequence of arylboration and C-B bond transformation, elaboration of readily available starting materials can deliver diverse saturated nitrogen heterocycles.
RESULTS AND DISCUSSION: MECHANISM
Based on our prior observations, a Ni(I)-Ni(III) catalytic cycle was proposed (Scheme 11).12 While both Ni(II) or Ni(0) pre-catalysts can be used, the formation of Ni(I)-complexes can occur through a comproportionation pathway.24,25 The key step in the Ni(I)-Ni(III) cycle is the formation of a [Ni(I)]-Bpin complex followed by addition across an alkene. The resulting alkyl-[Ni(I)]-complex then undergoes reaction with the arylbromide, perhaps via an oxidative addition/reductive elimination sequence, to generate the product and regenerate [Ni(I)]-Br. The formation of the 1,1-arylboration adduct likely arises from β-hydride elimination of the alkyl-[Ni(I)]-complex followed by reinsertion and capture with the arylbromide.
Scheme 11.

Proposed Catalytic Cycle
Additional data gathered in this study corroborates this mechanistic hypothesis. Key experiments are outlined in Scheme 12. 1) Radical intermediates are unlikely to be involved in the reaction pathway, as the reactions are stereospecific and ring opening of cyclopropane only occurs in minor quantities (Scheme 12A). 2) A pathway in which a bimetallic transmetalation between two different Ni species is unlikely as substrates in which the alkene and arylbromide units are tethered delivered the intramolecular arylboration product in good yield (Scheme 12B).26 For a bimetallic reaction pathway of this type to function on these substrates, two different Ni-centers must react with the same molecule of substrate, which is highly unlikely. Moreover, crossover products that would arise from a bimetallic pathway via the intermediate shown in Scheme 12B were not observed in these intramolecular reactions in the presence of bromobenzene. 3) A linear free energy relationship between relative rate of reaction of various arylbromides and σ was observed (Scheme 3C). Due to the large positive ρ-value observed (2.6), electrophilic capture of the alkyl-Ni-intermediate occurs (either via oxidative addition or σ-bond metathesis).27 4) Determination of 13C KIE at natural abundance revealed small, but statistically significant values for both carbons of the alkene (Scheme 12D).28 The similar values and magnitude of the KIE suggests an early transition state with similar degrees of bond formation at both carbons of the alkene.
Scheme 12.

Mechanistic Investigations
A catalytic cycle involving Ni(0) and Ni(II) intermediates was also considered (Scheme 13A). In this process, Ni(0) would undergo oxidative addition with the arylbromide to generate an Ar-[Ni(II)]-Br complex. Transmetalation with (Bpin)2 would occur to provide Ar-[Ni]-Bpin which upon migratory insertion and reductive elimination would generate the product. The formation of the 1,1-arylboration adduct would arise from a similar β-hydride elimination sequence as shown in Scheme 11.
Scheme 13.
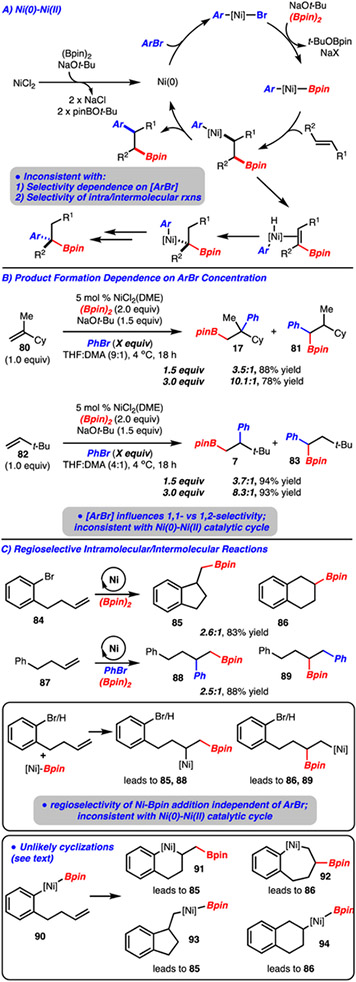
Unlikely Ni(0)-Ni(II) Catalytic Cycle
However, this catalytic cycle is inconsistent with two experiments outlined in Scheme 13. The first involves varying the equivalents of arylbromide (Scheme 13B). In particular, it was observed that the 1,1-arylboration regioisomer (e.g., 81 and 83) could be suppressed with increased quantities of arylbromide. In the Ni(0)-Ni(II) catalytic cycle, the relative rates for the formation of the 1,1- and 1,2-arylboration products should be independent of arylbromide concentration (i.e., arylbromide is only involved in oxidative addition, which precedes the divergence point for formation of the 1,1- and 1,2-arylboration products). In contrast, the data shown in Scheme 13B is consistent with the Ni(I)-Ni(III) catalytic cycle illustrated in Scheme 11, in which increased concentration of arylbromide results in more rapid capture of the alkyl-[Ni]-complex relative to β-hydride elimination and thus results in less 1,1-arylboration product formation. The second experiment examines the regioselectivity of intra vs. intermolecular arylboration reaction (Scheme 13C). For reaction of 84 and 87, similar regioselectivity is observed, suggesting the formation of similar alkyl-[Ni]-intermediates prior to capture with an arylbromide, which is fully consistent with the Ni(I)-Ni(III) catalytic cycle. However, this is inconsistent with the Ni(0)-Ni(II) catalytic cycle in which the regioselectivity of migratory insertion from the Ar-[Ni]-Bpin complex 90 should be influenced by relative rate of ring forming reactions to form 91 and 92 and thus the regioselectivity should be different for reaction of substrates 84 and 87. Furthermore, if the Ni(0)-Ni(II) pathway were operative it would seem likely that 90 would undergo insertion via a well established 5-exo-trig pathway to result in exclusive formation of 85 via 9329 (6-endo-trig cyclization, such as 94, are very rare and only occur with specialized substrates).30 However, since a significant amount of product 86 was observed, [Ni]-Ar migratory insertion is less likely than initial [Ni]-Bpin migratory insertion occurring first, followed by intramolecular trapping of the [Ni]-alkyl species with the arylbromide.
The experimental investigations outlined above have elucidated the overall features of the Ni(I)-Ni(III) catalytic cycle; however, several details remained unclear, specifically: 1) What is the nature of the [Ni]-Bpin and the alkyl-[Ni]-complexes? 2) What is the basis for the regioselectivity of migratory insertion? 3) How does the alkyl-[Ni]-complex undergo reaction with arylbromide? To address these questions, a computational investigation of the reaction was undertaken at the UM06/SDD–6-311+G(d,p)/SMD(THF)//UB3LYP/LANL2DZ–6-31G(d) level of theory.
The reaction between model substrate 2-methylprop-1-ene 95 and bromobenzene to afford product 96 was used as the model reaction in the calculations (Scheme 14). Under the experimental conditions with THF/DMA cosolvents, several possible ligands can potentially bind to the Ni(I)–Bpin, an active species in the catalytic cycle proposed in Scheme 11. These possible structures of the Ni-Bpin complex were probed computationally and a three-coordinate Ni-Bpin bound with a DMA and an alkene (INT-1, Figure 1) was found to be the most favorable structure (see Supporting Information for details). Alkene migratory insertion from INT-1 occurred to generate the tertiary alkyl-Ni-complex INT-2. At this stage the regioselectivity of the reaction is determined, since the calculations indicate the alkene migratory insertion is irreversible. The computed regioselectivity (ΔΔG‡ = 2.7 kcal/mol for TS1 vs. TS5) is consistent with the experimental data (>20:1 regioselectivity) in that the insertion occurs to generate the less stable tertiary-Ni-complex INT-2 as opposed to the more stable primary alkyl-Ni-complex INT-11. The primary factor in controlling the regioselectivity appears to be minimization of an unfavorable steric repulsion between the B-atom and the two methyl groups on the disubstituted alkene 95 in TS5, thus favoring TS1. It should also be noted that the computed geometry of TS1 is consistent with the observed 13C KIE, as it is a synchronous four-membered cyclic transition state with nearly equal bond formation with the two alkene carbons. The insertion step was also determined to be turnover-limiting, with a barrier of ΔG‡ = 7.4 kcal/mol, which is consistent with a reaction that occurs readily at 4 °C.
Scheme 14.

Model Reaction Used in the Computational Studies
Figure 1:

Computed Energy Profile of the Arylboration Reaction.
It was found that alkyl-Ni-intermediate INT-2 is stabilized by a chelating coordination with the neighboring oxygen of the Bpin unit. We speculate that formation of a favorable five-membered chelation and occupying a coordination site are the primary factors in governing the stability of this unusual tertiary alkyl-Ni-complex. Reaction of INT-2 with bromobenzene occurred readily through an oxidative addition/reductive elimination sequence via Ni(III)-complex INT-4, corroborating the observed linear free energy relationship (Scheme 12C). The rapid reductive elimination of this intermediate is a likely explanation for the formation of minimal byproducts in these reactions. Finally, turnover of the catalyst can occur through a series of ligand exchanges and transmetallation assisted by the alkoxide base via TS4. Additionally, the competition experiment between different arylbromides with bromobenzene was investigated computationally (Figure 2) and a good correlation was obtained between the experimental and DFT-computed relative rate data (R2 = 0.873). Overall, the calculated catalytic cycle is wholly consistent with the experimental data.
Figure 2.

Comparison of Predicted and Experimental LFER
CONCLUSIONS
In summary, a strategy for the functionalization of a wide range of alkenes through arylboration is presented. Key to the development of a broadly applicable process was identification of dimethylacetamide as a key additive, which functions as a Lewis base to suppress side reactions. Notably, this method allows for the synthesis of quaternary carbons by cross coupling of readily available alkenes, arylbromides, and diboron reagents. The reactions proceed with high levels of regioselectivity and diastereoselectivity across a broad range of substrates. In addition, the reaction involves a rare cross coupling of a tertiary alkyl-[Ni], with control of stereochemistry in many cases. These advances allowed for development of a new strategy for the synthesis of saturated nitrogen-containing heterocycles, which are synthetically and pharmacologically valuable. Finally, the mechanistic details of this process were evaluated with computational and experimental techniques and provide insight into the details of this unique reaction.
Supplementary Material
ACKNOWLEDGMENT
We thank Indiana University and the NIH (R01GM114443 and R35GM128779) for financial support. This project was partially funded by the Vice Provost for Research through the Research Equipment Fund and the NSF (CHE1726633). We also thank Dr. Kaitlyn M. Logan for informative initial studies and scientific discussion. Dr. Frank Gao’s assistance with acquisition of 13C NMR spectra for KIE analysis is greatly appreciated. We acknowledge supercomputer resources provided by the Center for Research Computing at the University of Pittsburgh and the Extreme Science and Engineering Discovery Environment (XSEDE) supported by the NSF.
Footnotes
Supporting Information
Experimental procedures and analytical data for all compounds. This information is available free of charge via the Internet at http://pubs.acs.org/
The authors declare no competing financial interest.
REFERENCES
- 1.For reviews, see:Jana R; Pathak TP; Sigman MS Advances in Transition Metal (Pd,Ni,Fe)-Catalyzed Cross-Coupling Reactions Using Alkyl-Organometallics as Reaction Partners. Chetn. Rev 2011, 111 (3), 1417–1492.Swift EC; Jarvo ER Asymmetric Transition Metal-Catalyzed Cross-Coupling Reactions for the Construction of Tertiary Stereocenters. Tetrahedron 2013, 69 (29), 5799–5817.Wang C-Y; Derosa J; Biscoe MR Configurationally Stable, Enantioenriched Organometallic Nucleophiles in Stereospecific Pd-Catalyzed Cross-Coupling Reactions: an Alternative Approach to Asymmetric Synthesis. Chem. Sci 2015, 6 (9), 5105–5113.Cherney AH; Kadunce NT; Reisman SE Enantioselective and Enantiospecific Transition-Metal-Catalyzed Cross Coupling Reactions of Organometallic Reagents to Construct C-C Bonds. Chem. Rev 2015, 115 (17), 9587–9652.
- 2.For selected recent examples with tertiary alkyl metals/metaloids, see: Ni-catalysisJoshi-Pangu A; Wang C-Y; Biscoe MR Nickel-Catalyzed Kumada Cross-Coupling Reactions of Tertiary Alkylmagnesium Halides and Aryl Bromides/Triflates. J. Am. Chem. Soc 2011, 133 (22), 8478–8481.Lohre C; Droge T; Wang C; Glorius F Nickel-Catalyzed Cross-Coupling of Aryl Bromides with Tertiary Grignard Reagents Utilizing Donor-Functionalized N-Heterocyclic Carbenes (NHCs). Chem. Enr. J 2011, 17 (22), 6052–6055.Ren P; Stern L-A; Hu X Copper-Catalyzed Cross-Coupling of Functionalized Alkyl Halides and Tosylates with Secondary and Tertiary Alkyl Grignard Reagents. Angew. Chem. Int. Ed 2012, 51 (36), 9110–9113.Ando S; Mawatari M; Matsunaga H; Ishizuka T An N-Heterocyclic Carbene-Based Nickel Catalyst for the Kumada-Tamao-Corriu Coupling of Aryl Bromides and Tertiary Alkyl Grignard Reagents. Tetrahedron Letters 2016, 57 (30), 3287–3290.Primer DN; Molander GA Enabling the Cross-Coupling of Tertiary Organoboron Nucleophiles Through Radical-Mediated Alkyl Transfer. J. Am. Chem. Soc 2017. 139 (29). 9847–9850.
- 3.For selected recent examples with tertiary alkyl electrophiles, see:Zhou Q; Cobb KM; Tan T; Watson MP Stereospecific Cross Couplings to Set Benzylic, All-Carbon Quaternary Stereocenters in High Enantiopurity. J. Am. Chem. Soc 2016, 138 (37), 12057–12060.Ariki ZT; Maekawa Y; Nambo M; Crudden CM Preparation of Quaternary Centers via Nickel-Catalyzed Suzuki-Miyaura Cross-Coupling of Tertiary Sulfones. J. Am. Chem. Soc 2017. 140 (1). 78–81.Wang X; Ma G; Peng Y; Pitsch CE; Moll BJ; Ly TD; Wang X; Gong H Ni-Catalyzed Reductive Coupling of Electron-Rich Aryl Iodides with Tertiary Alkyl Halides. J. Am. Chem. Soc 2018, 140 (43), 14490–14497.Chen T-G; zhang H; Mykhailiuk PK; Merchant RR; Smith CA; Qin T; Baran PS Quaternary Centers by Nickel-Catalyzed Cross-Coupling of Tertiary Carboxylic Acids and (Hetero)Aryl Zinc Reagents. Angew. Chem. Int. Ed 2019, 58 (8), 2454–2458.
- 4.For an alternative approach, see:Bonet A; Odachowski M; Leonori D; Essafi S; Aggarwal VK Enantiospecific sp2–sp3 coupling of secondary and tertiary boronic esters Nat. Chem 2014, 6 584–589.Odachowski M; Bonet A; Essafi S; Conti-Ramsden P; Harvey JN; Leonori D; Aggarwal VK Development of Enantiospecific Coupling of Secondary and Tertiary Boronic Esters with Aromatic Compounds J. Am. Chem. Soc 2016. 138 9521–9532.
- 5.For an alternative approach that involved hydroarylation, see: Green SA; Vásquez-Céspedes S; Shenvi RA Iron–Nickel Dual-Catalysis: a New Engine for Olefin Functionalization and the Formation of Quaternary Centers. J. Am. Chem. Soc 2018, 140 (36), 11317–11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6 (a).Giri R; KC S Strategies toward Dicarbofunctionalization of Unactivated Olefins by Combined Heck Carbometalation and Cross-Coupling J. Org. Chem 2018, 83 3013–3022. [DOI] [PubMed] [Google Scholar]; (b) Derosa J; Tran VT; van der Puyl VA; Engle KM Carbon-carbon π-bonds as Conjunctive Reagents in Cross-couping. Aldrichimica Acta 2018, 51 21–32 [Google Scholar]
- 7 (a).Smith KB; Logan KM; You W; Brown MK Alkene Carboboration Enabled by Synergistic Catalysis. Chem. Enr. J 2014, 20 (38), 12032–12036. [DOI] [PubMed] [Google Scholar]; (b) Logan KM; Smith KB; Brown MK Copper/Palladium Synergistic Catalysis for the Syn- and Anti-Selective Carboboration of Alkenes. Angew. Chem 2015, 127 (17), 5317–5320. [DOI] [PubMed] [Google Scholar]; (c) Logan KM; Brown MK Catalytic Enantioselective Arylboration of Alkenylarenes. Angew. Chem 2016, 129 (3), 869–873. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smith KB; Brown MK Regioselective Arylboration of Isoprene and Its Derivatives by Pd/Cu Cooperative Catalysis. J. Am. Chem. Soc 2017, 139 (23), 7721–7724. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sardini SR; Brown MK Catalyst Controlled Regiodivergent Arylboration of Dienes. J. Am. Chem. Soc 2017, 139 (29), 9823–9826. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Bergmann A; Dorn S; Smith K; Logan K; Brown MK Catalyst-Controlled 1,2- and 1,1-Arylboration of A-Alkyl Alkenylarenes. Angew. Chem. Int. Ed 2018, 1719–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.For reviews, see:Shimizu Y; Kanai M Recent Progress in Copper-Catalyzed Difunctionalization of Unactivated Carbon-carbon Multiple Bonds. Tetrahedron Letters 2014, 55 (28), 3727–3737.Semba K; Fujihara T; Terao J; Tsuji Y Copper-Catalyzed Borylative Transformations of Non-Polar Carbon-Carbon Unsaturated Compounds Employing Borylcopper as an Active Catalyst Species. Tetrahedron 2015, 71 (15), 2183–2197.Lazreg F; Nahra F; Cazin CSJ Copper–NHC Complexes in Catalysis. Coordination Chemistry Reviews 2015, 293-294 48–79.Neeve EC; Geier SJ; Mkhalid IAI; Westcott SA; Marder TB Diboron(4) Compounds: From Structural Curiosity to Synthetic Workhorse. Chem. Rev 2016, 116 (16), 9091–9161.Hemming D; Fritzemeier R; Westcott SA; Santos WL; Steel PG Copper-Boryl Mediated Organic Synthesis. Chem. Soc. Rev 2018, 47(19), 7477–7494.
- 9.Sandford C; Aggarwal VK Stereospecific Functionalizations and Transformations of Secondary and Tertiary Boronic Esters. Chem. Commnn 2017. 53 (40). 5481–5494. [DOI] [PubMed] [Google Scholar]
- 10 (a).Semba K; Nakao Y Arylboration of Alkenes by Cooperative Palladium/Copper Catalysis. J. Am. Chem. Soc 2014, 136 (21), 7567–7570. [DOI] [PubMed] [Google Scholar]; (b) Jia T; Cao P; Wang B; Lou Y; Yin X; Wang M; Liao J A Cu/Pd Cooperative Catalysis for Enantioselective Allylboration of Alkenes. J. Am. Chem. Soc 2015, 137 (43), 13760–13763. [DOI] [PubMed] [Google Scholar]; (c) Semba K; Ohtagaki Y; Nakao Y Arylboration of 1-Arylalkenes by Cooperative Nickel/Copper Catalysis. Org. Lett 2016. 18 (16). 3956–3959. [DOI] [PubMed] [Google Scholar]; (d) Chen B; Cao P; Yin X; Liao Y; Jiang L; Ye J; Wang M; Liao J Modular Synthesis of Enantioenriched 1,1.2-Triarylethanes by an Enantioselective Arylboration and Cross-Coupling Sequence. ACS Catal. 2017, 7 (4). 2425–2429. [Google Scholar]
- 11.For Pd-catalyzed arylboration, see:Yang K; Song Q Palladium-Catalyzed Arylboration of Bicyclic Alkenes. J. Org. Chem 2015, 81 (3), 1000–1005.Yang K; Song Q Pd-Catalyzed Regioselective Arylboration of Vinylarenes. Org. Lett 2016, 18 (21), 5460–5463.Liu Z; Ni H-Q; Zeng T; Engle KM Catalytic Carbo- and Aminoboration of Alkenyl Carbonyl Compounds via Five- and Six-Membered Palladacycles. J. Am. Chem. Soc 2018, 140 (9), 3223–3227.Liu Z; Li X; Zeng T; Engle KM Directed, Palladium(II)-Catalyzed Enantioselective Anti- Carboboration of Alkenyl Carbonyl Compounds. ACS Catal. 2019, 9(4), 3260–3265.
- 12.Logan KM; Sardini SR; White SD; Brown MK Nickel-Catalyzed Stereoselective Arylboration of Unactivated Alkenes. J. Am. Chem. Soc. 2018. 140(1). 159–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.For carboboration of unactivated terminal alkenes, see:Kubota K; Yamamoto E; Ito H Copper(I)-Catalyzed Borylative exo-Cyclization of Alkenyl Halides Containing Unactivated Double Bond J. Am. Chem. Soc. 2013, 135 (7), 2635.Kageyuki I; Yoshida H; Takaki K Three-Component Carboboration of Alkenes under Copper Catalysis Synthesis 2014, 76(14), 1924.Su W; Gong T-J; Lu X; Xu M-Y; Yu C-G; Xu Z-Y; Yu H-Z; Xiao B; Fu Y Ligand-Controlled Regiodivergent Copper-Catalyzed Alkylboration of Alkenes Angew. Chem. Int. Ed 2015. 54 (39). 12957.
- 14.For other Ni-catalyzed borylation reactions, see:Elirano K; Yorimitsu H; Oshima K Nickel-Catalyzed β-Boration of α,β-Unsaturated Esters and Amides with Bis(pinacolato)diboron,. Org. Lett 2007, 9, 5031.Sumida Y; Yorimitsu H; Oshima, Nickel-Catalyzed Borylative Ring-Opening Reaction of Vinylcyclopropanes with Bis(pinacolato)diboron Yielding Allylic Boronates, K. Org. Lett 2008, 10 4677.Cho HY; Morken JP, Diastereoselective Construction of Functionalized Elomoallylic Alcohols by Ni-Catalyzed Diboron-Promoted Coupling of Dienes and Aldehydes, J. Am. Chem. Soc 2008, 130 16140.Crotti S; Bertolini F; Macchia F; Pineschi, Nickel-Catalyzed Borylative Ring Opening of Vinyl Epoxides and Aziridines, M. Org. Lett. 2009, 11 3762.A. S.; Fu GC Nickel-Catalyzed Coupling Reactions of Alkyl Electrophiles, Including Unactivated Tertiary Halides, To Generate Carbon–Boron Bonds, Dudnik, J. Am. Chem. Soc 2012, 134 10693.
- 15 (a).Gong T-J; Yu S-H; Li K; Su W; Lu X; Xiao B; Fu Y Copper-Catalyzed Alkynylboration of Alkenes with Diboron Reagents and Bromoalkynes. Chem. Asian J 2017, 12 (22), 2884–2888.Cheng F; Lu W; Huang W; Wen L; Li M; Meng F Cu-Catalyzed Enantioselective Synthesis of Tertiary Benzylic Copper Complexes and Their in Situ Addition to Carbonyl Compounds. Chem. Sci 2018, 9 (22), 4992–4998.Whyte A; Burton KI; Zhang J; Lautens M Enantioselective Intramolecular Copper-Catalyzed Borylacylation Angew. Chem. Int. Ed 2018. 57(42). 13927.ref 7f
- 16.For a (1,n) arylboration of unactivated monosubstittued alkene, see: Wang W; Ding C; Li Y; Li Z; Li Y; Peng L; Yin G Migratory Arylboration of Unactivated Alkenes Enabled by Nickel Catalysis. Angew. Chem. Int. Ed 2019, 58 (14), 4612–1616. [DOI] [PubMed] [Google Scholar]
- 17 a).Lovering F; Bikker J; Humblet C Escape From Flatland: Increasing Saturation as an Approach to Improving Clinical Success. J. Med. Chem 2009, 52 6752–6756. [DOI] [PubMed] [Google Scholar]; (b) Lovering F Escape From Flatland 2: Complexity and Promiscuity. Med. Chem. Commun 2013, 4 515–515. [Google Scholar]; Taylor RD; MacCoss M; Lawson ADG Rings in Drugs. J. Med. Chem 2014, 57 5845–5859. [DOI] [PubMed] [Google Scholar]; (d) Vitaku E, Smith DT, Njardarson JT, J. Med. Chem 2014, 57 10257–10274. [DOI] [PubMed] [Google Scholar]; (d) Vitaku E; Smith DT; Njardarson JT Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles Among U.S. FDA Approved Pharmaceuticals. J. Med. Chem 2014, 57 10257–10274. [DOI] [PubMed] [Google Scholar]
- 18.If LiOt-Bu is used in place of NaOt-Bu, slightly higher regioselectivity is observed (3:1 rr vs. 1.3:1 rr) from ref 12 and as shown in Scheme 2, respectively). A synergistic combination of NaOt-Bu and DMA is observed and is required to observe 18:1 rr (2:3) as shown in Scheme 3.
- 19.The difference between NMR yields and yields of isolated product is due to, at times, a difficult separation by silica gel column chromatography.
- 20.For reactions of 1,1-disubstiuted and trisubstituted alkenes, it was found that 9:1 THF:DMA was sufficient as opposed to 4:1 THF:DMA that was optimal for monosubstituted alkenes.
- 21.2-Substituted pyridyl bromides are required, likely to mitigate catalyst deactivation by coordination.
- 22.Arylboration reaction of 53 was previous reported and proceeds in similar yield. See ref 12.
- 23 (a).Yang C-T; Zhang Z-Q; Tajuddin H; Wu C-C; Liang J; Liu J-H; Fu Y; Czyzewska M; Steel PG; Marder TB; Liu L Alkylboronic Esters From Copper-Catalyzed Borylation of Primary and Secondary Alkyl Halides and Pseudohalides. Angew. Chem. Int. Ed 2011, 51 (2), 528–532. [DOI] [PubMed] [Google Scholar]; (b) Martin R; Buchwald SL Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling Reactions Employing Dialkylbiaryl Phosphine Ligands. Acc. Chem. Res 2008, 41 (11), 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24 (a).Fujita T; Arita T; Ichitsuka T; Ichikawa J Catalytic Defluorinative [3 + 2] Cycloaddition of Trifluoromethylalkenes with Alkynes via Reduction of Nickel(II) Fluoride Species. Dalton Trans. 2015, 44 (45), 19460–19463. [DOI] [PubMed] [Google Scholar]; (b) For a potential reductive elimination of MeOBpin from aNi(III)-complex, see: Xu H; Zhao C; Qian Q; Deng W; Gong H Nickel-Catalyzed Cross-Coupling of Unactivated Alkyl Halides Using Bis(pinacolato)diboron as Reductant. Chem. Sci 2013, 4 (10), 4022–4029. [Google Scholar]
- 25.For a review regarding Ni(I)-complexes, see: Lin C-Y, Power PP Complexes of Ni(I): a “rare” oxidation state of growing importance, Chem. Soc. Rev 2017. 46 5347–5399. [DOI] [PubMed] [Google Scholar]
- 26.For an example of a bimetallic Transmetalation, see: Yu P; Morandi B, Nickel-Catalyzed Cyanation of Aryl Chlorides and Triflates Using Butyronitrile: Merging Retro-hydrocyanation with Cross-Couping, Angew. Chem. Int. Ed. 2017, 56 15693. [DOI] [PubMed] [Google Scholar]
- 27.Hammett studies regarding oxidative addition of arylbromides with Ni(0) complexes generally describe positive ρ-values; however, studies can be complicated due to ligand exchange processes,Foa M; Cassar L Oxidative Addition of Aryl Halides to Tris(triphenylphosphine)nickel(0). J. Chem. Soc., Dalton Trans, 1975, 2572–2576.Tsou T; Kochi J Mechanism Of Oxidative Addition. Reaction of nickel(0) Complexes With Aromatic Halides. J. Am. Chem. Soc, 1979, 101, 6319–6332.Bajo S; Laidlaw G; Kennedy A; Sproules S; Nelson D Oxidative Addition of Aryl Electrophiles to a Prototypical Nickel(0) Complex: Mechanism and Structure/Reactivity Relationships. Organometallics, 2017, 36, 1662–1672.
- 28.Singleton DA; Thomas AA High-Precision Simultaneous Determination of Multiple Small Kinetic Isotope Effects at Natural Abundance. J. Am. Chem. Soc, 1995. 117, 9357–9358. [Google Scholar]
- 29.For select examples, see:Cong H; Fu GC Catalytic Enantioselective Cyclization/Cross-Coupling with Alkyl Electrophiles. J. Am. Chem. Soc 2014, 136 (10), 3788–3791.Wang K; Ding Z; Zhou Z; Kong W Ni-Catalyzed Enantioselective Reductive Diarylation of Activated Alkenes by Domino Cyclization/Cross-Coupling. J. Am. Chem. Soc 2018, 140 (39), 12364–12368.Jin Y; Wang C Ni-Catalysed Reductive Arylalkylation of Unactivated Alkenes. Chem. Sci 2019, 10 (6), 1780–1785.
- 30.For example, see: Dankwardt JW; Flippin LA Palladium-Mediated 6-Endo-Trig Intramolecular Cyclization of N-Acryloyl-7-Bromoindolines. a Regiochemical Variant of the Intramolecular Heck Reaction. J. Org. Chem 1995, 60 (8), 2312–2313. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.