

SUMMARY
Although elevated levels of reactive oxygen species have been observed in cancer cells and cancer cells aberrantly proliferate, it is not known whether the level of reactive oxygen species and the accumulation of oxidative damage to macromolecules vary across the cell cycle. Here, we measure the prevalence of reactive oxygen species and of biomolecule oxidation across the cell cycle in freely-cycling cancer cells. We report that reactive oxygen species vary during the cell cycle, and peak in mitosis resulting in mitotic accumulation of oxidized protein cysteine residues. Prolonged mitotic arrest further increased the levels of ROS and the abundance of oxidatively damaged biomolecules, including cysteine sulfenic acid-containing proteins and 8-oxoguanine. These finding suggest that mitotic arrest agents may enhance the effects of ROS-dependent anticancer therapies.
Graphical Abstract
eTOC Blurb
We studied connections between ROS and the cell cycle in unsynchronized cancer cells and using multiple independent assays found that ROS and oxidative damage to biomolecules is highest in mitosis and can be further enhanced by mitotic arrest.
INTRODUCTION
Due to the increased metabolic demands of sustained proliferation (Hanahan and Weinberg, 2011; Vander Heiden and DeBerardinis, 2017), cancer cells have elevated production of reactive oxygen species (ROS) relative to non-cancer cells (Szatrowski and Nathan, 1991) and are more reliant on the processes that neutralize ROS and prevent an accumulation of oxidatively damaged biomolecules (DeNicola et al., 2011; Gorrini et al., 2013; Sullivan and Chandel, 2014). Modest increases in levels of ROS appear to accelerate the proliferation rate of cancer cells (Irani et al., 1997), activate growth factor signaling pathways through inhibition of phosphatases by oxidation of catalytic cysteine residues (Finkel, 2012; Meng et al., 2002; Rhee, 2006) and activate stress response pathways that provide resistance to chemotherapy. Due to its highly reactive and damaging nature, however, high concentrations of ROS result in oxidative damage to proteins (proteotoxic stress, (Dickinson and Chang, 2011)) and nucleic acids (Cooke et al., 2003) resulting in apoptosis and cancer cell death. Manipulating the balance between ROS and antioxidants is therefore particularly important in cancer cells, and might represent a potential therapeutic target (Watson, 2013).
ROS, oxidative stress, and the redox state in general, may play an important role in controlling certain stages of the cell cycle. Indeed, the concept that redox state, ROS production, and progression into mitosis might be intertwined is an old one (Kawamura, 1960; Mazia, 1958), but the connection remains poorly understood, paradoxical, and sometimes conflicting. For example, Cdc25B and C, two canonical regulators of CyclinB/Cdk1 that drive mitotic entry from G2, are inactivated by oxidation (Buhrman et al., 2005; Savitsky and Finkel, 2002) while, conversely, Cyclin B/Cdk1 itself seems to enhance mitochondrial oxidative metabolism (Wang et al., 2014) and concomitant ROS leakage at the G2/M transition. ROS levels have been observed to be elevated in mitotic cells in some studies (Havens et al., 2006; Lim et al., 2015), while others have reported that mitotic cells are in a more reduced state (Conour et al., 2004). Similarly, drugs that result in mitotic arrest (antimitotics) have been reported to increase oxidative stress (Alexandre et al., 2007; Ramanathan et al., 2005), while oxidative stress itself has been reported both to either induce mitotic arrest (Wang et al., 2017), or instead to override the mitotic spindle checkpoint resulting in mitotic exit and aneuploidy (D’Angiolella et al., 2007). Clearly, a better understanding of ROS and redox state during specific stages of the cell cycle is needed to address these controversies. The small number of previously published studies in this area have largely relied on drug treatments, analyzed cells only following synchronization, or did not distinguish among all four stages of the cell cycle (Goswami et al., 2000; Havens et al., 2006; Lim et al., 2015). To overcome these limitations, here we used multiple independent assays in freely cycling cancer cells of various origins to assess ROS levels and protein and nucleotide oxidation as a function of cell cycle stage, and in response to a treatment with a variety of antimitotic agents.
RESULTS
ROS levels are elevated in freely cycling mitotic cells and enhanced by mitotic arrest.
To examine the link between the cell cycle and oxidative stress in non-synchronized cells, we utilized the dye CM-H2DCFDA as an intracellular probe for ROS (Oparka et al., 2016). Upon reaction with oxidants, the molecule displays green fluorescence and can be used to quantitatively measure ROS. Since ROS reporting by CM-H2DCFDA requires live cells, while accurate assessment of cell cycle stage requires fixation, we developed a two-step approach. First, a freely cycling culture of cells was labeled with CM-H2DCFDA, and living cells were then flow-sorted into populations with a high, medium, or low degree of CM-H2DCFDA fluorescence relative to their cell size as judged by forward scatter (FSC) (Figs. 1A and S1A, top panels). These populations were then immediately fixed, stained for DNA content using DAPI, and an antibody against the mitotic marker phospho-histone H3 (pHH3), and analyzed by flow cytometry (Figs. 1A and S1A, lower three panels). We performed this analysis using the suspension-phase hematopoietic cell lines Ramos and U937 (B-cell lymphoma and acute myeloid leukemia, respectively) to avoid any potential confounding effects on ROS/oxidation resulting from trypsinization of adherent cells (Halliwell and Whiteman, 2004). As quantified in Figure 1B, the population that had a high CM-H2DCFDA relative to their FSC was clearly enriched for cells in the G2 and M phases of the cell cycle. Conversely, the population of cells with low CM-H2DCFDA relative to FSC was highly enriched for G1 cells. These findings are consistent with increased oxidative stress as cells progress through the cell cycle. Furthermore, treatment with a variety of antimitotic drugs, including the microtubule stabilizer docetaxel, the microtubule depolymerizer nocodazole, the Plk1 inhibitor BI2536 (Steegmaier et al., 2007) and the Aurora A kinase inhibitor alisertib (Manfredi et al., 2011) at concentrations that cause strong mitotic arrest (or G2/M arrest in the case of alisertib) in these cell lines (Figure S1B) increased the level of FSC-normalized CM-H2DCFDA fluorescence in the entire arrested cell population (Fig. 1C).
Figure 1. ROS increases in mitotic cancer cells.
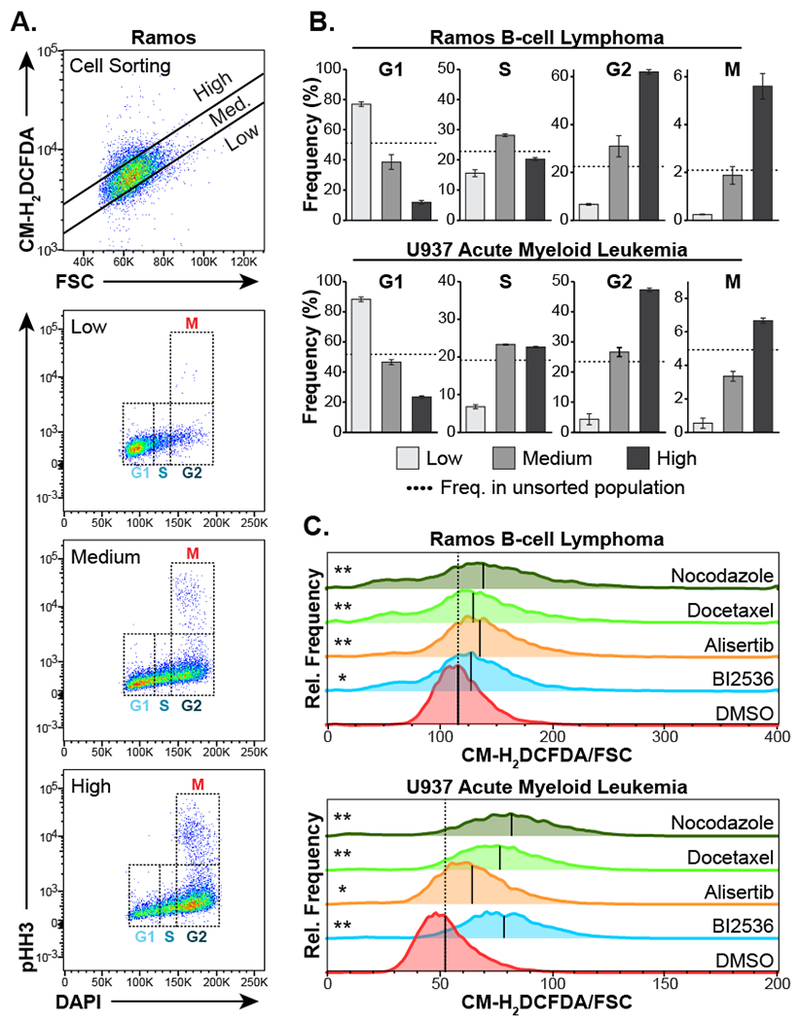
A, Ramos B-cell lymphoma cells were loaded with the ROS-responsive dye CM-H2DCFDA, then physically separated into populations with a low, medium, or high CM-H2DCFDA fluorescence relative to forward scatter (FSC), comprising approximately 15%, 70% and 15% of the total population respectively, using fluorescence-activated cell sorting (top panel). These cell populations were immediately fixed, stained for pHH3 and DNA content (DAPI) and analyzed by flow cytometry to determine the relative proportion of cells in various stages of the cell cycle (bottom three panels).
B, Quantification of cell cycle stage in cells sorted according to CM-H2DCFDA/FSC-low, medium, and high populations for both Ramos and U937 cells. Mean ± SEM of three independent experiments are shown. Dotted lines indicate the frequency of each cell cycle stage in the unsorted cell population.
C, Ramos and U937 cells were treated with the indicated antimitotic drugs for 16 hours, then immediately stained with CM-H2DCFDA and analyzed by flow cytometry as in panel A above. Shown are representative histograms of FSC-normalized CM-H2DCFDA staining with black lines indicating the median of each population. Dashed vertical lines indicate the median CM-H2DCFDA/FSC value seen in the DMSO control population. The mean CM-H2DCFDA/FSC ratio measurements from triplicate independent experiments were used in a two-tailed Students t-test versus DMSO control (* p < 0.05, ** p < 0.01).
Oxidative damage of proteins and nucleotides peaks in mitosis and after mitotic arrest.
The increase in ROS during G2 and M phases might be compensated for by various antioxidant systems in cancer cells. To determine if elevated levels of ROS in mitotic cancer cells resulted in increased oxidative damage to biomolecules, we measured total levels of protein cysteine oxidation during the distinct phases of the cell cycle. This was performed using dimedone labeling, a robust chemical method that takes advantage of dimedone’s irreversible reactivity with oxidized cysteine (cysteine sulfenic acid) in live cells, to generate a dimedone thioether-modified cysteine adduct. Following washing, fixation and permeabilization, this covalently modified cysteine can be recognized by a specific antibody (Seo and Carroll, 2009). By combining dimedone labeling with flow cytometric analysis using DAPI and pHH3, we simultaneously measured, both total cysteine oxidation and cell cycle stage in individual cells from otherwise unperturbed asynchronous cultures of the Ramos and U937 cells used above, as well as in adherent C4-2 castration-resistant prostate cancer (CRPC) cells, HeLa cervical cancer cells, and U2OS osteosarcoma cells.
As shown in Figures 2A, B, and C and Figure S2A, dimedone labeling was consistently elevated in mitotic cells relative to all other stages of the cell cycle in both suspension and adherent cells, even after normalizing for cell size. This elevation of cysteine oxidation in mitotic cells was statistically significant in all of the cell lines examined (Fig. 2C). Furthermore, the addition of antimitotic drugs, including docetaxel, nocodazole, BI2536 and alisertib all resulted in a further elevation of oxidized cysteines relative to non-arrested mitotic cells (Figs. 2D and S2B). Finally, mitotic arrest also resulted in enhanced cell staining of C4-2 cells with an antibody against the oxidized nucleotide 8-oxoguanine (Fig. 2E), consistent with similar findings reported by others (Crea et al., 2011). These independent measures of oxidative stress lend additional support to the notion that the elevated levels of endogenous ROS in mitotic cancer cells result in biomolecule oxidation, and that mitotic arrest further increases the degree of oxidative damage.
Figure 2. Oxidative damage of biomolecules increases in mitotic cancer cells.
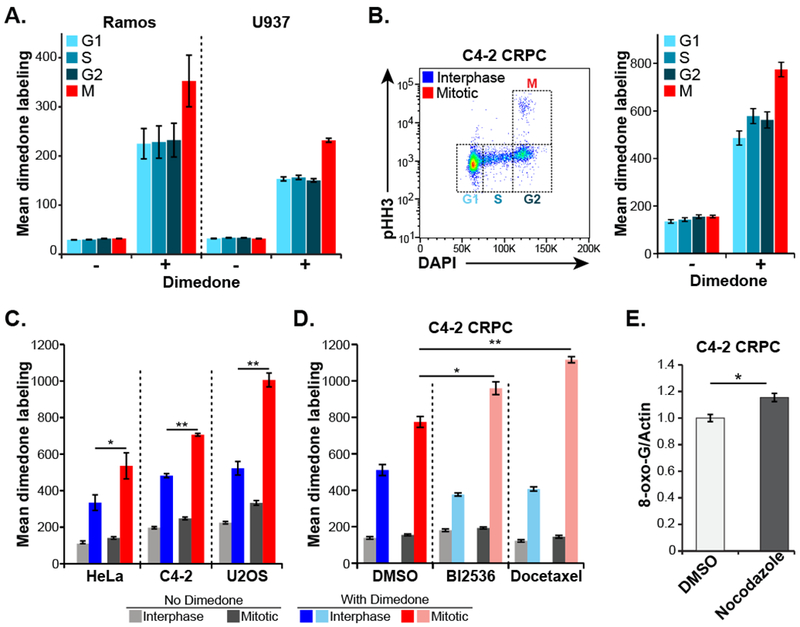
A, Live asynchronous Ramos and U937 hematopoietic cells were labeled on oxidized protein cysteine residues with dimedone, then washed, fixed, stained for pHH3 and DNA content (DAPI) and analyzed by flow cytometry to determine cell cycle stage. “Mean dimedone labeling” was defined as the mean dimedone channel signal normalized to FSC on an individual cell basis. Mean ± SEM of three independent experiments are shown.
B, C4-2 CRPC cells were labeled and analyzed as in panel A. Shown is an example scatter plot with cell cycle distribution (left panel) and quantification of dimedone labeling (right panel) in each stage of the cell cycle.
C, Flow cytometry and dimedone labeling analysis was performed as in panel B on the indicated adherent cell lines. Gating was performed to separate interphase (G1, S and G2) cells from mitotic cells, and the data analyzed and plotted as above. Mean ± SEM of three independent experiments are shown. (* p < 0.05 ** p < 0.01 by two-tailed paired Student’s t-test).
D, Dimedone labeling analysis was performed in C4-2 CRPC cells treated with the indicated antimitotic drugs (BI2536, 10 nM; docetaxel, 2.5 nM) or DMSO vehicle for 24 hours. These cells were then analyzed for cell cycle distribution and cysteine oxidation levels, and plotted as in panel B. Mean ± SEM of three independent experiments are shown. (* p < 0.05 ** p < 0.01 using a two-tailed Student’s t-test).
E, Levels of 8-oxoguanine were assessed in C4-2 cells after a 16-hour treatment with nocodazole or DMSO control followed by drug washout and 24-hour recovery. Cells were fixed, stained with antibodies raised against 8-oxoguanine and normalized to actin. Mean ± SEM of three independent experiments are shown. (* p = 0.0195 using a two-tailed Student’s t-test).
DISCUSSION
We have made three observations that support the notion of a progressive increase in endogenous oxidants and consequent oxidative stress as cells transit though the cell cycle. First, cell populations that exhibited elevated fluorescence of an oxidative stress-dependent dye were highly enriched in mitotic cells. Second, in freely cycling cells the levels of cysteine oxidation was significantly enhanced in mitotic compared to interphase cells, and could be further elevated by the addition of antimitotic drugs. Third, we observed elevated levels of 8-oxo-G in cells following mitotic arrest, similar to Crea et al. (2011). Consistent with these findings, elevated ROS levels have been observed in mitotic cells (Goswami et al., 2000; Havens et al., 2006), and in cancer cells in general (DeNicola et al., 2011; Szatrowski and Nathan, 1991).
It is interesting to note that while we observed high ROS levels in both G2-phase and mitosis, we only saw the consequences of oxidative damage to proteins and nucleotides in mitosis. This may simply reflect the time necessary to induce measurable damage, but could alternatively indicate cell cycle-dependent regulation of cellular mechanisms for detecting, detoxifying, or otherwise mitigating oxidative damage. In either case, the dependence of biomolecule oxidation on cell cycle stage suggests the possibility that it may serve as an important signal during mitotic progression, an idea concordant with the finding that pericentrosomal hydrogen peroxide enhances mitotic entry by inactivating the phosphatase Cdc14B (Lim et al., 2015) .
The elevated levels of ROS that we observe in mitotic cells may allow development of new treatments that kill cancer cells through ROS- or oxidative stress-dependent toxicities (Watson, 2013; Yun et al., 2015). Specifically our observations of increased biomolecule oxidation following mitotic arrest suggest that antimitotic drugs could sensitize cancer cells to drugs that potentiate the toxicity of oxidative DNA or protein damage.
STAR METHODS
CONTACTS FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Michael Yaffe (myaffe@mit.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines were purchased from ATCC (Ramos, HeLa, U937, and U2OS) or the Characterized Cell Line Core Facility at MD Anderson (C4-2). Ramos, U937, and C4-2 are male cell lines. HeLa and U2OS cells are female. All cell lines were incubated at 37°C in a humidified incubator supplied with 5% CO2, maintained in subconfluent culture, and used for ≤ 20 passages. All media was supplemented with 10% fetal bovine serum (FBS) and 2 mM glutamine. U-2 OS and HeLa cells were grown in DMEM medium. Ramos, U937, and C4-2 cells were grown in RPMI-1640 medium.
METHOD DETAILS
Flow cytometry measurement of CM-H2DCFDA oxidation and cell cycle stage
For all CM-H2DCFDA experiments, cells were collected by centrifugation, washed with PBS, incubated with 5 μM CM-H2DCFDA in PBS for 30 minutes in the dark at 37°C, collected by centrifugation, washed with PBS and then analyzed within 15 minutes. To determine the cell cycle distribution of cells in populations with high, medium or low CM-H2DCFDA fluorescence relative to forward scatter (FSC), exponentially growing cells were treated as above, immediately sorted using the BD™ FACSAria IIIu (Becton Dickson) and collected into tubes containing prechilled 100% ethanol. Sorted cells were then collected by centrifugation and stored at −20°C in 70% ethanol 30% PBS. Fixed cells were then washed with cold PBS containing 1% bovine serum albumin (PBS-BSA), permeabilized by a 15 minute incubation in PBS 0.25% Triton X-100 on ice and washed with PBS-BSA. Cells were then incubated overnight with anti-phospho S10 histone H3 diluted 1:100 in PBS-BSA, washed twice with PBS-BSA, incubated for 1 hour with fluorescent dye-conjugated secondary antibody diluted 1:200 (Alexa Fluor®, Molecular Probes), washed twice with PBS-BSA and resuspended in PBS containing 1 μg/ml 4,6-diamidino-2-phenylindole (DAPI, Molecular Probes). Flow cytometry was performed using a BD™ LSRII flow cytometer (Becton Dickson) and analyzed using the FlowJo® software package (FlowJo, LLC). To examine CM-H2DCFDA oxidation during mitotic arrest Ramos cells were treated with 200 nM nocodazole, 20 nM docetaxel, 400 nM alisertib, or 40 nM BI2536. U937 cells were treated with the same BI2536 and alisertib concentrations, but 800 nM nocodazole or 40 nM docetaxel. These doses were chosen as those beyond which further increase had no appreciable effect on mitotic arrest after a 24-hour treatment (Figure S1B). These cells were then CM-H2DCFDA treated as above and immediately analyzed by flow cytometry using a BD™ LSRII flow cytometer (Becton Dickson). During the analysis with the FlowJo® software package, CM-H2DCFDA/FSC ratios in each experiment were multiplied by 100 to produce numbers in a more easily interpretable range.
Dimedone labeling based measurement of cysteine sulfenic acid
Freely cycling cells were treated with 2.5 mM dimedone for 30 minutes and then washed twice to remove all remaining dimedone prior to trypsinization. For experiments involving treatment with antimitotic drugs, cells were treated with drug for 24 hours, labeled with dimedone for 30 min, washed as above. The media and wash solutions were combined with the trypsinized cell pellet to retain any detached cells. Cells were fixed for 15 minutes with 4% formaldehyde in PBS containing freshly-prepared 30 mM iodoacetamide to prevent further oxidation/dimedone labeling during fixation (Kaplan et al., 2011). Fixed cells were then washed with PBS-BSA, resuspended in −20°C methanol, washed twice with PBS-BSA containing 0.1% Tween-20, and incubated with primary antibodies overnight at 4°C. Cells were then washed with PBS-BSA 0.1% Tween-20 prior to a 1 hour room temperature incubation with fluorescent dye-conjugated secondary antibodies diluted 1:200 (Alexa Fluor®, Molecular Probes). Finally, cells were washed twice with PBS-BSA 0.1% Tween-20 and resuspended in PBS containing 1 μg/ml DAPI, and analyzed by flow cytometry as above. The dimedone-labeling values were derived by dividing the signal intensity from the dimedone channel by the FSC on an individual event basis, and multiplying by 10,000 to obtain a reasonable scale.
In-cell western assays of 8-oxoguanine
In-cell western assays were used to measure 8-oxoguanine in C4-2 cells following the published method of (Crea et al., 2011). After the indicated treatment, cells were fixed with methanol at −20°C for 20 minutes, washed, and blocked for 60 minutes. Blocking, washes, and antibody incubations were performed using Odyssey™ blocking buffer at room temperature. Primary antibodies including anti-8-oxoguanine and anti-β-actin (diluted 1:200 and 1:1000, respectively) were incubated overnight. Appropriate infrared-dye conjugated secondary antibodies (diluted 1:500) for 2 hours. Finally, cells were washed, wells were filled with PBS, and signal intensity assessed using an Odyssey™CLx scanner.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless otherwise indicated, data is presented as the mean of triplicate experiments, and bars represent standard error of the mean.
DATA AND SOFTWARE AVAILABILITY
None.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho S10 histone H3 [3H10] | EMD Millipore | Cat#05-806 |
| Cysteine sulfenic acid (dimedone) | EMD Millipore | Cat#07-2139 |
| 8-oxoguanine clone 483.15 | EMD Millipore | Cat#MAB3560 |
| β-actin (rabbit) | Cell Signaling Technologies | Cat#4967 |
| Bacterial and Virus Strains | ||
| None | ||
| Biological Samples | ||
| None | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM | Corning CellGro™ | Cat#10-017-CV |
| RPMI-1640 | Gibco™ | Cat#11875-093 |
| Fetal Bovine Serum (FBS) | Seradigm | Cat#1500-500 |
| chloromethyl 2′,7′-dichlorodihydrofluorescein diacetate (CM-H2DCFDA) | Invitrogen | Cat#C6827 |
| BI2536 | Selleck Chemicals | Cat#S1109 |
| Alisertib | Selleck Chemicals | Cat#S1133 |
| Nocodazole | Sigma Aldrich | Cat#M1404 |
| 5,5-dimethyl-1,3-cyclohexanedione (dimedone) | Sigma Aldrich | Cat#38490 |
| iodoacetamide | Sigma Aldrich | Cat#I-1149 |
| Docetaxel | LC Laboratories | Cat#D-1000 |
| 4,6-diamidino-2-phenylindole (DAPI) | Molecular Probes | Cat#D1306 |
| Critical Commercial Assays | ||
| None | ||
| Deposited Data | ||
| None | ||
| Experimental Models: Cell Lines | ||
| Ramos | ATCC | CRL-1569 |
| U937 | ATCC | CRL-1593.2 |
| C4-2 | MD Anderson | N/A |
| U-2 OS | ATCC | HTB-96 |
| HeLa | ATCC | CCL-2 |
| Experimental Models: Organisms/Strains | ||
| None | ||
| Oligonucleotides | ||
| None | ||
| Recombinant DNA | ||
| None | ||
| Software and Algorithms | ||
| FlowJo version 7.6.5 | FlowJo, LLC | |
| Seaborn statistical data visualization version 0.7.1 | http://seaborn.pydata.org | |
| Other | ||
| None | ||
Highlights.
ROS varies with the cell cycle in freely-cycling cancer cells
ROS levels peak in G2 and mitosis
Oxidation of biomolecules is maximal in mitosis
Mitotic arrest further enhances ROS and oxidative damage to proteins and nucleotides
ACKNOWLEDGEMENTS
We thank Karl Merrick, Ian Cannell, Yi Wen Kong, and other members of the Yaffe and Lauffenburger laboratories, and Forest White for helpful discussion and advice. We also thank the Flow Cytometry Core Facility of the Swanson Biotechnology Center. This research was supported by grants from the National Institute of Health (R01-GM104047 (M.B.Y), R01-ES015339 (M.B.Y.), R35-ES028374 (M.B.Y.), U54-CA112967 (D.A.L. and M.B.Y.), and U54-CA217377 (D.A.L.)), DoD Peer Reviewed Medical Research Program, Contract Number W81XWH-16-1-0464 (M.B.Y.), the Charles and Marjorie Holloway Foundation (M.B.Y.), the MIT Center for Precision Cancer Medicine, and an American Cancer Society Post-Doctoral Fellowship to J.C.P. Support was also provided in part by the Koch Institute Support Grant (P30-CA14051) from the National Cancer Institute and the Center for Environmental Health Support Grant (P30-ES002109).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Michael B. Yaffe is a member of the Divisions of Surgical Oncology and Trauma and Critical Care, Department of Surgery, Beth Israel Deaconess Medical Center, Harvard Medical School, Boston, MA, and the Chief Academic Editor of the journal Science Signaling.
REFERENCES
- Alexandre J, Hu Y, Lu W, Pelicano H, and Huang P (2007). Novel action of paclitaxel against cancer cells: bystander effect mediated by reactive oxygen species. Cancer Res 67, 3512–3517. [DOI] [PubMed] [Google Scholar]
- Buhrman G, Parker B, Sohn J, Rudolph J, and Mattos C (2005). Structural mechanism of oxidative regulation of the phosphatase Cdc25B via an intramolecular disulfide bond. Biochemistry 44, 5307–5316. [DOI] [PubMed] [Google Scholar]
- Conour JE, Graham WV, and Gaskins HR (2004). A combined in vitro/bioinformatic investigation of redox regulatory mechanisms governing cell cycle progression. Physiol Genomics 18, 196–205. [DOI] [PubMed] [Google Scholar]
- Cooke MS, Evans MD, Dizdaroglu M, and Lunec J (2003). Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J 17, 1195–1214. [DOI] [PubMed] [Google Scholar]
- Crea F, Duhagon Serrat MA, Hurt EM, Thomas SB, Danesi R, and Farrar WL (2011). BMI1 silencing enhances docetaxel activity and impairs antioxidant response in prostate cancer. Int J Cancer 128, 1946–1954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Angiolella V, Santarpia C, and Grieco D (2007). Oxidative stress overrides the spindle checkpoint. Cell Cycle 6, 576–579. [DOI] [PubMed] [Google Scholar]
- DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, et al. (2011). Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature 475, 106–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson BC, and Chang CJ (2011). Chemistry and biology of reactive oxygen species in signaling or stress responses. Nat Chem Biol 7, 504–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T (2012). From sulfenylation to sulfhydration: what a thiolate needs to tolerate. Sci Signal 5, pe10. [DOI] [PubMed] [Google Scholar]
- Gorrini C, Harris IS, and Mak TW (2013). Modulation of oxidative stress as an anticancer strategy. Nat Rev Drug Discov 12, 931–947. [DOI] [PubMed] [Google Scholar]
- Goswami PC, Sheren J, Albee LD, Parsian A, Sim JE, Ridnour LA, Higashikubo R, Gius D, Hunt CR, and Spitz DR (2000). Cell cycle-coupled variation in topoisomerase Ilalpha mRNA is regulated by the 3′-untranslated region. Possible role of redox-sensitive protein binding in mRNA accumulation. J Biol Chem 275, 38384–38392. [DOI] [PubMed] [Google Scholar]
- Halliwell B, and Whiteman M (2004). Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? Br J Pharmacol 142, 231–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Havens CG, Ho A, Yoshioka N, and Dowdy SF (2006). Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species. Mol Cell Biol 26, 4701–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irani K, Xia Y, Zweier JL, Sollott SJ, Der CJ, Fearon ER, Sundaresan M, Finkel T, and Goldschmidt-Clermont PJ (1997). Mitogenic signaling mediated by oxidants in Ras-transformed fibroblasts. Science 275, 1649–1652. [DOI] [PubMed] [Google Scholar]
- Kaplan N, Urao N, Furuta E, Kim SJ, Razvi M, Nakamura Y, McKinney RD, Poole LB, Fukai T, and Ushio-Fukai M (2011). Localized cysteine sulfenic acid formation by vascular endothelial growth factor: role in endothelial cell migration and angiogenesis. Free Radic Res 45, 1124–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura N (1960). Cytochemical and quantitative study of protein-bound sulfhydryl and disulfide groups in eggs of Arbacia during the first cleavage. Exp Cell Res 20, 127–138. [DOI] [PubMed] [Google Scholar]
- Lim JM, Lee KS, Woo HA, Kang D, and Rhee SG (2015). Control of the pericentrosomal H2O2 level by peroxiredoxin I is critical for mitotic progression. J Cell Biol 210, 23–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manfredi MG, Ecsedy JA, Chakravarty A, Silverman L, Zhang M, Hoar KM, Stroud SG, Chen W, Shinde V, Huck JJ, et al. (2011). Characterization of Alisertib (MLN8237), an investigational small-molecule inhibitor of aurora A kinase using novel in vivo pharmacodynamic assays. Clin Cancer Res 17, 7614–7624. [DOI] [PubMed] [Google Scholar]
- Mazia D (1958). SH compounds in mitosis. I. The action of mercaptoethanol on the eggs of the sand dollar Dendraster excentricus. Exp Cell Res 14, 486–494. [DOI] [PubMed] [Google Scholar]
- Meng TC, Fukada T, and Tonks NK (2002). Reversible oxidation and inactivation of protein tyrosine phosphatases in vivo. Mol Cell 9, 387–399. [DOI] [PubMed] [Google Scholar]
- Oparka M, Walczak J, Malinska D, van Oppen L, Szczepanowska J, Koopman WJH, and Wieckowski MR (2016). Quantifying ROS levels using CM-H2DCFDA and HyPer. Methods 109, 3–11. [DOI] [PubMed] [Google Scholar]
- Ramanathan B, Jan KY, Chen CH, Hour TC, Yu HJ, and Pu YS (2005). Resistance to paclitaxel is proportional to cellular total antioxidant capacity. Cancer Res 65, 8455–8460. [DOI] [PubMed] [Google Scholar]
- Rhee SG (2006). Cell signaling. H2O2, a necessary evil for cell signaling. Science 312, 1882–1883. [DOI] [PubMed] [Google Scholar]
- Savitsky PA, and Finkel T (2002). Redox regulation of Cdc25C. J Biol Chem 277, 20535–20540. [DOI] [PubMed] [Google Scholar]
- Seo YH, and Carroll KS (2009). Profiling protein thiol oxidation in tumor cells using sulfenic acid-specific antibodies. Proc Natl Acad Sci U S A 106, 16163–16168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steegmaier M, Hoffmann M, Baum A, Lenart P, Petronczki M, Krssak M, Gurtler U, Garin-Chesa P, Lieb S, Quant J, et al. (2007). BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol 17, 316–322. [DOI] [PubMed] [Google Scholar]
- Sullivan LB, and Chandel NS (2014). Mitochondrial reactive oxygen species and cancer. Cancer Metab 2, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szatrowski TP, and Nathan CF (1991). Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res 51, 794–798. [PubMed] [Google Scholar]
- Vander Heiden MG, and DeBerardinis RJ (2017). Understanding the Intersections between Metabolism and Cancer Biology. Cell 168, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang GF, Dong Q, Bai Y, Yuan J, Xu Q, Cao C, and Liu X (2017). Oxidative stress induces mitotic arrest by inhibiting Aurora A-involved mitotic spindle formation. Free Radic Biol Med 103, 177–187. [DOI] [PubMed] [Google Scholar]
- Wang Z, Fan M, Candas D, Zhang TQ, Qin L, Eldridge A, Wachsmann-Hogiu S, Ahmed KM, Chromy BA, Nantajit D, et al. (2014). Cyclin B1/Cdk1 coordinates mitochondrial respiration for cell-cycle G2/M progression. Dev Cell 29, 217–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson J (2013). Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol 3, 120144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, Roper J, Chio II, Giannopoulou EG, Rago C, et al. (2015). Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 350, 1391–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
None.