

Abstract
Background
Gastrointestinal stromal tumors (GISTs) represent the most frequent mesenchymal tumor of the gastrointestinal tract. Less than 5% of them seem to be hereditary, being succinate dehydrogenase complex (SDHx) deficient disorders and neurofibromatosis type 1 the more related inherited conditions. Wild type (WT) KIT and PDGFRα GISTs constitute a clue for a hypothetical underlying germline condition.
Case presentation
We present a case of a 20 years old female diagnosed of a gastric WT GIST who developed hepatic metastases during her clinical course. No significant or typical phenotypic features suggestive of a specific syndrome were detected by physical examination. Also, her family history seemed to be irrelevant, since no other cases of GISTs, paragangliomas or pheochromocytomas were reported. Her paternal grandfather died as a consequence of a pituitary adenoma. In light of the age of tumor presentation and somatic features of gastric GIST, we performed genetic testing of SDHx genes. Analysis obtained from peripheral blood sample revealed the presence, in heterozygous state, of the c.1A > C; p.(Met1?) pathogenic variant in the SDHA.
Conclusions
To the best of our knowledge, this is the first published report in which the c.1A > C; p.(Met1?) pathogenic variant in the SDHA is associated with a GIST. SDHA pathogenic variants increase the risk of paraganglioma, pheochromocytoma, GIST, pituitary adenoma and renal cancer in an autosomal dominant inherited condition named paraganglioma syndrome type 5. The absence of family history of tumors in SDHA pathogenic variants carriers could be related to its low penetrance. All patients diagnosed with WT GISTs should be referred to a hereditary cancer genetic counseling unit regardless of the age at presentation or the absence of a suspicious family history.
Keywords: GIST, Hereditary, SDH, Paraganglioma, KIT, PDGFRα, Cancer genetic counseling
Background
Gastrointestinal stromal tumors (GISTs) constitute the most frequent mesenchymal tumor of the gastrointestinal tract [1]. About 70% of GISTs develop in the stomach and 20% in small intestine, being the rest of the gastrointestinal tract or other abdominal organs less frequently affected [2]. The mean age at presentation of GISTs is 60 years old, but in rare cases affects young adults and children [3]. These tumors are driven mostly by CD117 (KIT) and platelet-derived growth factor receptor alpha (PDGFRα) gain of function mutations [4] but, about a 15% [5] are KIT and PDGFRα wild type (WT). These WT GISTs are predominantly somatic succinate dehydrogenase (SDH) deficient and have a predilection for gastric location [6]. The younger the patient diagnosed with a GIST is, the higher is the probability of harboring a SDH-deficient tumor [7].
Less than 5% of GISTs have an underlying hereditary or syndrome related condition, with neurofibromatosis type 1 (NF1) the most frequent related disease [8]. We can classify these hereditary GISTs in two main groups: SDH-competent and SDH-deficient GISTs [9]. The first group includes KIT-mutated and PDGFRα-mutated syndromic GISTs, with germline mutations of KIT, and PDGFRα respectively. Until now few cases of families harboring KIT or PDGFRα germline mutations have been published [10, 11]. NF1 constitutes the main hereditary entity related to GIST in the SDH-competent group [12] and it is estimated that about 7% of NF1 patients will develop this tumor [13]. GISTs constitute the main gastrointestinal feature in NF1 affected patients. In NF1 GISTs are almost always KIT and PDFGRα WT, they tend to be multifocal and have predilection for the small bowel [14]. The second group corresponds to the SDH-deficient GISTs and they are related to germline pathogenic variants in succinate dehydrogenase complex (SDHx) genes or SDHC promoter hypermethylation [15]. In WT GISTs related to SDHx, the stomach is the main organ of presentation, usually in the gastric antrum, as multifocal disease of epithelioid variant [16]. SDH-deficiency represents the most important WT GIST subgroup [17]. These WT GISTs tumors have a higher rate of lymphovascular invasion and liver metastases and they are not expected to respond to tyrosine kinase inhibitor imatinib as they lack KIT or PDGFRα oncogenic mutations [18]. Paradoxically these SDHx deficient GIST tumors seem to be more indolent even in the presence of advanced disease compared to KIT or PDGFRα dependent GISTs [19].
The SDHx genes or mitochondrial complex 2 (SDHA, SDHB, SDHC, SDHD) encode the subunits of the mitochondrial SDH enzyme, that are essential in the conversion of succinate to fumarate in the Krebs cycle (tricarboxylic acid or citric acid cycle), playing a critical role in mitochondrial respiratory and metabolic functions [20]. If any component of this complex is affected, the entire SDH complex becomes unstable and SDHB immunohistochemistry (IHC) becomes negative because it is rapidly degraded in the cytoplasm. As a consequence, loss of expression of SDHB in a tumor specimen could be used as a suspicion of a germline SDHx mutation [21]. SDH deficiency generates an accumulation of intracellular succinate which causes activation of different cellular pathways, favoring tumorigenesis in last instance [22].
SDHx germline mutations have been associated to different kind of tumors, mainly paraganglioma and pheochromocytoma. SDHA (5p15.33) heterozygous pathogenic variants are related to an autosomal dominant inherited condition named paraganglioma syndrome type 5 (PGL5, OMIM 614165) associated to an increase in the risk of developing paragangliomas, pheochromocytomas, GIST and pituitary adenomas [23]. The Carney-Stratakis syndrome (CSS) also known as GIST-paraganglioma dyad, (OMIM 606864) characterized by the presence of paraganglioma and GIST in the same individual, is related to germline mutations in SDHB, SDHC and SDHD, but its association with SDHA pathogenic mutations has been rarely recognized [24]. Carney triad (CT, OMIM 604287), first described in 1977, is characterized by the presence of gastric GISTs, paragangliomas and pulmonary chondromas, it’s almost always not inherited and it’s related to SDHC promoter hypermethylation [25]. Lastly, homozygous and compound heterozygous mutations in the SDHA have been occasionally related to the Leigh syndrome [26], a rare recessive disease characterized by neurodegenerative mitochondrial encephalomyopathy that becomes apparent mostly in the first year of life.
Case presentation
A 20 year old female was referred to our Medical Oncology department after a gastric antrum GIST resection. A subtotal gastrectomy revealed a pathologic specimen compatible with an epithelioid GIST variant localised to the muscularis propia. The diameter of the tumor was 1.8 cm and the mitotic count rate was of 18 per 50 HPF (high-power field). IHC assays revealed that the cells presented a strong positive expression of CD117 (KIT) and DOG1, which supported a GIST diagnosis. The tumor was classified as a pT1pN0 (0/1) with a high mitotic rate - stage II – according to the eighth edition of TNM classification. Armed Forces Institute of Pathology (AFIP) criteria calculate the risk of this tumor relapsing and/or progressing as zero, due to the low number of published cases. The mutational analysis revealed that the tumor was KIT and PDGFRα WT. After complete resection, a whole body computed tomography (CT) scan with contrast was performed, revealing no signs of metastases. Two years after the initial diagnosis, during the follow-up, a CT scan, a magnetic resonance imaging (MRI) of the liver and a positron emission tomography (PET) were performed. These procedures revealed the presence of multiple hepatic metastases. After confirmation of resectability, she underwent surgical resection of the hepatic metastases and postsurgical image studies confirmed no evidence of disease so, in accordance with current medical evidence in patients with complete resection of WT GIST, adjuvant treatment with imatinib was not delivered. At present, the patient is 26 years old and she has no evidence of active disease.
Because of the age at presentation and the molecular features of her tumor, the patient was sent to our Hereditary Cancer Genetic Counseling Unit for further investigations. Complete physical examination was irrelevant and no typical phenotypic features suggestive of a specific syndrome were detected. The family history of the patient seemed to be unremarkable: she has a 19 years old healthy brother. Her parents, 51 years old, had no history of any diseases. The maternal family history was anodyne but the paternal family history included a grandfather who had died at the age of 52 as a consequence of a pituitary adenoma. He was diagnosed when he was 30 years of age and he received radiotherapy treatment. There was no history of other tumors (paraganglioma, pheochromocytoma, GIST or pulmonary chondromas) or other significant diseases in maternal or paternal lines.
On the basis of the described molecular features of diagnosed GIST, age at presentation and in a spite of an apparently irrelevant family history, we decided to perform SDHx germline analysis in peripheral blood, after performing pretest counseling and obtaining informed consent.
Methodology
Analysis of exons 9, 11, 13 and 17 of KIT and exons 12, 14 and 18 of PDGFRα were performed in tumor samples using amplification of the exons of interest by polymerase chain reaction (PCR) followed by direct sequencing (Sanger method) of amplification products.
Sequence analysis of coding exons and flanking intronic regions of SDHD (NM_003002.2), SDHB (NM_003000.2), SDHC (NM_003001.3) and SDHA (NM_004168.2) was undertaken in germline DNA using standard PCR and direct sequencing reactions, avoiding SDHC and SDHA pseudogenes (BigDye v3.1, Applied Biosystems, Foster City, CA). (Primer sequences and PCR conditions are available on request). Sequencing reactions were analyzed using a 3500 Genetic Analyzer and the data were processed by Sequencing Analysis and Variant Reporter software (all from Applied Biosystems), Detection of large rearrangements in these genes was performed by multiplex ligation-dependent probe amplification (MLPA) analysis, using a commercially available kit (SALSA MLPA P226 kit, MRC-Holland, Amsterdam), according to the manufacturer’s instructions.
Results
Germline genetic analysis revealed the presence of a heterozygous c.1A > C; p.(Met1?) pathogenic variant in the SDHA. This missense variant is predicted to be pathogenic [27, 28] because it affects to the initiation codon AUG (methionine), which is responsible for translation initiation from the messenger RNA (mRNA). This variant is absent in gnomAD (genome aggregation database). Figure 1 shows an electropherogram with the SDHA c.1A > C variant.
Fig. 1.

Electropherogram showing the SDHA c.1A > C variant
Subsequently, we performed predictive tests to her relatives. Her brother was found not to be a carrier of the variant; her father and paternal uncle were carriers (confirming paternal inheritance) and her paternal grandmother was also negative for this variant. These results suggest that the paternal grandfather, who died as a consequence of a pituitary adenoma, could be obligate carrier of the c.1A > C; p.(Met1?) pathogenic variant in the SDHA. Figure 2 shows the family pedigree.
Fig. 2.
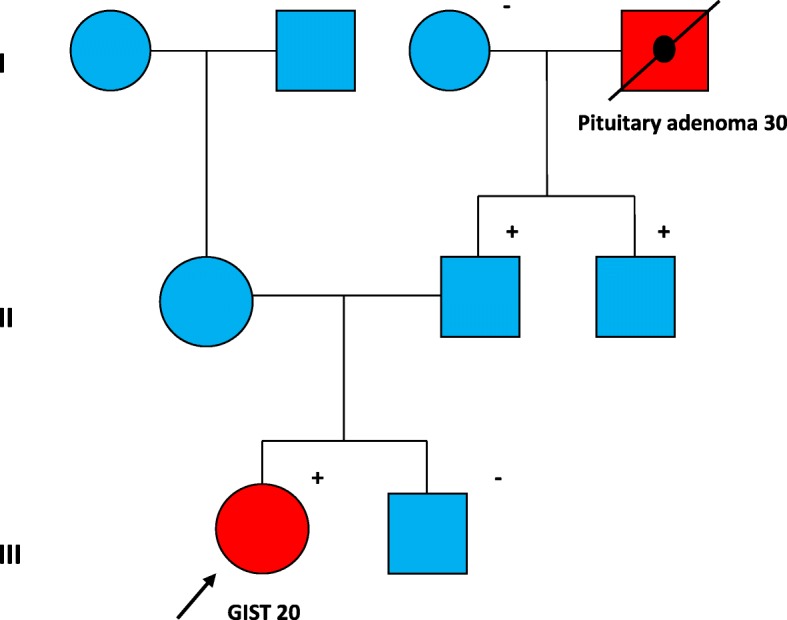
Family pedigree. Unaffected female: blue circle. Unaffected male: blue square. Affected female: red circle. Affected male: red square Plus (+) and minus (−) signs represent carrier status of tested family members
Discussion and conclusions
An exhaustive review of databases and the medical literature was undertaken in order to assess the pathogenicity of this variant. The c.1A > C; p.(Met1?) variant has previously been reported in a patient diagnosed with Leigh syndrome, in which genetic analysis revealed the presence of a compound heterozygote mutation in the SDHA. Second allele mutation, in trans with the other SDHA pathogenic variant, corresponded to a heterozygous A to C substitution which changed the methionine translation initiation codon to a leucine. Additional functional studies confirmed a quantitative decrease and instability of the corresponding mRNA, which supports its causality [28]. On the basis of this study, ClinVar database (http://www.ncbi.nlm.nih.gov/clinvar/) describes the c.1A > C; p. (Met1?) variant in SDHA as pathogenic [29].
In another recent report which includes 972 patients with paraganglioma-pheochromocytoma [30], a 66 year old patient diagnosed of thoracic paraganglioma, with negative family history of other malignancies, carried the c.1A > C pathogenic variant in the SDHA. Another germline pathogenic variant in the same codon (c.2 T > C) has been reported in a 23 year old patient with a WT GIST and a renal chromophobe cell tumor [31].
SDHA heterozygous pathogenic variants are related to a different spectrum of tumours. Germline SDHA variants have been quite recently reported to be associated to paragangliomas and the disease penetrance among carriers is estimated to be low [32]. As a consequence the family history of the carriers can go unnoticed. PGL5 is associated with an increase in the risk of developing paragangliomas, pheochromocytomas, GISTs and pituitary adenomas, and it’s compatible with our clinical suspicion on the basis of this patient’s GIST, her paternal grandfather pituitary adenoma [33] and the co-segregation of the c.1A > C pathogenic variant in the SDHA gene with the disease. The CSS, characterized by the presence of paraganglioma and GIST in the same individual is principally related to germline mutations in SDHB, SDHC and SDHD, but its association with SDHA pathogenic mutations is more exceptional. Strictly, we cannot rule out a hypothetical clinical diagnosis of CSS in our patient, but at the moment no paragangliomas have been detected by radiological studies. We also emphasize that her paternal grandfather (obligate carrier of SDHA pathogenic variant) died as a consequence of a pituitary adenoma, which is also associated with SDHA pathogenic variants, but it’s not a typical feature of CSS. SDHx deficiency related diseases and their phenotypes frequently overlap. In fact, currently, in an era in which genetic analyses are more accurate and performed more extensively, patients with a previous clinical diagnosis of CT can show SDHx mutations [34]. The clinical course of our patient and of the c.1A > C; p.(Met1?) SDHA pathogenic variant carriers in her paternal family could help define better their clinical diagnoses in the spectrum of SDHA inherited related conditions, but we must take into consideration that SDHA variants have a reduced disease penetrance compared to other components of SDHx complex, especially SDHB and SDHD [35].
GISTs constitute the most frequent mesenchymal tumor of the gastrointestinal tract [1]. WT GISTs are predominantly somatic SDH-deficient and have predilection for gastric location [6]. The younger the diagnosis of GIST is, the higher the probability of harboring a SDH-deficient tumor [7]. As previously mentioned, these SDH-deficient GISTs seem to be more indolent even in the presence of advanced disease compared to KIT or PDGFRα onco-addicted GISTs [19]. Our patient represents a clear example of the more indolent biological behaviour of the SDHx-deficient GISTs in the context of advanced disease: having survived six years since her initial diagnosis with no evidence of recurrent disease. In addition, the clinical case reflects the need for further studies in those GISTs with an early age at presentation, even in the absence of clear family history or other pathognomonic features.
To the best of our knowledge this is the first published report in which the c.1A > C; p.(Met1?) pathogenic variation in the SDHA seems to be associated to a gastric GIST in the context of PGL5. We must emphasize that SDH screening in patients with GIST, especially in those diagnosed at early ages, should be undertaken. All patients diagnosed of WT GISTs should be referred to a hereditary cancer genetic counseling unit.
Acknowledgements
Not applicable.
Abbreviations
- CSS
Carney-Stratakis syndrome
- CT
Carney triad
- GIST
Gastrointestinal stromal tumor
- KIT
KIT proto-oncogene receptor tyrosine kinase
- PDGFRα
Platelet-derived growth factor receptor alpha
- PGL
Paraganglioma
- SDH
Succinate dehydrogenase
- WT
Wild type
Authors’ contributions
CS and BE have written the report. All the authors have reviewed the literature and contribute to the report. All authors read and approved the final manuscript.
Funding
Not applicable. No funding.
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Written informed consent for publication of their clinical details was obtained from the patient. A copy of the consent form is available for review by the Editor of this journal.
Competing interests
The authors declare that they have not competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Sergio Carrera, Phone: +34946006333, Email: sergio.carrerarevilla@osakidetza.eus.
Elena Beristain, Email: elena.beristainmendizabal@osakidetza.eus.
Aintzane Sancho, Email: aintzane.sanchogutierrez@osakidetza.eus.
Eluska Iruarrizaga, Email: eluska.iruarrizagaovejas@osakidetza.eus.
Pilar Rivero, Email: mariapilar.riverosobreviela@osakidetza.eus.
Juan Manuel Mañe, Email: juanmanuel.manemartinez@osakidetza.eus.
Guillermo López Vivanco, Email: guillermo.lopezvivanco@osakidetza.eus.
References
- 1.Mietinnen M, Lasota J. Gastrointestinal stromal tumors-definition, clinical, histological, immunohistochemical, and molecular genetic features and differential diagnosis. Virchow Archiv: Int J Pathol. 2001;438:1):1–1)12. doi: 10.1007/s004280000338. [DOI] [PubMed] [Google Scholar]
- 2.Mietinnen M, Majidi M, Lasota J. Pathology and diagnostic criteria of gastrointestinal stromal tumors (GISTs): a review. Eur J Cancer. 2002;38(Suppl 5):S39–S51. doi: 10.1016/S0959-8049(02)80602-5. [DOI] [PubMed] [Google Scholar]
- 3.Mietinnen M, Lasota J. Gastrointestinal stromal tumors: a review on morphology, molecular pathology, prognosis and differential diagnosis. Arch Pathol Lab Med. 2006;130(10):1466–1478. doi: 10.5858/2006-130-1466-GSTROM. [DOI] [PubMed] [Google Scholar]
- 4.Lasota J, Mietinnen M. Clinical significance of oncogenic KIT and PDGGFRA mutations in gastrointestinal stromal tumors. Histopathology. 2008;53(3):245–266. doi: 10.1111/j.1365-2559.2008.02977.x. [DOI] [PubMed] [Google Scholar]
- 5.Reichard P, Hogendoorn PC, Tamborini E, Loda M, Gronchi A, Poveda A, et al. Gastrointestinal stromal tumors 1: pathology, pathobiology, primary therapy an surgical issues. Semin Oncol. 2009;36(4):290–301. doi: 10.1053/j.seminoncol.2009.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Martin-Broto J, Rubio I, Alemany R, Lopez-Guerrero JA. Clinical implications of KIT and PDGFRA genotyping in GIST. Clin Transl Oncol. 2010;12(10):670–676. doi: 10.1007/s12094-010-0576-7. [DOI] [PubMed] [Google Scholar]
- 7.Mietinnen M, Lasota J. Succinate dehydrogenase deficient gastrointestinal stromal tumors (GISTs) – a review. Int J Biochem Cell Biol. 2014;53:514–519. doi: 10.1016/j.biocel.2014.05.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gopie P, Mei L, Faber AC, Grossman SR, mith SC, Boikos SA. Classification of gastrointestinal stromal tumor syndromes. Endocr Relat Cancer. 2018;25(2):R49–R58. doi: 10.1530/ERC-17-0329. [DOI] [PubMed] [Google Scholar]
- 9.Ricci R. Syndromic gastrointestinal stromal tumors. Hered Cancer Clin Pract. 2016;14:15. doi: 10.1186/s13053-016-0055-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nishida T, Hirota S, Taniguchi M, Hashimoto K, Isozaki K, Nakamura H, et al. Familial gastrointestinal stromal tumours with germline mutation of the KIT gene. Nat Genet. 1998;19:323–324. doi: 10.1038/1209. [DOI] [PubMed] [Google Scholar]
- 11.Carney JA, Stratakis CA. Stromal, fibrous, and fatty gastrointestinal tumors in a patient with a PDGFRA gene mutation. Am J Surg Pathol. 2008;32:1412–1420. doi: 10.1097/PAS.0b013e31816250ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ricci R, Dei Tos AP, Rindi G. GISTogram: a graphic presentation of the growing GIST complexity. Virchows Arch. 2013;463:481–487. doi: 10.1007/s00428-013-1467-4. [DOI] [PubMed] [Google Scholar]
- 13.Zöller ME, Rembeck B, Odén A, Samuelsson M, Angervall L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer. 1997;79(11):2125–2131. doi: 10.1002/(SICI)1097-0142(19970601)79:11<2125::AID-CNCR9>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 14.Wada R, Arai H, Kure S, Peng WX, Naito Z. “Wild type” GIST: Clinicopathological features and clinical practice. Pathol Int. 2016;66(8):431–437. doi: 10.1111/pin.12431. [DOI] [PubMed] [Google Scholar]
- 15.Settas N, Faucz FR, Stratakis CA. Succinate dehydrogenase (SDH) deficiency, Carney triad and the epigenome. Mol Cell Endocrinol. 2018;469:107–111. doi: 10.1016/j.mce.2017.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Smyrk TC, Young WF, Stratakis CA, Carney JA. Gastric stromal tumors in Carney triad are different clinically, pathologically, and behaviorally from sporadic gastric gastrointestinal stromal tumors: findings in 104 cases. Am J Surg Pathol. 2010;34(1):53–64. doi: 10.1097/PAS.0b013e3181c20f4f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gill AJ. Succinate dehydrogenase (SDH)-deficient neoplasia. Histopathology. 2018;72(1):106–116. doi: 10.1111/his.13277. [DOI] [PubMed] [Google Scholar]
- 18.Nishida T. Therapeutic strategies for wild-type gastrointestinal stromal tumor: is it different from KIT or PDGFRA-mutated GISTs? Transl Gastroenterol Hepatol. 2017;2:92. doi: 10.21037/tgh.2017.11.05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kays JK, Sohn JD, Kim BJ, Goze K, Koniaris LG. Approach to wild-type gastrointestinal stromal tumors. Transl Gastroenterol Hepatol. 2018;3:92. doi: 10.21037/tgh.2018.10.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gill AJ. Succinate dehydrogenase (SDH) and mitochondrial driven neoplasia. Pathology. 2012;44(4):285–292. doi: 10.1097/PAT.0b013e3283539932. [DOI] [PubMed] [Google Scholar]
- 21.van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EM, et al. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009;10(8):764–771. doi: 10.1016/S1470-2045(09)70164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, et al. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005;7(1):77–85. doi: 10.1016/j.ccr.2004.11.022. [DOI] [PubMed] [Google Scholar]
- 23.Benn DE, Robinson BG, Clifton-Bligh RJ. 15 YEARS OF PARAGANGLIOMA: clinical manifestations of paraganglioma syndromes types 1-5. Endocr Relat Cancer. 2015;22(4):T91–T103. doi: 10.1530/ERC-15-0268.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carney JA, Stratakis CA. Familial paraganglioma and gastric stromal sarcoma: a new syndrome distinct from the Carney triad. Am J Med Genet. 2002;108:132–139. doi: 10.1002/ajmg.10235. [DOI] [PubMed] [Google Scholar]
- 25.Haller F, Moskalev EA, Faucz FR, Barthelmeß S, Wiemann S, Bieg M, et al. Aberrant DNA hypermethylation of SDHC: a novel mechanism of tumor development in Carney triad. Endocr Relat Cancer. 2014;21(4):567–577. doi: 10.1530/ERC-14-0254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Horváth R, Abicht A, Holinski-Feder E, Laner A, Gempel K, Prokisch H, et al. Leigh syndrome caused by mutations in the flavoprotein (Fp) subunit of succinate dehydrogenase (SDHA) J Neurol Neurosurg Psychiatry. 2006;77(1):74–76. doi: 10.1136/jnnp.2005.067041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.NGS in PPGL (NGSnPPGL) Study Group. Toledo RA, Burnichon N, Cascon A, Benn DE, Bayley JP, Welander J, et al. Consensus statement on next-generation-sequencing-based diagnostic testing of hereditary phaeochromocytomas and paragangliomas. Nat Rev Endocrinol. 2017;13(4):233–247. doi: 10.1038/nrendo.2016.185. [DOI] [PubMed] [Google Scholar]
- 28.Parfait B, Chretien D, Rötig A, Marsac C, Munnich A, Rustin P. Compound heterozygous mutations in the flavoprotein gene of the respiratory chain complex II in a patient with Leigh syndrome. Hum Genet. 2000;106(2):236–243. doi: 10.1007/s004390051033. [DOI] [PubMed] [Google Scholar]
- 29.National Center for Biotechnology Information. ClinVar; Variation ID 8744, https://preview.ncbi.nlm.nih.gov/clinvar/variation/8744 (Accessed 31 Mar 2019).
- 30.Bausch B, Schiavi F, Ni Y, et al. Clinical characterization of the Pheochromocytoma and Paraganglioma susceptibility genes SDHA, TMEM127, MAX, and SDHAF2 for gene-informed prevention. JAMA Oncol. 2017;3(9):1204–1212. doi: 10.1001/jamaoncol.2017.0223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang Q, Zhang Y, Zhou YH, et al. A novel germline mutation in SDHA identified in a rare case of gastrointestinal stromal tumor complicated with renal cell carcinoma. Int J Clin Exp Pathol. 2015;8(10):12188–12197. [PMC free article] [PubMed] [Google Scholar]
- 32.Casey RT, Ascher DB, Rattenberry E, Izatt L, Andrews KA, Simpson HL, et al. SDHA related tumorigenesis: a new case series and literature review for variant interpretation and pathogenicity. Mol Genet Genomic Med. 2017;5:237–250. doi: 10.1002/mgg3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dwight T, Mann K, Benn DE, Robinson BG, McKelvie P, Gill AJ, et al. Familial SDHA mutation associated with pituitary adenoma and pheochromocytoma/paraganglioma. J Clin Endocrinol Metab. 2013;98(6):E1103–E1108. doi: 10.1210/jc.2013-1400. [DOI] [PubMed] [Google Scholar]
- 34.Boikos SA, Xekouki P, Fumagalli E, et al. Carney triad can be (rarely) associated with germline succinate dehydrogenase defects. Eur J Hum Genet. 2015;24(4):569–573. doi: 10.1038/ejhg.2015.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maniam P, Zhou K, Lonergan M, Berg JN, Goudie DR, Newey PJ. Pathogenicity and penetrance of germline SDHA variants in Pheochromocytoma and Paraganglioma (PPGL) J Endocr Soc. 2018;2(7):806–816. doi: 10.1210/js.2018-00120. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.