

Summary
Accumulation of senescent cells during aging contributes to chronic inflammation and age-related diseases. While senescence is associated with profound alterations of the epigenome, a systematic view of epigenetic factors in regulating senescence is lacking. Here, we curated a library of short hairpin RNAs for targeted silencing of all known epigenetic proteins and performed a high-throughput screen to identify key candidates whose downregulation can delay replicative senescence of primary human cells. This screen identified multiple new players including the histone acetyltransferase p300 that was found to be a primary driver of the senescent phenotype. p300, but not the paralogous CBP, induces a dynamic hyper-acetylated chromatin state and promotes the formation of active enhancer elements in the non-coding genome, leading to a senescence-specific gene expression program. Our work illustrates a causal role of histone acetyltransferases and acetylation in senescence, and suggests p300 as a potential therapeutic target for senescence and age-related diseases.
Keywords: chromatin, epigenetics, senescence, p300, enhancers
eTOC blurb
Epigenetic dysregulation is a hallmark of senescence/aging. In this article, Sen et al. have identified the histone acetyltransferase p300 as a key protein regulating senescence from a high-throughput screen. p300 induces formation of new super enhancers that drive senescence-related gene expression. p300 is an attractive candidate for anti-aging therapy.
Graphical Abstract
Introduction
Cellular senescence is a state of durable cell cycle arrest induced by stressors such as DNA damage, telomere erosion and oncogene activation(He and Sharpless, 2017). Senescent cells express high levels of β-galactosidase (SA-β-gal), p16, hypo-phosphorylated Rb, inflammatory cytokines (known as the senescence-associated secretory phenotype or SASP), and chromatin changes(Coppe et al., 2010). The latter include senescence-associated heterochromatic foci (SAHF)(Narita et al., 2003), inappropriate euchromatin(Shah et al., 2013) and chromatin fragmentation with cytoplasmic extrusion(Dou et al., 2017; Dou et al., 2015; Ivanov et al., 2013). The anti-proliferative property and acute SASP of senescent cells serves a tumor suppressive function, however, chronic SASP contributes to inflammation, tissue aging and disease(Coppe et al., 2010). Recent evidence points to the causal roles of senescent cells in decreasing healthspan and lifespan. For example, (1) aging human tissues show accumulation of senescent cells(van Deursen, 2014), (2) cells isolated from human patients suffering from premature aging syndromes show premature senescence in culture(Aliper et al., 2015), (3) targeted elimination of senescent cells extends healthspan and lifespan in mice(Baker et al., 2016; Baker et al., 2011), (4) “senolytic” compounds that induce senescent cell death show lifespan extension and therapeutic benefits in several age-associated diseases(Kirkland and Tchkonia, 2017) and (5) SASP mediates inflammation and cancer(Coppe et al., 2010).
Senescent cells accumulate in aged tissues in part due to replicative exhaustion and/or chronic telomere shortening in cells capable of division(Hornsby, 2002). This phenomenon, called replicative senescence (RS), can be recapitulated in vitro by repeated passage of cells in culture(Campisi, 1997). RS cells show profound chromatin reorganization with changes in both DNA and histone modifications(Sen et al., 2016). RS cells show a general loss of all canonical histones(O’Sullivan et al., 2010), upregulation of an alternative pool of histones and histone variants (including H3.3)(Rai et al., 2014), downregulation of lamin B1 and/or loss of nuclear-lamina associated heterochromatin(Dou et al., 2015; Shah et al., 2013). Concomitantly, a new chromatin landscape is formed that exerts a strong anti-proliferative, pro-senescence cellular phenotype. Our previous studies revealed broad areas of enrichment (mesas) in trimethylated lysine 4 on histone H3 (H3K4me3) and H3K27me3 over lamin B1-associated domains and depletion (canyons) of H3K27me3 in putative enhancers(Shah et al., 2013). Repressive H4K20me3 is increased in SAHFs and repeat elements(Nelson et al., 2016). These changes indicate a general reorganization of chromatin domains outside coding regions and predict consequential changes in gene expression.
Interest in the non-coding genome has recently surfaced with accelerated emphasis in the past few years(Li et al., 2016). Transcription factors (TFs) bind to enhancers and mediate RNA initiation from distal transcriptional start sites (TSS) of genes. Epigenome sequencing of human and mouse during development has revealed an enormous number of enhancers, which provide exquisite tuning of gene regulation(Atlasi and Stunnenberg, 2017; Rada-Iglesias et al., 2011). Enhancers are activated by relaxation of compact chromatin, which occurs by a poorly understood sequence of events. Histone modifying enzymes mediate deposition of marks such as H3K4me1 or H3K27ac, and bidirectional RNAs are transcribed by RNA polymerase II, which together, with additional known and unknown mechanisms, culminate in looping to the target promoter and initiation of mRNA transcription over gene bodies(Hnisz et al., 2013).
The functional importance of enhancers has been implicated by their mutations in diseases. For example, ENCODE sequencing of DNA revealed an unanticipated large number of mutations in enhancers acquired in disease states(Consortium et al., 2007). Sequencing of human genomes from cancer-derived tissues reveal that key enhancers tend to harbor mutations that disrupt binding of TFs(Hnisz et al., 2013). While the role of enhancers has been demonstrated in development and cancer, there is limited understanding of enhancer biology in senescence and aging. Enhancers are abundantly decorated with histone acetylation such as H3K27ac. Histone acetylation has long been implicated in yeast and Drosophila aging with an overall model wherein histone hypoacetylation prolongs lifespan by promoting autophagy, while suppressing oxidative stress and necrosis(Peleg et al., 2016) through (1) inhibition of acetyl-CoA producing enzymes, (2) spermidine supplementation or (3) inactivation of histone acetyltransferases (HATs). However, the role of histone acetylation in cellular senescence is not clearly understood.
In this work, we systematically screened epigenetic proteins to discover potential roles in RS, with the ultimate goal of identifying “druggable” targets and pathways to ameliorate age-related disease. We focused on the HAT p300, for further investigation given the importance of acetylation in aging and the important role of p300 at enhancers(Vo and Goodman, 2001). Our results suggest that p300 serves a specific role in senescence, and has the potential to be a novel anti-aging target.
Results
A high-throughput screen for epigenetic factors regulating RS
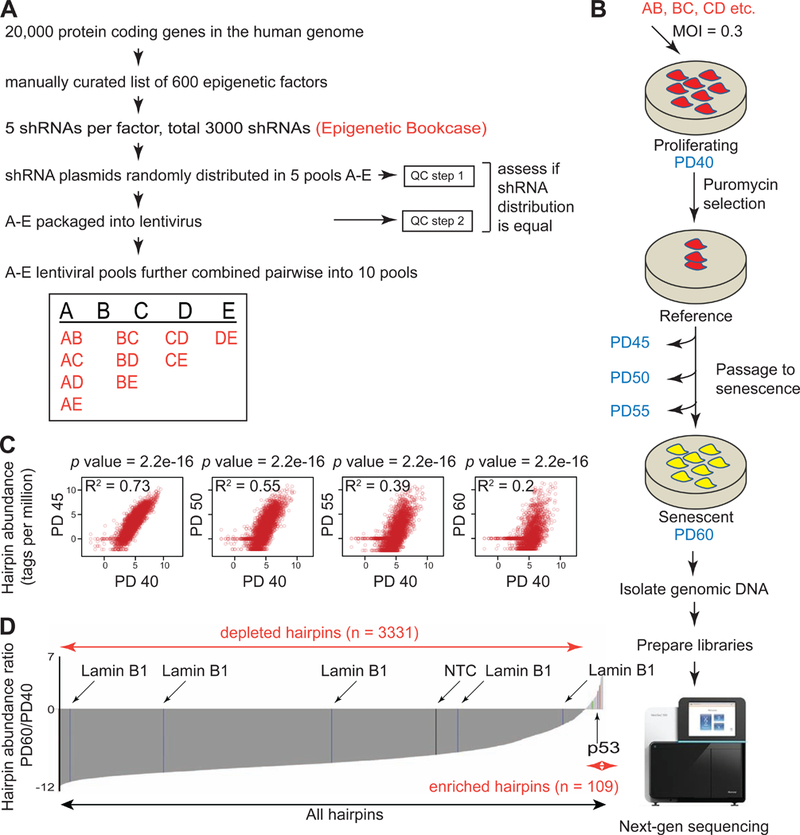
To discover chromatin-related pathways impacting senescence, we devised an epigenetics-focused pooled RNAi screen(Sims et al., 2011). A list of ~600 epigenetic factors was manually curated based on lists from Zuber (Zuber et al., 2011), Thermo Fisher, Dharmacon and Qiagen. A Gene Ontology (GO) analysis of the targets confirmed their multifaceted role in chromatin regulation (Figure S1A).
For the screen, 5 short hairpin RNAs (shRNAs) per target from The RNAi Consortium (total 3000 hairpins) were organized into 5 random pools A-E. Each pool included additional control hairpins verified to accelerate (lamin B1)(Shah et al., 2013) or delay (p53)(Qian and Chen, 2010) senescence, as well as a non-targeting control (NTC). Lentiviral supernatants were generated from these pools (see Methods) and combined into all possible pairwise combinations to form 10 pools that were used to infect 10 plates of IMR90 cells (Figure 1A). The combinatorial method was advantageous because each major pool is represented in quadruplicate in a single experiment, and each specific hairpin is present in different pools, reducing the likelihood of it being dependent on, or outcompeted by others.
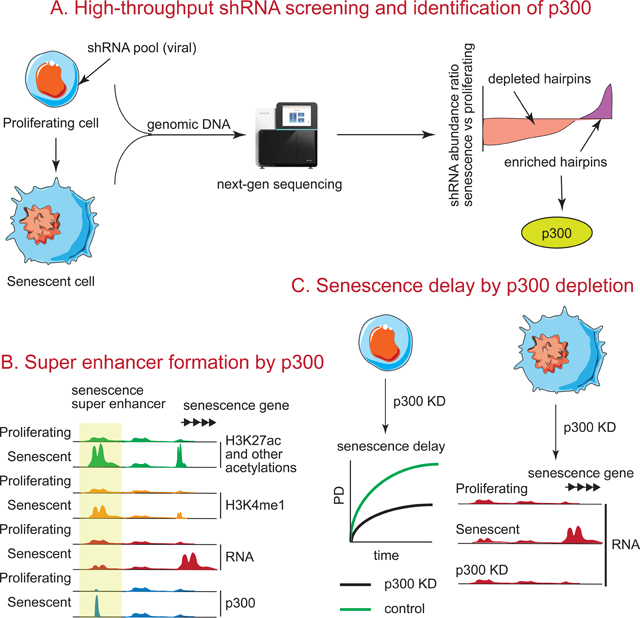
Figure 1: RNAi screen for identifying epigenetic factors that regulate RS.
(A) Workflow showing the construction of shRNA library. (B) Schematic of the RNAi screen performed in (A). (C) Scatter plot of shRNA abundances during passage to senescence relative to PD40. p values from Wilcoxon tests are indicated. (D) Abundance ratios (PD60 vs PD40) of all shRNAs used in the screen. Non-targeting (NTC), negative (Lamin B1) and positive (p53) control hairpins are indicated.
To confirm equal distribution of shRNAs across the pools, we tested their abundance in the plasmid and virus pool. The plasmid pool was either tested at this stage, or was amplified 15–20 cycles by PCR to assess amplification bias. Unamplified or amplified hairpins were digested with XhoI to generate ~300bp fragments containing the half hairpin sequence that could subsequently be used for sequencing without confounding issues due to secondary structure formation (see Methods). The same strategy was used for viral pools, except that viral RNA was converted to cDNA before PCR amplification.
The histogram (Figure S1B and TS1) summarizes tag distribution in the plasmids and viruses after sequencing. The main conclusions derived were: (1) the distribution was unimodal and did not change upon PCR amplification (blue and red) or packaging (green) relative to the total number of tags sequenced, (2) most of the hairpins were represented equally with the difference in copy numbers not exceeding 2.5 fold (considering 25th and 75th percentiles of the distribution) and, (3) 50% of the hairpins were present in at least ~220 copies (median tags per million). These values compared favorably to other commercial libraries such as the Decipher (Cellecta) or Mission (Sigma). The results thus confirmed equal representation of shRNA sequences in our custom pools and any enrichment or depletion in senescence would likely be due to the assay and not to an artifact of pool representation.
Proliferating IMR90 cells at population doubling (PD) 40 were infected with the 10 lentiviral pools (AB, BC, etc.; Figure 1B). At least 1000 cells per hairpin were infected to ensure quantitative estimation of sequences. To prevent multiple integrations and complex phenotypic effects, viruses were transduced at low multiplicity of infection of 0.3. After selection, cells were harvested and labeled as “reference” or “PD40”. The remaining cells were passaged, until RS was established (“senescent” or “PD60”) as assayed by SA-β-gal, growth cessation and absence of EdU incorporation (Figure S1D and data not shown). The replicative lifespan (RLS) curves of all the pools used in the screen are shown in Figure S1C. There was no difference noted in the overall RLS of any pool or the NTC, ruling out handling errors or biased pool composition. Genomic DNA was isolated from an aliquot of each of the 10 pools at every 5 passages (PD40–60), and was incorporated into sequencing libraries (Figure S1E-F).
To determine whether a specific hairpin promoted or delayed senescence, we quantified the abundance at the start (PD40) and end (PD60) of the senescence assay using next-generation sequencing. The sequenced DNA was aligned to a custom hairpin tag library. Alignment percentages across each pool and PD were similar (Figure S1G and TS2). Hairpins enriched in PD60 are predicted to delay senescence, while depleted hairpins, are predicted to cause either premature senescence, cell death, or an anti-proliferative response. Figure 1C shows scatter plots of shRNA abundance during the senescence assay. The progressively reduced R2 values at higher PDs are indicative of dramatic changes in the relative abundance of the shRNAs as cells establish RS. A Wilcoxon test on each time-point relative to PD40, showed that the ratios drastically deviate from 1, suggesting the distributions are significantly skewed with every tested passage. In Figure 1D, the enrichment profile for each shRNA is represented as a log2 ratio (PD60/PD40). The majority of the hairpins were depleted in RS cells, but a group of 109 hairpins corresponding to 100 protein targets (including positive control p53) was enriched and thus represented positive candidates for further investigation (TS2).
Depletion of the HAT p300 delays senescence
Top hits from the screen were chosen based on specific criteria. Initially, we focused on strongly enriched or depleted hairpins, which potentially delay or accelerate senescence respectively. We prioritized further investigation of enriched hairpins. To generate a rigorous list of true candidates, only those targets for which at least 2 shRNAs scored in the enriched category were selected for further validation. The selected shRNAs were tested for their ability to efficiently deplete (≥30%) their target by qPCR (Figure 2D and Figure S2E, 2K, 2Q and 2W) following which repeat RLS assays were performed in biological replicates. By imposing these criteria, we identified and validated several targets (Figure S2; all shRNAs used for individual knockdowns [KDs] are listed in the Key Resources Table). To maximize the power of our longitudinal assay while correcting for statistical dependence between time points, we adopted a repeat measure linear model (see Methods). By a generalized least squares regression, we found that there was a significant time-dependent increase in RLS upon treatment with p300, H2A.Z and MEN1-targeting shRNAs (time:treatment, Figure 2A-F and Figure S2M-X). DDB1 and RPA1 KD did not pass the statistical test in the validation experiments (Figure 2F and Figure S2A-L) despite an effective reduction of DDB1 and RPA1 at the RNA level (Figure S2E and S2K). We also estimated cell viability (based on trypan blue exclusion) throughout the assay to rule out increased cell death (Figure 2E and Figures S2F, S2L, S2R and S2X). Overall viability remained high throughout the assay except during puromycin selection.
Figure 2: Validation studies with p300 hairpins that emerged as potential candidates from the RNAi screen.
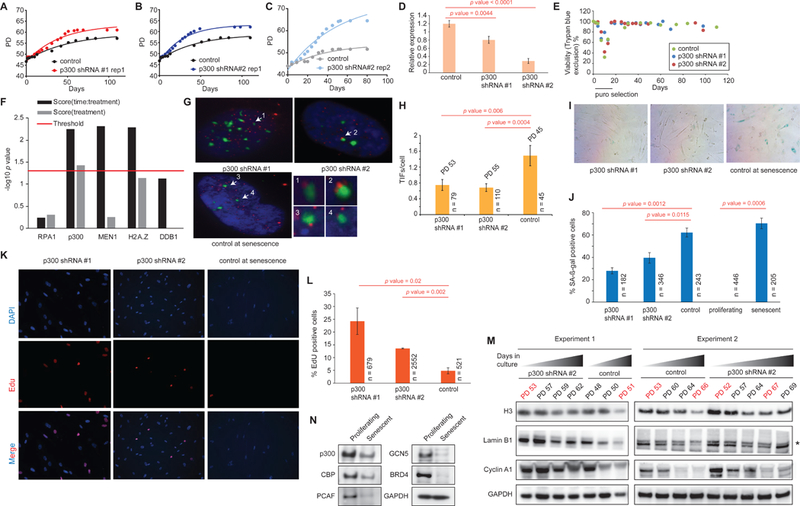
(A-C) RLS curves of cells harboring hairpins targeting p300 or a control. (D) qPCR showing KD efficiency of p300 hairpins relative to a housekeeping gene. (E) Representative viability plot during an RS assay with cells harboring p300 KD using trypan blue exclusion. (F) Plot showing p values of lifespan experiments in (A-C) and Figure S2A-X using a repeat measure analysis. The red line indicates the threshold for significance (p=0.05). (G) IF images showing TIF formation in cells harboring a control or p300 hairpin. TIFs (arrow) are assigned if there is co-localization of 53BP1 (green) and Cy3-labeled telomere end (red). Insets show a magnified view of the TIFs. (H) The number of TIFs per cell are counted and plotted. (I) Representative SA-β-gal staining in cells harboring control or p300 hairpins. (J) The number of β-gal positive cells represented as a percentage of total counted cells. (K) IF images showing EdU incorporation in cells harboring a control or p300 hairpin. (L) The percentage of EdU incorporation is estimated by counting the percentage of cells (DAPI, blue in K) that are stained with EdU (red in I) and plotted as a bar plot. (M) Western blots from 2 independent RLS experiments showing the downregulation of histone H3, lamin B1 and cyclin A1 during passage to senescence of cells harboring either p300 or control hairpin. Note that at comparable PDs (indicated in red), cells that are depleted in p300 retain more H3, lamin B1 and cyclin A1 compared to control. (N) Western blots showing protein levels of multiple HATs in proliferating and senescent cells including CBP and p300. n = number of cells scored, p value estimates are from unpaired t-tests.
Further extensive analyses focused on p300. p300, and paralog CBP, are the major HATs in multicellular eukaryotes regulating enhancers, and have been implicated in numerous human diseases associated with aging(Valor et al., 2013). A range of compounds targeting p300 and CBP (such as small molecules C646, EML425 and other natural products) are available for therapeutic interventions(Dancy and Cole, 2015).
Validation experiments confirmed that cells harboring p300 hairpins underwent ~6–7 more PDs than the control (~32–64-fold increase in cumulative cell number) before reaching RS (Figure 2A-C) accompanied by a significant time delay in RS establishment (Figure 2G-L). In our hands, shRNA #2 most robustly and reproducibly knocked down p300 at the protein and RNA levels and was therefore preferred for downstream experiments. Using four independent assays, we estimated the time delay at a point when cells with the control hairpin had ceased to grow, while those harboring p300 KD were still slowly proliferating: (1) we estimated the total number of Telomere dysfunction-Induced Foci (TIFs) which form when DNA damage proteins bind to uncapped, short telomeric ends at RS (Takai et al., 2003). We detected and quantified TIFs per cell by immunofluorescence (IF) studies where 53BP1 (green) was co-localized to Cy3 (red) labeled telomere probes (Figure 2G arrows, enlarged in inset). The quantification revealed that cells harboring p300 KD had approximately 2-fold fewer TIFs per cell (Figure 2G-H). (2) A semi-quantitative SA-β-gal staining also showed a significantly lower percentage of β-gal positive cells in the p300 KD (Figure 2I-J). (3) We quantified the percentage of EdU positive cells, as low EdU incorporation index (≤5%) implies full establishment of senescence (Wei and Sedivy, 1999). In our assay, cells harboring the control hairpin had ~4.8% EdU positive cells, those with p300 hairpins had ~2.8 to 5-fold more EdU positive cells, supporting senescence delay (Figure 2K-L). (4) We measured the kinetics of lamin B1, Cyclin A1 and histone H3 downregulation by western blot in a time-course experiment (O’Sullivan et al., 2010). While cells harboring the control hairpin progressively downregulated Lamin B1, Cyclin A1 and H3 with increased passages, cells harboring p300 hairpin only slightly downregulated these protein levels despite being at a higher PD (Figure 2M). Taken together, these results confirmed that reducing p300 levels in cells significantly delayed the onset of RS without any evidence of immortalization. Additionally, we investigated the status of p300 protein levels in RS by Western blotting. As comparison, we also probed the levels of other well-known HATs such as CBP, PCAF, GCN5 and BRD4. Our results show that overall, the levels of all HATs decline in RS but comparatively, p300 levels decrease modestly (approximately 2-fold; Figure 2N).
We speculated that if depletion of p300 delayed RS, overexpression of p300 will cause premature senescence. Thus, we overexpressed p300 in IMR90 cells and passaged them to senescence. p300 overexpression was evident (~2.3 fold) and specific (with no evidence of CBP being overexpressed; Figure S2Y), and we observed a slight (Figure S2Z-AA) but significant (Figure S2AH left) decrease in RLS. This decrease in RLS is more notable when the data are plotted in cumulative cell numbers rather than PDs (Figure S2AG left). Viability remained high throughout the time-course (Figure S2AB). In contrast, overexpression of CBP (Figure S2AC), did not lead to significant decrease in RLS (Figure S2AD-AF, 2AH right). These data imply that increased levels of p300 (but not CBP) slightly decreases RLS, but is insufficient to cause robust premature senescence, and likely requires additional changes in TFs, modifications, or proper chromatin environment.
Senescent cells contain hyper-acetylated histones that are putative targets of p300
The role of p300 in promoting senescence, led us to examine RS cells for enrichment of acetylated histones, which are the best characterized targets of p300. We took a proteomics approach, first, isolating nuclei and then acid-extracting total histones. Using a peptide-centric analysis via mass spectrometry with label-free quantification (see Methods), we measured the relative abundance of all detectable modifications in canonical and variant histones (TS3). As proof-of-principle we quantified the relative ratio of H3.3 to H3 and the relative abundance of H4K20me3, both predicted to increase in senescent cells(Nelson et al., 2016; Rai et al., 2014). As expected, we noticed significant enrichment of these positive controls (Figure 3A and 3G). With this assurance, we plotted the relative abundance of various acetylated peptides. Notably, among all modified H3 and H4 peptides, acetylated peptides in general showed a higher enrichment in senescent cells (Figure 3B-E) with only 4 acetylated peptides being relatively less abundant in senescence. Importantly, these latter peptides were detected at <2% abundance (Figure 3E) and therefore cannot be confidently quantified. Moreover, 3 of the 4 acetylated peptides that were downregulated in senescence were mono-acetylated; it is expected that an increase in peptides harboring multiple acetylations co-occurs with a decrease in mono-acetylated forms. While not all of these changes reached significance across three biological replicates (Figure 3D), the overall abundance of acetylated histones in RS was highly reproducible within each individual experiment (three independent biological replicates, Figure 3C). Acetylated peptides from H3.1/2 (Figure 3F-H), H3.3 (Figure 3H) and H4 (Figure 3I) were then individually assessed. Interestingly, a number of well-known p300 targets (H3Kl22ac, H3K18acK23ac on H3.1 and H3K27acK36ac on H3.3) were enriched in senescent cells(Weinert et al., 2018). There was also a significant decrease in the abundance of the H4 tail peptide (H4 4–17, with 4 possible acetylation sites at K5/8/12/16) acetylated on a single lysine (H4–1ac) which was also the only highly abundant acetylation quantified with confidence as decreasing in senescence. Instead, there was a marked shift towards multiple acetylated forms (H4–2ac, 3ac and 4ac; Figure 3I). In conclusion, these results demonstrate that senescent cells are enriched in hyper-acetylated histones that are likely targets of p300.
Figure 3: Histone peptides are hyper-acetylated in senescence.
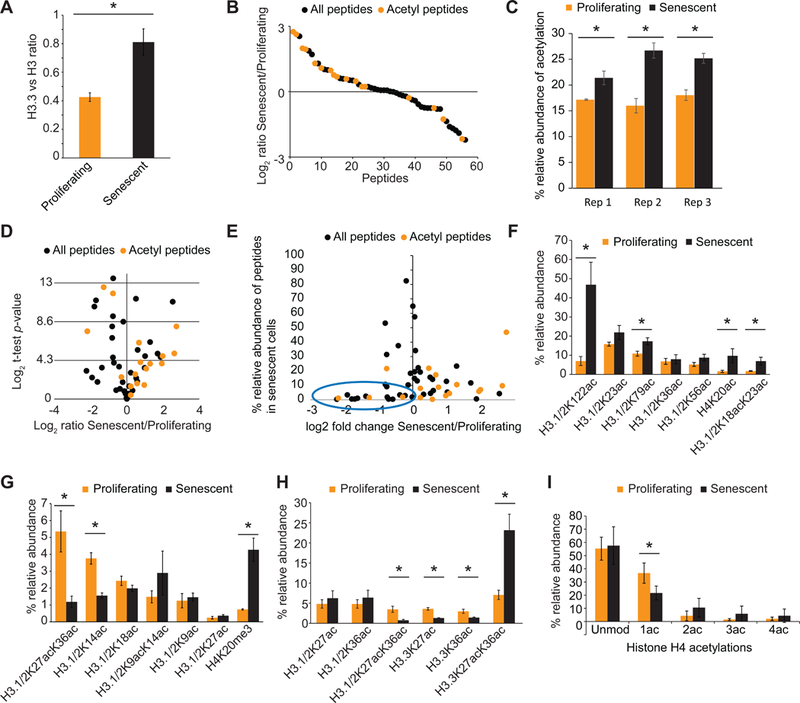
(A) Ratio of H3.3 to H3 increases in senescence as previously reported. (B) Ranked enrichment of all histone peptides detected by mass spec are plotted. Many acetylated peptides (red) are enriched in senescence compared to other peptides (black). (C) The relative abundance of acetylated peptides in RS is significantly higher than in proliferating cells in all 3 biological replicate samples. (D) Volcano plot (fold enrichment vs significance) of peptides in 3 replicate mass spec experiments. Peptides above y = 4.3 represent significantly altered peptide abundances. (E) Fold change vs abundance graph showing that acetylated peptides that are decreased in senescence have very low abundance (blue oval). (F) Relative abundance of several acetylated H3.1 peptides that increase in RS. (G) Relative abundance of acetylated H3.1/H3.2 peptides that decrease in RS. H4K20me3 is a positive control. (H) Relative abundance of several acetylated H3.1/3.2 and H3.3 peptides that are altered in RS. Note that H3.1/H3.2K27acK36ac decreases while H3.3K27acK36ac increases significantly upon senescence establishment. (I) Plot showing the abundance of single and multiple acetylations on the H4 4–17 peptide. Note that single acetylations decrease significantly with a shift towards multiple acetylated forms. Asterisk indicates a p<0.05 as assessed by unpaired t-test across 3 biological replicate experiments. Proliferating cells were at PD 26, 31 and 34 while senescent cells at PD 75, 75 and 78.
Histone acetylation is enriched over the non-coding genome during RS establishment
To assess the genome-wide distribution of acetylated histones in RS cells, we performed ChIP-seq of select histone acetyl marks identified in mass spec studies (H3K18ac, H3K23ac, H3K27ac, H3K122ac and H4K5ac). Antibodies raised against the C-terminus of H3 and H4 were used as total histone controls and IgG as a specificity control. Reads from two independent biological samples were combined for calling peaks (TS7). Notably, in both proliferating and RS cells, the peaks were primarily intergenic or intronic suggesting they mark non-coding regulatory elements in the genome (Figure S3A). With the exception of H3K23ac, the majority of other acetyl marks were located ≥5Kb away from an annotated TSS (Figure S3B). The enhancer landscape was visualized by mapping both H3K4me1 and H3K27ac(Creyghton et al., 2010). We called approximately similar numbers of H3K27ac peaks in RS and proliferating cells (~185K and ~142K respectively; Figure S3C).
To further classify the different types of peaks, the previous definition of “super” enhancers (SEs) and “typical” enhancers (TEs) based on Mediator occupancy was slightly modified (see Methods) to accommodate accurate enhancer calling using the H3K27ac mark(Whyte et al., 2013). Briefly, we stitched peaks that were within 12.5Kb of each other, determined their width (Kb) and normalized tag density (RPM/bp). Peaks were designated as SEs if their width was ≥3Kb and mean tag density ≥2RPM/bp (Figure S3D and S4B). Using these criteria, we captured an approximate point in the intensity versus peak rank plot where the H3K27ac signal increased rapidly (Figure S4A red dot). With this cutoff, we called ~4300 SEs in proliferating cells, ~2800 SEs in RS cells, ~137K TEs in proliferating cells and ~183K TEs in RS cells (Figure S3C and S3E and TS7). However, despite the comparable peak numbers, senescent cells had ~5 fold more activated SEs (11 versus 55) and ~2.5 fold more activated TEs (10852 versus 27571). These “Senescence-Activated” peaks were defined based on stringent criteria: at least 4 fold higher signal over proliferating and a p<0.0001 (Figure S4C-D and TS7) based on stringent criteria reported previously(Tasdemir et al., 2016). Heatmap/metaplot of H3K27ac (Figure 4A-B) and individual browser track views of H3K4me1 and numerous acetylation marks (Figure 4C) of these Senescence-Activated enhancer regions showed strong enrichment, confirming that our bioinformatic analysis accurately captured putative SEs and TEs licensed during RS. Importantly, 54 out of the 55 Senescence-Activated SEs and 17407 out of 27571 Senescence-Activated TEs have increased H3K4me1 (Figure S5A). We noticed that SEs as defined by H3K27ac enrichment (n = 55), tended to be enriched with other acetylation marks such as H3K18ac, H3K122ac and H4K5ac (Figure S5A). Enrichments in multiple acetylation marks was not evident for all TEs (n = 27571; Figure S5B) or an equal number (n = 55) of randomly selected TEs (Figure S5C). However, the top 55 TEs showed an enrichment of multiple acetylation marks much like SEs (Figure S5D). This top subset of TEs had features that resembled SEs such as an overall larger mean width. In conclusion, large regions of the non-coding genome decorated with multiple acetylations were activated in senescence.
Figure 4: New enhancers are licensed during RS establishment.
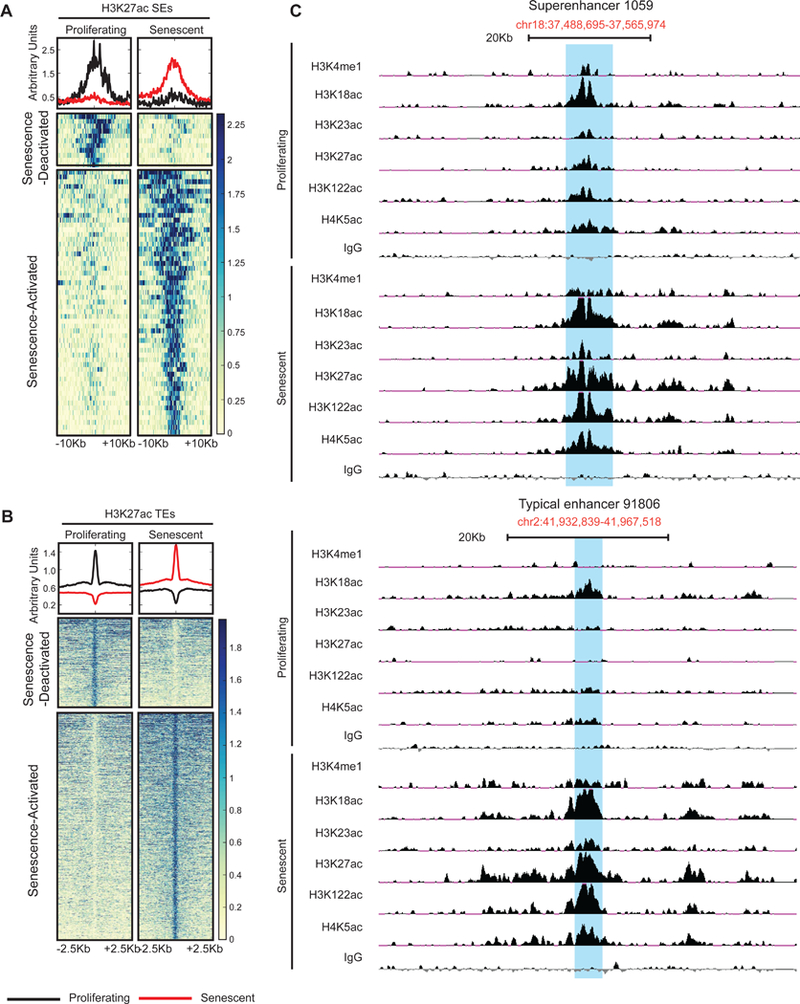
(A-B) Metaplot and heatmap of Senescence-Activated and -Deactivated SEs (A) and TEs (B) based on H3K27ac signal. (C) Browser track view of an SE (top) and TE (bottom). For histone acetylation and H3K4me1 ChIP-seq, proliferating cells were at PD29–36, and senescent cells at PD75–79 (see TS4 for details).
Senescence-Activated SEs actively transcribe eRNAs
SEs are known to be proximal to genes that determine cell type specificity and master regulators of cell fate decisions. For example, in embryonic stem cells SEs form near pluripotency genes(Whyte et al., 2013); in cancer cells SEs are enriched near oncogenes(Chapuy et al., 2013; Hnisz et al., 2013; Loven et al., 2013); in oncogene-induced senescence (OIS), SEs are near SASP genes(Tasdemir et al., 2016). In addition, SEs are highly enriched in trait-associated variants and disease-associated SNPs(Hnisz et al., 2013). Thus, we investigated whether our Senescence-Activated SEs were functionally active and whether they correlated with senescence-associated gene expression. For this purpose, we generated total (ribo-depleted) and run-on (PRO-seq) RNA-seq libraries from proliferating and RS cells (Figure 5A). As opposed to total RNA-seq which measures steady state stable RNA levels, PRO-seq measures nascent transcripts at base-pair resolution using a global adaption of nuclear run-on assay(Mahat et al., 2016). Thus, PRO-seq is able to quantitatively capture low abundance, unstable transcripts such as enhancer RNAs (eRNAs) providing an independent measure of enhancer activity(Li et al., 2016). We defined target genes with the nearest annotated TSS and envision that the enhancer loops over to the gene to regulate its expression (Figure 5B).
Figure 5: Senescence-Activated SEs correlate with senescence-related gene expression.
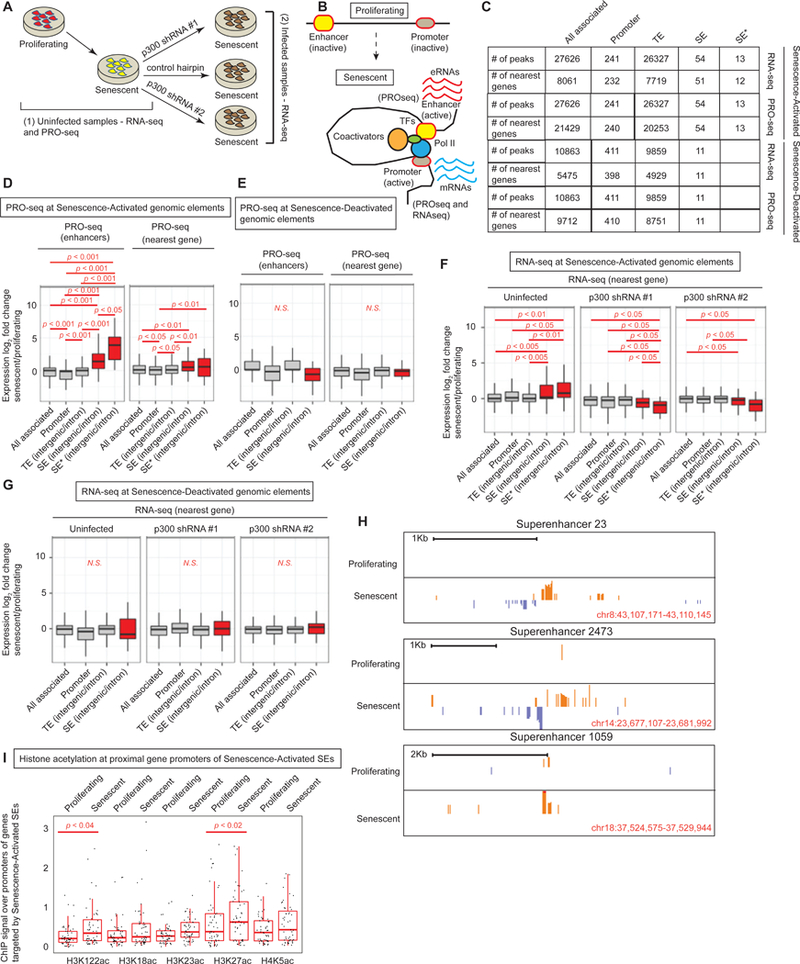
(A) Schematic showing samples for RNA- and PRO-seq libraries; (1) from proliferating and senescent uninfected cells and (2) senescent cells harboring control or p300-targeting hairpins. (B) Schematic showing the licensing of new enhancers in senescence with chromatin looping, engagement of transcription machineries and production of eRNAs and mRNAs. (C) Table showing the numbers of genomic elements (all, promoter, TE, SE and SE*) and their target genes used to draw box plots in D-G in proliferating and senescence conditions. SE* represents SEs with multiple acetylations. (D) Box plot of fold change of PRO-seq signal (senescence vs proliferating) across different genomic elements in senescence (TE, SE and SE*) measured at the enhancers (left) and the nearest target gene (right). (E) Same as (D) except the genomic elements are called in proliferating cells. (F) Box plot of fold change of RNA-seq signal (senescence vs proliferating or control vs KD) across different genomic elements called in senescence. (G) Box plot of fold change of RNA-seq signal (senescence vs proliferating) across different genomic elements called in proliferating cells. (H) Browser track views of PRO-seq signal at 3 SEs. 2-tailed Mann-Whitney-Wilcoxon Test was used to compare bins within and between boxplots. (I) ChIP signal of various histone acetylations at the promoters of the nearest genes identified as targets of the Senescence-Activated SEs. For PRO-seq, proliferating cells were at PD 20 and senescent cells at PD 76. For RNA-seq, proliferating cells were at PD 29 and 34 while senescent cells were at PD 78 and 79.
In order to assess activity of Senescence-Activated enhancers, we tested for eRNA production. There were significant increases in the PRO-seq signal at SEs (H3K27ac enriched) and SE*s (multiple acetyl marks enriched; numbers of these elements in the genome shown in Figure 5C) compared to “all-associated” (representing all genomic elements with H3K27ac enrichments; Figure 5D). Interestingly, PRO-seq signal at SE* elements showed a more pronounced increase in eRNA (Figure 5D left and TS7). Senescence-Activated TEs did not show this general trend which is probably linked to their general lack of histone acetylation at multiple sites compared to SEs. These data indicate that eRNA production is related to histone acetylation diversity and dosage. As a control, Senescence-Deactivated SEs did not show an increased PRO-seq signal (Figure 5E left and TS7). Browser track views of PRO-seq signal over some Senescence-Activated SEs can be visualized in Figure 5H. Taken together, these data imply that Senescence-Activated enhancers are not merely enrichments in histone acetylation but that they are active and functional regulatory elements.
Senescence-Activated SEs correlate with senescence-specific transcriptome profiles
In addition to eRNA production, we tested for nascent (PRO-seq) and steady state mRNA (total RNA-seq) generation at proximal gene targets of the Senescence-Activated enhancers. Our analyses revealed that genes proximal to Senescence-Activated SEs and SE*s correlated strongly with senescence-associated gene expression measured by both PRO-seq (Figure 5D right, compare SE and SE* to all-associated) and total RNA-seq (Figure 5F left). Interestingly, this positive correlation was not evident with other genomic elements such as promoters and TEs. As an additional control, we correlated Senescence-Deactivated SEs to gene expression in senescent cells and no correlation was found (Figure 5E right and Figure 5G left). Furthermore, to impart confidence in our assignment of target genes (i.e. genes most proximal to the SE element), we tested histone acetylation status of the proximal gene promoter. We noted that H3K122ac and H3K27ac was significantly increased at these locations (Figure 5I). Overall, these data demonstrate that SEs best predict the expression of target genes in senescence.
p300 depletion attenuates expression of SE target genes
To determine whether SE and SE* activation of target genes in senescence is directly dependent on p300, we depleted p300 in cells that had already established RS (Figure 5A). This was a critical strategy - to deplete p300 in senescent cells instead of in proliferating cells - to minimize expression differences due to unequal passage number and to evaluate senescence genes other than those that alter the cell cycle. We used 2 different shRNAs targeting p300 and a control hairpin. After KD and selection, we isolated total RNA and performed RNA-seq on ribo-depleted samples. Correlation of senescence-associated gene expression (as identified from the uninfected proliferating and senescent samples; Figure 5F left) in cells harboring p300 hairpins showed a significant downregulation if the genes were near SE or SE* (Figure 5F middle and right) but not in other genomic elements such as promoter or TE. Additionally, this downregulation was not evident for genes located near Senesence-Deactivated SEs (Figure 5G middle and right). These results demonstrate at the genome-wide level, that target genes proximal to Senescence-Activated SEs not only best predict gene expression, but importantly, are regulated by p300.
TEs with high density of histone acetylation behave like SEs
Given our previous observation that the top 55 TEs showed an enrichment of multiple acetylation marks much like SEs, we wanted to probe the overall lack of correlation of gene expression to all TEs. To dissect TE function in greater resolution, we divided TEs into 14 unequal-sized quantiles based on H3K27ac signal in RS and compared the fold change in gene expression. As expected, we found a strong positive correlation of the top Senescence-Activated TEs with both total (Figure 6A) and run-on (Figure 6C) transcription at the target genes and at the enhancer sites (Figure 6E) in senescence. The correlation was particularly strong in the top TE quantile as evidenced by lower p values (TS6). As expected, Senescence-Deactivated TEs had opposite effects, in that their expression was negatively correlated to senescence-associated gene expression (Figure 6B, D, F and TS6). These data suggest an association of senescence-related gene expression to histone acetylation dosage at specific sites in the non-coding genome.
Figure 6: TEs highly enriched with histone acetylation signal also correlate with senescence-related gene expression.
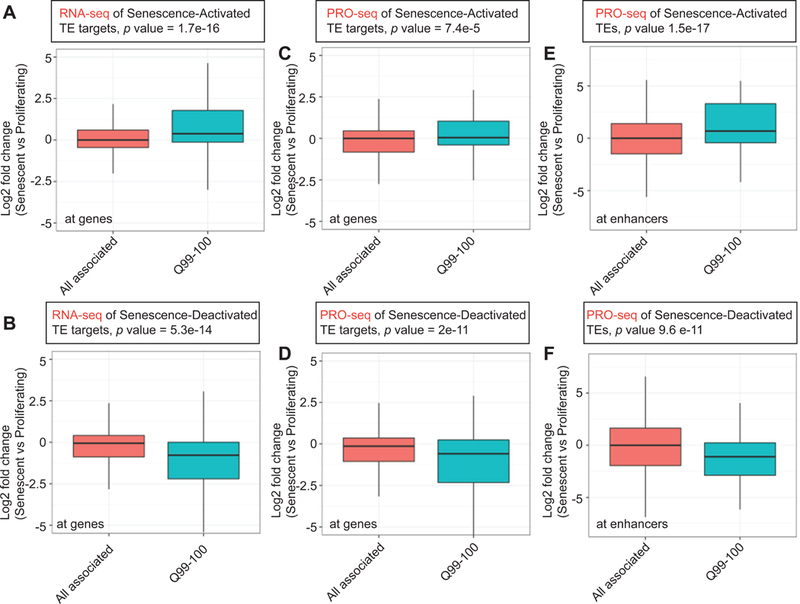
(A) Senescence-Activated TEs were partitioned into 14 unequal sized quantiles and the fold change of RNA-seq signal at target genes plotted. p values are recorded from t-tests comparing “all associated” to Q99–100 (Figure) or each quantile (TS6). (B) Same as in (A) except RNA-seq signal at Senescence-Deactivated TEs are plotted. (C-D) Same as in (A and B) except fold change of PRO-seq signal at the gene targets is plotted. (E-F) Same as in (C and D) except fold change of PRO-seq signal at the TEs (not target genes) is plotted.
Senescence-Activated SEs drive the expression of membrane-bound genes that regulate metabolic pathways
To gain insight into the functional pathways activated by p300-mediated acetylation at Senescence-Activated SEs, we performed GO and Ingenuity Pathway Analysis on the list of proximal target genes. Despite the small gene list, we noted significant pathways related to superoxide production, to integral membrane components including the peroxisome, and to NAD salvage and nucleotide metabolism pathways (Figure S6A-C). For example, NOX4 (NADPH oxidase 4) generates reactive oxygen species (ROS) and is capable of inducing nuclear DNA damage(Lener et al., 2009). FAR2, is a peroxisomal protein that also binds NADP and catalyzes the reduction of saturated but not unsaturated C16 or C18 fatty acyl-CoA to fatty alcohols(Cheng and Russell, 2004). PEX2 is a protein involved in peroxisome biogenesis and implicated in the negative regulation of fibroblast proliferation(Shimozawa et al., 1992). Overall, these terms indicate that SEs in RS drive the expression of genes that produce metabolic byproducts contributing to senescence. These metabolic pathways contrast with a previous study investigating OIS where most Senescence-Activated SEs drive the expression of SASP(Tasdemir et al., 2016).
Senescence-Activated SEs are occupied by p300
Our initial findings in the RNAi screen revealed p300, but not CBP, as a candidate driving RS (Figure 2). We thus explored the connection between SEs that drive gene expression in senescence and occupancy by p300 and CBP in proliferating and RS cells. Both p300 and CBP are known to occupy promoter and gene distal enhancer regions(Visel et al., 2009). Additionally, p300 and CBP are largely considered to be redundant in their genome-wide occupancies and functional targets(Vo and Goodman, 2001). We were intrigued therefore to note that only p300 depletion delayed RS in our initial screen, and further probed the distinct contribution of CBP and p300.
We pooled reads from two ChIP-seq experiments to investigate p300 and CBP binding in proliferating and RS cells. Overall, we called ~28K and ~23K peaks for CBP and ~32K and ~36K peaks for p300 in proliferating and RS cells, respectively. Systematic compartment analyses confirmed previous reports of p300 and CBP binding primarily at intergenic/intronic regions (Figure S6D), being mostly 5->100Kb away from an annotated TSS (Figure S6E) and within 5Kb of an enhancer (proximal) with a small proportion >50+Kb (distal) away in both proliferating and senescent cells (Figure S6F-I). Upon closer investigation, the latter set of distal CBP peaks appeared to be weak and located primarily in intergenic or intronic locations (data not shown).
We then investigated whether Senescence-Activated SEs are occupied by p300 and/or CBP. Normalized tag densities of p300 and CBP were measured at the 55 SEs of interest (see Methods for identifying p300/CBP peaks overlapping with an SE). A dot plot (Figure 7A) shows the distribution of enrichments under proliferating and senescent conditions. Although these SEs gained both CBP and p300, we noted that 36 of the 55 SEs gained p300 binding in senescence (26 of which are new binding sites not present in proliferating), and among 27 SEs that gained CBP, 23 of them gained p300 at the same time. This result suggested that p300, rather than CBP, is the major driver of senescence-related acetylation at these sites. In contrast, the top 55 TEs (based on H3K27ac signal in senescence) did not gain any significant amount of either CBP or p300 in senescence (Figure 7B) but rather retained a basal amount of binding. Conversely, SEs (Figure 7C) and TEs (Figure 7D-E) that are deactivated in senescence, showed a preference for cBp rather than p300 occupancy in proliferating cells. Genome browser snapshots of p300 and CBP binding at some Senescence-Activated SEs are shown in Figure 7F-I, and illustrate the greater accumulation of p300 relative to CBP. Taken together, these data suggest that p300 is a major driver of de novo enhancer formation in senescence.
Figure 7: p300 but not CBP binding drives new SE formation in senescence.
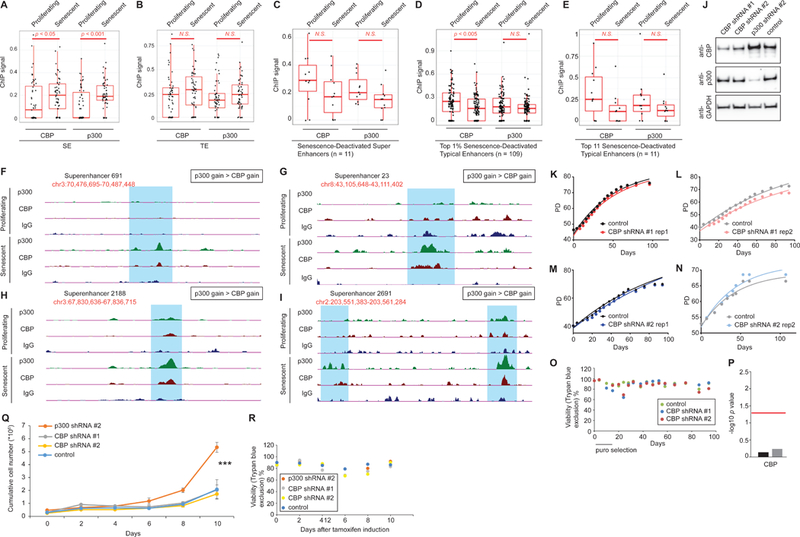
(A) Dot plot showing the enrichment of p300 and CBP over 55 Senescence-Activated SEs. (B) Dot plot showing the enrichment of p300 and CBP over op 55 Senescence-Activated TEs. (C-E) CBP and p300 occupancy at all Senescence-Deactivated SEs (C), top 1% Senescence-Deactivated TEs (D) and top 11 Senescence-Deactivated TEs (E). (F-I) Browser track views of p300/CBP signal at 4 SEs. (J) Western blot showing the shRNA-mediated depletion of p300 and CBP in cells used for RS assays in (K-N). (K-N) RLS curves of cells harboring shRNAs targeting CBP in multiple biological replicate experiments. (O) Representative viability plot during an RS assay with cells harboring CBP KD. (P) Plot showing p values of RLS experiments in (K-N) using a repeat measure analysis. The red line indicates the threshold for significance (p=0.05). (Q) Plot showing cumulative cell numbers in an oncogene-induced senescence assay with cells harboring p300, CBP or control hairpins. Asterisk indicates a p<0.0001 in a 2-way ANOVA test. (R) Representative viability plot during the oncogene-induced senescence assay shown in (Q). For p300/CBP ChIP-seq, proliferating cells were at PD 30 and 25, and senescent cells at pD 78 and 76.
CBP depletion does not delay RS
To more definitively compare the importance of p300 vs CBP function in senescence establishment, we tested hairpins targeting CBP individually in 2 models of senescence. To ensure reproducibility and integration of multiple copies for robust KD, we tested several hairpins and a few viral titrations. The hairpins/titrations that knocked down CBP successfully by western blot are shown (Figure 7J). Figure 7J also shows the specificity of the antibodies we used, as p300 was not depleted in cells harboring a CBP shRNA and vice versa. We then tested the RLS of these cells harboring the correct CBP hairpins but failed to see any delay compared to a control hairpin (Figure 7K-N). Despite the high viability of cells throughout the experiment (Figure 7O), rigorous statistical testing using a repeat measure model confirmed that the RLS changes with CBP KD were not significant (Figure 7P) as compared to clear lifespan reduction with p300 KD (see Figure 2A-F). Using an independent tamoxifen-inducible ER-Ras senescence induction model(Young et al., 2009), we tested whether p300 (but not CBP) KD delays the onset of oncogene-induced senescence (OIS). We observed that cells with KD of p300 were able to maintain a mitotic phase (evident as significantly higher cumulative cell numbers at the end of the assay) for a longer time compared to control (Figure 7Q). In clear contrast, CBP KD failed to show this delay in senescence establishment (Figure 7Q). Viability of cells was high throughout the 10 day cell sampling period (Figure 7R).
We also investigated the effect on RLS upon CBP overexpression because p300 overexpression showed significant decrease of cell proliferation (see above). Similar to Figure S2Y-AA, CBP was overexpressed in proliferating cells and passaged to senescence. CBP overexpression was robust (~4.2 fold; Figure S2AC) but no significant time-dependent change in RLS was noted (Figure S2AD-AE, 2AG right and 2AH) and viability remained high throughout the assay (Figure S2AF).
Additionally, CBP KD in cells that were in stable senescence (compare to Figure 5A sample 2, except with CBP hairpins) failed to significantly lower senescence-specific gene expression (Figure S7) – in clear contrast to p300 KD strongly reducing gene expression (Figure 5F). Collectively, we conclude that p300, and not CBP, drives new SEs which in turn elevate senescence-associated gene expression.
Discussion
Our results provide evidence that the HAT p300, is a primary driver of the RS phenotype, linked to its ability to license new SEs. New Senescence-Activated SEs are enriched in H3K4me1 and multiple histone acetyl marks (Figure 4 and Figure S5A), actively transcribe eRNAs (Figure 5D and 5H), gain p300 binding (Figure 7), and direct senescence-specific gene expression (Figure 5F left). Importantly, depletion of p300 alone was sufficient to downregulate senescence genes (Figure 5F middle and right) and extend RLS (Figure 2A-C and 2F). We additionally demonstrate non-redundancy of the role of p300 in senescence as depletion of its paralog CBP, is unable to phenocopy the senescence delay in both RS and OIS models (Figure 7K-R). We also confirm differential recruitment of p300 and CBP at licensed enhancers with a predominant bias towards p300-mediated regulation (Figure 7A). To our knowledge, this is the first evidence of a unique functional role for p300 despite previous speculation based on distinct occupancy and unique disease profiles of p300 and CBP(Vo and Goodman, 2001).
We discovered the role of p300 in senescence in an unbiased shRNA screen focused on epigenetic factors. A major advantage of sub-library screens is the higher representation of individual shRNAs compared to whole genome libraries(Kampmann et al., 2015). This increased representation tremendously improves the signal to noise ratio in screens and increases the sensitivity of assays to distinguish true candidates from false positives. Additionally, sub-libraries minimize the scale of an experiment, which assists technical execution. Screens of focused libraries with genes of defined function can thus lead to actionable secondary assays. This was evident in successful identification of several candidate genes from our screen that were independently validated (Figure 2 and Figure S2).
Our senescence induction method of choice was replicative exhaustion, although p300 KD was also able to delay the onset of OIS (Figure 7Q-R). RS has been implicated in tissues with high turnover such as skin, gut and blood(Hornsby, 2002), and likely the few mitotic cells in the body, such as adult stem cells(Liu and Rando, 2011). Senescent cells that spuriously appear early in life due to acute stresses not related to replicative exhaustion, serve an anti-tumor role and are cleared by the immune system. In support, OIS enhancers are proximal to SASP genes(Tasdemir et al., 2016) whose expression attracts immune cells for rapid clearance. However, RS cells likely appear later in life, compromise cellular fitness, tissue integrity, and tumor suppression(Campisi, 1997). In support, we found enhancers in RS are proximal to genes related to nucleotide metabolic dysfunction and peroxisomal genes that likely promote oxidative stress and DNA damage (Figure S6A-C).
A potential negative aspect of our senescence delay strategy, given its primordial role in tumor suppression and wound healing, may be a propensity for cancer. However, a depletion of p300 protein levels (rather than bi-allelic mutation or CRISPR knockout) did not show immortalization at least in the tested cell type (Figure 2A-C). Furthermore, we detected fewer TIFs in cells harboring p300 hairpin (Figure 2G-H). This result is particularly important because p300 mutations have been implicated in cancer where TIFs are more likely(Iyer et al., 2004). Thus, our result demonstrates that a modest depletion of p300, while delaying senescence, does not have a pro-cancer effect, at least not in IMR90 cells. Importantly, cells with p300 depletion eventually establish senescence as assayed by growth cessation (Figure 2A-C) and SA-p-gal activity (Figure 2I-J), also supporting the notion that p300 depletion does not lead to acquisition of cancer characteristics.
We conclude, that in combination with safe senolytics, senescence delay by modest depletion of p300 may be a beneficial future therapeutic strategy in treating age-related diseases in the future. In addition to implicated roles in cancer, homozygous mutations in p300 or CBP are early embryonic lethal as are CBP/p300 double heterozygotes suggesting critical roles in development(Iyer et al., 2004). Thus, p300 conforms to the antagonistic pleiotropy theory of aging whereby a single gene controls multiple antagonistic traits, in this case, development and aging(Moskalev et al., 2014).
Finally, our findings also provide major mechanistic insights into the role of HATs, histone acetylation, enhancer licensing and target gene regulation in senescence. We dissected the role of p300 at SEs and contrast it to the paralog CBP, using pooled screening methodology, proteomic, and global genomic measurements. Run-on sequencing mapped active and functional enhancers licensed during senescence establishment. These mechanistic studies lay the foundation for translational discoveries probing the in vivo role of p300 in senescence, tissue, and organismal aging. However, we acknowledge that the distinct roles of p300 and CBP need further exploration, particularly because validation via simultaneous knock out and rescue in IMR90 primary cells was not feasible. Additionally, differences in shRNA-mediated KD efficiency and antibody-based immunoprecipitation may confound our conclusions. A pertinent future question is the basis of altered p300 function in senescent cells, for example whether post-translational modifications augment p300 catalytic activity in senescence. With commercial availability of inhibitors and potential of genome editing, we anticipate p300 as a novel therapeutic target against aging and age-associated diseases.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shelley Berger (bergers@pennmedicine.upenn.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
IMR90 cells (ATCC CCL-186) which are female primary lung fibroblasts were obtained from Coriell Institute for Medical Research, Camden, NJ, and grown in standard tissue culture medium (DMEM with 10% FBS and 1% penicillin/streptomycin; Invitrogen) at 3% oxygen. Senescence was induced in one of 2 ways; (1) cells were either replicatively passaged until growth ceased, (2) induced to express oncogenic Ras. Markers such as p16 expression and β-gal were tested to confirm senescence. For RS, senescent cells were maintained in dishes for 2 weeks to ensure growth termination. Briefly, for each passage, cells were washed with PBS, trypsinized at 37 °C for 5 minutes, and plated on fresh 10 cm plates at 0.5–1e6/plate or fresh 15cm plates at 1–2e6/plate. Cells were counted with a Countess automated cell counter (Life Technologies), and the numbers were recorded for growth curve generation. Cell viability record was maintained via Trypan Blue staining. For OIS, proliferating ER-Ras cells(Young et al., 2009) were first infected with viruses harboring p300, CBP or control hairpins, selected with puromycin and then induced with 100nM tamoxifen (Sigma). Cells were then counted every 2 days for a period of 10 days while maintaining Ras expression and splitting when confluent. For KD experiments, midlife or senescent IMR90 cells were infected with lentiviral hairpin constructs (The RNAi Consortium or TRC) designed against selected targets in medium containing 8ug/ml polybrene. After 24 hours, the cells were replaced with polybrene-free media and after another 24 hours, selection was initiated with addition of 0.5ug/ml puromycin. After selection was complete, the media was changed back to standard medium. For overexpression experiments, expression vectors were packaged into lentiviruses and used to infect midlife IMR90 cells. Overexpression was confirmed by western blotting and RLS curves were generated.
METHOD DETAILS
Preparation of pooled plasmid and lentivirus for RNAi screen
shRNA clones (3455) for 717 epigenetic targets were cherry-picked from bacterial glycerol stocks of the human TRC v1.0 shRNA library (Open Biosystems) and sub-cultured as arrayed clones in 96 deep well plates containing Terrific Broth (American Bioanalytical) supplemented with 50ug/ml carbenicillin (Amresco). Bacterial cultures were incubated for 14–16 hours at 37°C/300 rpm until OD 600 nm achieved 1.0–1.6. Bacterial glycerol stocks of the newly arrayed shRNA clones were prepared. The cherry-pick was later validated by sequencing plasmid DNA prepared from 2 wells of each new glycerol stock plate (i.e. A02 and H11). The remaining bacterial culture was combined into 6 pools (A-F, ~125 gene targets/640 shRNA clones per pool), respectively, and centrifuged at 5000g for 15 minutes at 4°C. Plasmid DNA was isolated from the pooled bacterial pellets using the Qiafilter plasmid megakit (Qiagen). The plasmid pools of shRNA clones were packaged into lentivirus using second generation packaging plasmids (i.e. pSPAX2 (addgene 12260) and pMD2.G (addgene 12259)) and transfection into HEK293T/T17 cells with Lipofectamine 2000 (Invitrogen). Cell supernatant was harvested 24 hours post-transfection, clarified by centrifugation at 1000g for 15 min and filtered through a 0.45 micron filter to remove cellular debris. Concentrated vector was prepared by ultracentrifugation at 47,000g for 2 hours at 18°C in a SW28 (Beckman) swinging bucket rotor. The supernatant was carefully decanted and viral pellets were suspended in 50–100 ul sterile PBS. The concentrated virus was aliquoted (5ul) and stored at −80°C. Viral titer was assayed both qualitatively (crystal violet staining) and quantitatively (p24 ELISA assay). For qualitative estimation, 2e5 cells were infected with a serial dilution of concentrated virus. The media was replaced with fresh media containing 2ug/ml puromycin 3 days post-infection and 5. Eight days post-infection, cells were fixed with 4% formaldehyde and stained with crystal violet for 20 minutes. The stain was aspirated and repeatedly rinsed with water. Quantitative p24 ELISA assay was performed with the HIV-1 p24 Elisa Assay Kit (XpressBio). Viral titers were determined to be 0.5e9–1e10 TU/ul.
Library preparation for high throughput screening of hairpins
3 types of materials were used to make libraries; (1) plasmids, (2) cDNA from viral RNA and (3) genomic DNA. Viral RNA was isolated using the QiaAmp Viral RNA miniprep kit (Qiagen) and converted to cDNA using the High Capacity cDNA to RNA kit (Thermo Scientific) before amplification. Genomic DNA was isolated by standard phenol chloroform and isopropanol precipitation method. To assess equal distribution of shRNAs in the plasmid and viral pools, we used the TruSeq HT DNA sample prep kit (Illumina) with dual index adapters to prepare libraries that were then sequenced on the Illumina MiSeq platform. Tags were aligned to our custom shRNA sequence database and only the best alignments were scored. For the actual RNAi screen, genomic DNA from reference and senescent samples were extracted, PCR amplified around the hairpin region, digested with XhoI to generate half hairpins and processed using the NEBNext Ultra kit (New England Biolabs) kit. The 96-plex capability of the kit allowed us to sequence 50 libraries in one lane using dual index adapters. All libraries were checked for quality and quantity by Bioanalyzer (Agilent) and qPCR (Kapa Biosystems). Equal pooling was confirmed by performing a test run on the MiSeq platform before sequencing on the NextSeq 500 (Illumina).
Validation of targets from the screen
Via growth curve assessment: Individual shRNA constructs were packaged into lentiviruses and used to infect proliferating IMR90 cells. Cells were passaged to senescence over a period of 2–3 months and cell counts recorded at every split. PDs were recorded against time in days to generate growth curves.
Via TIF assay: Cells were grown overnight on multiwell slides, fixed with 4% paraformaldehyde (PFA) for 10 min at room temperature, permeabilized with 0.2% triton X-100 in PBS for one minute, and then washed with PBS 3 times. For primary antibody staining, cells were blocked in PBST (PBS+0.2%Tween 20) containing 4% BSA for 30 min at 37°C in a humidified chamber, then probed with rabbit anti-53BP1 antibody (Novus, NB100–304) in PBST containing 1% BSA at 1:175 dilution at 37°C for 2 hours. After 3 × 5 minute washes with PBST, Alexa-488 conjugated goat anti-rabbit (1:500) secondary antibody was added in PBST with 1% BSA for 1 hour at 37°C. After 3 × 5 minute PBST washes, cells were fixed with 4% PFA in PBS for 20 minutes at room temperature, washed with 0.25 mM glycine in PBS, and dehydrated with a graded ethanol series before air drying. 5ul of 5uM Cy3-labeled telomere probe (Cy3-OO-(CCCTAA)3; PNA Bio Inc.) was added to 95ul of hybridization mix [70ul freshly deionized formamide, 15ul PNA hybridization buffer (80 mM Tris-Cl pH 8, 33 mM KCl, 6.7 mM MgCl2, 0.0067% Triton X-100) and 10ul 25 mg/ml acetylated BSA] and the cells were covered with a glass coverslip, being careful to avoid air bubbles. The slides were denatured at 85°C for 5 min on a heating block and hybridized in a dark and humidified chamber overnight at room temperature. Slides were then washed 3 × 10 min in 70% formamide / 2X SSC (Saline Sodium Citrate) solution, 10 min in 2X SSC and 10 min in PBS and blocked with blocking buffer. For tertiary staining, the slides were probed with Alexa-488-conjugated donkey anti-goat antibody (Invitrogen A11055) at 1:500 dilution in PBST with 1% BSA for one hour at 37°C. Following this, the slides were washed in PBS, stained with DAPI in PBS and mounted with ProLong Gold Antifade reagent (Invitrogen, P36934) and saved in the dark at 4°C. Confocal images were obtained with a Nikon Ti-U inverted microscope with CSU-10 spinning disk confocal head (Spectral Boralis) using a Nikon Plan APO60x/1.4 lens and Hamamatsu Orca-ER camera. The Cy3 laser was held at a constant intensity to capture all images. Overlapping 53BP1 and Cy3 signals were scored as TIFs, which was performed independently by two investigators and in a blinded fashion.
Via EdU assay: After senescence establishment in cells harboring control hairpins, they were subjected to EdU assays (Click-iT™ EdU Alexa Fluor™ 488 Imaging Kit, Thermo Scientific) to measure the percentage of cycling cells together with cells harboring hairpins targeting p300. Briefly, 2e5 cells were plated on coverslips [treated with 0.1% poly-L-lysine (P8920 Sigma)] in a 24-well plate and labeled with 1X EdU solution overnight (24 hours). The cells were fixed with 4% PFA and permeabilized using 0.5% Triton X-100 in PBS. EdU labeled cells are then detected by a copper-catalyzed “click” reaction with a small molecule conjugated to Alexa Fluor followed by DAPI staining. The coverslips were mounted with ProLong Gold Antifade reagent (Invitrogen, p36934) and EdU incorporation calculated as a percentage of DAPI-stained cells also showing Alexa-fluor signal imaged by a fluorescent microscope (Nikon Eclipse 80i).
Via Western blotting: Cell pellets collected during passage to senescence were Western blotted as described below and probed for H3 (to assess the kinetics of H3 downregulation in senescence) and GAPDH (loading control).
SA-β-gal assay
SA-β-gal assays were performed using a cellular senescence assay kit (Chemicon KAA002), according to the manufacturer’s protocol. Cells were incubated with β-gal detection solution at 37 °C overnight, and imaged by light microscopy.
Quantitative PCR (qPCR)
To estimate gene expression by RNA, qPCR was performed. Total RNA was isolated using the RNeasy kit (Qiagen) with on-column DNase I treatment to remove genomic DNA. 500ng of purified RNA was converted to cDNA using the high capacity cDNA to RNA kit (Thermo Fisher). qPCR was performed using FastSYBR (Thermo Fisher) with standards generated with a dilution series of “plus RT” reactions. “Minus RT” controls were included to rule out genomic DNA contamination.
Western blotting
Cells were lysed in buffer containing 50 mM Tris pH 7.5, 0.5 mM EDTA, 150 mM NaCl, 1% NP40, 1% SDS, supplemented with 1X Halt Protease inhibitor cocktail (Thermo Scientific). The lysates were briefly sonicated, and cleared by centrifugation at max speed for 10 minutes at 4°C. The supernatants were quantified using the BCA kit ((Pierce) and subjected to electrophoresis using NuPAGE 12% Bis-Tris for histones or 3–8% Tris acetate precast gels (Life Technologies) for p300/CBP. The proteins were transferred to a 0.2 micron nitrocellulose membrane using a wet transfer apparatus (BioRad) for 1.5 hours at 100V (4°C). 5% milk in TBS supplemented with 0.1% Tween 20 (TBST) was used to block the membrane at room temperature (~25 °C) for 1 h. Primary antibodies were diluted in 5% BSA in TBST, and incubated at 4 °C overnight. The list of antibodies used in this study is listed in the Key Resources Table. The membrane was washed 3 times with TBST, each for 10 minutes, followed by incubation with HRP-conjugated secondary antibodies (BioRad) at room temperature for 1 h, in 5% milk/TBST. The membrane was washed again 3 times, and imaged by a Fujifilm LAS-4000 imager.
Histone preparation for mass spectrometry
Histone purification was performed from ~9e6 proliferating and senescent cells as previously described(Sidoli et al., 2016). Briefly, nuclei were isolated by suspending cells into nuclei isolation buffer (15mM Tris–HCl (pH 7.5), 15mM NaCl, 60mM KCl, 5mM MgCl2, 1mM CaCl2, 250mM sucrose, 0.2% NP-40) including the following inhibitors; 1mM DTT, 0.5mM AEBSF and 10mM sodium butyrate. Nuclei were separated by centrifugation (1000g for 10 minutes) and 2ml of cold 0.4N H2SO4 was added to the nuclei pellet. Nuclei were incubated at 4°C with shaking for 2 hours, pelleted at 3400g for 5 minutes, and proteins were precipitated from the supernatant with 33% TCA (w/v). Purified histones were resuspended in 30ul of 50mM NH4HCO3, pH 8.0. Derivatization and digestion was performed using the bottom-up strategy described previously(Sidoli et al., 2016). Briefly, derivatization of the lysine residues side chains was performed by preparing a mix of pure propionic anhydride:acetonitrile (1:3); 15ul of this mix was added to the histone sample, followed by immediate addition of 7ul of ammonium hydroxide to adjust the pH to 8.0 after the introduction of propionic anhydride to the mixture. The reaction was performed twice. Digestion was performed using trypsin at an enzyme:sample ratio of 1:20, overnight at room temperature. The derivatization reaction was repeated to derivatize peptide N-termini. Samples were then desalted using Stage-tipping.
Bottom-up nanoLC-MS/MS and data analysis
Each of the 3 biological replicate samples were run in three technical replicates. Samples were analyzed by using a nanoLC-MS/MS setup. The nanoLC was configured with a 75 μm ID × 17 cm Reprosil-Pur C18-AQ (3um; Dr. Maisch GmbH, Germany) nano-column using an EASY-nLC nanoHPLC (Thermo Fisher Scientific, San Jose, CA, USA). The HPLC gradient was 0–28% solvent B (A = 0.1% formic acid; B = 95% acetonitrile, 0.1% formic acid) over 45 min and from 28% to 80% solvent B in 5 minutes at a flow-rate of 300nl/min. The nanoLC was coupled with an Orbitrap Elite (Thermo Fisher Scientific, San Jose, CA, USA) mass spectrometer. Spray voltage was set at 2.3 kV and capillary temperature was set at 275°C. Full scan MS spectrum (m/z 300–1100) was performed in the Orbitrap with a resolution of 120,000 (at 200 m/z) with an AGC target of 10e6. The instrument operated in a data-independent acquisition (DIA) mode, as previously described(Sidoli et al., 2015). Fragmentation and MS/MS detection was performed in the ion trap using DIA windows of 50 m/z with CID normalized collision energy of 35 and an AGC target of 10e4. The intensity of isobaric peptides, i.e. peptides with the same mass but with PTMs on different positions on the amino acid sequence, was determined using fragment ions. Briefly, the intensity of the precursor ion signal was split between the isobaric forms according to the relative intensities of the fragment ion signals. Peak area was extracted from raw files by using our in-house software EpiProfile(Yuan et al., 2015), which includes a pre-compiled list of peptides for quantification. The relative abundance of a given PTM was calculated by dividing its intensity by the sum of all modified and unmodified peptides sharing the same sequence. For statistical analysis, we applied a 2-tails homoscedastic t-test (significant if p<0.05).
Chromatin immunoprecipitation-sequencing (ChlP-seq)
ChIP was performed as described previously from ~12e6 cells(Shah et al., 2013). p300 and CBP ChIP was performed using the SimpleChIP plus sonication kit (CST #56383). Briefly, cross-linked cells were lysed and the chromatin was sheared using a Covaris Ultrasonicator to achieve an average size of <500 bp. 2ug of sonicated chromatin (estimated by DNA concentration) was used per immunoprecipitation reaction (see Key Resources Table for a list of antibodies), and 10% of the amount was saved as input. Immunoprecipitation was performed using protein A Dynabeads (Thermo Fisher). ChIP-qPCR was performed against standards made from a dilution series of an input DNA mix. Enrichment was ascertained by calculating signal over IgG. Primer sequences are available on request. For ChIP-seq, 1ng of DNA from immunoprecipitation and input was used to prepare libraries using the NEBNext Ultra II kit (New England Biolabs). The libraries were checked for quality and quantity by Bioanalyzer (Agilent) and qPCR (Kapa Biosystems) respectively. Multiplexed libraries were sequenced on a single lane of the NextSeq 500 platform (Illumina).
RNA-sequencing (RNA-seq)
Total RNA was isolated from frozen pellets of IMR90 cells using the RNeasy kit (Qiagen) following instructions. 1ug was used as input in the Scriptseq Complete kit (Epicentre). Briefly, total RNA was ribodepleted using a Ribozero magnetic protocol for human, mouse and rat (Epicentre, Illumina). The ribodepleted RNA was then ethanol precipitated, fragmented, and tagged at both ends for stranded library preparation using the Scriptseq v2 library preparation protocol. The PCR amplification step was used to index the libraries and multiplexed libraries were quantified by Bioanalyzer (Agilent) and qPCR (Kapa Biosystems). The RNA-seq run was performed on the NextSeq 500 platform (Illumina).
Precision Run-On-sequencing (PRO-seq)
PRO-seq was performed on proliferating and senescent IMR90 cells as described previously(Mahat et al., 2016). Briefly, ~12e6 trypsinized cells were permeabilized and stored in storage buffer (10mM Tris-HCl pH 8.0, 25% glycerol, 5mM MgCl2, 0.1mM EDTA, 5mM DTT) at −80°C until use. The permeabilized cells were then resuspended in 1X nuclear run-on reaction mix containing biotin-11-rCTP and unlabeled rATP, rGTp and rUTP for 3 minutes at 37C. RNA was extracted, fragmented and subjected to biotin enrichment. The enriched nascent RNA population was then ligated to a 3’ adapter sequence and subjected to a second round of biotin enrichment. The 5’ ends of the RNA were decapped, phosphorylated, ligated to 5’ adapter and subjected to a third biotin enrichment. The RNA was then reverse transcribed and PCR amplified with appropriate index primers. The libraries were purified by extraction from an 8% polyacrylamide gel, quantified by Bioanalyzer (Agilent) and qPCR (Kapa Biosystems). The PRO-seq run was performed on the NextSeq 500 platform (Illumina).
Information on replicates and cell passage numbers used in genome-wide experiments
The number of replicate experiments, PD numbers and other QC parameters are included in the TS2-5.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNAi screen bioinformatics analysis
Hairpin tags were trimmed to remove the first 6 bases and all bases after the 27th, producing the hairpin-complementary region and removing the excision site. Trimmed tags were then aligned using bowtie v1.0.1 to a custom index of hairpin sequences and control sequences (parameters --best). Aligned sequence tags were merged across pools and per-hairpin tag counts at each time point were RPM-adjusted and log-transformed, then time points after t = PD40 were plotted as a function of t = PD40.
Lifespan analysis
To assess the effect of shRNA KD (or overexpression) on lifespan, a generalized least squares (GLS) model was employed using the gls() function in the R library nlme. The data were binned by averaging into eight time-points (t0, 0–10 days, 11–20 days, 21–30 days, 31–40 days, 41–50 days, 81–100 days, and tfinal) in order to minimize the effect of missing measurements; remaining time points with missing measurements were removed from consideration. A GLS linear model of the form PD ~ time + shRNA treatment + time : shRNA treatment was fitted. Variance-covariance structure was tested over 4 models (compound symmetry, unstructured, autoregressive, and autoregressive with heterogenous variance) and Akaike Information Criterion (AIC)(Akaike, 1974) scores were used to determine that autoregressive variance was the best fit. The significance of the interaction term under this model is reported. For other shRNA lifespan assays, either the autoregressive or autoregressive with heterogeneous variance model was used depending on the AIC score.
ChIP-seq bioinformatics analysis
ChIP-seq libraries were prepared as previously described and sequenced using an Illumina NextSeq 500 platform (75bp, single-end sequencing). For each ChIP experiment, replicates from two independent sequencing runs were pooled together to yield an average of 79 million reads per sample (TS4). Raw reads were mapped to the reference human genome assembly GRCh37 (hg19) using Bowtie2, allowing up to 1 mismatch in the seed alignment. The alignment result was further processed by SAMtools to remove low quality alignments (a minimum mapping quality of 10), duplicate reads, reads mapped to mitochondria and reads mapped to ENCODE blacklisted genomic regions. For H3K122ac, H3K18ac, H3K23ac, H4K5ac and H3K27ac, MACS2 was used to identify ChIP-Seq enriched regions (or peaks), requiring peak calling FDR lower than 0.01. For H3K4me1, SICER was used with recommended parameters (redundancy threshold = 1, window size = 200, fragment size = 150, effective genome fraction = 0.74, gap size = 600 and FDR cutoff = 0.01). For CBP and p300, MACS2 was used with a less stringent FDR cutoff of 0.05 and the broad peak calling configuration (“--nomodel --extsize 150 – broad”).
Identification of TEs and SEs
H3K27ac ChIP-seq peaks in proliferating and senescent conditions were analyzed separately. For either condition, peaks within a distance of 12.5Kb were stitched together using HOMER suite (“mergePeaks -d 12500”). Tag density in the sample were computed for each stitched and isolated peak region, using HOMER suite (“makeTagDirectory” and “annotatePeaks -size given”, by default normalized to 10 million reads per sample). If a region was wider than 3Kb and the average tag density was greater than 2 read per million mapped tags per base pair (RPM/bp), the region was classified as a SE, otherwise the peak region was classified as a TE. Stitched regions that did not meet the tag density requirement were unstitched to the original isolated peak regions and classified as TEs. In a similar approach, peaks from H3K122ac, H3K18ac, H3K23ac and H4K5ac samples were classified into TEs and SEs, for proliferating and senescent conditions respectively (TS4).
Identification of Senescence-Activated and Senescence-Deactivated peaks
Tag density in each sample was computed for each peak region (regardless of TE or SE). To control for histone density, background noise and non-specific binding, the final tag density for each histone modification ChIP experiment was computed as the tag density in the histone modification ChIP minus the H3-ChIP or H4-ChIP, minus the tag density difference between IgG ChIP and the input control, i.e. ( Histone-modification-ChIP – H3-/H4-ChIP ) – ( IgG-ChIP – Input ). For each TE and SE in a proliferating sample, we used an Exact Poisson Test to examine whether its tag density in the proliferating sample is greater than its tag density in the corresponding senescent sample. Significant TEs and SEs (p-value lower than 10e-4 and a fold change of 4) were considered as Senescent-Deactivated. Similarly, significant TE and SE in a senescent sample compared to the corresponding proliferating sample were considered as Senescent-Activated. Differential analysis was performed for H3K122ac, H3K18ac, H3K23ac, H4K5ac and H3K27ac experiments respectively (TS4).
PRO-seq bioinformatics analyses
PRO-Seq data were aligned to the NCBI v37 assembly of the human genome (hg19) using ProSeqMapper (Danko lab, https://github.com/Danko-Lab/tutorials/blob/master/PRO-seq.md) with default parameters and a BWA index (TS5). The Danko lab’s PRO-seq alignment pipeline contains processing steps for adapter trimming, quality control read filtering, read alignment, and track generation. The internal alignment is done with bwa 0.7.10. We compared alignment parameters using ProSeqMapper and STAR. However, with standard STAR alignment pipeline for RNA-seq, we saw only half of the alignments we got with the PRO-seq pipeline. For this reason, we decided to use the PRO-seq pipeline for all PRO-seq and PRO-cap alignments. PRO-seq reads over H3K27ac identified enhancers are counted using featureCounts, with “-s 0” to count reads in an unstranded way. Similarly, PRO-seq reads over RefSeq annotated genes (whole gene body including introns) are counted using featureCounts, with “-s 2” to count reads in a stranded way. To generate boxplots, log2 fold change of read counts per million assigned reads in senescence over that in proliferating cells is used. A pseudocount of 10e-5 was added to avoid division by zero.
Track visualization & generating metaplots and heatmaps
Genome-wide tag density was computed using BEDtools and UCSC command line utilities to generate tracks for visualization, for which purpose the visualization tracks were normalized to 10 million reads per sample. Tracks were subtracted by deepTools, to generate tracks of histone modification ChIP minus H3- or H4-ChIP (corresponding to the peak calling approach used), and tracks of histone modification ChIP minus H3- or H4-ChIP then minus the difference between IgG-ChIP and the input control (corresponding to the differential analysis approach). Metaplots and heatmaps of tag density near TEs and SEs were also generated by deepTools.
Overlap between H3K27ac peaks and H3K122ac/H4K5ac/CBP/p300 Peaks
H3K27ac-defined TEs and SEs were considered as overlapping with H3K122ac or H4K5ac peaks if they overlap by at least 1bp (“bedtools intersect”). Differently, H3K27ac-defined TEs and SEs were considered as overlapping with CBP or p300 if a binding site (or peak) was found within 3.5Kb of the TE or SE. In heatmaps where normalized tag density for H3K27ac-defined TEs and SEs was represented, the tag density was summarized over the entire TE/SE region for histone modifications, but only summarized over binding sites within 3.5Kb for CBP/p300.
Correlating peaks to nearest genes
Gene target for each peak was identified as gene with nearest TSS to the peak, using HOMER suite (“annotatePeaks -size given”). Gene official symbols were used to correlate ChIP-seq peaks with the expression dataset (RNA-seq) later on. The HOMER annotation process also performed compartment analysis, separating the peaks into 4 categories: Intergenic, Intron, Promoter-TSS (−1Kb to +100bp of TSS) and Others.
RNA-seq expression analysis
RNA-seq libraries were prepared as previously described and sequenced using an Illumina NextSeq 500 platform (only forward reads in the paired-end sequencing were used, 48bp). Approximately 50–60 million reads were acquired per replicate (TS5). Sequencing reads were trimmed using Trimmomatic to remove leading and trailing low quality base calls (Phred < 3). The resulting reads were then aligned to GRCh37.75(hg19) using STAR with default parameters, and alignments with a mapping score lower than 10 were discarded using SAMtools. FeatureCounts was used to generate a matrix of mapped fragments per RefSeq annotated gene. Analysis for differential gene expression was performed using edgeR R package with the cutoff of FDR < 0.05, and pathway/upstream regulator analysis was performed using Ingenuity Pathway Analysis (IPA).
Correlating ChIP-seq with expression data
Gene expression fold changes were acquired from RNA-seq differential analysis. Gene official symbols were used to correlate a gene targeted by TE or SE (defined by the nearest gene as described in the above section) and the gene’s expression fold change. Boxplots were generated for different groups of TEs and SEs, and for different RNA-seq experiments (wild type and p300 KD experiment). 2-tailed Mann-Whitney-Wilcoxon Test was used to compare bins within and between boxplots.
GO analysis and IPA
GO analysis was performed using DAVID Bioinformatics Resources
(http://david.abcc.ncifcrf.gov/home.jsp). Pathway analyses for enrichment were performed using Ingenuity Pathway Analysis (IPA).
Supplementary Material
TS1 (Related to Figure 1): Tag distribution parameters in plasmid and viral pools
TS2 (Related to Figure 6): p values for TE quantiles correlated to RNA-seq at target genes, PRO-seq at target genes and PRO-seq at enhancer location
TS3 (Related to Figure 1): Alignment statistics and counts of hairpin tag library
TS4 (Related to Figure 3): Relative abundance of histone peptides from mass spec analysis
TS5 (Related to Figure 4 and 7): Alignment statistics, peak information and classification of enhancers from ChIP-seq libraries
TS6 (Related to Figure 5): PRO-seq and RNA-seq alignment statistics
TS7 (Related to Figure 4, 5 and 7): Peak location of “all” enhancers, senescence-activated enhancers (green font) and senescence-deactivated enhancers (red font)
Highlights.
High-throughput screening identifies key epigenetic proteins driving senescence
Depletion of the histone acetyltransferase p300 delays senescence
p300 induces the formation of de novo super enhancers in senescence
Depletion of p300 suppresses senescence-related gene expression
Acknowledgements
We thank Christopher Lord, Katherine Alexander, Digbijay Mahat, Ronen Marmorstein and Dan Bose for suggestions related to the screen, PRO-seq and general information on p300/CBP. We thank Gabor Egervári for statistical analysis and Raffaella Nativio for discussions. This work was supported by NIH grants P01AG031862 to S.L.B., P.D.A and F.B.J.; CA196539, GM110174 and AI11 8891 to B.A.G.; CPRIT award R1306 and Ted Nash Long Life Foundation grant to W.D.; American Heart Association grant 15POST21230000 and AFAR Irene Diamond Transition Award DIAMOND 17113 to P.S.
Footnotes
Declaration of Interests
The authors declare no competing interests.
DATA AND SOFTWARE AVAILABILITY
All software used in this manuscript is listed in the Key Resources Table. Additional dedicated scripts developed for this work are available upon request.
Description:
The RNAi screen, ChIP-Seq, RNA-seq and PRO-seq data have been deposited in the GEO under ID code GSE105937.
Description:
https://chorusproject.org/1375
All MS raw files were uploaded in the public repository Chorus (https://chorusproject.org), project number: 1375.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akaike H (1974). A new look at the statistical model identification. IEEE Transactions on Automatic Control 19, 716–723. [Google Scholar]
- Aliper AM, Csoka AB, Buzdin A, Jetka T, Roumiantsev S, Moskalev A, and Zhavoronkov A (2015). Signaling pathway activation drift during aging: Hutchinson-Gilford Progeria Syndrome fibroblasts are comparable to normal middle-age and old-age cells. Aging (Albany NY) 7, 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlasi Y, and Stunnenberg HG (2017). The interplay of epigenetic marks during stem cell differentiation and development. Nat Rev Genet. [DOI] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. (2016). Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, and van Deursen JM (2011). Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J (1997). The biology of replicative senescence. Eur J Cancer 33, 703–709. [DOI] [PubMed] [Google Scholar]
- Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, et al. (2013). Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell 24, 777–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JB, and Russell DW (2004). Mammalian wax biosynthesis. I. Identification of two fatty acyl-Coenzyme A reductases with different substrate specificities and tissue distributions. J Biol Chem 279, 37789–37797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium EP, Birney E, Stamatoyannopoulos JA, Dutta A, Guigo R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, et al. (2007). Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447, 799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppe JP, Desprez PY, Krtolica A, and Campisi J (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol 5, 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dancy BM, and Cole PA (2015). Protein lysine acetylation by p300/CBP. Chem Rev 115, 2419–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Ghosh K, Vizioli MG, Zhu J, Sen P, Wangensteen KJ, Simithy J, Lan Y, Lin Y, Zhou Z, et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou Z, Xu C, Donahue G, Shimi T, Pan JA, Zhu J, Ivanov A, Capell BC, Drake AM, Shah PP., et al. (2015). Autophagy mediates degradation of nuclear lamina. Nature 527, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, and Sharpless NE (2017). Senescence in Health and Disease. Cell 169, 1000–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-Andre V, Sigova AA, Hoke HA, and Young RA (2013). Super-enhancers in the control of cell identity and disease. Cell 155, 934–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornsby PJ (2002). Cellular senescence and tissue aging in vivo. J Gerontol A Biol Sci Med Sci 57, B251–256. [DOI] [PubMed] [Google Scholar]
- Ivanov A, Pawlikowski J, Manoharan I, van Tuyn J, Nelson DM, Rai TS, Shah PP, Hewitt G, Korolchuk VI, Passos JF, et al. (2013). Lysosome-mediated processing of chromatin in senescence. J Cell Biol 202, 129–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer NG, Ozdag H, and Caldas C (2004). p300/CBP and cancer. Oncogene 23, 4225–4231. [DOI] [PubMed] [Google Scholar]
- Kampmann M, Horlbeck MA, Chen Y, Tsai JC, Bassik MC, Gilbert LA, Villalta JE, Kwon SC, Chang H, Kim VN, et al. (2015). Next-generation libraries for robust RNA interference-based genome-wide screens. Proc Natl Acad Sci U S A 112, E3384–3391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland JL, and Tchkonia T (2017). Cellular Senescence: A Translational Perspective. EBioMedicine 21, 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lener B, Koziel R, Pircher H, Hutter E, Greussing R, Herndler-Brandstetter D, Hermann M, Unterluggauer H, and Jansen-Durr P (2009). The NADPH oxidase Nox4 restricts the replicative lifespan of human endothelial cells. Biochem J 423, 363–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Notani D, and Rosenfeld MG (2016). Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nat Rev Genet 17, 207–223. [DOI] [PubMed] [Google Scholar]
- Liu L, and Rando TA (2011). Manifestations and mechanisms of stem cell aging. J Cell Biol 193, 257–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI, and Young RA (2013). Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell 153, 320–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahat DB, Kwak H, Booth GT, Jonkers IH, Danko CG, Patel RK, Waters CT, Munson K, Core LJ, and Lis JT (2016). Base-pair-resolution genome-wide mapping of active RNA polymerases using precision nuclear run-on (PRO-seq). Nat Protoc 11, 1455–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskalev AA, Aliper AM, Smit-McBride Z, Buzdin A, and Zhavoronkov A (2014). Genetics and epigenetics of aging and longevity. Cell Cycle 13, 1063–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita M, Nunez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, and Lowe SW (2003). Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113, 703–716. [DOI] [PubMed] [Google Scholar]
- Nelson DM, Jaber-Hijazi F, Cole JJ, Robertson NA, Pawlikowski JS, Norris KT, Criscione SW, Pchelintsev NA, Piscitello D, Stong N, et al. (2016). Mapping H4K20me3 onto the chromatin landscape of senescent cells indicates a function in control of cell senescence and tumor suppression through preservation of genetic and epigenetic stability. Genome Biol 17, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan RJ, Kubicek S, Schreiber SL, and Karlseder J (2010). Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol 17, 1218–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peleg S, Feller C, Ladurner AG, and Imhof A (2016). The Metabolic Impact on Histone Acetylation and Transcription in Ageing. Trends Biochem Sci 41, 700–711. [DOI] [PubMed] [Google Scholar]
- Qian Y, and Chen X (2010). Tumor suppression by p53: making cells senescent. Histol Histopathol 25, 515–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rai TS, Cole JJ, Nelson DM, Dikovskaya D, Faller WJ, Vizioli MG, Hewitt RN, Anannya O, McBryan T, Manoharan I, et al. (2014). HIRA orchestrates a dynamic chromatin landscape in senescence and is required for suppression of neoplasia. Genes Dev 28, 2712–2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen P, Shah PP, Nativio R, and Berger SL (2016). Epigenetic Mechanisms of Longevity and Aging. Cell 166, 822–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah PP, Donahue G, Otte GL, Capell BC, Nelson DM, Cao K, Aggarwala V, Cruickshanks HA, Rai TS, McBryan T, et al. (2013). Lamin B1 depletion in senescent cells triggers large-scale changes in gene expression and the chromatin landscape. Genes Dev 27, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimozawa N, Tsukamoto T, Suzuki Y, Orii T, Shirayoshi Y, Mori T, and Fujiki Y (1992). A human gene responsible for Zellweger syndrome that affects peroxisome assembly. Science 255, 1132–1134. [DOI] [PubMed] [Google Scholar]
- Sidoli S, Bhanu NV, Karch KR, Wang X, and Garcia BA (2016). Complete Workflow for Analysis of Histone Post-translational Modifications Using Bottom-up Mass Spectrometry: From Histone Extraction to Data Analysis. Journal of visualized experiments : JoVE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidoli S, Simithy J, Karch KR, Kulej K, and Garcia BA (2015). Low Resolution Data-Independent Acquisition in an LTQ-Orbitrap Allows for Simplified and Fully Untargeted Analysis of Histone Modifications. Analytical chemistry 87, 11448–11454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims D, Mendes-Pereira AM, Frankum J, Burgess D, Cerone MA, Lombardelli C, Mitsopoulos C, Hakas J, Murugaesu N, Isacke CM, et al. (2011). High-throughput RNA interference screening using pooled shRNA libraries and next generation sequencing. Genome Biol 12, R104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, and de Lange T (2003). DNA damage foci at dysfunctional telomeres. Curr Biol 13, 1549–1556. [DOI] [PubMed] [Google Scholar]
- Tasdemir N, Banito A, Roe JS, Alonso-Curbelo D, Camiolo M, Tschaharganeh DF, Huang CH, Aksoy O, Bolden JE, Chen CC, et al. (2016). BrD4 Connects Enhancer Remodeling to Senescence Immune Surveillance. Cancer Discov 6, 612–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valor LM, Viosca J, Lopez-Atalaya JP, and Barco A (2013). Lysine acetyltransferases CBP and p300 as therapeutic targets in cognitive and neurodegenerative disorders. Curr Pharm Des 19, 5051–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM (2014). The role of senescent cells in ageing. Nature 509, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Blow MJ, Li Z, Zhang T, Akiyama JA, Holt A, Plajzer-Frick I, Shoukry M, Wright C, Chen F, et al. (2009). ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature 457, 854–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo N, and Goodman RH (2001). CREB-binding protein and p300 in transcriptional regulation. J Biol Chem 276, 13505–13508. [DOI] [PubMed] [Google Scholar]
- Wei W, and Sedivy JM (1999). Differentiation between senescence (M1) and crisis (M2) in human fibroblast cultures. Exp Cell Res 253, 519–522. [DOI] [PubMed] [Google Scholar]
- Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Scholz C, Hamilton WB, Zucconi BE, Wang WW, Liu WR, et al. (2018). Time-Resolved Analysis Reveals Rapid Dynamics and Broad Scope of the CBP/p300 Acetylome. Cell 174, 231–244 e212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whyte WA, Orlando DA, Hnisz D, Abraham BJ, Lin CY, Kagey MH, Rahl PB, Lee TI, and Young RA (2013). Master transcription factors and mediator establish super-enhancers at key cell identity genes. Cell 153, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young AR, Narita M, Ferreira M, Kirschner K, Sadaie M, Darot JF, Tavare S, Arakawa S, Shimizu S, Watt FM, et al. (2009). Autophagy mediates the mitotic senescence transition. Genes Dev 23, 798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZF, Lin S, Molden RC, Cao XJ, Bhanu NV, Wang X, Sidoli S, Liu S, and Garcia BA (2015). EpiProfile Quantifies Histone Peptides With Modifications by Extracting Retention Time and Intensity in High-resolution Mass Spectra. Molecular & cellular proteomics : MCP 14, 1696–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. (2011). RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 478, 524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TS1 (Related to Figure 1): Tag distribution parameters in plasmid and viral pools
TS2 (Related to Figure 6): p values for TE quantiles correlated to RNA-seq at target genes, PRO-seq at target genes and PRO-seq at enhancer location
TS3 (Related to Figure 1): Alignment statistics and counts of hairpin tag library
TS4 (Related to Figure 3): Relative abundance of histone peptides from mass spec analysis
TS5 (Related to Figure 4 and 7): Alignment statistics, peak information and classification of enhancers from ChIP-seq libraries
TS6 (Related to Figure 5): PRO-seq and RNA-seq alignment statistics
TS7 (Related to Figure 4, 5 and 7): Peak location of “all” enhancers, senescence-activated enhancers (green font) and senescence-deactivated enhancers (red font)