

Abstract
T cells expressing chimeric antigen receptors (CART) have shown significant promise in clinical trials to treat hematologic malignancies, but their efficacy in solid tumors has been limited. Oncolytic viruses have the potential to act in synergy with immunotherapies due to their immunogenic oncolytic properties and the opportunity of incorporating therapeutic transgenes in their genomes. Here, we hypothesized that an oncolytic adenovirus armed with an EGFR-targeting, bispecific T-cell engager (OAd-BiTE) would improve the outcome of CART-cell therapy in solid tumors. We report that CART cells targeting the folate receptor alpha (FR-α) successfully infiltrated preestablished xenograft tumors but failed to induce complete responses, presumably due to the presence of antigen-negative cancer cells. We demonstrated that OAd-BiTE-mediated oncolysis significantly improved CART-cell activation and proliferation, while increasing cytokine production and cytotoxicity, and showed an in vitro favorable safety profile compared with EGFR-targeting CARTs. BiTEs secreted from infected cells redirected CART cells toward EGFR in the absence of FR-α, thereby addressing tumor heterogeneity. BiTE secretion also redirected CAR-negative, nonspecific T cells found in CART-cell preparations toward tumor cells. The combinatorial approach improved antitumor efficacy and prolonged survival in mouse models of cancer when compared with the monotherapies, and this was the result of an increased BiTE-mediated T-cell activation in tumors. Overall, these results demonstrated that the combination of a BiTE-expressing oncolytic virus with adoptive CART-cell therapy overcomes key limitations of CART cells and BiTEs as monotherapies in solid tumors and encourage its further evaluation in human trials.
Introduction
Adoptive cell therapy (ACT) with T cells expressing a chimeric antigen receptor (CART) has shown exceptional success in treating hematopoietic tumors, leading to sustained remissions in patients with refractory or relapsed B-cell malignancies (1–5). A major challenge facing the field is to reproduce this clinical success in solid tumors (6).
A key limitation of CART therapy in solid tumors is the immunosuppressive tumor microenvironment (7–9), which preferentially recruits regulatory T cells, tumor-associated macrophages (TAM), and myeloid-derived suppressor cells (MDSC; refs. 10, 11), which can mediate CART-cell inhibition. Malignant cells can directly inhibit T-cell function through expression of the cognate ligands of surface inhibitory receptors such as CTLA-4, PD-1, LAG-3, and TIM-3 expressed on tumor-infiltrating lymphocytes (TIL; refs. 12–14). These conditions can lead to T-cell hypofunction, restricting CART-cell persistence within the tumor and limiting the benefits of engineered T-cell therapies (15).
Another important hurdle encountered with CART cells is tumor immune evasion due to antigen loss. CD19-negative relapses have emerged as a major problem in patients with hematologic malignancies treated with CD19-directed immunotherapies (5, 16). The possibility of tumor escape is increased in solid tumors, which are generally composed of heterogeneous populations of cells with variable antigen expression (17–19). Tumor cells use a variety of mechanisms to escape immune recognition, including antigen mutation, downregulation or deletion of target antigens, and selective survival of antigen-negative tumor cells (20). Therefore, developing strategies to revert tumor immunosuppression while overcoming antigen loss would represent a significant advance in the field.
Oncolytic adenoviruses (OAd) may mitigate these challenges to T-cell therapy in the tumor microenvironment. By selectively infecting and replicating in malignant cells, OAds may provide the dual benefit of debulking the tumor through selective lysis and providing a viral danger signal that could create a more appropriate environment for T-cell expansion and functionality (21, 22). OAds can be genetically modified to selectively express a therapeutic transgene in the tumor microenvironment (23–26).
Bispecific T-cell engagers (BiTE) are immunotherapeutic molecules consisting of an anti-CD3 single-chain variable fragment (scFv) fused to an antitumor-associated antigen scFv via a flexible linker. Blinatumomab, a BiTE targeting CD19, was approved by the FDA for treatment of acute lymphoblastic leukemia, and several other BiTEs targeting various antigens are currently under clinical investigation (27). However, exogenous administration of BiTEs to treat solid tumors has several drawbacks, including limited capacity to penetrate into the tumor and a short serum half-life, thus requiring continuous systemic infusions that can lead to increased toxicities (28).
We have reported the generation of ICO15K-cBiTE, an OAd that secretes an EGFR-targeting BiTE (OAd-BiTE) upon infection of malignant cells (25). We showed that the OAd-BiTE induces robust and specific T-cell activation and proliferation upon infection of cancer cells, enhancing the antitumor efficacy of the virus in mouse xenograft models of cancer.
Here, we tested the hypothesis that combining CART cells targeting folate receptor alpha (FR-α) with OAd-BiTE could improve CART-cell therapy. We showed that the dual treatment resulted in increased T-cell activation, proliferation, and cytotoxicity in vitro and enhanced antitumor efficacy due to improved T-cell activation in xenograft mouse models. Therefore, the combination therapy of CART cells and an OAd-BiTE has the potential to overcome the limitations of CARs and BiTEs as monotherapies in solid tumors.
Materials and Methods
Cells
SKOV3, HCT116, Panc-1, and NCI-H226 were obtained from ATCC and were authenticated in 2016 by the University of Arizona Genetics Core using the PowerPlex 16 system (Promega). C30 were kindly provided by Daniel Powell (Ovarian Cancer Research Center, Center for Cellular Immunotherapies, University of Pennsylvania, Philadelphia, PA). All cells were regularly validated to be Mycoplasma free. Primary human keratinocytes and fibroblasts were purchased from the Dermatology Core Facility at the University of Pennsylvania.
SKOV3, NCI-H226, and C30 cells were maintained in RPMI-1640 (Gibco) supplemented with 10%FBS (Gibco), andHCT116 and Panc-1 cells were maintained in DMEM-F12 supplemented with 10% FBS.
Generation of CART cells
Primary lymphocytes from normal donors were provided by the University of Pennsylvania Human Immunology Core. T cells were maintained in RPMI 1640 media supplemented with 10% FBS, penicillin (100 U/mL; Gibco), streptomycin sulfate (100 mg/mL; Gibco), and 10 mmol/L Hepes (Gibco). Tcells were stimulated, expanded, and transduced as previously described (29). Briefly, CD4+ and CD8+ T cells were cultured separately with CD3/CD28-activating Dynabeads (Invitrogen) at a bead-to-cell ratio of 3. Approximately 24 hours after activation, T cells were transduced with lentiviral vectors at an MOI of 5, by adding the virus supernatant to T-cell cultures. For CD8+ T cells, human IL2 (Prometheus) was added every other day to a final concentration of 50 IU/mL. Beads were removed from cultures at day 5, and T cells were counted and fed every day after day 5. T cells were cryopreserved when returning to the resting state, as determined by decreased growth kinetics and cell size.
For bioluminescence assays, control cells were transduced with lentiviral vectors (pTRPE) encoding GFP and Click Beetle Red (CBR) luciferase, connected by a T2A signal peptide (GFP-T2A-CBR), as previously described (30). CART cells expressing CBR were obtained by cotransduction with concentrated lentiviral supernatants containing CAR and GFP-T2A-CBR. CARs targeting CD19 (CD19-BBz) and FR-α (FR-CART) have been described previously (31, 32). Cetux-CAR was generated through de novo synthesis of a codon-optimized scFv derived from the variable light and variable heavy domains of the 225 mAb (cetuximab) and linked by a (GGGGS) 3× linker (Supplementary Table S1). Subcloning of the 225 scFv was performed by BamHI and BspEI digestion and subsequent ligation into a pTRPE-BBz CAR plasmid backbone.
Oncolytic adenoviruses
The OAds ICOVIR15K and ICOVIR15K-cBiTE (OAd-BiTE) were generated and titered as previously described (25, 33). Briefly, OAds were propagated in A549 cells and double purified by cesium chloride gradient centrifugation. Physical titer (viral particles (VP)/mL) was determined by optical absorbance at 260 nm after dissociation of viral particles in lysis buffer (10 mmol/L Tris, 1 mmol/L EDTA, 0.1% SDS, pH 8.0) for 5 minutes at 56°C.
Flow cytometry
For all experiments, T-cell suspensions were stained with fixable live/dead violet stain (Invitrogen), followed by surface antibody staining. The Foxp3/Transcription Factor Staining Buffer set (Thermo Fisher Scientific) was used for intracellular staining according to the manufacturer’s instructions. The following antibodies against human molecules were used: anti-CD45–PerCpCy5.5 (cat. #45–9459-42), anti-CD4-PE (cat. #12–0048-42), anti-TIM3-PerCp-eF710 (catalog # 46–3109-42), and anti-Ki67-FITC (cat. #11–5699-42) from Thermo Fisher/eBioscience; anti-CD4-BV510 (cat. #317444), anti-EGFR–PE (cat. #352904), anti–PD-1–BV711 (cat. #329928), anti-INFγ–PE (cat. #502509), and anti-TNFα-AF700 (cat. #502928) from BioLegend; anti-CD45–APC-H7 (cat. # 560178), anti-EpCam–BV605 (cat. #563410), and anti-CD25–PECy7 (cat. # 335789) from BD Bioscience; anti-CD8–APC-H7 (cat. # 560179), anti-FOLR1–AF700 (cat. #FAB5646N) from R&D; and anti–LAG-3–FITC (cat. #ALX-804–806F-C100, Enzo Life Sciences). An antibody against mouse CD45-BV650 (cat. #563410, BD Bioscience) was used to exclude mouse immune cells from analysis. Expression of CAR proteins was detected using biotinylated goat anti-mouse IgG (Jackson ImmunoResearch, cat. #115–035-003) followed by streptavidin-APC (BD Bioscience; cat. #554067). All experiments were conducted on a BD LSRFortessa flow cytometer (Becton Dickinson) and analyzed with FlowJo v9.7 (Tree Star).
In vitro coculture experiments
All coculture experiments were performed with a CD4+ cell-to-CD8+ cell ratio of 1, with 50% CAR+ population. Tumor cells (1 × 105) were seeded in 48-well plates and infected with OAd or OAd-BiTE at an MOI of 1, with the exception of SKOV3, which were infected at an MOI of 10. After 24 hours, 3 × 105 T cells were added at an effector-to-target ratio of 3. For cytokine production assays, supernatants were collected 48 hours after coculture and assessed for human IFNγ using the DuoSet ELISA Development Kit (R&D Systems), as per the manufacturer’s protocol. For T-cell activation assays, cells were collected at 48 hours and stained for CD45, CD4, CD8, CAR, and CD25 as described above. For T-cell proliferation assays, cells were collected before coculture and after 5 days of incubation with tumor cells and stained for CD45, CD4, CD8, and CAR as described above. Absolute T-cell counts were determined using CountBright fluorescent beads (Invitrogen).
Cytotoxicity assays
Cytotoxic killing of target cells was assessed using the xCELLi-gence Real-Time Cell Analyzer System (ACEA Biosciences). Target tumor cells were plated (1 × 104 cells/well) and infected with OAd or OAd-BiTE (MOI = 10 for SKOV3 and MOI = 1 for all other cell lines). After overnight cell adherence, T cells were added at the indicated E:T ratios. Cell index (relative cell impedance) was monitored every 20 minutes for 5 days and normalized to the maximum cell index value immediately prior to effector-cell plating. Shaded lines reflect the mean of triplicate wells ± SD. Cytotoxic killing of keratinocytes was analyzed by a flow cytometry-based assay as previously described (31). Briefly, target cells (2 × 105 keratinocytes in 24 well/plates and 1 × 105 SKOV3 in 48 well/plates) were infected with OAd-BiTE at an MOI of 10. After 24 hours, control T cells or CART cells were added at the indicated E:T ratios. Eighteen hours after coculture, supernatants and adherent cells were collected and stained with live/dead violet and anti-human CD45. Live tumor cells (i.e., CD45–) were counted using CountBright absolute counting beads (Thermo Fisher Scientific).
Gene-expression analysis by RT-PCR
Quantitative (q)RT-PCR was used to analyze expression of FR-α and EGFR. RNA from frozen cell pellets was extracted with RNea-syMini kit (Qiagen) and reverse transcribed with the High Capacity RNA-to-cDNA Kit (Applied Biosystems), according to the manufacturer’s instructions. qRT-PCR was performed using TaqMan Universal PCR Master Mix in a ViiA 7 Real Time PCR system. The following probes were purchased from Applied Biosystems: FOLR1 (Hs01124179_g1), EGFR (Hs01076090_m1), and GAPDH (Hs02786624_g1). Target gene expression was calculated by the 2ΔΔCr method for relative quantification after normalization to Gapdh expression.
Xenograft models
The University of Pennsylvania Institutional Animal Care and Use Committee approved all animal experiments. NOD/scid/IL2rγ–/– (NSG) mice were purchased from The Jackson Laboratory and bred and housed in the vivarium at the University of Pennsylvania in pathogen-free conditions. Xenograft tumors were established by subcutaneous injection of indicated tumor cells into the flanks of NSG mice. For Panc-1, a solution of 50% Matrigel (BD Biosciences) and 50% PBS was used. After mean tumor volume reached 100 mm3, mice were treated with one intratumoral injection of OAd-BiTE (1 × 109vp) or PBS. After 3 or 5 days, mice were treated with one intravenous injection of 1 × 107 CART cells (50% CAR+, 1:1 CD4:CD8). In HCT116 tumors, a second CART-cell injection was performed after one week. Tumors were measured once or twice a week with calipers, and volumes were calculated as V = ½ × length (L) × width (W) × W. The endpoint was established at a tumor volume ≥1,500 mm3.
Bioluminescent imaging
T cells expressing CBR luciferase were used to detect trafficking of the T cells to the tumor. Anesthetized mice were imaged using a Xenogen Spectrum system and Living Image v4.2 software. Mice were given an intraperitoneal injection of 150 mg/kg D-luciferin (Caliper Life Sciences, Hopkinton, MA) and imaged at the peak of photon emission (5 minutes). Tumor radiance was measured by drawing a region of interest around the tumor contour.
Immunohistochemistry and RNA in situ detection
Tumors were harvested at the experimental endpoint and embedded in paraffin. For CD8 and adenovirus detection, sections were stained with anti-CD8 (Thermo Fisher Scientific; RB-9009-PO) and anti-OAd (Santa Cruz Biotechnology; SC-430), followed by a biotinylated anti-rabbit secondary (Vector Labs, BA-1000). For RNA in situ detection, RNAscope (ACD Bio) was used according to the manufacturer’s instructions. Stained slides were digitally scanned at 20× magnification on an Aperio CS-O slide scanner (Leica Biosystems). The proportion of cells staining positive for CD8 was analyzed using the Nuclear Staining Algorithm (Leica Biosystems).
Ex vivo studies
Panc-1 tumors were harvested at day 12 (for CD19-CART) or 15 (for FR-CART) after T-cell injection. Single-cell suspensions from tumors were prepared by mechanical disaggregation in RPMI without serum using the gentleMACS Dissociator (Miltenyi) and the gentleMACS C tubes at 1,200 rpm for 59 seconds. Cell suspensions were filtered with 100 mm Falcon cell strainers (Thermo Fisher Scientific) to ensure a single-cell suspension and stained for viability, followed by staining for murine CD45 and human CD45, CD4, CD8, EpCAM, CD25, Ki67, PD-1, LAG-3, and TIM-3, as already described.
Statistical analyses
All statistics were performed using GraphPad Prism v6. For comparisons of two groups, two-tailed unpaired t tests were used. One-way ANOVA with Tukey post hoc test was used for comparison of three or more groups in a single condition. Statistical analysis for tumor volume was performed using two-way repeated-mea-sure ANOVA. Kaplan-Meier survival data were analyzed using a log rank (Mantel-Cox) test.
Results
CART-cell therapy of solid tumors induces tumor escape
To assess the killing capacities of CART cells in vitro, we cocultured control cells (untransduced, UTD) or FR-α-specific CART cells (Fig. 1A) with a panel of cancer cell lines with variable levels of FR-α expression. SKOV3 (ovarian), HCT116 (colon), and Panc-1 (pancreatic) cells showed high, intermediate, and low FR-α expression, respectively, whereas NCI-H226 (lung squamous carcinoma) and C30 (ovarian) were FR-α-negative (Fig. 1B). CART cells exhibited dose-dependent and antigen-specific cytotoxicity. However, total tumor cell clearance was observed only in the cell line expressing the highest FR-α, SKOV3 (Fig. 1C).
Figure 1.
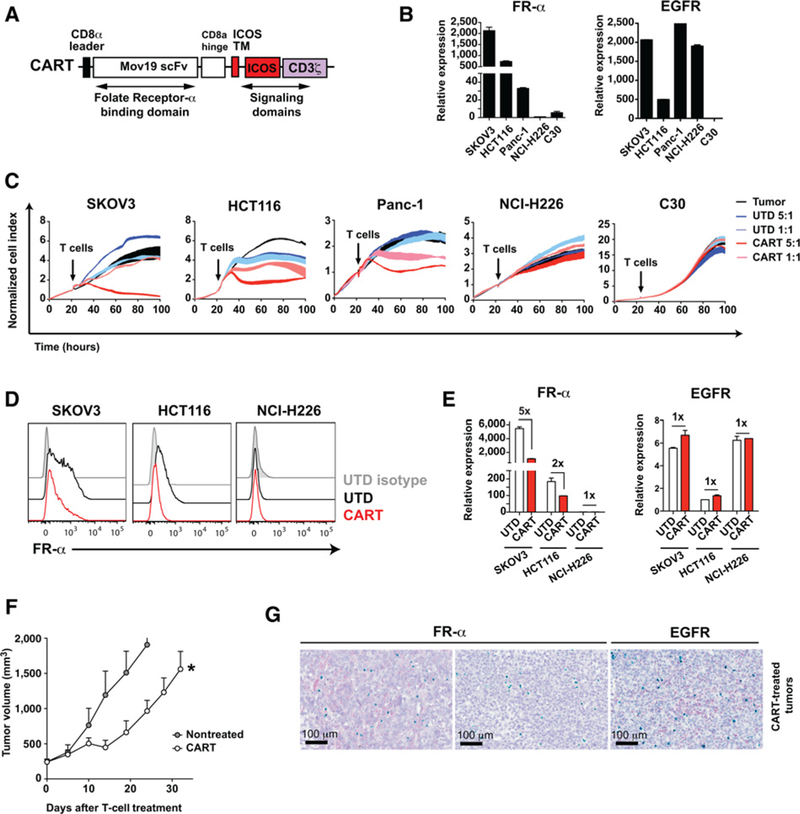
Tumors escape CART-cell therapy against FR-α by selection of antigen-negative populations. A, Schematic representation of the FR-CAR. The CAR consists of Mov19scFvthat binds to FR-α, the ICOS transmembrane (TM) and intracellular domains, and the human CD3Z chain. B, Expression of FR-α and EGFR was measured by qRT-PCR for the indicated cell lines. The mean ± SD of triplicates is shown. C, A real-time cytotoxicity assay (xCelligence) was used to evaluate the lysis of the indicated tumor cells when treated with control cells (UTD) or CART cells at the indicated E:T ratios over a 100-hour period. D and E, Tumor cells were cocultured with UTD or CART cells (E:T = 3:1). After 24 hours, FR-α and EGFR expression was analyzed in surviving tumor cells by (D) flow cytometry or (E) qRT-PCR to detect relative expression of FR-α and EGFR for the indicated cell lines in the presence or absence of CART cells. The mean ± SD from two independent experiments is shown. F and G, NSG mice bearing SKOV3 tumors were left untreated or treated with two intravenous injections of 4 × 106 CART cells (n = 5 per group). F, Tumor volume was monitored. The mean tumor volume ± SEM is shown. Representative results from two independent experiments are shown. *, P < 0.05 by two-way ANOVA with Tukey post hoc test. G, Tumors were obtained at the end point and FR-α (red), EGFR (red), and CD3 (green) expression in tumors was analyzed by RNAscope. Representative images from the CART cell-treated group are shown. Two different areas of the same tumor are shown for FR-α. EGFR expression in an area negative for FR-α expression in consecutive sections is shown.
We hypothesized that CART cells may kill tumor cells with high CAR-targeted antigen expression but would fail to eliminate cells with low FR-α densities. To test this, tumor cells were cocultured with UTD or FR-CART cells, and FR-α expression was assessed in surviving cells 24 or 72 hours after coculture. As a control, the expression of EGFR, a tumor-associated antigen not targeted by the CAR, was also evaluated. EGFR is expressed in SKOV3, HCT116, Panc-1, and NCI-H226 but is absent in C30 cells (Fig. 1B). SKOV3 and HCT116 cells that escaped CART-cell therapy showed lower FR-α expression compared with tumor cells cocultured with control T cells (Fig. 1D and E), indicating that tumor cells with reduced FR-α expression were not killed as effectively by FR-CART cells. No difference in EGFR expression was observed in surviving tumor cells after CART-cell treatment compared with UTD cell treatment (Fig. 1E).
We next addressed whether the heterogeneity of FR-α antigen expression would impact the in vivo efficacy of FR-CART cells. NSG animals bearing SKOV3 tumors were treated with two doses of CART cells or left untreated. CART-cell treatment significantly delayed tumor growth, but eventually, tumors grew exponentially (Fig. 1F). To understand why tumors escaped CART-cell therapy, tumors were resected 32 days after treatment and expression of FR-α, EGFR, and CD3 were determined by RNAscope (Fig. 1G). Several areas of the tumor expressed FR-α and contained large numbers of CD3+ T cells, suggesting that, despite successfully infiltrating the tumors, CART cells had become hypofunctional. We also found areas of the tumors with no FR-α expression in both treated and untreated tumors, suggesting that heterogeneity of antigen expression could also be a mechanism for tumor escape. EGFR was expressed in tumor areas with no FR-α expression, suggesting an opportunity to combine EGFR-and FR-α-targeting therapies to minimize tumor escape.
A BiTE-armed oncolytic adenovirus improves tumor killing of CART cells
We hypothesized that combining CART cells with a BiTE-armed oncolytic virus could enhance tumor killing by using a multimodal killing mechanism. To test this, FR-CART cells were used in combination with OAd-BiTE, an oncolytic adenovirus encoding an EGFR-targeting BiTE that can potentially replicate in any cancer cell with a deregulated retinoblastoma pathway. The EGFR-targeting BiTE gene is under the control of the major late promoter, securing BiTE expression only upon successful replication of the adenovirus in cancer cells (Fig. 2A).
Figure 2.
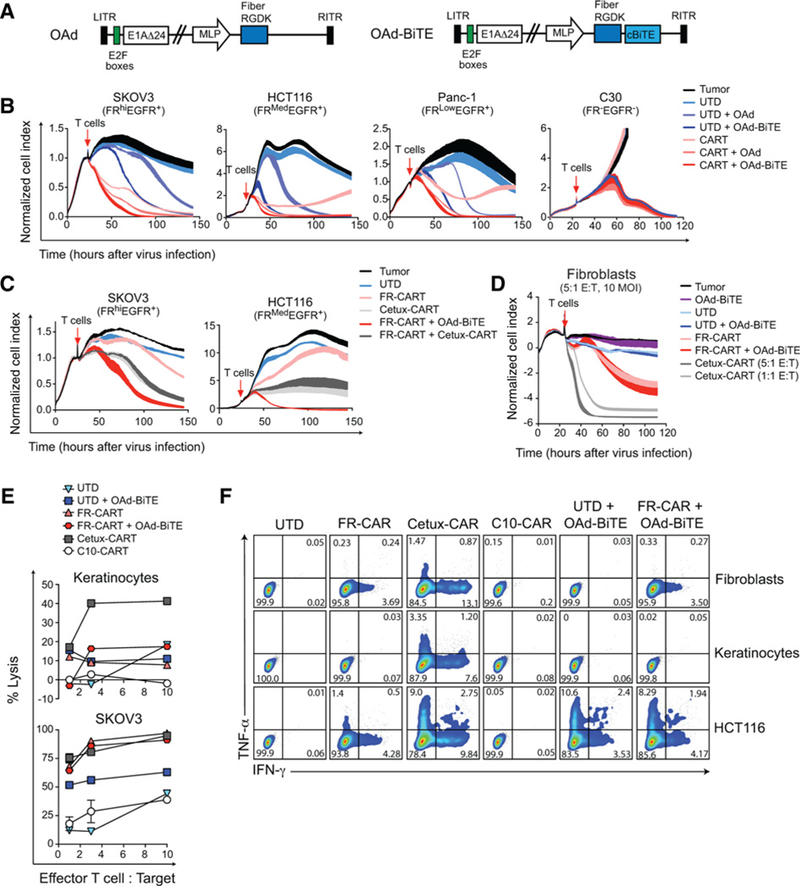
A BiTE-armed oncolytic adenovirus improves tumor killing of CART cells without compromising their safety profile. A, Schematic representation of the oncolytic viruses (OAd and OAd-BiTE). B-D, A cytotoxicity assay was used to evaluate the lysis of target cells over a 140-hour period. The mean ± SD of duplicates is shown. Representative of two to four independent experiments. E, Target cells were infected with virus and 24 hours later T cells were added at the indicated E:T ratios. Specific cytolysis was determined by flow cytometry 48 hours after T-cell addition to the cultures. F, Intracellular cytokine staining for TNFα and IFNγ in T cells after coculture with human primary or tumor cells for 20 hours.
To assess the efficacy of the combination of CART cells and OAd-BiTE, FR-α+/EGFR+ tumor cells (SKOV3, HCT116, and Panc-1) were infected with viruses at low MOI and cocultured with UTD or CART cells. C30 tumor cells were used as controls, and the parental virus (OAd; Fig. 2A) was used as a control for virus oncolysis. Similarto Fig. 1C, CART cells alone reduced tumor cell growth but, except for SKOV3 cells, tumor cell lines were able to escape therapy and proliferate (Fig. 2B). OAd-BiTE in combination with UTD cells improved the overall killing of EGFR+ cells compared with the combination of the parental virus and UTD cells. The combination of OAd-BiTE with CART cells reduced the time of killing of all FR-α+/EGFR+ cells compared with all other tested treatments. C30 cells were eliminated in the presence of both viruses, but no T cell-mediated killing was observed in these cells, indicating that both CART-and BiTE-mediated killing of cancer cells were antigen-specific.
Several studies have addressed the benefit of simultaneously targeting two antigens with pooled or bispecific CART cells (34–36). We, therefore, compared the killing kinetics of the OAd-BiTE with FR-CAR cells to that of cetuximab-based EGFR-targeting CART cells (Cetux-CART) pooled with FR-CART cells. Cetux-CART cells efficiently killed cancer cells, but pooling cetux-CARTs with FR-CARTs did not enhance the overall efficacy of the former (Fig. 2C; Supplementary Fig. S1A and S1B). The combination of OAd-BiTE and FR-CART cells outperformed the killing kinetics of single-targeting or pooled CART cells.
One of the main challenges of CART cells for the treatment of solid tumors is the selection of cancer-specific targets to avoid the on-target, off-tumor side effects observed in clinical trials (37). Because EGFR is expressed in a wide variety of healthy tissues, we compared the safety profile of both strategies in vitro. To this end, primary cells (fibroblasts and keratinocytes) and tumor cell lines were left uninfected or infected with OAd-BiTE and cocultured with control T cells or with EGFR-or FR-α-targeting CART cells. Cytotoxicity and T-cell activation were evaluated. Low-affinity anti-EGFR CART cells (C10-CART) were included as a control for improved safety profile (38). As previously demonstrated (39), cetux-CART cells eradicated normal primary cells and tumor cell lines and produced type I cytokines in response to endogenous levels of EGFR in normal tissues (Fig. 2D–F; Supplementary Fig. S1). C10-CART cells were not activated by primary cells, although this came at the expense of antitumor potency, as C10-CART cells could only kill EGFRhi-expressing Panc-1 cells (Supplementary Fig. S1A–S1C). The OAd-BiTE did not increase UTD-or FR-CART cell–mediated lysis of primary cells. Similarly, T cells were not activated by OAd-BiTE-infected fibroblasts or keratinocytes but produced cytokines in the presence of infected cancer cells (Fig. 2F; Supplementary Fig. S1F). Overall, the combination of OAd-BiTE and FR-CART cells resulted in improved killing of cancer cells and showed a favorable safety profile.
OAd-BiTE–mediated oncolysis enhances activation and proliferation of CART cells
CART-cell preparations used in clinical trials contain a high percentage (50%–95%) of CAR- (i.e., untransduced) cells that do not recognize tumor antigens but could be redirected toward tumor cells in the presence of the OAd-BiTE. To investigate whether OAd-BiTE-mediated oncolysis of FR-α+EGFR+ tumor cells could enhance the effector function of CART-cell preparations, HCT116, SKOV3, and Panc-1 tumor cells were left untreated (mock) or infected with either OAd or OAd-BiTE at low MOI, and cocultured with either UTD or CART cells. The combination therapy increased activation (measured by %CD25+) of both CD4+ and CD8+ T cells after 48 hours compared with CART-cell preparations alone or in combination with the OAd (Fig. 3A). This increased activation was primarily due to the BiTE-mediated activation of the UTD cells within the CART-cell preparation (Fig. 3B).
Figure 3.
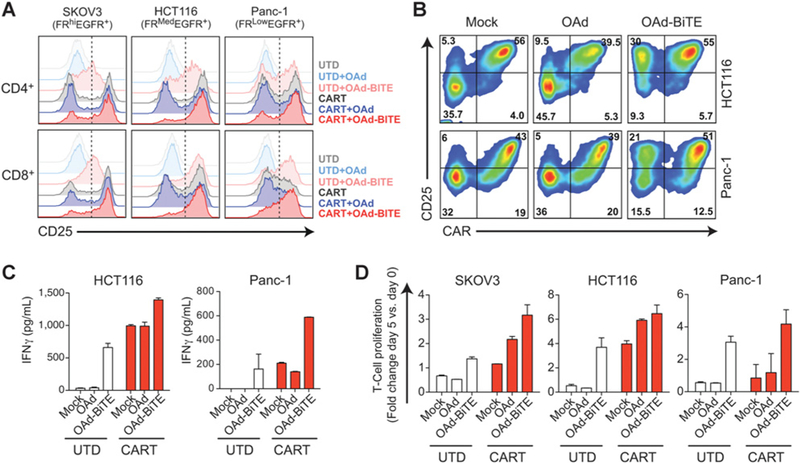
OAd-BiTE-mediated oncolysis enhances the activation and proliferation of CART-cell preparations. FR-α+EGFR+ tumor cells were infected with OAd, OAd-BiTE, or left uninfected (mock), and after 24 hours, CART or UTD cells were added (E:T = 2). A and B, T cells were collected 48 hours after T-cell coculture and stained with antibodies against CD45, CD4, CD8, CAR, and CD25. B, CAR and CD25 expression on CART-cell preparations after coculture with tumor cells. C, Supernatants were obtained 48 hours after coculture, and IFNγ production was analyzed by ELISA. D, T-cell proliferation following coculture with tumor cells. The mean ± SD is shown in C and D. Representative of one of three independent experiments.
Next, we assessed cytokine production and proliferation after coculture with tumor cells. Infection of tumor cells with the parental virus had no effect on the cytokine production or proliferation of UTD or CART cells (Fig. 3C and D). UTD cells cocultured with OAd-BiTE-infected tumor cells had similar proliferation rates and IFNγ secretion to CART cells alone, indicating that both CARs and BiTEs can redirect T cells toward tumor antigens efficiently. We also observed a significant increase in IFNγ production by CART preparations exposed to OAd-BiTE-infected tumor cells, as well as an increase in T-cell proliferation. Overall, these results indicated that OAd-BiTE–mediated oncolysis of FR-α+/EGFR+ tumor cells enhance effector T-cell function of CART-cell preparations, in part, by activation of the CAR–T-cell fraction.
OAd-BiTE–mediated oncolysis activates CART cells in the absence of FR-α
We next hypothesized that BiTEs secreted from adenovirus-infected cells could address FR-α expression heterogeneity by redirecting CART cells to EGFR-expressing tumor cells. To demonstrate this, NCI-H226 cells were used as a model for FR-α loss while EGFR expression still occurs, and C30 cells were used as an antigen-negative control. Infection of NCI-H226 cells with OAd-BiTE induced activation of both CAR+ and CAR– T cells, as evidenced by CD25 expression (Fig. 4A and B) and the presence of the BiTE-induced IFNγ production and proliferation of CART-cell preparations in the absence of FR-α expression on tumor cells (Fig. 4C and D). When T cells were cocultured with C30 cells, no activation or proliferation were observed in any of the coculture groups. This demonstrated that the T-cell activation observed in the absence of the CAR-targeted antigen was EGFR-specific and that the dual-antigen targeting of the combination therapy could lead to activated T cells in the absence of one antigen.
Figure 4.
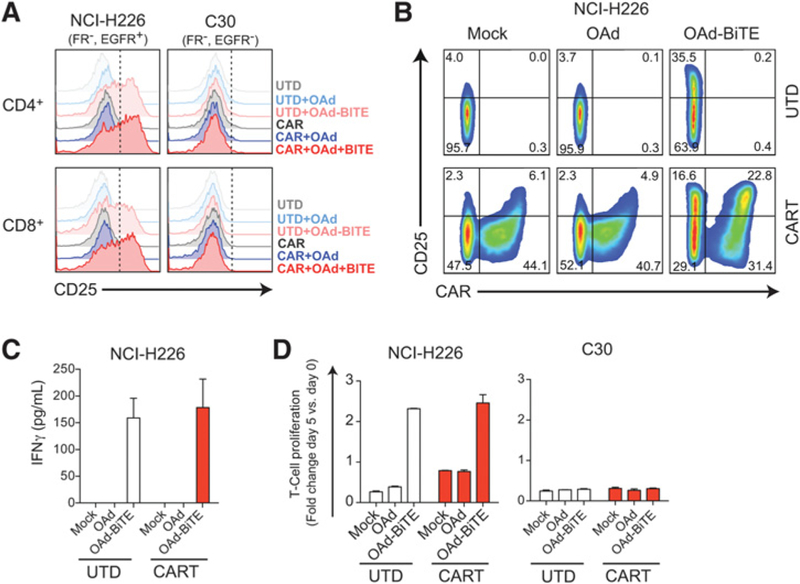
OAd-BiTE-mediated oncolysis activates CART cells in the absence of FR-α. NCI-H226 and C30 tumor cells were infected with OAd, OAd-BiTE, or left uninfected, and after 24 hours, CART cells were added (E:T = 2). A and B, T cells were collected 48 hours after T-cell coculture and stained with antibodies against CD45, CD4, CD8, CAR, and CD25. B, CAR and CD25 expression on UTD or CART-cell preparations after coculture with NCI-H226. C, Supernatants were obtained 48 hours after coculture, and cytokine production was analyzed by ELISA. D, T-cell proliferation following coculture with tumor cells. The mean ± SD is shown in C and D. Representative of one of two independent experiments.
Combined CART cells and OAd-BiTE enhances antitumor effects in vivo
To determine whether OAd-BiTE improves the antitumor efficacy of CART cells in vivo, NSG mice bearing subcutaneous HCT116 tumors were treated with OAd-BiTE or PBS (Fig. 5). Three and 7 days later, animals received an intravenous injection of 1 × 107 Tcells (50% CAR+). In this fast-growing tumormodel, treatment with OAd-BiTE showed no antitumor effect compared with saline-treated animals. CART cells alone resulted in moderate antitumor effect, as tumors initially decreased in size but eventually reoccurred and grew aggressively (Fig. 5A). The combination of OAd-BiTE and CART cells significantly decreased tumor volume when compared with single-agent therapies and significantly improved survival, as all animals treated with CART cells and OAd-BiTE survived until the experimental endpoint of 41 days, in contrast to treatment with OAd-BiTE alone (median survival 20 days) or CART cells alone (median survival 38 days, Fig. 5B).
Figure 5.
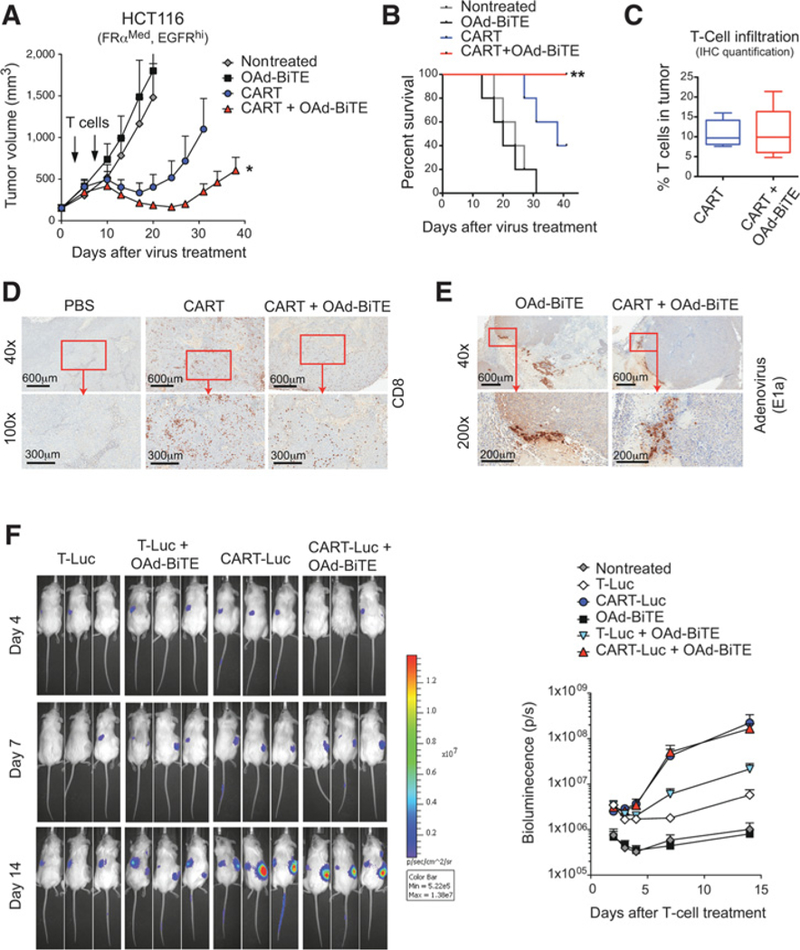
Combination of OAd-BiTE and CART cells shows synergistic antitumor effects in vivo. A–E, NSG mice bearing 15-day established HCT116 tumors were treated with OAd-BiTE (1 × 109vp) or PBS. 3 and 7 days after virus treatment, animals were treated with 10 × 106 CART cells. A, The mean tumor volume ± SEM (n = 5 per group) is plotted. *, P< 0.05 by two-way ANOVA with Tukey post hoc test B, Kaplan-Meier survival curves of the experiment described in A. Representative of two independent experiments. **, P < 0.01 by long-rank (Mantel-Cox) test. C, The immune infiltrate in the tumors described in A was evaluated by immunohistochemistry for human CD8+ T cells at day 41 after treatment. The percentage of nuclei that stained positive for CD8+ T cells in the tumor was quantified. Representative images for (D) CD8 and (E) adenovirus immunohistochemical stains are shown. F, T-cell accumulation in HCT116 tumors. NSG mice bearing HCT116 tumors were treated with OAd-BiTE (1 × 109vp) or PBS. Three and 10 days after virus treatment, animals were left untreated or treated with an intravenous injection of 10 × 106 control T-Luc cells (40% CBG+) or CART-Luc cells (50% CAR+, 40% CBG+). A total of 5 mice per group were used for the indicated treatments. Bioluminiscence was analyzed at the indicated time points.
To investigate the mechanism behind the synergistic antitumor effects of the combined treatment, the presence of OAd-BiTE and T cells in the tumor was evaluated by histologic analysis. All tumors treated with CART cells had high T-cell infiltration (Fig. 5C and D), and no differences in the levels of tumor-infiltrating CD8+ T cells in the tumors treated with CART cells alone or CART cells and OAd-BiTE were observed (Fig. 5C). Isolated virus replication foci were observed around necrotic areas in tumors treated with OAd-BiTE, indicating that virus oncolysis had some effect but was not the main mechanism behind the enhanced antitumor effect observed with the combination therapy (Fig. 5E).
In order to confirm these results, we performed a biodistribution assay using T cells modified to express CBR luciferase and the FR-CAR (CART-Luc). As control cells, CBR-expressing T cells without the CAR (T-Luc) were used. Quantification of bioluminescence in tumors indicated that treatment with OAd-BiTE significantly enhanced T-cell accumulation in the tumors treated with control T cells, as we have previously demonstrated (ref. 25; Fig. 5F). CART-Luc cells accumulated in the tumors significantly better than control T-Luc cells, indicating that tumor specificity is key for T-cell persistence. However, the presence of OAd-BiTE did not enhance the accumulation of FR-CART cells in the tumors, confirming the histology results observed in the antitumor efficacy experiment. Overall, these results indicated that in this aggressive tumor model, OAd-BiTE worked synergistically with CART cells to control tumor growth without affecting CART-cell accumulation.
Combined CART cells and OAd-BiTE improves T-cell activation in vivo
To test whether the combination therapy could further enhance the effect of CART cells as a monotherapy on tumors expressing lower CAR-targeted antigen, we used a xenograft mouse model of pancreatic cancer. NSG mice bearing Panc-1 tumors were treated with a single dose of OAd-BiTE or PBS intratumorally, followed by a single dose of FR-CART cells 3 days later. Treatment with FR-CART cells showed an initial antitumor effect, but it was insufficient to induce tumor regression, and eventually, tumors progressed rapidly (Fig. 6A). In contrast, the combination of CART cells with OAd-BiTE induced a robust antitumor effect, with 100% of the tumors regressing after treatment. In this slow-growing tumor, treatment with OAd-BiTE as a single agent was able to control tumor growth. However, 80% of the treated tumors were progressing by the end of the experiment. To better understand the contribution of each element of the combination therapy to the observed antitumor effect, we repeated this experiment, including control CD19-CART cells and the parental virus (OAd). CD19 is not expressed in Panc-1 tumors, and thus this control mimics a lack of antigen expression due to tumor heterogeneity or antigen loss. Panc-1 tumors grew exponentially in untreated animals and those treated with CD19-CART cells. Treatment with the parental virus in combination with CD19-CART cells induced a modest but significant delay in tumor growth compared with nontreated tumors (Fig. 6B; Supplementary Fig. S2). The combination of OAd-BiTE with CD19-CART cells delayed tumor growth at a similar rate to FR-CART cells, indicating that in the absence of the targeted-tumor antigen, OAd-BiTE can redirect CAR-T cells toward EGFR and efficiently mediate tumor control. Combining FR-CART cells with OAd further enhanced antitumor effects, although differences were not significant compared with CART cells alone. The combination of CAR-T cells with the OAd-BiTE further increased the antitumor activity, and it was the only treatment leading to tumor regression. These results indicated that each element of the combination therapy (i.e., CAR, BiTE, and oncolysis) contributed to tumor regression.
Figure 6.
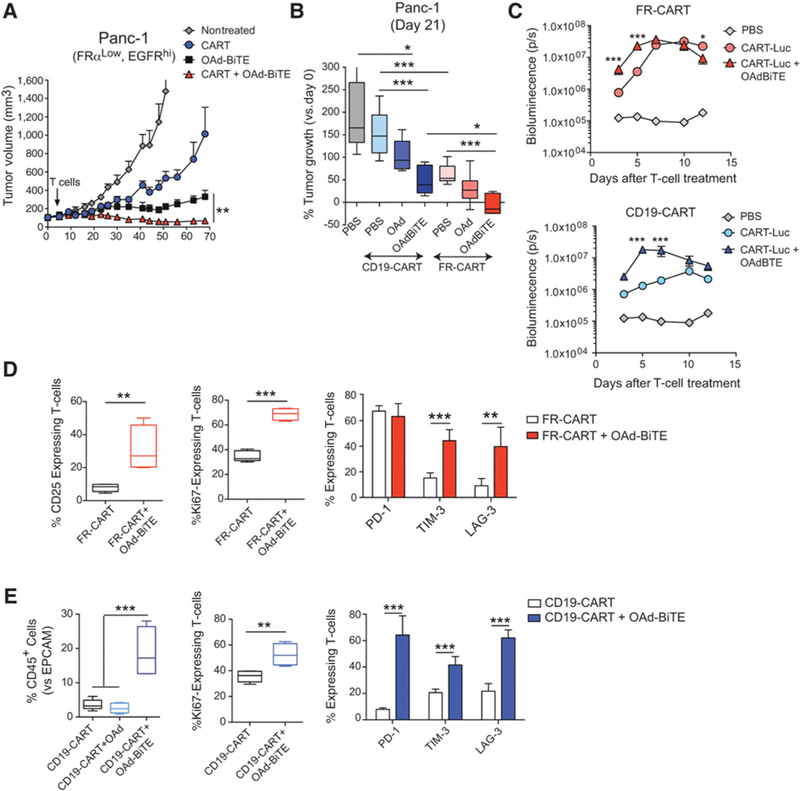
Combination of OAd-BiTE and CART cells shows enhanced antitumor effects with improved T-cell activation in vivo. NSG mice bearing 30-day established Panc-1 tumors were treated with OAd-BiTE (1 × 109vp) or PBS. Five days later, animals were treated with an intravenous injection of 1 × 107 FR-CART cells. A, The mean tumor volume ± SEM (n = 5 per group) is plotted. *, P < 0.05 by two-way ANOVA with Tukey post hoc test. B, Percentage of Panc-1 tumor growth indicated as the change in tumor volume on day 21 versus baseline (n = 5–7 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001 by one-way ANOVA with Tukey post hoc test. C, T-cell accumulation in Panc-1 tumors as measured by bioluminescence. Mice bearing Panc-1 tumors were treated with OAd-BiTE or PBS. Three days later, animals were treated with 1 × 107 FR-CART-Luc or CD19-CART-Luc cells. Five animals were used for each indicated group. *, P < 0.05; **, P < 0.01; ***, P < 0.001 by two-way ANOVA with Tukey post hoc test. D and E, Proliferation of tumor-infiltrating T-cells (as indicated by Ki67 staining) and T-cell phenotype were evaluated by flow cytometry at day 15 (D) or 12 (E) after treatment. **, P < 0.01; ***, P < 0.001 by unpaired t test (for comparison of two groups) or one-way ANOVA with Tukey post hoc test (for comparison of more than two groups).
We next assessed whether the treatment with OAd-BiTE could enhance T-cell trafficking to tumors with low expression of the target antigen (Panc-1). Biodistribution studies in mice showed enhanced FR-CART–cell accumulation (>6-fold) at the tumor site in the presence of OAdBiTE at early time points after injection (Fig. 6C). In the absence of CART-cognate antigen expression, the OAd-BiTE treatment further increased the accumulation of CART cells in the tumor, as the bioluminescence signal in tumors treated with CD19-CART cells increased 14-fold in the presence of OAd-BiTE.
Finally, we hypothesized that the enhanced antitumor effect observed in the combination therapy could also be due to increased activation of the T cells infiltrating the tumors in the presence of the OAd-BiTE. To test this, Panc-1 tumor–bearing mice were treated with FR-CART cells in the presence or absence of OAd-BiTE, and tumor-infiltrating T cells were isolated from tumors 15 days after T-cell injection. T cells isolated from the tumors treated with the combination therapy showed increased expression of Ki67, a proliferation marker, and of the activation markers CD25, TIM-3, and LAG-3 (Fig. 6D). This in vivo experiment was repeated using CART cells targeting CD19 to mimic CART cells unable to find their cognate antigen, and the OAd was used as a control to better distinguish the role of the BiTE from the role of the oncolysis. Tumors treated with CD19-CART cells in the presence of OAd-BiTE showed an increased percentage of CD45+-infiltrating T cells versus EPCAM+ tumor cells when compared with tumors treated with OAd or without virus (Fig. 6E). These results indicate that in the absence of cognate antigen, the accumulation of CART cells can be enhanced by the presence of a BiTE targeting a different antigen. The phenotypic characterization of CAR TILs revealed a significantly higher frequency of Ki67, PD-1, LAG-3, and TIM-3 expression on TILs in the presence of OAd-BiTE when compared with CD19-CART cells alone, confirming and extending the results with FR-CART cells. The role of oncolysis in T-cell activation could not be analyzed due to the low numbers of T cells found in the tumors treated with the OAd.
The enhanced early T-cell accumulation at the tumor site of CART cells, including those unable to recognize their cognate antigen, and the enhanced T-cell activation in vivo explains, at least in part, the enhanced antitumor effect and prolonged survival observed with the combination therapy.
Discussion
CART cells face several hurdles in solid tumors, including tumor-antigen heterogeneity and an immunosuppressive tumor microenvironment (20). In this work, we aimed to address some of these limitations by combining anti–FR-α CART cells with an oncolytic adenovirus expressing a BiTE targeting the EGFR. Our studies demonstrate that the combined action of virus, BiTE, and CART cells leads to a synergistic control of the tumor growth and increased overall survival without compromising safety.
The combination of CART cells with OAd-BiTE enhanced the activation, proliferation, and killing activity of CART-cell preparations in vitro. These enhanced effector functions are due, in part, to the BiTE-mediated engagement of the untransduced (CAR-) T cells present in the CART-cell preparations. The MOI of lentiviral vectors that can be used to generate CART-cell products for clinical applications is restricted for safety reasons, resulting in preparations with 5% to 45% CAR+ T cells. Our results demonstrated that redirection of the UTD cells within CART-cell preparations toward tumor antigens can maximize the antitumor effects of the CART therapy. Our study reports a strategy that exploits the effector potential of CAR-negative cells found in CART-cell preparations. We also show that BiTE expression by an OAd can redirect CAR+ T cells toward a secondary tumor antigen (i.e., EGFR) in the event of loss or lack of expression of the CAR-targeted antigen (i.e., FR-α).
Consistent with the enhanced tumor cell killing shown in vitro, the combination of CART cells and OAd-BiTE also displayed an improved antitumor effect in vivo when compared with each monotherapy. We found that human CART cells alone were able to accumulate to high numbers in the tumor tissue and initially induced tumor regression. However, despite persisting in the tumor microenvironment, CART cells lost their capacity to control tumor growth during the course of the treatment. These findings are consistent with previous reports indicating that CART cells undergo rapid loss of functional activity in solid tumors as a result of a multifactorial process, which includes the expression of T-cell-inhibitory enzymes and surface inhibitory receptors and loss of CAR expression by the T cells (15, 40). Here, we demonstrated that in the presence of OAd-BiTE, T cells showed enhanced in vivo activation 15 days after T-cell administration. We observed an increased percentage of T cells expressing the proliferation marker Ki67. Different factors can account for this increased T-cell activation and proliferation. In the presence of the EGFR-targeting BiTE, CART cells can be activated through CD3 in the event of CAR expression loss by T cells or lack of FR-α expression by tumor cells. OAds can induce tumor debulking, which may create a more suitable environment for T-cell activation. In this regard, OAds can also activate the innate immune response through the release of tumor antigens, pattern recognition receptor ligands, and danger signals. One of the main limitations of this study is that the use of NSG mice precludes the study of the effects of the interaction between the OAd and the tumor immune cells, which could further enhance the antitumor effects of the combination therapy.
Solid tumors exhibit antigen heterogeneity, making it challenging to identify a universal target expressed throughout the whole tumor. Here, we showed that after exposure to CART cells targeting FR-α, cancer cells with low or undetectable FR-α expression but high EGFR expression were selected in vitro. Similarly, we found that tumor xenografts contained areas with low or undetectable FR-α expression, suggesting that tumor heterogeneity could have a key role in tumor escape. In agreement with this observations, Hedge and colleagues reports that exposure to HER2-specific CART cells select a tumor cell population with dim to undetectable HER2 expression, but with increased IL13Ra2 expression (36). In our study, we chose two different target antigens, FR-α and EGFR, which are often coexpressed in a high percentage of solid tumors. On top of targeting two antigens, the addition of an OAd increases the chances of targeting the entire range of tumor cells, as the OAd (ICO15K) can potentially kill any cancer cell with aderegulated retinoblastoma pathway. OAds kill cancer cells by oncolysis, adding a different mechanism of cell killing and reducing the chances of tumor escape.
Tumor cells expressing the CAR-targeted antigen can also escape T-cell therapy by losing the expression of the antigen, especially when targeting nondriving or nononcogenic proteins. Various mutations in the CD19 locus, in addition to alternative splice variants of CD19, have been associated with the development of CART19-resistant acute lymphocytic leukemia (ALL; ref. 16). Diminished CD22 surface expression on B-cell ALL cells has also been recently identified as a mechanism for relapse following CD22-CAR therapy (41). The simultaneous targeting of different tumor antigens has been reported as a promising solution for antigen loss in hematologic malignancies and solid tumors, with dual and bispecific CART cells providing superior potency than pooled combination of CART cells (34, 36, 41). A main drawback of these approaches and other targeted therapies is that selecting safe solid tumor targets can be challenging due to on-target, off-tumor side effects (42). One advantage of our therapy is the restricted BiTE expression from the oncolytic virus in cancer cells. The combination of OAd-BiTE with FR-CART showed a favorable in vitro safety profile in keratinocytes and fibroblasts compared with EGFR-targeting CART cells alone or in combination with FR-CARTs. Thus, the localized BiTE expression in the tumor microenvironment adds a level of selectivity which is lacking in CART cells or BiTEs as monotherapies.
Oncolytic viruses have gained attention in the past years due to their synergistic potential with existing immunotherapies (22, 43). OAds have recently been evaluated in combination with CART cells in preclinical studies. Nisho and colleagues showed that the combination of CART cells targeting GD2 with a non-armed OAd have transient beneficial effects, but that arming the virus with therapeutic transgenes is needed to increase the antitumor effects (44). Specifically, OAd-mediated delivery of RANTES and IL15 increases T-cell accumulation in the tumor and prolongs survival of tumor-bearing animals. Another study demonstrates that a helper-dependent adenovirus expressing a PD-L1–blocking mini-body (CAd-VECPDL1) in combination with an OAd improves the antitumor effects of anti-HER2 CART cells (45).
In summary, we have shown that anti-FR-α CART cells and a BiTE-expressing OAd displayed synergistic antitumor effects in vivo by enhancing T-cell activation, which resulted in prolonged survival of tumor-bearing mice. These data provide the rationale to test this combination therapy in human trials in patients with solid tumors.
Supplementary Material
Acknowledgments
This work was supported in part by the Penn Skin Biology and Diseases Resource-based Center (P30-AR069589). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This research was supported by the Parker Institute for Cancer Immunotherapy. The investigators A.D. Posey Jr, C.H. June, and S. Guedan are members of the Parker Institute for Cancer Immunotherapy, which supports the University of Pennsylvania Cancer Immunotherapy Program.
The authors thank Daniel Powel for the gift of Mov19 vectors and C30 cells. We thank Alba Rodriguez-Garcia for technical assistance and helpful discussions. We also thank the Perelman School of Medicine StemCell and Xenograft Core, the Human Immunology Core from the University of Pennsylvania, and the Pathology Core from CHOP.
Footnotes
Disclosure of Potential Conflicts of Interest
A.D. Posey reports receiving a commercial research grant from Tmunity and has ownership interest (including patents) in ImmVet Biotherapeutics. C. H. June reports receiving commercial research grants from Novartis and Tmunity, has ownership interest (including patents) in Tmunity, and is a consultant/advisory board member for Tmunity, Immune Design, and Celldex Therapeutics. No potential conflicts of interest were disclosed by the other authors.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Note: Supplementary data for this article are available at Cancer Immunology Research Online (http://cancerimmunolres.aacrjournals.org/).
C.H. June and S. Guedan share senior authorship of this article.
References
- 1.Brentjens RJ, Davila ML, Riviere I, Park J, Wang X, Cowell LG, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med 2013;5:177ra38–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kochenderfer JN, Wilson WH, Janik JE, Dudley ME, Stetler-Stevenson M, Feldman SA, et al. Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 2010;116: 4099–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med 2014;371:1507–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Porter DL, Levine BL, Kalos M, Bagg A, June CH. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 2011; 365:725–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grupp SA, Kalos M, Barrett D, Aplenc R, Porter DL, Rheingold SR, et al. Chimeric antigen receptor-modified T cells for acute lymphoid leukemia. N Engl J Med 2013;368:1509–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilham DE, Debets R, Pule M, Hawkins RE, Abken H. CAR-Tcells and solid tumors: tuning T cells to challenge an inveterate foe. Trends Mol Med 2012;18:378–85. [DOI] [PubMed] [Google Scholar]
- 7.Gajewski TF, Woo SR, Zha Y, Spaapen R, Zheng Y, Corrales L, et al. Cancer immunotherapy strategies based on overcoming barriers within the tumor microenvironment. Curr Opin Immunol 2013;25:268–76. [DOI] [PubMed] [Google Scholar]
- 8.Anderson KG, Stromnes IM, Greenberg PD. Obstacles posed by the tumor microenvironment to T cell activity: A case for synergistic therapies. Cancer Cell 2017;31:311–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol 2011;29:235–71. [DOI] [PubMed] [Google Scholar]
- 10.Lavin Y, Kobayashi S, Leader A, Amir ED, Elefant N, Bigenwald C, et al. Innate immune landscape in early lung adenocarcinoma by paired single-cell analyses. Cell 2017;169:750–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, et al. An immune atlas of clear cell renal cell carcinoma. Cell 2017;169:736–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ahmadzadeh M, Johnson LA, Heemskerk B, Wunderlich JR, Dudley ME, White DE, et al. Tumor antigen-specific CD8 T cells infiltrating the tumor express high levels of PD-1 and are functionally impaired. Blood 2009; 114:1537–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumor-specific CD8+ T cells in metastases from melanoma patients. J Clin Invest 2011;121:2350–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crespo J, Sun H, Welling TH, Tian Z, Zou W. T cell anergy, exhaustion, senescence, and stemness in the tumor microenvironment. Curr Opin Immunol 2013;25:214–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moon EK, Wang LC, Dolfi DV, Wilson CB, Ranganathan R, Sun J, et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clin Cancer Res 2014;20:4262–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, et al. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov 2015; 5:1282–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013;501: 338–45. [DOI] [PubMed] [Google Scholar]
- 18.McGranahan N, Swanton C. Biological and therapeutic impact of intra-tumor heterogeneity in cancer evolution. Cancer Cell 2015;27:15–26. [DOI] [PubMed] [Google Scholar]
- 19.O’Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD, et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol 2004;22:329–60. [DOI] [PubMed] [Google Scholar]
- 21.Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol 2012;30:658–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Keller BA, Bell JC. Oncolytic viruses—immunotherapeutics on the rise. J Mol Med 2016;94:979–91. [DOI] [PubMed] [Google Scholar]
- 23.Guedan S, Rojas JJ, Gros A, Mercade E, Cascallo M, Alemany R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol Ther 2010;18:1275–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guedan S, Grases D, Rojas JJ, Gros A, Vilardell F, Vile R, et al. GALV expression enhances the therapeutic efficacy of anoncolytic adenovirus by inducing cell fusion and enhancingvirus distribution. Gene Ther 2011;19: 1048–57. [DOI] [PubMed] [Google Scholar]
- 25.Fajardo CA, Guedan S, Rojas LA, Moreno R, Arias-Badia M, de Sostoa J, et al. Oncolytic adenoviral delivery of an EGFR-targeting T-cell engager improves antitumor efficacy. Cancer Res 2017;77:2052–63. [DOI] [PubMed] [Google Scholar]
- 26.Cody JJ, Douglas JT. Armed replicating adenoviruses for cancer virotherapy. Cancer Gene Ther 2009;16:473–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klinger M, Benjamin J, Kischel R, Stienen S, Zugmaier G. Harnessing T cells to fight cancer with BiTE® antibody constructs-past developments and future directions. Immunol Rev 2016;270:193–208. [DOI] [PubMed] [Google Scholar]
- 28.Klinger M, Brandl C, Zugmaier G, Hijazi Y, Bargou RC, Topp MS, et al. Immunopharmacologic response of patients with B-lineage acute lymphoblastic leukemia to continuous infusion of T cell-engaging CD19/CD3-bispecific BiTE antibody blinatumomab. Blood 2012;119: 6226–33. [DOI] [PubMed] [Google Scholar]
- 29.Carpenito C, Milone MC, Hassan R, Simonet JC, Lakhal M, Suhoski MM, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci 2009;106:3360–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Barrett DM, Liu X, Jiang S, June CH, Grupp SA, Zhao Y. Regimen-specific effects of RNA-modified chimeric antigen receptor T cells in mice with advanced leukemia. Hum Gene Ther 2013;24:717–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Guedan S, Chen X, Madar A, Carpenito C, McGettigan SE, Frigault MJ, et al. ICOS-based chimeric antigen receptors program bipolar TH17/TH1 cells. Blood 2014;124:1070–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Milone MC, Fish JD, Carpenito C, Carroll RG, Binder GK, Teachey D, et al. Chimeric receptors containing CD137 signal transduction domains mediate enhanced survival of T cells and increased anti leukemic efficacy in vivo. Mol Ther 2009;17:1453–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rojas JJ, Gimenez-Alejandre M, Gil-Hoyos R, Cascallo M, Alemany R. Improved systemic antitumor therapy with oncolytic adenoviruses by replacing the fiber shaft HSG-binding domain with RGD. Gene Ther 2011;19:453–7. [DOI] [PubMed] [Google Scholar]
- 34.Ruella M, Barrett DM, Kenderian SS, Shestova O, Hofmann TJ, Perazzelli J, et al. Dual CD19 and CD123 targeting prevents antigen-loss relapses after CD19-directed immunotherapies. J Clin Invest 2016;126:3814–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hegde M, Mukherjee M, Grada Z, Pignata A, Landi D, Navai SA, et al. Tandem CART cells targeting HER2 and IL13Ra2 mitigate tumor antigen escape. J Clin Invest 2016;126:3036–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hegde M, Corder A, Chow KK, Mukherjee M, Ashoori A, Kew Y, et al. Combinational targeting offsets antigen escape and enhances effector functions of adoptively transferred T cells in glioblastoma. Mol Ther 2013;21:2087–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing Erbb2. Mol Ther 2010;18:843–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC, et al. Affinity-tuned ErbB2 or EGFR chimeric antigen receptor T cells exhibit an increased therapeutic index against tumors in mice. Cancer Res 2015; 75:3596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Johnson LA, Scholler J, Ohkuri T, Kosaka A, Patel PR, McGettigan SE, et al. Rational development and characterization of humanized anti-EGFR variant III chimeric antigen receptor T cells for glioblastoma. Sci Transl Med 2015;7:275ra22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang E, Wang LC, Tsai CY, Bhoj V, Gershenson Z, Moon E, et al. Generation of potent T-cell immunotherapy for cancer using DAP12-based, multichain, chimeric immunoreceptors. Cancer Immunol Res 2015;3:815–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fry TJ, Shah NN, Orentas RJ, Stetler-Stevenson M, Yuan CM, Ramakrishna S, et al. CD22-targeted CART cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med 2017;24:20–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol 2013;31:999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ribas A, Dummer R, Puzanov I,VanderWalde A, Andtbacka RHI, Michielin O, et al. Oncolytic virotherapy promotes intratumoral T cell infiltration and improves anti-PD-1 immunotherapy. Cell 2017;170:1109–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V, et al. Armed oncolytic virus enhances immune functions of chimeric antigen receptor-modified T cells in solid tumors. Cancer Res 2014;74:5195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tanoue K, Rosewell Shaw A, Watanabe N, Porter C, Rana B, Gottschalk S, et al. Armed oncolytic adenovirus-expressing PD-L1 mini-body enhances antitumor effects of chimeric antigen receptor T cells in solid tumors. Cancer Res 2017;77:2040–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.