

Abstract
Metastasis of cancer cells to distant organ systems is a complex process that is initiated with the programming of cells in the primary tumor. The formation of distant metastatic foci is correlated with poor prognosis and limited effective treatment options. We and others have correlated Mouse double minute 2 (Mdm2) with metastasis; however, the mechanisms involved have not been elucidated. Here it is reported that shRNA-mediated silencing of Mdm2 inhibits epithelial-mesenchymal transition (EMT) and cell migration. In vivo analysis demonstrates that silencing Mdm2 in both post-EMT and basal/triple negative breast cancers resulted in decreased primary tumor vasculature, circulating tumor cells, and metastatic lung foci. Combined, these results demonstrate the importance of Mdm2 in orchestrating the initial stages of migration and metastasis.
Keywords: Mdm2, EMT, p53, Metastasis, Migration
Introduction
Metastatic foci arise after primary tumor cells successfully transition through various phases of the metastatic process. These phases include invasive migration from the primary tumor, intravasation into the blood stream, extravasation into a distant organ, and colonization in conducive tissues. In response to avascular hypoxic conditions, primary tumor cells may produce vascular endothelial growth factor (VEGF), which stimulates angiogenesis, recruits endothelial cells for neovascularization, and/or permeabilizes existing vasculature through which tumor cells can intravasate. The dissemination of tumor cells to distant organs can be initiated through an intracellular program called epithelial-mesenchymal-transition (EMT). The programming of tumor cells to migrate requires the upregulation of transcription factors such as Snail Family Zinc Finger 1 (SNAI1), and increased levels of adhesion proteins and filaments including N-cadherin and vimentin. Conversely, E-cadherin is decreased as cells transition into a mesenchymal phenotype. Moreover, we have shown that the oncogene Mouse Double Minute 2 (Mdm2) is also positively correlated with EMT (1–5). Clinically, mdm2 gene amplification occurs in approximately 10% of all human cancers, yet more importantly, several studies have detected the Mdm2 protein in 40–80% of high-grade human tumors (6,7). Increased levels of Mdm2 are correlated with poor prognosis, especially in breast cancer (8), and several correlative studies have inferred its involvement in metastasis (9–12). Similarly, we have previously shown that Mdm2 is elevated in 73% of breast carcinomas with known metastases (1).
Although several reports have asserted that Mdm2 promotes metastasis, they were either correlative studies using human patient samples (9,10,12), or in vitro studies with or without a correlative component (1,3–5). Only two studies thus far have explored the importance of Mdm2 in metastasis in vivo. One study, conducted in pancreatic carcinoma cells, found that silencing Mdm2 resulted in decreased proliferation (2). This study, along with another that used tail vein injection of transiently transfected KHT-C murine cells (11), employed approaches that did not directly assess the initiation or other phases of metastasis to define the role of Mdm2. The authors of both studies attributed the differences in metastasis to the ability of Mdm2 to downregulate wild type p53. Early reports show that Mdm2 can facilitate the destabilization of p53 in response to genotoxic stress, however, 20–40% of breast cancers subtypes (13), and 80% of basal like breast cancers (14) maintain mutant p53 (mutp53). Considering the gain-of-function properties of mutant p53, it might be expected that any downregulation of mutant p53 would result in decreased metastasis. Regardless, unlike p53, mutp53 is unableto induce mdm2 gene expression. We recently demonstrated that the TGFß1-Smad3/4 axis transcriptionally activates the mdm2 gene independently of p53. Furthermore, the activated TGFß1 pathway and elevated Mdm2 levels were correlated with human invasive/metastatic breast cancer specimens (1). Here we show that Mdm2 is necessary for upregulating essential markers of EMT, and consequently migration/invasion. Additionally, we show that Mdm2 is important in upregulating MMP-2 (Matrix Metalloproteinase 2), an indicator of highly metastatic cancers that colonize the lung. Our orthotopic experiments show that Mdm2 is a key mediator of metastatic lung foci through regulation of angiogenesis and intravasation. These events are independent of mutp53 and demonstrate a distinct role for Mdm2 at specific stages of the metastatic process.
MATERIAL AND METHODS
Cell lines, culture conditions, induction of EMT and western blotting
shMdm2 was obtained in pLKO from open biosystems (TRCN0000003380 which targets exon 12). Additional cell lines expressing shMdm2 constructs (TRCN0000003376 and TRCN0000003377) were also examined to confirm the phenotypes were due to knockdown of Mdm2. Previously described vectors include shp53 (15,16), MCHERRY (17), and EGFP (17). Lentivirus was produced as previously described (16). Briefly, Lentiviral vectors were packaged in 293T cells using second-generation packaging constructs: pCMV-dR8.74 and pMD2G. Supernatant media containing virus were collected at 36 to 48 h, supplemented with 4 μg/mL polybrene, filtered through a 0.45-μm filter, and added to MDA-MB-468 (R273H p53) and TMD231 cells (a derivative of MDA-MB-231 cells which metastasize to the lung (18) and have R280K p53). Cells were selected for puromycin (2 μg/ml) resistance. All cells were grown as described (1). Cells were incubated under normoxia (21% oxygen in complete DMEM), hypoxia (1% oxygen in complete DMEM/F-12 1:1 HyClone), and/or with 10ng/ml TGFß1 for the times indicated. Western blot analysis was performed as described by Waning et al (19) using antibodies: Mdm2 [IF2 (Ab-1; OP46), and 4B2 (OP145) from CalBiochem and 2A10 (ab16895) from Abcam; N-Cadherin (610920) from BDBioSci]; p53 (DO-1; sc-126), GAPDH (6C5; sc-32233), Vinculin (N-19; sc-7649 or E1E9V-13901p from cell signaling), E-Cadherin (G-10; sc-8426 and 67A4; sc-2179), Vimentin (RV202; sc-32322), VEGF (147; sc-507), Tubulin (TU-02; sc-8035), and PAI (H-135-sc-8979) from Santa Cruz Biotechnology; Snail (F.31.8-MA5-14801 from ThermoSci); Actin (A1978) Sigma; HIF1α (NBP1–02160) from Novus and MMP2 (MAB-3308) from Millipore.
Cell line verification: all cells were tested for mycoplasma with the Mycoplasma Detection Kit (InvivoGen) during the experiments and before injection into animals; TMD231 cells have a mesenchymal morphology and MDA-MB-468 have a epithelial-like morphology which was verified by microscopy. Molecularly, TMD231 do not express E-Cadherin, and MDA-MB-468 express E-Cadherin which are verified by western blot analysis. The TMD231 cells were generated in-house from MDA-MB-231 cells by Dr. Nakshatri. MDA-MB-468 cells were obtained from ATCC. All cells were passaged for no more than 3 months between thawing and use in experiments.
Wound healing and invasion assays
Migration of MDA468 cells were assessed using culture inserts (IBIDI) according to manufacturer’s instructions. Briefly, 2.45×104 MDA468 shControl cells and 4.9×104 MDA468 shMdm2 cells were placed into both wells of the culture insert (separate insert for each cell line). After a 48 h incubation, inserts were removed and images were obtained at the indicated time points. Gap area was quantified using the wound healing tool for image J (http://dev.mri.cnrs.fr/projects/imagej-macros/wiki/Wound_Healing_Tool). Inserts were manufactured to result in a 500 μm gap. Time 0 h was imaged at 5X magnification, and all subsequent time points were imaged at 10X magnification.
TMD231 mCherry shControl and GFP shMdm2 cells were plated onto coverslips in a 35 mm plate (64,000 cells/plate). Coverslips were then transferred to a 60 mm plate and secured with paramount the next day. Images were obtained immediately and at the times indicated; the number of cells of each color in three separate fields of view were counted and averaged.
Boyden chamber assays were conducted with BD Matrigel Invasion chamber 8.0 Micron pore size according to manufacturer’s instruction. Briefly, 2.5×104 cells were seeded into each well of matrigel chambers with 5% serum used as the chemoattractant. After a 22 h incubation under normal growth conditions, the media was removed and the surface of the membrane was wiped with a cotton tip swab to remove non-invading cells. Membranes were then removed from the chambers and stained by incubating in methanol for 2 min and then 1% toludine blue for 2 min. Cells were counted in three random fields at a magnification of 20X and the experiment was done in triplicate. Percent invasion was calculated by dividing the mean number of cells invading through the matrigel insert membrane by the mean number of cells migrating through the control insert membrane.
Orthotopic implantation in mice and assessment
All animal studies have been approved by Indiana University School of Medicine IACUC (protocol #11190) and were performed by the Indiana University In Vivo Therapeutics Core. 7.5×105 TMD 231 shGFP, or shMdm2 cells or 1×106 MDA468 shControl or shMdm2 cells were injected into the fourth mammary fat pad of NOD-SCID Gamma mice (n=7 per group) obtained from the In Vivo Therapeutics Core of the Indiana University Simon Cancer Center. Tumor growth was calculated by volumetric analysis using calipers. Tumors were allowed to grow to an average volume of 600 mm3 (TMD231 and MDA468 shControl), 200mm3 (MDA468 shMdm2), or 900 mm3 (TMD231 in the shp53 experiment).
Circulating tumor cell analysis
Cardiac punctures with 1ml TB syringe and a 27G x ½ needle (BD Biosciences) were performed at necropsy and temporarily stored in vacutainers coated with sodium heparin (BD Biosciences) prior to peripheral blood mononuclear cells (PBMC) processing. PBMCs were isolated by Ficoll-paque™ Plus (GE Healthcare) separation and cells were stained with 1μg phycoerythrin (PE) conjugated mouse anti-human EGF Receptor antibody (#555997; BD Pharmigen) per 1× 106 cells. FACS analysis was conducted and data analysis was performed using FlowJo 10.0.8r1. The percentage of CTC population of each shControl and shMdm2 sample was determined by positive human-specific EGFR staining of CTCs in mouse blood.
Tissue Processing and Immunostaining
Tissues were fixed overnight at room temperature in 10% neutral buffered formalin after which they were transferred through graded concentrations of alcohol to xylene inside a Leica Automated Vacuum Tissue Processor. Samples were embedded in paraffin before being cut into five micron thick sections, mounted onto positively charged slides, and baked at 60 °C.
The Indiana University Pathology Core performed all tumor preparation, hematoxylin & eosin (H&E) and CD31 staining, and quantitation. Slides were then deparaffinized in xylene and rehydrated through graded alcohols to water. Antigen retrieval was performed by immersing the slides in Target Retrieval Solution (DAKO) for 20 min at 90 °C, cooling at room temperature for 10 min., washing in water and then proceeding with immunostaining. Slides were blocked with protein blocking solution (Dako) for 30 minutes. All subsequent staining steps were performed using the Dako FLEX SYSTEM on an automated Immunostainer; incubations were done at room temperature and Tris buffered saline plus 0.05% Tween 20, pH 7.4 (TBS - Dako Corp.) was used for all washes and diluents. Thorough washing was performed after each incubation. Standard H&E staining was then performed. Alternatively, slides were stained with anti-mouse CD31 (DIA 310; Dianova). Control sections were treated with an isotype control using the same concentration as primary antibodies to verify the staining specificity.
Image Capturing
Aperio’s whole slide digital imaging system, ScanScope CS, was used for imaging (360 Park Center Drive Vista, CA 92081). All slides were imaged at 20x. Digital images were obtained using Aperio’s software, ImageScope. For CD31 analysis, whole primary tumor cross sections were analyzed, with the exception of the excluded necrotic tissue. Vessels less than 125 μm and/or having a diameter less than 30 μm were considered immature. Data from 5 independent hand counts per tumor were confirmed by a pathologist, averaged and presented as the average percent of immature vessels (the average number of immature vessels divided by the average total vessels in each primary tumor).
Lung Metastases Quantification
H&E evaluation of lung metastases was performed by a Pathologic Assistant. All hand counts matched Pathological Reads performed by a board certified pathologist. Metastases from all lungs were classified as Small (1–25 cells), Medium (26–100 cells), Large (101–500 cells), or Very Large (501+). Represented values are averages from each corresponding metastatic size group.
The entire lung of each mouse was scored for metastases and divided by the area of each lung.
Results:
The EMT program is regulated by Mdm2
To confirm that Mdm2 is required for metastasis, MDA468 and TMD231 cells were transduced with control and Mdm2-specific shRNA lentiviruses. MDA468 cells, a basal–like cell line, can undergo EMT while TMD231 cells, a derivative of MDA231 cells, are post-EMT and resemble mesenchymal cells. Once the cell lines were confirmed for decreased levels of Mdm2 (Fig. 1a and Fig.S1), we analyzed levels of mutp53 since Mdm2 can regulate p53 and many studies have demonstrated that mutp53 promotes tumorigenesis and metastasis (20–38). Interestingly, knock-down of Mdm2 in the MDA468 cells resulted in an increase in mutp53 levels, while no discernable difference was observed when TMD231 shMdm2 and shControl cells were compared (Fig. 1a). We next explored whether shMdm2 cell lines would undergo EMT. The mesenchymal marker, SNAI1, was decreased in both shMdm2 cell lines (Fig. 1b). EMT markers in shControl and shMdm2 MDA468 cells were further examined and shMdm2 cells were found to molecularly resemble epithelial cells with regards to E-Cadherin, N-Cadherin, and vimentin (Fig. 1c and Fig.S1). Silencing Mdm2 also resulted in a more epithelial-like morphology compared to the shControl cells (Fig, 1d), which is consistent with the low level of EMT markers. MDA468 cells were treated with TGFβ1 and hypoxia to induce EMT. While TGFβ1 treatment of shControl cells resulted in increased levels of various mesenchymal markers and a more mesenchymal morphology, it did not affect the markers or alter the epithelial-like morphology of shMdm2 cells. TGFβ1 treatment of TMD231 cells did not cause EMT as these cells are morphologically (spindle-shaped) and molecularly (no E-Cadherin, high snail, vimentin, N-cadherin) mesenchymal-like (Fig. S1). Interestingly, despite having increased mutp53 levels, shMdm2 MDA468 cells did not have a mesenchymal phenotype, suggesting a mutant p53-independent role for Mdm2 in EMT. Taken together, these results indicate that Mdm2 plays an important role in regulating key intracellular programing for EMT.
Figure 1. Silencing of Mdm2 results in decreased levels of EMT markers and mesenchymal morphology.
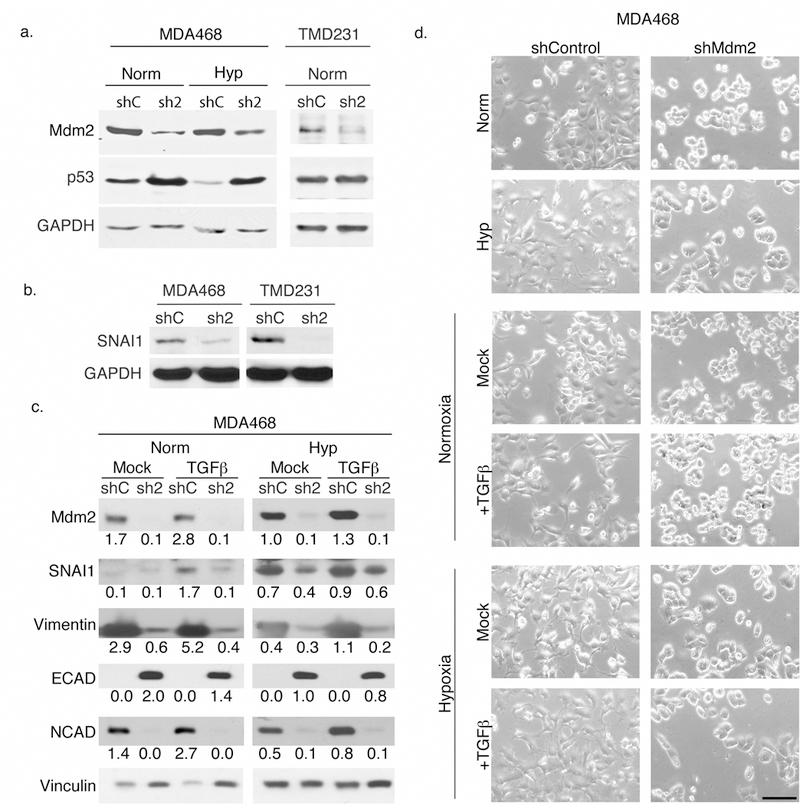
(a) Knock-down of Mdm2 was confirmed by western blot in MDA468 cells and resulted in increased p53 levels. Although silencing Mdm2 in TMD231 cells resulted in decreased Mdm2 levels, p53 levels were not affected. (b) SNAI1 levels were also decreased in the shMdm2 cell lines. (c) SNAI1, Vimentin, and N-cadherin levels were correlated with Mdm2 protein levels while E-cadherin (an epithelial marker) was inversely correlated with Mdm2 levels in MDA468. TGFß treatment (for 48 h with normoxia and 72 h with hypoxia) resulted in increased EMT marker levels in control cells, but not in shMdm2 MDA468 cells. (d) Phase contrast images showing the morphology of MDA468 cells after 48 h TGFß treatment and with or without hypoxia as indicated. (Scale bar represents 100 ⎧m). All panels: shControl = shC; shMdm2= sh2; Norm= 21% oxygen; Hyp = 1% oxygen.
Migration and invasion are impaired in cells with decreased Mdm2
Since migration is coupled with EMT, we tested whether Mdm2 affected cellular migration. Initially, we conducted traditional wound healing assays and quantified gap closure (Fig. 2a). Gap closure was dramatically delayed in shMdm2 MDA468 cells (50% at 70h) compared to shControl cells (completed at 40h). To confirm these data, we also conducted a slight variation of the wound healing assay by examining the migration of fluorescent cells off of coverslips. A GFP-shMdm2 plated coverslip and a mCherry shControl plated coverslip were transferred and fixed on a new plate. Cells that migrated into the void between the coverslips were then counted. This latter technique allowed us to compare shMdm2 cells and shControl cells under the same conditions. shControl cells were more effective at migration than shMdm2 cells (Fig. 2b). Lastly, using a Boyden Chamber assay we demonstrated that shControl cells were able to invade through matrigel more effectively than shMdm2 cells (Fig. 2c). Thus, using three independent methods, our results show that migration and invasion are diminished with decreased Mdm2.
Figure 2. shMdm2 cells have impaired migration and invasion.
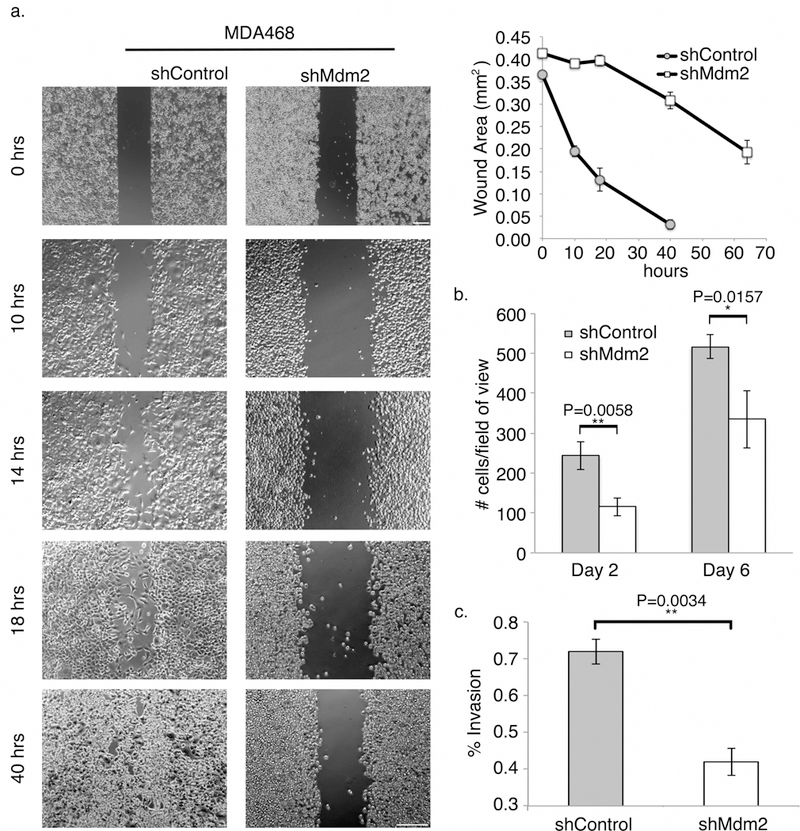
(a) Traditional wound healing assay showing that MDA468 shControl cells (left) were able to close the gap in less time compared to shMdm2 cells (right). Note: 0 h time points were imaged at 5X; all following time points were imaged at 10X (Scale bars = 200 ⎧m). Gap area was measured using a wound healing macro for ImageJ (bottom panel). (b) mCherry fluorescent shControl TMD231 cells and GFP fluorescent shMdm2 TMD231 cells were plated on coverslips and transferred to a new plate. Migrating cells of each color were counted in three separate fields of view and averaged. (c) shControl and shMdm2 cells were plated in 8 μm Matrigel chambers and allowed to invade for 22 h using serum as a chemoattractant. Cells were counted in three random 25X magnification fields and experiments were done in triplicate. Percent invasion was determined as defined in Materials and Methods. All panels: Error bars represent ± SEM (n=3). Significances of differences between shControl and shMdm2 values were determined using unpaired, 2-tailed, student’s t tests (*P<0.05; **P<0.01).
MDA468 and TMD231 shMdm2 cells have delayed tumor growth
Since cell growth and migration can be inter-related, and shMdm2 cells exhibit a defect in migration, we performed cell cycle analysis by Fluorescence-Activated Cell Sorting (FACS). Interestingly, while both cell lines have mutp53 (elevated in shMdm2 MDA468), there were no differences in cell cycle distribution between shControl and shMdm2 (Table S1). These data eliminate the role of Mdm2 in regulating gain of function mutp53 in cell cycle and migration. To garner insight into the role of Mdm2 in in vivo tumor growth, we orthotopically injected shControl or shMdm2 MDA468 or TMD231 into the mammary fat pad of 6 week old NSG female mice (n=7 for each group). The average tumor volumes and weights of shMdm2 MDA468 tumors were lower than shControl tumors (Fig. 3a and 3b left panels), despite allowing them to develop for 30 days longer. This result is notable because mutp53 is thought to enhance tumorigenicity, and its levels are higher in the shMdm2 MDA468 cell line. There were no significant differences between the final TMD231 tumor volumes or weight at the time of euthanasia (Fig. 3a and 3b, right panels).
Figure 3. Knock-down of Mdm2 slows tumor growth.
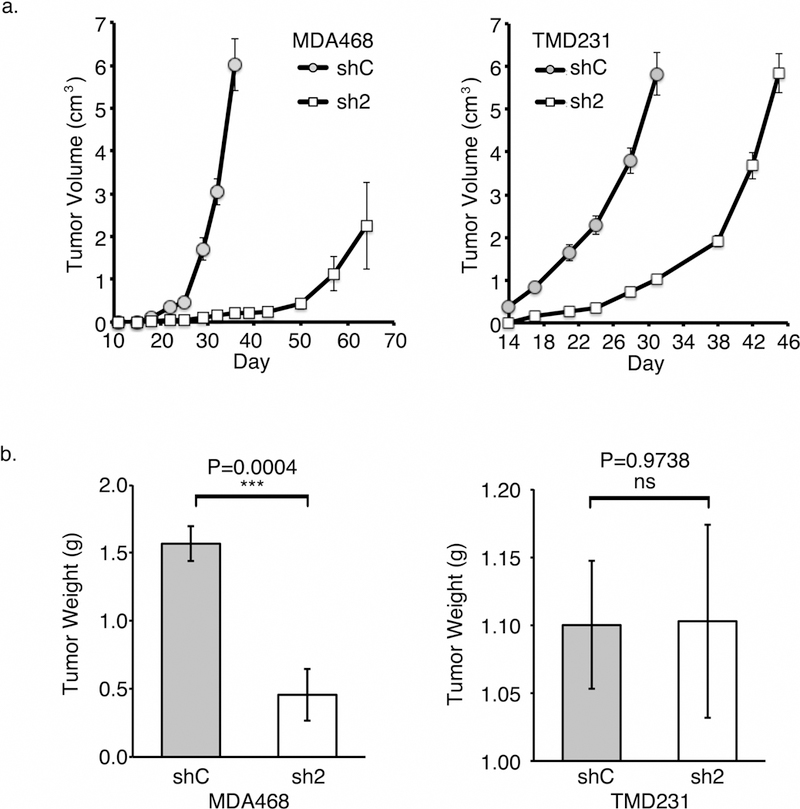
(a) Tumor volumes of MDA468 (left panel) and TMD231 (right panel) shControl orthotopically implanted animals increased faster than that of shMdm2 implanted mice. (b) The final tumor weights of MDA468 shControl tumors were significantly greater than those in the shMdm2 orthotopically implanted animals (left panel), while the TMD231 set was not significantly different (right panel). All panels: Error bars represent ± SEM (n=7). Significances of differences between shControl (shC) and shMdm2 (sh2) values were determined using unpaired, 2-tailed, student’s t tests (ns = not significant; ***P<0.001).
Mdm2 promotes vascularization
Considering the delay in tumor growth in both TMD231 and MDA468 shMdm2 cells, we examined in vitro levels of key factors associated with angiogenesis. We and others have shown that Mdm2 can play a role in stabilizing Hypoxia Inducible Factor 1α (HIF1α) and up-regulating VEGF (39–41). Mdm2 can also stabilize VEGF mRNA levels to promote angiogenesis (42). In accordance with these published findings, there was a substantial decrease in the levels of HIF1α and VEGF in both the cultured TMD231 and MDA468 lines grown under normoxia or hypoxia when Mdm2 was knocked-down compared to the shControl (Fig. 4a). Based on these in vitro results, primary tumors were sectioned and the endothelial cells were stained with cluster of differentiation 31 (CD31). In the presence of Mdm2, the percentage of newly formed, immature vessels in both MDA468 and TMD231 shControl tumors was significantly higher than shMdm2 tumors (Fig. 4b). We next analyzed lysates from MDA468 and TMD231 shMdm2 tumor sets to confirm the in vitro results. In agreement with the levels seen in cell culture (Fig.4a), Fig. 4c demonstrates diminished HIF1α and VEGF levels in shMdm2 tumors compared to shControl tumors. Taken together, our in vitro and in vivo data are congruent in showing that Mdm2 is integrated into the angiogenesis pathway.
Figure 4. Mdm2 is important for angiogenesis.
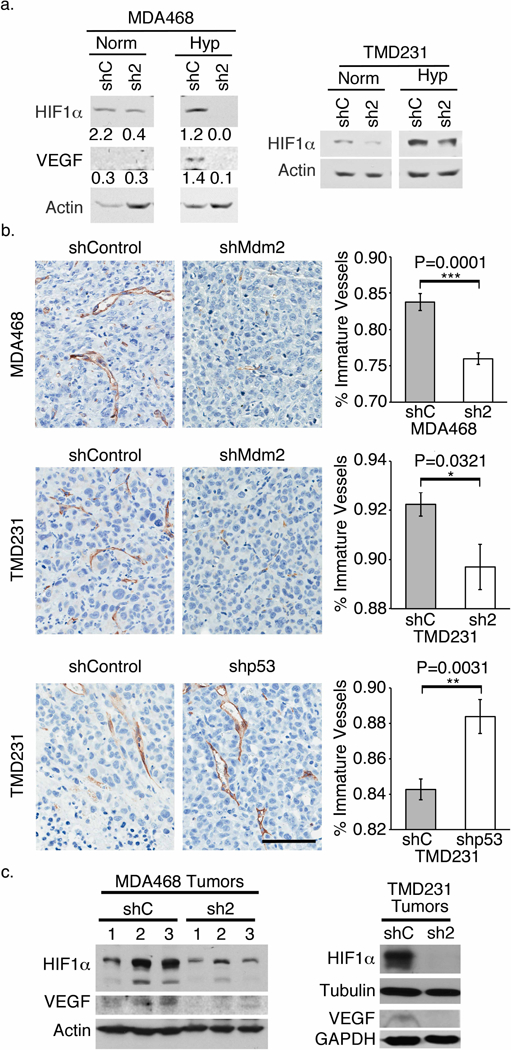
(a) Western blots of cell lysates from cells grown in either normoxia or hypoxia showing that despite culture conditions, when Mdm2 is knocked down, HIF1α and VEGF levels are decreased. (b) Representative images of tumor sections stained brown with CD31 (PECAM-1). The percentages of immature vessels in tumors of both shMdm2 cell lines were significantly lower compared to the respective shControl tumors. The percentages in shp53 tumors, on the other hand, were significantly higher than the shControl tumors. (Scale bar = 100 ⎧m). (c) HIF1α and VEGF levels were lower in the shMdm2 tumors compared to shControl tumors as assessed by western blot analysis. All panels: shControl = shC; shMdm2= sh2; Norm= 21% oxygen; Hyp = 1% oxygen. Error bars represent ± SEM (n=7). Significances of differences were determined using unpaired, 2-tailed, student’s t tests (*P<0.05; **P<0.01; ***P<0.001).
While our data suggest Mdm2 is an important driver of angiogenesis, previous reports implicate mutp53 is also important for this phenomenon (27, 43–45). Considering that our cells maintain mutp53, we tested the importance of this protein in angiogenesis by implanting TMD231 cells that had been transduced with either shControl or shp53 plasmid (Fig. S2). Surprisingly, shp53 tumors had a significantly higher percentage of immature vessels compared to shControl tumors (Fig. 4b). Analysis of tumor extracts revealed that Mdm2 levels were slightly elevated in shp53 tumors (Fig. S2). Thus, Mdm2 promotes vascularization of the tumor via regulation of the HIF1α-VEGF axis and possibly permits intravasation of tumor cells into the blood stream.
Abrogation of Mdm2 results in diminished Metastasis
Tumor cells undergo intracellular reprograming to signal for cellular migration. To assess if these changes were evident in our in vivo metastasis models we examined EMT markers from shMdm2 MDA468 tumor extracts. The results support our in vitro findings of decreased levels of N-Cadherin and SNAI1, and increased levels of E-Cadherin compared to the shControl tumors (Fig. 5a). Two additional proteins that are associated with metastasis, Plasminogen Activator Inhibitor-1 (PAI-1), an important tumor angiogenic factor involved in metastasis (46), and Matrix Metalloproteinase-2 (MMP2), a protein that acts to break down extracellular matrix and is associated with metastatic lung foci (47) were examined by western blot analysis of tumor extracts. Both PAI-1 and MMP2 were found to be decreased in shMdm2 primary tumors (Fig. 5a).
Figure 5. Mdm2 is ultimately critical for metastasis.
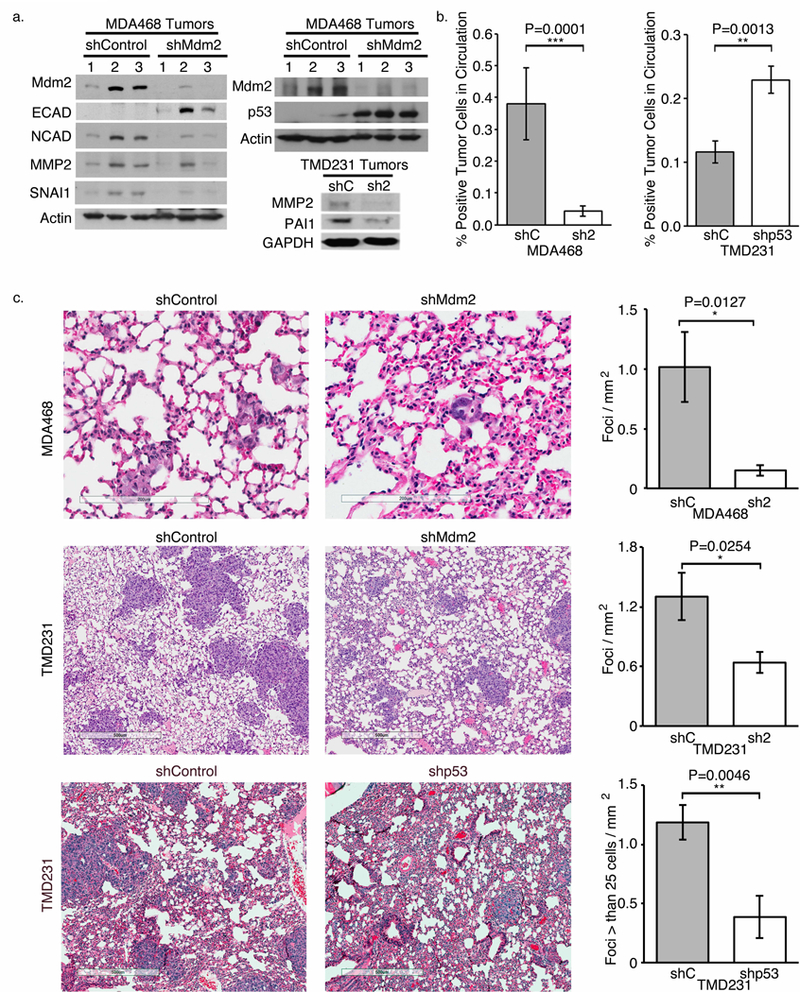
(a) Western blots showing that EMT markers and MMP2 are higher in lysates from shControl tumors compared to shMdm2 tumors. E-cadherin and p53 are more abundant in shMdm2 MDA468 tumors compared to shControl tumors. (b) FACs analysis using human-specific EGFR revealed that there was less shMdm2 and more shp53 CTC compared to the respective controls. (c) H&E stained lung sections (left) demonstrating decreased metastasis with the shMdm2 implanted mice compared to those implanted with shControl cells. Only the foci composed of greater than 25 cells were counted for the TMD231 shControl and shp53 implanted animals. All panels: shControl = shC; shMdm2= sh2. Error bars represent ± SEM (n=7). Significances of differences were determined using unpaired, 2-tailed, student’s t tests (ns= not significant; *P<0.05; **P<0.01; ***P<0.001).
These data indicate that the tumors cells are primed to intravaste and, with our angiogenesis data in Figure 4, suggest that shControl tumor cells would enter into the blood stream. We extracted blood from the euthanized animals harboring shControl and shMdm2 tumors, and stained for human EGFR to detect circulating tumor cells (CTC) by FACS analysis. We found a greater than four-fold increase in CTC in animals implanted with MDA468 shControl cells compared to those implanted with the shMdm2 cells (Fig. 5b). Conversely, a greater number of shp53 CTC versus shControl CTC was observed. This result is in accordance with the vasculature data shown in Figure 4, but contradicts published findings that suggest mutant p53 is driving metastasis (20–23,25–37,48–50). Thus, in the presence of Mdm2, tumor cells are priming the vasculature and entering the blood stream.
After establishing that the numbers of tumor cells entering the blood stream was dependent on Mdm2, we determined if a corresponding change in lung metastatic foci was evident. MDA468 shControl cells readily metastasized to the lung with an average of 1 foci/mm2, while the shMdm2 cells averaged only 0.15 foci in the same area. When Mdm2 was silenced, an 85% decrease in lung foci was observed even with elevated mutp53 (Fig. 5c top). Similarly, the TMD231 shControl cells were significantly more likely to metastasize to the lung than shMdm2 cells (Fig. 5c middle). Surprisingly, considering the higher number of CTC in shp53 TMD231 cells, there were less metastatic foci in the lungs compared to control animals (P=0.0046) (Fig. 5c bottom). These results are supported by the majority of publications implicating a role for mutp53 in metastasis (20–23,25–37,48–50); however, mutp53 is dispensible for metastatic initiation which requires Mdm2.
Discussion:
Here, we demonstrate that Mdm2 is extremely important in breast cancer metastases to the lung. Specifically, we show that Mdm2 is important to promote cancer invasiveness via cell migration, angiogenesis, and intravasation (Fig. 6). Molecularly, Mdm2 stabilizes HIF1α which increases VEGF and transcription factors associated with intracellular metastatic programming (such as SNAI1)(11). This intracellular pathway increases angiogenesis, and the decrease of vasculature observed with decreased Mdm2 provides one possible explanation for the small tumor size in the MDA468 shMdm2 tumors as well as the delay in tumor growth of TMD231 shMdm2 tumors. Therefore, the abundance of CTC, and lung metastatic foci is reliant on the presence of Mdm2. Several studies have reported a role for Mdm2 in migration or EMT in vitro. In contrast, our experiments were conducted in vivo, which is significant in light of recent publications delineating the importance of the microenvironment in tumor growth and invasion.
Figure 6. Mdm2 and p53 impact metastasis in distinct ways.
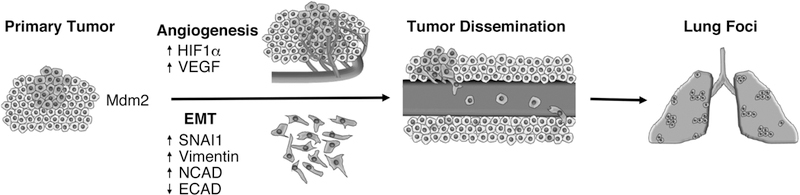
Schematic representing major steps of metastasis and the involvement of Mdm2.
In addition to being essential in early metastasis, Mdm2 may also be important in later stages as well. While extravasation and colonization was not directly assessed, at least one study has shown that tail vein injection (bypassing the initial stages of metastasis) of transiently overexpressing Mdm2 in murine cells results in increased metastatic potential. This suggests that Mdm2 may have a role in extravasion and/or foci formation. Not surprisingly, this study also demonstrated increased metastatic potential with tail vein injected murine cells that were transiently knocked down for p53. The main difference between this study and our study is that we knocked down gain-of-function p53 and not wild type p53. The only other in vivo study that demonstrated a role for Mdm2 in metastasis was also in a wild type p53 background and credited the decreased metastasis on the activation of p53 genes. Furthermore, the knock down of Mdm2 by RNAi resulted in decreased proliferation and tumor growth, which also contributed to the decrease in metastasis in these wild type p53 cells.
Our results provide a significant advancement in our understanding of how Mdm2 is regulating the cellular programs to promote tumor metastasis independent of gain of function mutp53. If mutp53 was driving tumorigenesis, one would expect the MDA468 shMdm2 tumors (which have significantly more mutp53 than the controls) to be increased in size. However, these cells grew much slower in the implanted animals. Furthermore, when mutp53 was knocked down, the tumors actually had increased neovascularization than the controls. Importantly, we show that the animals implanted with the shp53 cells had higher numbers of CTCs, suggesting that mutp53 is actually a disadvantage in the early stages of metastasis. However, the abundance of shp53 CTC did not correlate with increased numbers of metastatic foci, signifying that mutp53 is important for extravasation or other steps of metastasis. Lastly, knocking-down mutp53 by shRNA in shMdm2 cells did not alter EMT-related responses demonstrating that the effects of Mdm2 knock-down are independent of mutp53 levels (Fig. S3). Thus, we show there are independent and distinct yet dynamic roles of mutp53 and Mdm2 during the different phases of metastasis.
A major road block in the ability to treat metastatic disease is delineating the basic signaling pathways involved in the genesis, survival, drug-resistance, migration, and growth of metastatic cells. The effectiveness of new therapies will require a comprehensive approach to target multiple pathways, preferably those that are important before tumor cells reach circulation. Based on our work, future drugs should be designed to interrupt the p53 independent functions of Mdm2.
Supplementary Material
Implications:
Mdm2 is the major factor in the initiation of metastasis.
Acknowledgements:
In Vivo Therapeutics core for implantation and collection of in vivo data.
Grant Support: This study was supported by NIH/NCI CA172256 and a diversity supplement to this RO1, and Riley Children’s foundation (LDM). NIH support: In Vivo Therapeutics Core IUSCC P30 CA082709, T32HL007910 (DJO), and T35HL110854 (RMC).
References:
- 1.Araki S, Eitel JA, Batuello CN, Bijangi-Vishehsaraei K, Xie XJ, Danielpour D, et al. TGF-beta1-induced expression of human Mdm2 correlates with late-stage metastatic breast cancer. The Journal of clinical investigation 2010;120:290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shi W, Meng Z, Chen Z, Hua Y, Gao H, Wang P, et al. RNA interference against MDM2 suppresses tumor growth and metastasis in pancreatic carcinoma SW1990HM cells. Molecular and cellular biochemistry 2014;387:1–8 [DOI] [PubMed] [Google Scholar]
- 3.Chen X, Qiu J, Yang D, Lu J, Yan C, Zha X, et al. MDM2 promotes invasion and metastasis in invasive ductal breast carcinoma by inducing matrix metalloproteinase-9. PloS one 2013;8:e78794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Polanski R, Warburton HE, Ray-Sinha A, Devling T, Pakula H, Rubbi CP, et al. MDM2 promotes cell motility and invasiveness through a RING-finger independent mechanism. FEBS letters 2010;584:4695–702 [DOI] [PubMed] [Google Scholar]
- 5.Yang JY, Zong CS, Xia W, Wei Y, Ali-Seyed M, Li Z, et al. MDM2 promotes cell motility and invasiveness by regulating E-cadherin degradation. Molecular and cellular biology 2006;26:7269–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Onel K, Cordon-Cardo C. MDM2 and prognosis. Molecular cancer research : MCR 2004;2:1–8 [PubMed] [Google Scholar]
- 7.Rayburn E, Zhang R, He J, Wang H. MDM2 and human malignancies: expression, clinical pathology, prognostic markers, and implications for chemotherapy. Curr Cancer Drug Targets 2005;5:27–41 [DOI] [PubMed] [Google Scholar]
- 8.Turbin DA, Cheang MC, Bajdik CD, Gelmon KA, Yorida E, De Luca A, et al. MDM2 protein expression is a negative prognostic marker in breast carcinoma. Mod Pathol 2006;19:69–74 [DOI] [PubMed] [Google Scholar]
- 9.Ladanyi M, Cha C, Lewis R, Jhanwar SC, Huvos AG, Healey JH. MDM2 gene amplification in metastatic osteosarcoma. Cancer research 1993;53:16–8 [PubMed] [Google Scholar]
- 10.Ladanyi M, Lewis R, Jhanwar SC, Gerald W, Huvos AG, Healey JH. MDM2 and CDK4 gene amplification in Ewing’s sarcoma. The Journal of pathology 1995;175:211–7 [DOI] [PubMed] [Google Scholar]
- 11.Zhang L, Hill RP. Hypoxia enhances metastatic efficiency by up-regulating Mdm2 in KHT cells and increasing resistance to apoptosis. Cancer research 2004;64:4180–9 [DOI] [PubMed] [Google Scholar]
- 12.Zhang P, Wu SK, Wang Y, Fan ZX, Li CR, Feng M, et al. p53, MDM2, eIF4E and EGFR expression in nasopharyngeal carcinoma and their correlation with clinicopathological characteristics and prognosis: A retrospective study. Oncology letters 2015;9:113–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tennis M, Krishnan S, Bonner M, Ambrosone CB, Vena JE, Moysich K, et al. p53 Mutation analysis in breast tumors by a DNA microarray method. Cancer Epidemiol Biomarkers Prev 2006;15:80–5 [DOI] [PubMed] [Google Scholar]
- 14.Cancer Genome Atlas N Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science 2002;296:550–3 [DOI] [PubMed] [Google Scholar]
- 16.Patton JT, Mayo LD, Singhi AD, Gudkov AV, Stark GR, Jackson MW. Levels of HdmX expression dictate the sensitivity of normal and transformed cells to Nutlin-3. Cancer research 2006;66:3169–76 [DOI] [PubMed] [Google Scholar]
- 17.Shannon HE, Fishel ML, Xie J, Gu D, McCarthy BP, Riley AA, et al. Longitudinal Bioluminescence Imaging of Primary Versus Abdominal Metastatic Tumor Growth in Orthotopic Pancreatic Tumor Models in NSG Mice. Pancreas 2015;44:64–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Burnett RM, Craven KE, Krishnamurthy P, Goswami CP, Badve S, Crooks P, et al. Organ-specific adaptive signaling pathway activation in metastatic breast cancer cells. Oncotarget 2015;6:12682–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waning DL, Lehman JA, Batuello CN, Mayo LD. c-Abl phosphorylation of Mdm2 facilitates Mdm2-Mdmx complex formation. The Journal of biological chemistry 2011;286:216–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arjonen A, Kaukonen R, Mattila E, Rouhi P, Hognas G, Sihto H, et al. Mutant p53-associated myosin-X upregulation promotes breast cancer invasion and metastasis. The Journal of clinical investigation 2014;124:1069–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Coffill CR, Muller PA, Oh HK, Neo SP, Hogue KA, Cheok CF, et al. Mutant p53 interactome identifies nardilysin as a p53R273H-specific binding partner that promotes invasion. EMBO Rep 2012;13:638–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dong P, Karaayvaz M, Jia N, Kaneuchi M, Hamada J, Watari H, et al. Mutant p53 gain-of-function induces epithelial-mesenchymal transition through modulation of the miR-130b-ZEB1 axis. Oncogene 2013;32:3286–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dong P, Tada M, Hamada J, Nakamura A, Moriuchi T, Sakuragi N. p53 dominant-negative mutant R273H promotes invasion and migration of human endometrial cancer HHUA cells. Clin Exp Metastasis 2007;24:471–83 [DOI] [PubMed] [Google Scholar]
- 24.Dong P, Xu Z, Jia N, Li D, Feng Y. Elevated expression of p53 gain-of-function mutation R175H in endometrial cancer cells can increase the invasive phenotypes by activation of the EGFR/PI3K/AKT pathway. Mol Cancer 2009;8:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hsiao M, Low J, Dorn E, Ku D, Pattengale P, Yeargin J, et al. Gain-of-function mutations of the p53 gene induce lymphohematopoietic metastatic potential and tissue invasiveness. Am J Pathol 1994;145:702–14 [PMC free article] [PubMed] [Google Scholar]
- 26.Kogan-Sakin I, Tabach Y, Buganim Y, Molchadsky A, Solomon H, Madar S, et al. Mutant p53(R175H) upregulates Twist1 expression and promotes epithelial-mesenchymal transition in immortalized prostate cells. Cell death and differentiation 2011;18:271–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lenfert E, Maenz C, Heinlein C, Jannasch K, Schumacher U, Pantel K, et al. Mutant p53 promotes epithelial-mesenchymal plasticity and enhances metastasis in mammary carcinomas of WAP-T mice. International journal of cancer Journal international du cancer 2015;136:E521–33 [DOI] [PubMed] [Google Scholar]
- 28.Mak AS. p53 in cell invasion, podosomes, and invadopodia. Cell Adh Migr 2014;8:205–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morton JP, Timpson P, Karim SA, Ridgway RA, Athineos D, Doyle B, et al. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proceedings of the National Academy of Sciences of the United States of America 2010;107:246–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muller PA, Caswell PT, Doyle B, Iwanicki MP, Tan EH, Karim S, et al. Mutant p53 drives invasion by promoting integrin recycling. Cell 2009;139:1327–41 [DOI] [PubMed] [Google Scholar]
- 31.Muller PA, Vousden KH, Norman JC. p53 and its mutants in tumor cell migration and invasion. J Cell Biol 2011;192:209–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neilsen PM, Noll JE, Mattiske S, Bracken CP, Gregory PA, Schulz RB, et al. Mutant p53 drives invasion in breast tumors through up-regulation of miR-155. Oncogene 2013;32:2992–3000 [DOI] [PubMed] [Google Scholar]
- 33.Noll JE, Jeffery J, Al-Ejeh F, Kumar R, Khanna KK, Callen DF, et al. Mutant p53 drives multinucleation and invasion through a process that is suppressed by ANKRD11. Oncogene 2012;31:2836–48 [DOI] [PubMed] [Google Scholar]
- 34.Subramanian M, Francis P, Bilke S, Li XL, Hara T, Lu X, et al. A mutant p53/let-7i-axis-regulated gene network drives cell migration, invasion and metastasis. Oncogene 2015;34:1094–104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weissmueller S, Manchado E, Saborowski M, Morris JPt, Wagenblast E, Davis CA, et al. Mutant p53 drives pancreatic cancer metastasis through cell-autonomous PDGF receptor beta signaling. Cell 2014;157:382–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeudall WA, Vaughan CA, Miyazaki H, Ramamoorthy M, Choi MY, Chapman CG, et al. Gain-of-function mutant p53 upregulates CXC chemokines and enhances cell migration. Carcinogenesis 2012;33:442–51 [DOI] [PubMed] [Google Scholar]
- 37.Adorno M, Cordenonsi M, Montagner M, Dupont S, Wong C, Hann B, et al. A Mutant-p53/Smad complex opposes p63 to empower TGFbeta-induced metastasis. Cell 2009;137:87–98 [DOI] [PubMed] [Google Scholar]
- 38.Lang GA, Iwakuma T, Suh YA, Liu G, Rao VA, Parant JM, et al. Gain of function of a p53 hot spot mutation in a mouse model of Li-Fraumeni syndrome. Cell 2004;119:861–72 [DOI] [PubMed] [Google Scholar]
- 39.Bardos JI, Chau NM, Ashcroft M. Growth factor-mediated induction of HDM2 positively regulates hypoxia-inducible factor 1alpha expression. Molecular and cellular biology 2004;24:2905–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Carroll VA, Ashcroft M. Regulation of angiogenic factors by HDM2 in renal cell carcinoma. Cancer research 2008;68:545–52 [DOI] [PubMed] [Google Scholar]
- 41.LaRusch GA, Jackson MW, Dunbar JD, Warren RS, Donner DB, Mayo LD. Nutlin3 blocks vascular endothelial growth factor induction by preventing the interaction between hypoxia inducible factor 1alpha and Hdm2. Cancer research 2007;67:450–4 [DOI] [PubMed] [Google Scholar]
- 42.Zhou S, Gu L, He J, Zhang H, Zhou M. MDM2 regulates vascular endothelial growth factor mRNA stabilization in hypoxia. Molecular and cellular biology 2011;31:4928–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kieser A, Weich HA, Brandner G, Marme D, Kolch W. Mutant p53 potentiates protein kinase C induction of vascular endothelial growth factor expression. Oncogene 1994;9:963–9 [PubMed] [Google Scholar]
- 44.Koura A, Vangolen K, Tsan R, Radinsky R, Price J, Ellis L. Regulation of genes associated with angiogenesis, growth, and metastasis by specific p53 point mutations in a murine melanoma cell line. Oncol Rep 1997;4:475–9 [DOI] [PubMed] [Google Scholar]
- 45.Perryman LA, Blair JM, Kingsley EA, Szymanska B, Ow KT, Wen VW, et al. Over-expression of p53 mutants in LNCaP cells alters tumor growth and angiogenesis in vivo. Biochemical and biophysical research communications 2006;345:1207–14 [DOI] [PubMed] [Google Scholar]
- 46.Chen H, Peng H, Liu W, Sun Y, Su N, Tang W, et al. Silencing of plasminogen activator inhibitor-1 suppresses colorectal cancer progression and liver metastasis. Surgery 2015;158:1704–13 [DOI] [PubMed] [Google Scholar]
- 47.Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, et al. Genes that mediate breast cancer metastasis to lung. Nature 2005;436:518–24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ali A, Shah AS, Ahmad A. Gain-of-function of mutant p53: mutant p53 enhances cancer progression by inhibiting KLF17 expression in invasive breast carcinoma cells. Cancer Lett 2014;354:87–96 [DOI] [PubMed] [Google Scholar]
- 49.Roger L, Jullien L, Gire V, Roux P. Gain of oncogenic function of p53 mutants regulates E-cadherin expression uncoupled from cell invasion in colon cancer cells. J Cell Sci 2010;123:1295–305 [DOI] [PubMed] [Google Scholar]
- 50.Wang SP, Wang WL, Chang YL, Wu CT, Chao YC, Kao SH, et al. p53 controls cancer cell invasion by inducing the MDM2-mediated degradation of Slug. Nature cell biology 2009;11:694–704 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.