

Abstract
Techniques that allow single cell analysis are gaining widespread attention and most of these studies utilize genomics-based approaches. While nanofluidic technologies have enabled mass spectrometric analysis of single cells, these measurements have been limited to metabolomics and lipidomic studies. Single cell proteomics has the potential to improve our understanding of intercellular heterogeneity. However, this approach has faced challenges including limited sample availability, as well as a requirement of highly sensitive methods for sample collection, clean-up and detection. We present a technique to overcome these limitations by combining a micropipette (pulled glass capillary) based sample collection strategy with offline sample preparation and nanoLC-MS/MS to analyze proteins through a bottom-up proteomic strategy. This study explores two types of proteomics data acquisition strategy namely data-dependent (DDA) and data-independent acquisition (DIA) strategy. Results from the study indicate DIA to be more sensitive enabling analysis of >1600 proteins from ~130 μm Xenopus laevis embryonic cells containing <6 nL of cytoplasm. The method was found to be robust in obtaining reproducible protein quantifications from single cells spanning 1 to 128 cell stage of development. Furthermore, we used micropipette sampling to study intercellular heterogeneity within cells in a single embryo and investigate embryonic asymmetry along both animal-vegetal and dorsal-ventral axes during early stages of development. Investigation of the animal-vegetal axis led to discovery of various asymmetrically distributed proteins along the animal-vegetal axis. We have further compared the hits found from our proteomic data sets with other studies and validated few hits using an orthogonal imaging technique. This study forms the first report of vegetal enrichment of the germ plasm associated protein DDX4/VASA in Xenopus embyos. Overall, the method and data presented here holds promise to enable important leads in developmental biology.
Cell-to-cell variability is a well-known phenomenon and has been a subject of intense investigation over the past decade.1,2 A classic example is the striking morphological, genetic, and proteomic variability among cancer cells.3 Genomic and transcriptomic level analysis of single cells is currently most common, owing to the ease of DNA amplification reactions such as polymerase chain reaction (PCR).4 Although a recent reports describes sophisticated sample handling strategy to enable proteomics from as few as ten mammalian cells and single plant cell,5–7 single cell metabolomic and proteomic studies have severely lagged behind due to an inability to amplify signal. Transcriptomic level information does not always correlate with proteome level information.8 Therefore, our goal was to develop a method that enables single cell proteomics through mass spectrometry based analyses.
Mass spectrometry based proteomics enables unbiased analysis of thousands of proteins in a single shot in contrast to other targeted antibody and fluorescence based techniques like cyTOF.9,10 Nanofluidic techniques are necessary to isolate single cells but a tool that can be utilized by the research community in a straightforward manner with minimal nanotechnology expertise remains unfulfilled as most methods require sophisticated micro-and nanofabrication. 11–13 To address this, pulled capillary based devices called micro-and nanopipettes were developed. 14,15 Fabrication of micropipettes is fairly simple and inexpensive and makes use of laser-based pipette pullers typically utilized to create nanoLC fused silica capillary columns. These pipettes have been applied to study small molecule metabolites, lipids and more recently proteins from single cells.11,14,16–20
Amphibian embryos such as Xenopus are one of the few model vertebrate systems in which the fate map is established at fertilization, and hence serves as an ideal model to study intercellular heterogeneity and to interrogate embryonic asymmetry as pioneered by several groups.21–24 Additionally, these embryos are larger than mammalian cells and deemed ideal for study of single cell proteomics by several groups.18,21,25–27 Traditionally, various dissection strategies have been utilized to isolate single cells which leads to sacrificing adjacent cells and severely complicates the study of cellular heterogeneity among adjacent Xenopus embryonic cells.21,25 On the contrary, pipette-based sampling has been found to be minimally invasive by Baker and Nemes groups for studying single cells. 14,15,18,28 Employing micropipettes, we investigated Xenopus embryonic cells from 1-cell stage to 128-cell stage of development.
A recent manuscript by Lombard-Banek et. al that was published when current manuscript was under review employed pulled capillary based devices to investigate proteins by capillary electrophoresis-electrospray ionization-mass spectrometry (CE-ESI-MSMS) and have investigated single cells from Xenopus (1–128 cell stage).18 Although, both the studies have combined the micropipette based sample collection strategy with offline sample preparation to analyze proteins through a bottom-up proteomic strategy, key differences between our study and that of Lombard-Banek et. al lies in proteomics data acquisition. Our manuscript utilizes easily accessible nanoLC-MS/MS coupled to a mass spectrometer for bottom-up proteomics and investigates both DDA and DIA MS acquisition strategies to conclude that DIA is more sensitive to analyze small amounts of sample harvested from single cells. DIA has enabled us to obtain deepest proteome coverage from the smallest X. laevis embryonic cells to date (>1650 protein from a single cell at 128-cell stage). A more comprehensive proteomic study of >4000 proteins was reported for 1-cell stage embryos by Dovichi and co-workers, and > 6000 proteins was reported by Kirschner and co-workers from Xenopus eggs. These studies report most comprehensive single cell proteomics data to-date.29,30
Xenopus eggs are divided into animal (heavily pigmented) and vegetal (unpigmented) hemispheres, with substantial cellular and molecular asymmetry along this axis.31 In fact, cellular determination during early development is caused by this molecular gradient which we investigated by employing micropipettes to perform localized sampling. The study lead to identification of several proteins that are differentially enriched along the animal-vegetal axis. While previous studies of animal-vegetal asymmetry through proteomics mostly focused on tentative hits through large-scale proteomics, we took an additional step to validate our findings using an orthogonal analytical approache. Some of the interesting hits were validated by immunofluorescence imaging of sectioned embryos to confirm differential expression of 20S CP subunits and DDX4/VASA across animal-vegetal axis. Proteasome subunits were found to be enriched in the animal hemisphere32 whereas VASA/DDX4 was found to be vegetally localized in Xenopus. This forms the first report showing vegetal enrichment of Vasa enabled by localized sampling and LC-MS/MS. Furthermore, comparison of the proteomic data sets between our study and that of Lombard-Banek et. al as well as Sindelka et. al was performed to reveal similarities as well as interesting trends in differential protein expression that can potentially be followed-up for interesting findings in developmental biology.
MATERIALS AND METHODS
Xenopus laevis were purchased from Nasco (Fort Atkinson, WI) and used for egg collection on an Institutional Animal Care and Use Committee (IACUC)-approved animal study protocol. Xenopus embryos were obtained by in-vitro fertilization and cultured at room temperature in 0.1X MMR as described by Sive et al.33 Micropipettes (nanofluidic sample collection devices) were fabricated from borosilicate glass capillaries purchased from Sutter Instruments using a micropipette puller Sutter Instrument P97 (Novato, CA). The position of the micropipette was controlled with a Micro Instruments (Oxford, UK) manual micromanipulator and individual cells in an embryo were targeted by observation under a MZ8 Leica stereoscope (San Jose, CA). The micropipettes were held under a slight positive pressure to prevent ingress of media during sampling process and negative pressure was applied only after puncturing the membrane of the target cell. The sampling was controlled pneumatically via a Harvard Apparatus (Holliston, MA) microinjector using the fill function as previously reported by Nemes and co-workers.15,34 Post-sampling, the samples were ejected into a small tube and prepared for nanoLC-MS/MS analysis via traditional bottom-up sample preparation steps. Bottom-up sample preparation steps were designed to keep the total volume as small as possible and minimize non-specific binding of proteins to the tube. Sample preparation step included addition of 2 μL of 25 mM dithiothreitol followed by 20 min of incubation, then 2μL of 40 mM iodoacetamide followed by 15 min incubation. Subsequently, the sample was treated with 0.5 μg of trypsin and digested at room temperature for 5 hours. Finally, the samples were acidified, de-salted on a small bed of C-18 to minimize sample loss and re-suspended in 0.1% formic acid for nanoLC-MS injection.
A reversed-phase high performance nLC with a two-component mobile phase system of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B) was used for separating the digested peptides. For all applications, 75 μm i.d. × 20 cm fused silica columns packed in-house with ReproSil-Pur 120 C18-AQ (3 μm) were used.
Data independent acquisitions: An UltimateTM 3000 RSLCnano System was coupled to a Q-Exactive™ HF-X Hybrid Quadrupole-Orbitrap™ Mass spectrometer (Thermo Scientific) for DIA samples. Peptides were eluted using a gradient of 5% to 30% B over 72 min followed by 30% to 45% B over a subsequent 18 minutes and a flow rate of 300 nL/min was employed. Data independent acquisition method used m/z quadrupole isolation windows of 20 Da. Full scans were performed at 120,000 FWHM resolving power (at 200 m/z) followed by sequential HCD MS/MS scans at stepped normalized collision energy of 25.5, 27, 30, and 30,000 FWHM resolution. A mass range of 350–1200 m/z was surveyed with a maximum injection time of 50 ms. AGC value was set to 3×106 for both MS1 and MS2 scans. DIA samples were spiked with iRT standards from Biognosys for retention time calibration and a peptide list was generated using Spectronaut™ 10 (Biognosys). Before DIA runs, we performed extensive DDA analysis to build a spectral library. Specifically, we took the Xenopus whole embryo lysate, digested the proteins from lysate to peptides as per protocols mentioned above, performed high pH fractionation of the peptides using C-18 spin columns and then subjected the ten fractions to DDA using the same gradient as with the DIA runs listed above. The DDA data files were searched in Proteome Discoverer™ software (Thermo Fisher Scientific). The search parameters for the DDA spectral library include a precursor mass tolerance of 10 ppm and product mass tolerance of 0.02 Da. Trypsin was chosen as the enzyme with 2 missed cleavages, and static modification of carbamidomethyl (Cys) with variable modification of oxidation (Met) were incorporated in the search. PSMs and protein false discovery rate were filtered for < 0.01. Next, we imported our identified spectra with retention time information into Spectronaut™ 10 to be used as our spectral library.
Data dependent acquisitions: For DDA samples, an Orbitrap Fusion™ Tribrid™ Mass Spectrometer (Thermo Scientific) coupled to Thermo Scientific™ Easy nLC™ 1000 UHPLC system was used. Chromatography gradient of 3–34% B was used over 120 min. MS scans were performed at 120,000 FWHM resolving power (at 200 m/z) followed by sequential HCD MS/MS scans at normalized collision energy of 30 and ion trap detection was used for MS/MS scans. A mass range of 300–1200 m/z was surveyed, with a maximum injection time of 60 ms and an AGC value of 5×105 for MS scans was used. For MS/MS scans maximum injection time was set to 80 ms and AGC target of 1.0×104 was used. The peptide list for DDA was generated using MaxQuant 1.6.0.3 and MS/MS spectra were analyzed by Andromeda search engine.35,36 The precursor mass tolerance was set to 10 ppm and product mass tolerance was set to 0.5 Da. Other search parameters include enzyme trypsin with 4 missed cleavages, static modifications of carbamidomethyl (C), and variable modification of oxidation (M) A label-free quantification method iBAQ37 (intensity based absolute quantification) was used for protein estimation. PSMs and protein false discovery rate were filtered for < 0.01.
All peptide searches were performed using Xenbase protein FASTA file (downloaded September 2017). iBAQ intensities were log2 transformed, normalized by the average of the protein abundance within each sample and statistical differences were estimated using the t-test. Normalization by average of the protein abundance within each sample was performed to account of variations in sampling volumes across conditions. All raw LC-MS/MS files can be found in Chorus repository, project ID 1543, 1556.
For immunofluorescence, embryos at the 8-cell stage were fixed with MEMFA (MgSO4, EGTA, MOPS, Formaldehyde solution) embedded in paraffin, and sectioned following standard protocols. Sections of embryos (5 μm) were incubated in blocking buffer (1% BSA, 0.1% Triton X-100, 0.05% tween 20 in PBS) with 2% goat serum for an hour at room temperature, and then stained with anti-proteasome 20S alpha+beta antibody (1:100) (ab22673, abcam) overnight at 4°C for 20S CP IF. Sections were washed with PBS with 0.1% tween-20 three times and stained with goat anti-rabbit IgG Alexa Fluor-594 antibody (1:500) (A11012, Thermo Fisher Scientific) for an hour at room temperature. Sections were washed again with PBS with 0.1% tween 20 three times and then mounted in mounting medium (H-1200, vector laboratories). Images were collected using Nikon ECLIPSE Ti confocal microscopy. For Vasa IF, rabbit anti-vasa (1:200) (sc-67185, Santa Cruz) was used as primary antibody and goat anti-rabbit 594 IgG Alexa Fluor-594 antibody (1:250) (A11012, Thermo Fisher Scientific) was used as secondary antibody. For western blot, 128-cell embryos were dissected into animal and vegetal halves and protein extracted from individual embryo halves for SDS-PAGE and immunoblotting with mouse anti-HSC70 (sc-7298, Santa Cruz; primary antibody dilution 1:10000) using sheep anti-mouse IgG HRP (NA931V, GE Healthcare; 1:10000) as the secondary Ab. Animal and vegetal halves from 6 embryos were analyzed individually; the Hsc70 band was scanned and quantitated using ImageJ and the fraction in the animal hemisphere relative to the total amount in both halves was used to determine the distribution of Hsc70.
RESULTS AND DISCUSSIONS
Throughout the study, pigmentation was utilized to distinguish between animal and vegetal hemispheres with the animal hemisphere being the heavily pigmented side. Furthermore, the pigmentation gradient of the animal hemisphere enabled us to distinguish between individual cells (blastomeres) at the 4 and 8-cell stages of development with dorsal blastomeres being less pigmented than ventral blastomeres. When comparing two presumptive dorsal blastomeres, the pigmentation was usually slightly asymmetric, and therefore we referred to the least pigmented cell as D1 and the other dorsal cell as D2. Ventral cells were similarly identified based on pigmentation, where the darkest cell was identified as V2.
Our first step was to establish the nanofluidic sampling method. Micropipette tips were fabricated and characterized as per previous work.14 Samples were collected inside the micropipette pneumatically as detailed in the methods. The aspiration pressure can be manipulated by tuning the tip diameter of the micropipette. To collect samples, the embryos were kept in Marc’s Modified Ringer’s solution, the micropipette was placed on to the cell of interest viewed through a dissecting microscope, the cell membrane was punctured, and cytoplasm was collected by application of negative pressure (Movie S1). Of note, at later cell stages (32–128 cell stage), a careful attention was paid to the adjacent cells and a successful sampling was only considered when no signs of adjacent cell deflation or membrane breakage was observed. The workflow for single cell proteomics is described through a schematic in Figure 1A and detailed in the methods section. Micropipette sampling was successfully employed to detect proteins from 1-cell to the 128-cell stage of Xenopus laevis development. Figure 1E represents the number of protein groups identified at various stages of development. Both data-dependent and data-independent MS acquisition strategies were explored in this study. Data-independent acquisition (DIA) proved to be superior for comprehensive detection and quantification of minute quantities of proteome samples from single cells. With data-dependent acquisition (DDA), a monotonic decrease in the number of protein IDs was observed across developmental time points which can be attributed to a decrease in the volume of cytoplasm. In DDA workflow, dynamic detection range is dependent on sequencing speed of the MS because of stochastic intensity based precursor selection strategy. Due to insufficient speed of current mass spectrometers, MS/MS spectra of all detectable precursor ions are not collected which hampers identification of low-abundance peptides. These problems was largely overcome by data-independent acquisition (DIA) workflows where predefined set of precursor ion masses independent of their signal intensities are used for MS/MS. This increased capability to identify low-abundance peptides and proteins over DDA and has been demonstrated on a large scale by several studies.38–40 Hence, DIA was explored for this study. DIA uses a spectral library generated by DDA runs and extracts with higher confidence the chromatographic profiles of each peptide present in the library. Details of spectral library generation can be found in the methods section. DIA reduces the number of missing values and enables more protein identifications at later time points. DDA enabled analysis of ~750 proteins which increased to ~1650 proteins for DIA from <6 nL of sample harvested from a single X. laevis blastomere at the 128-cell stage of development. At the 128-cell stage, individual cells are ~130 μm in diameter and estimated to contain ~40 ng of protein as per our estimation through bradford assay of earlier stages (quantity of protein per volume of cytoplasm and volume of cytoplasm aspirated at 128 cell stage was taken into consideration for this calculation). Our proteomics results describe the deepest proteome coverage from 128-cell stage X. laevis embryonic cells. Earlier studies have reported a smaller number of protein IDs (Figure S1a). Supporting information further details how prior data compares with the current study. Of note, DDA data from our study also reports similar number of proteins as reported by recent literature,18 DIA has enabled much deeper proteome coverage establishing that DIA is more sensitive for single cell proteomics.
Figure 1.
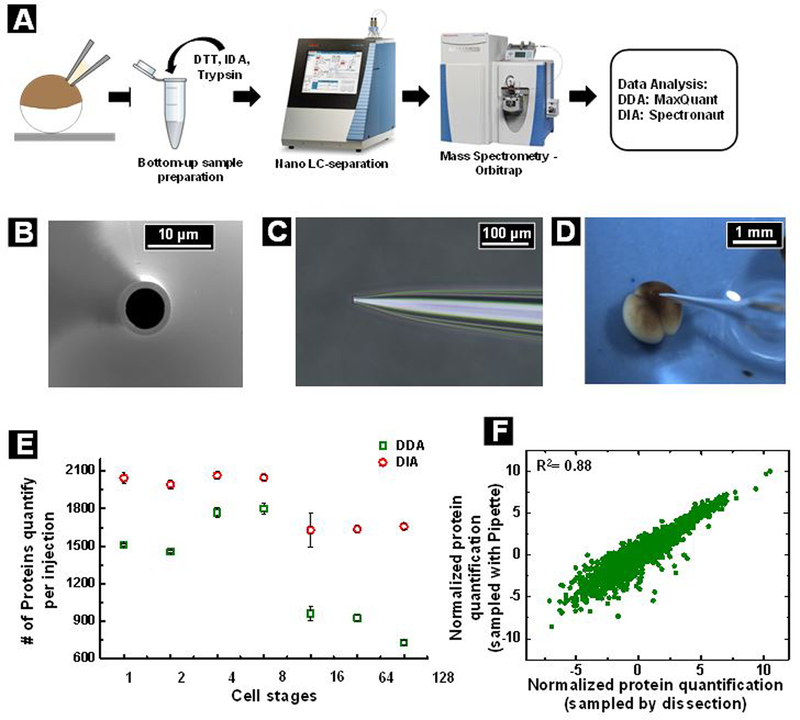
(A) Schematic showing the work-flow for single cell proteomics study, (B) scanning electron micrograph of a micropipette showing end-on view, pipette sputter coated with 4 nm iridium for image quality. (C) Optical micrograph of a micropipette showing side-on view, (D) optical micrograph showing a micropipette puncturing the embryo. (E) A plot showing number of proteins detected from single Xenopus laevis blastomeres at various stages of development. The cell sizes decrease progressively from stage 1 (single cell stage) to 7 (128 cell stage). n=3replicates were used for the study (F) A correlation plot showing log2 transformed and normalized protein quantification data from blastomeres (ventral, 8 cell stage) sampled with pipette (in y-axis) and physically separated by dissection (x-axis), n=3
Next, we evaluated the efficacy of the sampling process. Specifically, we determined whether micropipette sampling introduced any biases compared to using whole cell lysates. We compared protein quantification from micropipette sampled cells and whole cell lysates from individual cells dissected at the 8-cell stage (Figure 1F). Ventral cells (V2) at the 8-cell stage were used for this study. A correlation coefficient of 0.88 was achieved. A coefficient of 0.88 is a good correlation given that biological heterogeneity also contributes to measurement variation. Shown in Figure S2 are comparisons between two biological replicates with an R2 =0.88, demonstrating that our single-cell sampling technique achieves the same level of reproducibility as lysate-based analysis. We further verified sampling accuracy by assessing the coefficient of variation (CV) and missing values per run, as well as determining proteins and contaminant levels in dissected versus micropipette sampled cells (further detailed in Supporting Information). Overall, these studies indicated that no significant bias is introduced by our new methodology.
To test the robustness of this methodology in performing single cell proteomics, we repeatedly measured dorsal cells of Xenopus laevis embryos at the 4-cell stage. Three female frogs were used for this study and four embryos were sampled from each female to correlate twenty four measurements (two dorsal cells per embryo). The relative standard deviation (RSD) among individual proteins across replicates is depicted in a violin plot as shown in Figure 2a. The mean RSD across embryos from the same female was approximately 30% which is in line with the findings by Nemes and co-workers (~20% RSD in LFQ intensity).26 Typically, RSD among replicates in standard proteomics experiments are found to be <20% and obtained by averaging thousands of cells.41 The RSD was found to be higher than 50% when measurements from all females under study were correlated, which could be attributed to biological heterogeneity among females. These RSDs are in line with variations reported by Slavov and co-workers42 (60–85%) in single cell proteomics experiments.
Figure 2.
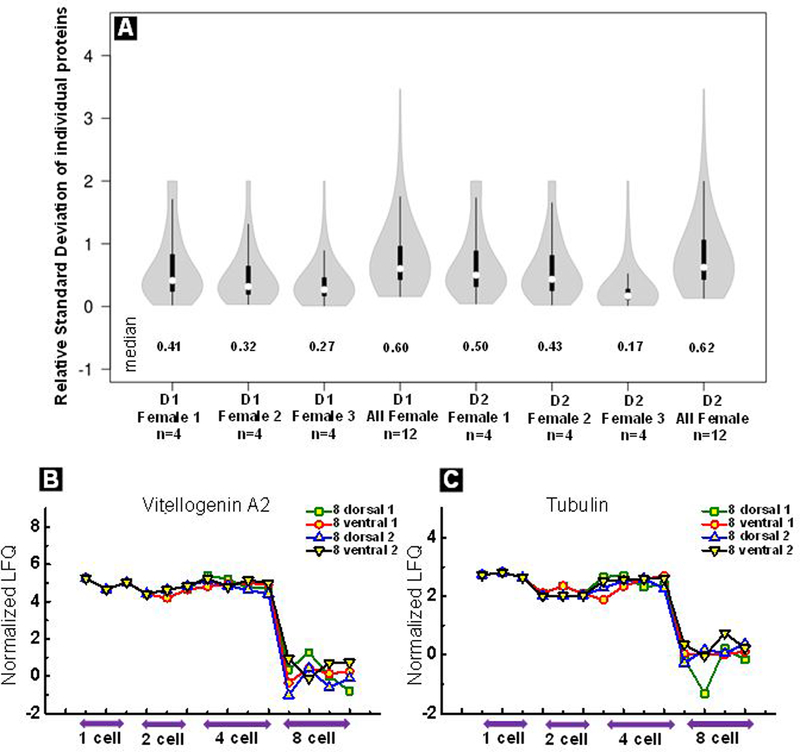
(A) A violin plot depicting the relative standard deviation in protein measurements (normalized iBAQ) across replicates belonging to same female and various females. Dorsal 1 (D1) and dorsal 2 (D2) cells from three females were used for the study. Median of each violin plot is listed in the figure. A time course plot showing decrease in abundance of vitellogenin A2 (B) and tubulin (C) with embryonic development. We see a sharp decrease in abundance of both proteins from 4 to 8 cell stage. Each trace in the plot represents a particular cell during 8 cell stage and its precursors at earlier stages. Each point in the plot (b and c) depicts one measurement, biological replicates are plotted separately (n=3 for 1-and 2 cell stage, n=4 for 4-and 8-cell stage).
Pipette sampling is minimally invasive and enables collection of cytoplasm from single embryonic cells without compromising the adjacent cells which is particularly beneficial for intercellular heterogeneity studies in whole embryos. A limitation of pipette sampling is that the method is capable of analyzing proteins solubilized in the cytoplasm and may not provide any information about membrane bound proteins. Preservation of neighboring cells enables the study of developmental asymmetries in Xenopus embryos as within the embryo there are critical differences in biomolecule population.43–45 First, we focused on the animal hemisphere of the embryo and sampled cytoplasm from all the cells at 2, 4 and 8-cell stage; shown in Figure S6a is a PCA plot demonstrating the segregation of proteomic data across developmental time points. A closer look at the data collected across all the time points revealed interesting trends. A decrease in relative abundance of vitellogenin proteins, the major protein in yolk, was observed in all 8-cell stage cells compared to the 4-cell stage (Figure 2B, S6b, S6c). This trend is compelling because yolk proteins are known to be more concentrated at the vegetal pole of the embryo.46 Cell division from the 4-cell to the 8-cell stage physically separates the animal and vegetal hemispheres of the embryo. As a result, when sampling animal hemisphere cells at the 8-cell stage, the access to yolk protein from the bottom half of the embryo is reduced. On the contrary, there is no physical separation of the animal and vegetal hemisphere up to the 4-cell stage which leads to aspiration of a greater amount of yolk protein at earlier stages. This observation further supports the applicability of our method to tease out authentic changes in protein composition during development.
Our next step was to investigate differences along the dorso-ventral axis, as the exact molecular nature of the heterogeneity between dorsal and ventral cells remains unclear. Recent attempts at understanding dorso-ventral asymmetry at the mRNA level during early stages of development (1-and 128-cell stage) yielded inconclusive results and further speculation that dorso-ventral asymmetry would be more readily observed at the protein level.47 The differential expression of proteins among cells can be visualized by the volcano plots as shown in Figure 3B, C and summarized by a histogram shown in Figure 3D. For these studies, 4 embryos from a single clutch (one male and one female frog was used for fertilization) were used as biological replicates to reduce variability. Although, the volcano plots were derived from analyzing 4 embryos from a single clutch, the same experiments were repeated with a different clutch of eggs from another female frog to validate the findings. At the 4-cell stage, greater heterogeneity was found among cells that are spatially distant from each other than between adjacent cells (Figure 3B). For example, 10 proteins were differentially expressed between D1 and V1 cells compared to 15 proteins between D2 and V1 cells. These results of differentially expressed proteins were filtered for a p-value of 0.05 and fold change of >2. This trend is enhanced at the 8-cell stage (Figure 3C) and 60 differentially expressed proteins were found between D2 and V1 cells, which are spatially the most distant cells. In contrast, 20 differentially expressed proteins were found between D1 and V1 cells. A higher number of differentially expressed proteins was found between cells at the 8-cell stage compared to 4-cell stage (Figure 3C), in keeping with the expectation of cellular differentiation across developmental time points.
Figure 3.
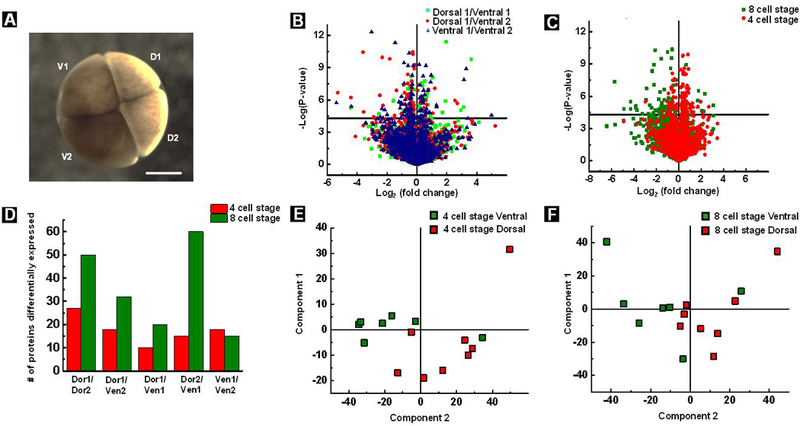
(A) Optical micrograph of an embryo at the 4-cell stage showing pigment asymmetry; the least pigmented cell is labeled dorsal 1 (D1) and the most pigmented cell is labeled ventral 2 (V2), scale: 0.5 mm (B) Significant differences in protein expression between two ventral cells and dorsal-ventral cells at the 4-cell stage is depicted in the volcano plot. (C) Volcano plots showing differences in protein composition between dorsal2 and ventral1 cell at both 4-and 8-cell stage. (D) A histogram depicting the number of differentially expressed proteins between cell types. Principal component analysis of proteome data at 4 (E) and 8 (F) cell stage, respectively.
While some interesting trends emerged from interrogation of dorsal and ventral cells, some well-established phenomena such as accumulation of β-catenin, Dvl, and GSK-3 in dorsal cells,48,49 could not be confirmed at the 4-and 8-cell stages of development. A lack of consistent protein quantification values for β-catenin, possibly because of rapid proteasomal degradation of cytosolic β-catenin, prevented this micropipette-based method from demonstrating the accepted dorsal-ventral variation in β-catenin abundance. We do not observe significant segregation in dorsal and ventral cells by principle component analysis (PCA) as shown in Figure 3E and F. The differences between dorsal and ventral cells is known to be established by a small number of low abundant proteins and hence could be a reason for this observation.
To investigate animal-vegetal asymmetry, we employed our micropipettes to collect cytoplasm from both animal and vegetal hemispheres of embryos at the 8-cell stage. This was performed by combining cytoplasm from four animal hemisphere cells into one sample and four vegetal hemisphere cells into another sample. The results of the study are shown in a volcano plot in Figure 4A. Interestingly, various subunits of the 20S proteasome core particle (20S CP) such as 20S proteasome alpha subunits (Psma 2–6) and 20S proteasome beta4 subunit (Psmb4) (indicated in red in Figure 4A) were enriched in the animal hemisphere at the 8-cell stage. Multiple subunits of the same complex enriching in a similar pattern motivated us to further validate these findings by immunofluorescence (Figure 4B). Immunofluorescence (IF) clearly indicates enrichment of the 20S CP in the animal hemisphere of 8-cell stage embryos, consistent with recent IF findings in fertilized eggs (Hwang et al).32 Asymmetric distribution of the proteasome during the oocyte to embryo transition in Xenopus may allow stabilization of vegetally localized proteins that are critical for germ cell development (Hwang et al.).32 Furthermore, the protein DDX4/Vasa (also known as Xenopus vasa-like gene 1) was localized to the vegetal hemisphere of the embryo (indicated in yellow in Figure 4A), in line with previous findings by Sindelka et.al.50 DDX4/Vasa belongs to the family of DEAD-box RNA helicases and is a marker for germ cell specification in diverse animal species. Vasa mRNA or protein is localized to germ plasm in oocytes and early embryos of Drosophila, C. elegans, sea urchins, and zebrafish.51 In Xenopus, germ plasm is localized to the vegetal hemisphere of the oocyte and early embryo52 but DDX4/Vasa mRNA has not been shown to be enriched in the vegetal hemisphere.50,53 To further address the localization of DDX4/VASA protein detected by MS in this study as well as reports from Sindelka et al50, we performed IF and confirmed that the protein is enriched in the vegetal hemisphere (Figure 4C), consistent with its known role in germ cell specification. The enrichment of DDX4/VASA protein in the vegetal hemisphere in the setting of equal distribution of the mRNA could arise through localized translation (as observed with other germ plasm components), higher proteasomal degradation rate in the animal hemisphere, or directed transport of the protein to the vegetal hemisphere; addressing these mechanisms would be the subject for future studies.
Figure 4.
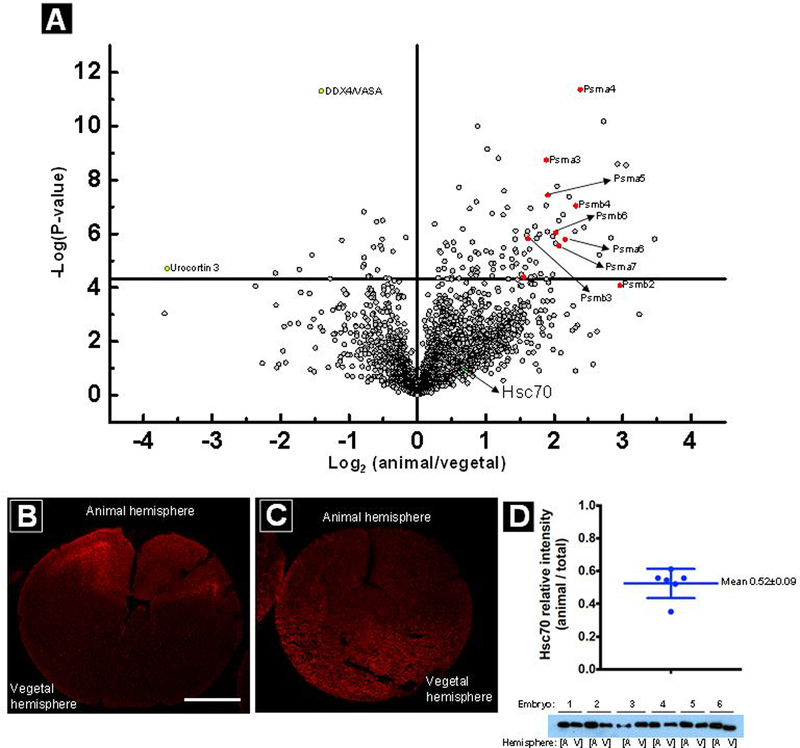
(A) A volcano plot showing differential localization of proteins across animal and vegetal hemispheres of embryos at the 8-cell stage (n=3). Marked in red are components of the 20S core particle that were found to be enriched in the animal hemisphere. In yellow are proteins enriched in the vegetal hemisphere and in green (also marked by arrow) is protein Hsc70 which is equally distributed across the animal and vegetal hemispheres. Enrichment of 20S CP in the animal hemisphere (B) and DDX4/VASA in the vegetal hemisphere (C) was further validated by immunofluorescence staining in sections of 8-cell embryos. Scale: 250 μm Equal distribution of Hsc70 protein was further validated by western blotting of protein extracted from individual animal vs. vegetal halves dissected from intact embryos (D). Western blot image from 6 embryos (lower panel) was scanned and quantitated by ImageJ, and fraction in each animal hemisphere relative to total for each embryo is plotted in the upper panel. l. The mean was found to be 0.52 with an error of 0.09 as depicted in the figure.
As a negative control, we dissected 128-cell stage embryos into animal and vegetal halves and performed western blot analysis of Hsc70, which our mass spectrometry data showed is uniformly distributed across the animal-vegetal axis.
Figure 4C confirms equal distribution of Hsc70 in animal and vegetal hemispheres, as shown by micropipette sampling and mass spectrometry.
A more detailed list of proteins asymmetrically distributed along the animal-vegetal axis can be found in the supporting information. A special mention about enrichment of thioredoxin reductase 2 in the animal hemisphere at is important considering that thioredoxin was found to be enriched in animal hemisphere at both 1-cell and 8-cell stage in our study. Sindelka el. al also reported thioredoxin to be enriched in the animal hemisphere at 8-cell stage. Thioredoxin reductase 2 was found to be 3-fold enriched in animal hemisphere (log2P=8.6) at 8 cell stage and 5-fold enriched at 1-cell stage (log2P=6.0) in our study. Additionally, Table 1 compares animal vs vegetal enrichment of few proteins between our studies, Sindelka et. al and Lombard-Banek et. al. Apart from various components of 20S CP that were found enriched at animal hemisphere and vitellogenins which were found to be vegetally enriched by both our study and Lombard-Banek et. al, we found few other proteins that had similar trends between studies. NADH dehydrogenase and ATP synthase were found to be mildly enriched (1.5 to 1.8 times) in animal hemisphere. Apparent fold changes of these proteins are not high from our study but further careful study of these proteins hold promise since they were found to be enriched by three studies.
Table 1.
List of proteins that were found to be differentially expressed in current study and by other groups at 1-cell stage of Xenopus laevis embryos
| Data from current study | Nemes group18 | Sindelka et. al50 | ||||
|---|---|---|---|---|---|---|
| Protein Names | Log2(animal/vegetal) | Log2(p-value) | Protein Names | Log2(animal/vegetal) | Protein Names | Localization |
| proteasome alpha 4 subunit | 3.35 | 7.21 | Proteasome subunit alpha type-4 | 3.00** | ||
| NADH dehydrogenase (ubiquinone) 1 alpha sub complex, 4 | 0.79 | 7.80 | NADH dehydrogenase [ubiquinone] 1 alpha sub complex subunit 4 | 0.50* | NADH dehydrogenase [ubiquinone] 1 alpha sub complex subunit 3, 5, 6,7 | Animal enrichment |
| Proteasome subunit alpha type-2 | 3.05 | 7.36 | proteasome subunit alpha 2 | 0.39* | ||
| NADH dehydrogenase (ubiquinone) 1 beta sub complex | 0.69 | 5.27 | NADH dehydrogenase [ubiquinone] 1 beta sub complex subunit 3 | 0.37* | NADH dehydrogenase [ubiquinone] 1 beta sub complex subunit 1 and 4 | Animal enrichment |
| ATP synthase, F1 complex | 0.58 | 7.03 | ATP synthase subunit f1 | 0.37* | ATP Synthase F1 subunit | Animal enrichment |
| 20S proteasome alpha5 subunit | 3.54 | 5.73 | Proteasome subunit alpha type-5 | 0.05* | ||
P-value <0.05,
P-value not available, peptides based calculations were used for fold change calculations
Although animal-vegetal asymmetry has been a subject of frequent investigation, utilizing technologies, such as mass spectrometry, that allow unbiased and high-throughput analysis has opened the field to new study. Further combination of mass spectrometry with nanofluidics enables localized measurements which can tease out important asymmetries in protein distribution. Local information obtainable by this technology can give rise to new knowledge in biology and trigger interesting investigations in the same way as we have shown with Vasa and 20S CP in this study.
CONCLUSION
A micropipette based nanofluidic device was used to study single cell proteomics from developing Xenopus laevis embryos. The method enabled sampling of cell cytoplasm from ~130 μm cells (128-cell stage) and detection of ~1650 proteins from individual cells. Intercellular heterogeneity among cells was studied at various stages of development which showed an increase in heterogeneity and hence an increase in the number of differentially expressed proteins from the 2-cell stage (Figure S8) to the 8-cell stage. These differences indicate gradual differentiation of cells at early stages of embryogenesis. Furthermore, the method was employed to look at embryonic asymmetry across the animal-vegetal and dorsal-ventral axes. Although variations in abundance of proteins such as cytosolic β-catenin were not captured by the micropipette sampling method, the cells spatially closer to each other were found to have fewer differentially expressed proteins as compared to spatially distant cells. Animal-vegetal asymmetry is more robust and was captured by micropipette sampling. Some of the lead hits were further validated by a more classical immunofluorescence approach to support the future applicability of this method in teasing out subtle heterogeneities among cells as well as perform localized measurements. Furthermore, we compare the results from our study with data available in literature to highlight interesting trends that can be followed-up in future. Overall, this study outlines a novel method to sample proteins from single Xenopus laevis embryonic cells that can be utilized to study local enrichment of biomolecule population in other systems. The simplicity of device fabrication as well as sample collection makes this method easily adoptable by researchers from various disciplines. Future applications of this method in investigating proteins from mammalian cells will require a more robust sample collection strategy that is feedback-controlled during approach of the pipette to the sample of interest 17,54 and dilution-free analysis of sample collected from single cells.28
Supplementary Material
ACKNOWLEDGMENT
The authors also appreciate the University of Pennsylvania, Singh Center for Nanotechnology for scanning electron microscope access and Dr. Yajamana Ramu for help with micropipette fabrication.
FUNDING
The authors gratefully acknowledge support from NIH grants CA196539, GM110174, AI118891 (BAG), NIH grants 1R01GM115517, 1R01MH100923, 1R01HL141759 (PSK) and NIH grant R01GM111816 to JY.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Supporting information (pdf) details additional studies that compare previous single cell proteomics data with the current study and assessment of micropipette sampling accuracy in comparison with dissected samples.
A movie demonstrating the sampling process (Movie S1-Quick Time Movie)
A spreadsheet of list of proteins (Microsoft Excel) can also be found in the supporting information.
DATA AVAILABILITY
All LC-MS/MS raw files were submitted to publically accessible Chorus repository, project ID 1543.
REFERENCES
- (1).Paolo A Small Methods 2018, 2, 1700300. [Google Scholar]
- (2).Heath JR; Ribas A; Mischel PS Nat. Rev. Drug Discov. 2015, 15, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Qian M; Wang DC; Chen H; Cheng Y Semin. Cell. Dev. Biol. 2017, 64, 143–149. [DOI] [PubMed] [Google Scholar]
- (4).Gawad C; Koh W; Quake SR Nat. Rev. Genet. 2016, 17, 175–188. [DOI] [PubMed] [Google Scholar]
- (5).Zhu Y; Piehowski PD; Zhao R; Chen J; Shen Y; Moore RJ; Shukla AK; Petyuk VA; Campbell-Thompson M; Mathews CE; Smith RD; Qian W-J; Kelly RT Nat. Commun. 2018, 9, 882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gong X; Zhao Y; Cai S; Fu S; Yang C; Zhang S; Zhang X Anal. Chem. 2014, 86, 3809–3816. [DOI] [PubMed] [Google Scholar]
- (7).Shao X; Wang X; Guan S; Lin H; Yan G; Gao M; Deng C; Zhang X Anal. Chem. 2018, 90, 14003–14010. [DOI] [PubMed] [Google Scholar]
- (8).Vogel C; Marcotte EM Nat. Rev. Genet. 2012, 13, 227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Li L; Yan S; Lin B; Shi Q; Lu Y In Advances in Cancer Research; Broome A-M, Ed.; Academic Press: 2018; Vol. 139, p 185–207. [DOI] [PubMed] [Google Scholar]
- (10).Spitzer MH; Nolan GP Cell 2016, 165, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Guillaume-Gentil O; Rey T; Kiefer P; Ibáñez AJ; Steinhoff R; Brönnimann R; Dorwling-Carter L; Zambelli T; Zenobi R; Vorholt JA Anal. Chem. 2017, 89, 5017–5023. [DOI] [PubMed] [Google Scholar]
- (12).Pan N; Rao W; Kothapalli NR; Liu R; Burgett AWG; Yang Z Anal. Chem. 2014, 86, 9376–9380. [DOI] [PubMed] [Google Scholar]
- (13).Onjiko RM; Portero EP; Moody SA; Nemes PJ Vis. Exp.2017. e56956.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Saha-Shah A; Weber AE; Karty JA; Ray SJ; Hieftje GM; Baker LA Chem. Sci. 2015, 6, 3334–3341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Onjiko RM; Portero EP; Moody SA; Nemes P Anal. Chem. 2017, 89, 7069–7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Saha-Shah A; Green CM; Abraham DH; Baker LA Analyst 2016, 141, 1958–1965. [DOI] [PubMed] [Google Scholar]
- (17).Yin R; Prabhakaran V; Laskin J Anal. Chem. 2018, 90, 7937–7945. [DOI] [PubMed] [Google Scholar]
- (18).Lombard-Banek C; Moody SA; Manzini MC; Nemes P Anal. Chem. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Aerts JT; Louis KR; Crandall SR; Govindaiah G; Cox CL; Sweedler JV Anal. Chem. 2014, 86, 3203–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Baxi AB; Lombard-Banek C; Moody SA; Nemes P ACS Chem. Neurosci. 2018, 9, 2064–2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Camille L-B; Moody A, Nemes S, P. Angew. Chem. Intl. Ed. 2016, 55, 2454–2458. [Google Scholar]
- (22).Onjiko RM; Moody SA; Nemes P Proc. Natl. Acad. Sci. 2015, 112, 6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Sun L; Dubiak KM; Peuchen EH; Zhang Z; Zhu G; Huber PW; Dovichi NJ Anal. Chem. 2016, 88, 6653–6657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Jorgensen P; Steen JAJ; Steen H; Kirschner MW Dev. 2009, 136, 1539–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Lombard-Banek C; Moody S; Nemes P In ABSTRACTS OF PAPERS OF THE AMERICAN CHEMICAL SOCIETY; AMER CHEMICAL SOC 1155 16TH ST, NW, WASHINGTON, DC 20036: USA: 2017; Vol. 254. [Google Scholar]
- (26).Lombard-Banek C; Reddy S; Moody SA; Nemes P Mol. cell. Proteomics 2016, 15, 2756–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Gupta M; Sonnett M; Ryazanova L; Presler M; Wuhr M Methods Molecular Biol. 2018, 1865, 175–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Saha-Shah A; Karty JA; Baker LA Analyst 2017, 142, 1512–1518. [DOI] [PubMed] [Google Scholar]
- (29).Sun L; Bertke MM; Champion MM; Zhu G; Huber PW; Dovichi NJ Sci. Rep. 2014, 4, 4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Wühr M; Freeman RM Jr.; Presler M; Horb ME; Peshkin L; Gygi S; Kirschner MW Curr. Biol. 2014, 24, 1467–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sindelka R; Sidova M; Abaffy P; Kubista M In Asymmetric Cell Division in Development, Differentiation and Cancer; Tassan J-P, Kubiak JZ, Eds.; Springer International Publishing: Cham, 2017, p 229–241. [Google Scholar]
- (32).Hwang H; Jin Z; Krishnamurthy VV; Saha A; Klein PS; Garcia B; Mei W; King ML; Zhang K; Yang J Dev. 2019, dev.172700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sive, H. L.; Grainger, R. M., Harland, R. M., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, N.Y. :, 2000.
- (34).Portero EP; Nemes P Analyst 2019, 144, 892–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Cox J; Mann M Nat. Biotechnol. 2008, 26, 1367–1372. [DOI] [PubMed] [Google Scholar]
- (36).Cox J; Neuhauser N; Michalski A; Scheltema RA; Olsen JV; Mann MJ Proteome Res. 2011, 10, 1794–1805. [DOI] [PubMed] [Google Scholar]
- (37).Schwanhäusser B; Busse D; Li N; Dittmar G; Schuchhardt J; Wolf J; Chen W; Selbach M Nature 2011, 473, 337. [DOI] [PubMed] [Google Scholar]
- (38).Schmidt A; Beck M; Malmström J; Lam H; Claassen M; Campbell D; Aebersold R Mol. Syst. Biol. 2011, 7, 510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Bauer M; Ahrné E; Baron AP; Glatter T; Fava LL; Santamaria A; Nigg EA; Schmidt AJ Proteome Res. 2014, 13, 5973–5988. [DOI] [PubMed] [Google Scholar]
- (40).Gillet LC; Navarro P; Tate S; Rost H; Selevsek N; Reiter L; Bonner R; Aebersold R Mol. cell. Proteomics 2012, 11, O111 016717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Searle BC; Pino LK; Egertson JD; Ting YS; Lawrence RT; MacLean BX; Villén J; MacCoss MJ Nat. Commun. 2018, 9, 5128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Budnik B; Levy E; Harmange G; Slavov N Genome Biol. 2018, 19, 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).King ML; Messitt TJ; Mowry KL Biol. Cell. 2005, 97, 19–33. [DOI] [PubMed] [Google Scholar]
- (44).Mowry KL; Cote CA FASEB 1999, 13, 435–445. [DOI] [PubMed] [Google Scholar]
- (45).Sidova M; Sindelka R; Castoldi M; Benes V; Kubista M Sci. Rep. 2015, 5, 11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Danilchik MV; Gerhart JC Dev. Biol. 1987, 122, 101–112. [DOI] [PubMed] [Google Scholar]
- (47).Flachsova M; Sindelka R; Kubista M Sci Rep 2013, 3, 2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).De Robertis EM; Kuroda H Annu. Rev. Cell Dev. Biol. 2004, 20, 285–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Larabell CA; Torres M; Rowning BA; Yost C; Miller JR; Wu M; Kimelman D; Moon RT The J Cell Biol. 1997, 136, 1123–1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Sindelka R; Abaffy P; Qu Y; Tomankova S; Sidova M; Naraine R; Kolar M; Peuchen E; Sun L; Dovichi N; Kubista M Sci. Rep. 2018, 8, 8315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Gustafson EA; Wessel GM BioEssays 2010, 32, 626–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Savage RM; Danilchik MV Dev. Biol. 1993, 157, 371–382. [DOI] [PubMed] [Google Scholar]
- (53).Komiya T; Itoh K; Ikenishi K; Furusawa M Dev. Biol. 1994, 162, 354–363. [DOI] [PubMed] [Google Scholar]
- (54).Yarger TJ; Yuill EM; Baker LA J. Am. Soc. Mass Spectrom. 2018, 29, 558–565. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.