

Abstract
G protein-coupled receptors (GPCRs) are capable of downstream signaling through distinct noncanonical pathways such as β-arrestins in addition to the canonical G protein-dependent pathways. GPCR ligands that differentially activate the downstream signaling pathways are termed functionally selective or biased ligands. A class of novel non-catechol G protein-biased agonists of the dopamine D1 receptor (D1R) was recently disclosed. We conducted the first comprehensive structure−functional selectivity relationship study measuring GS and β-arrestin2 recruitment activities focused on four regions of this scaffold, resulting in over 50 analogs with diverse functional selectivity profiles. Some compounds became potent full agonists of β-arrestin2 recruitment, while others displayed enhanced GS bias compared to the starting compound. Pharmacokinetic testing of an analog with an altered functional selectivity profile demonstrated excellent blood−brain barrier penetration. This study provides novel tools for studying ligand bias at D1R and paves the way for developing the next generation of biased D1R ligands.
Graphical Abstract
INTRODUCTION
As the largest superfamily of proteins in the druggable genome with functions that have been implicated in diverse biological processes, G protein-coupled receptors (GPCRs) represent an incredibly important target class in drug discovery.1,2 For decades, G proteins were believed to be the sole signaling effector molecules downstream of membrane-bound GPCRs. This canonical G protein-mediated signaling pathway is known to proceed upon ligand binding, which induces a conformational change in the GPCR, causing the release and dissociation of a heterotrimeric G protein that then drives downstream signaling processes within the cell.3–5
More recently, exciting discoveries in GPCR pharmacology have revealed that this receptor superfamily is capable of signaling through noncanonical G protein-independent pathways.6–9 This concept of “functional selectivity”, or biased signaling, now refers to the process by which a ligand with a given GPCR binding mode is capable of differentially activating distinct subsequent signaling cascades in the cell, including most notably those driven by G proteins or β-arrestins.10–16 In addition, recent studies have also shown that it is possible for a compound to differentially activate various G protein subtypes, such as GS/Golf, leading to subtype-selective biased agonism.17
Importantly, recent and ongoing studies on diverse classes of GPCRs have demonstrated that the direct therapeutic relevance of functional selectivity at these receptors as distinct signaling pathways can mediate divergent processes. In some cases, one downstream signaling pathway may even be responsible for the therapeutic benefit of the ligand, while the other pathway may underlie the observed adverse effects.18–25 For example, at the μ-opioid receptor (MOR), cellular processes related to analgesia, antinociception, and respiratory depression have been ascribed to differential signaling downstream of the receptor.20,22,26–28 Similarly, at the D2 receptor, biased ligands with various downstream functional activities have been shown to modulate diverse effects on motor, cognitive and antipsychotic processes.29–41 These findings provide a strong rationale for the discovery and characterization of functionally selective GPCR ligands with the potential to serve as valuable chemical tools with utility in dissecting the molecular pathways implicated in various pathophysiological processes.
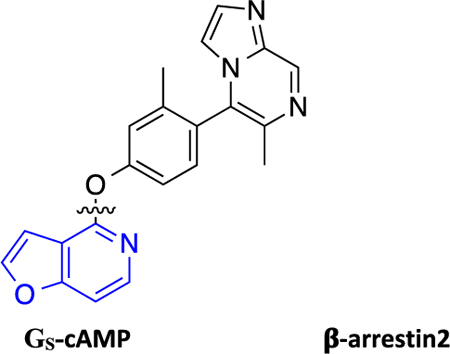
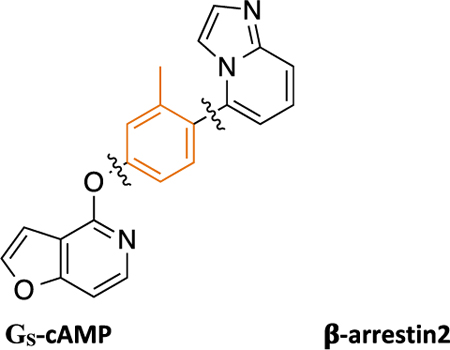
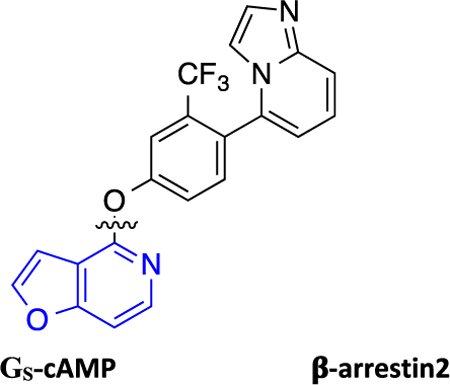
Recently, Gray et al. reported a novel non-catechol-containing D1 dopamine receptor (D1R) agonist scaffold with a putative orthosteric binding mode that differs in several important aspects from the binding mechanisms of virtually all catechol-containing agonists at aminergic GPCRs, including dopamine.42 As a direct result of this unique binding mechanism, the few reported compounds that were tested potently induced downstream cyclic adenosine monophosphate (cAMP) production, indicating stimulatory G protein (GS) pathway activation, while failing to recruit β-arrestin2.42 A subsequent study by Davoren et al. detailing the discovery of this scaffold via a high throughput screening campaign confirmed its potent induction of cAMP production, in addition to providing a detailed biophysical characterization of the atropisomerism that exists because of the locked biaryl ring system.43 Results from that study suggested that this property relates to the scaffold’s D1R high binding affinity and potent ability to stimulate cAMP production. Importantly, functional selectivity and β-arrestin2 recruitment activity were not considered in the study and all of the reported analogue ligands focused around altering the atropisomerism through modification of the locked biaryl ring system. In further studies, this non-catechol scaffold was also shown to be highly selective for D1R across a panel of class A aminergic GPCRs and neurotransmitter transporters.42,43 In an acute rodent model of Parkinson’s disease (PD), this agonist scaffold was shown to elicit a more sustained dopaminergic response in the animals in comparison with dopamine by virtue of its inability to recruit β-arrestin2, thereby resulting in diminished desensitization and tachyphylaxis after repeated dosing.42 In separate studies of downstream signaling at D1R in PD animal models, it has been suggested that the GS-mediated pathway may be responsible for the eventual development of Levodopa-induced dyskinesias (LIDs), while β-arrestin2-mediated signaling pathways may attenuate LIDs while maintaining locomotor improvements.18,44,45 We believe that such findings concerning D1R GS- and β-arrestin2-mediated signaling pathways make this a particularly interesting system for studying GPCR functional selectivity. As such, we saw a ripe opportunity to more completely characterize the structure-functional selectivity relationships (SFSR) of this unique scaffold and assess ligand structural determinants of D1R functional selectivity.
Herein, we report our SFSR study results that demonstrate certain robust trends in this distinctive scaffold that help confer its exquisite G protein functional selectivity at D1R. We describe the design, synthesis, and in vitro pharmacological assessment of novel derivatives that explore four regions of the scaffold represented by compound 1 (PF-6142), which was reported as a potent, highly selective, full agonist of the GS signaling pathway at D1R with minimal β-arrestin2 recruitment.42 These comprehensive SFSR studies provide critical information regarding the steric, electronic, and spatial determinants of G protein bias on this scaffold as well as an important framework for designing, characterizing, and optimizing compounds for ideal levels of functional selectivity at various GPCR targets.
RESULTS AND DISCUSSION
Traditional catechol D1R agonists are less CNS-penetrant and may be liable for metabolism to inactive metabolites, as observed in several well-known D1-selective catechol agonists.46–48 Non-catechol agonists, on the other hand, are more attractive for further development to avoid such extensive metabolism.42 Thus, we chose to study the SFSR of the scaffold represented by 1 (PF-6142), which features a neutral heterocyclic arylphenoxyaryl core with an interlocking biaryl ring system.
To determine whether modifying various structural elements of 1 could result in biased compounds favoring either cAMP or β-arrestin2 signaling, we divided the arylphenoxyaryl scaffold into four primary regions: (1) a right-hand side (RHS) aromatic heterocycle with various ring substitutions, (2) a substituted middle phenyl ring, (3) a linking heteroatom, and (4) a left-hand side (LHS) pyridine-containing heterocycle (Figure 1). We then investigated each region by making analogues with structural, spatial, and electronic changes isolated to that region and comparing their resulting potencies, efficacies, and functional selectivity profiles.
Figure 1.
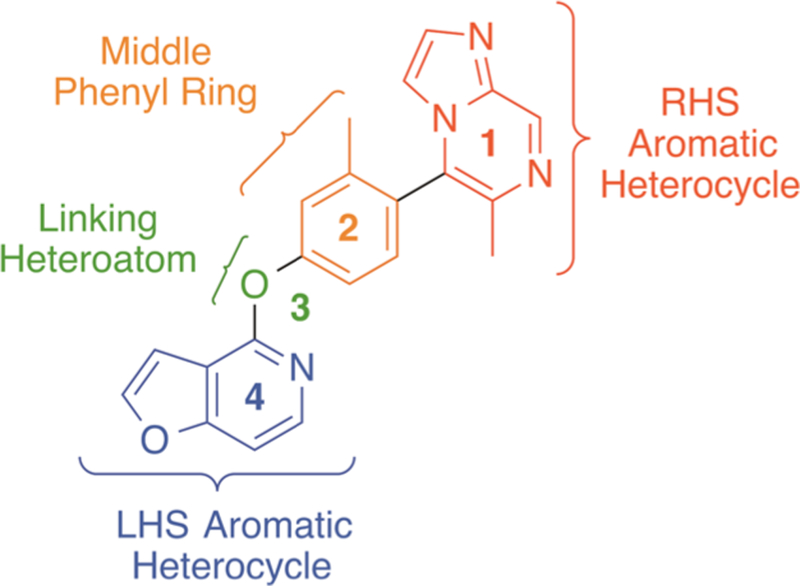
Four regions of the arylphenoxyaryl scaffold of compound 1 that were investigated for SFSR.
SFSR of the RHS Heterocycle.
To determine the effects of various heteroaromatic rings on the RHS, we designed and synthesized the compounds as presented in Table 1. We sought to create a diverse array of analogues of 1 with variously substituted bicyclic rings, including imidazopyridines, imidazopyrazines and pyrimidines, and monocyclic rings, including pyridines, to evaluate the ligand determinants of potency, efficacy, and functional selectivity.
Table 1.
SFSR of the RHS Moiety of Compound 1a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | RHS | EC50(nM) | pEC50 ± SEM | Emax ± SEM(%) | EC50(nM) | pEC50 ± SEM | Emax ± SEM(%) | Bias for cAMP accumulation |
| 1 |  |
0.25 | 9.61 ± 0.18 | 61 ± 3 | 6.94 ± 0.16 | 48±3 | 7.7 | |
| 4 | 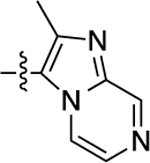 |
5.8 | 8.24 ± 0.11 | 63 ± 2 | 2,600 | 5.59 ± 0.10 | 89 ± 4 | 4.1 |
| 5 | 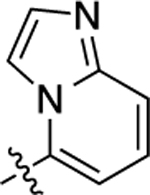 |
2.7 | 8.57 ± 0.03 | 95 ± 1 | 180 | 6.75 ± 0.09 | 63 ± 3 | 1.3 |
| 6 | 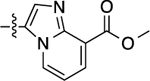 |
1,000 | 5.99 ± 0.07 | 44 ± 2 | N.A. | N.A. | N.A. | N.C |
| 7 | 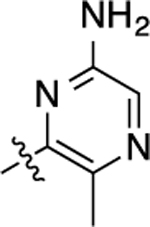 |
22 | 7.66 ± 0.04 | 91 ± 1 | >10,000 | <5.00 | N.D. | N.C |
| 8 | 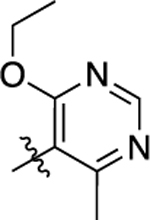 |
12 | 7.91 ± 0.12 | 67 ± 3 | >10,000 | <5.00 | N.D. | N.C |
| 9 | 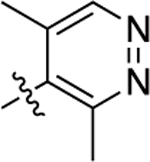 |
1.2 | 8.93 ± 0.15 | 61 ± 2 | N.A. | N.A. | N.A. | N.C |
| 10 | 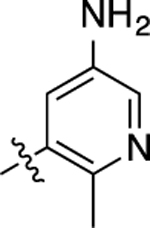 |
13 | 7.89 ± 0.09 | 67 ± 2 | 1,100 | 5.95 ± 0.08 | 64 ± 3 | 1.2 |
| 11 | 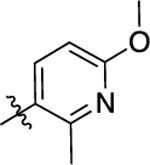 |
77 | 7.12 ± 0.06 | 80 ± 2 | >10,000 | <5.00 | N.D. | N.C |
| 12 | 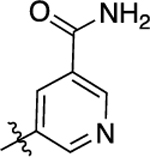 |
1,300 | 5.88 ± 0.08 | 59 ± 3 | N.A. | N.A. | N.A. | N.C |
| 13 | 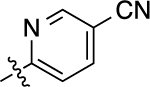 |
>10,000 | <5.00 | N.D. | N.A. | N.A. | N.A. | N.C |
EC50 and Emax values are the mean of at least three independent experiments performed in triplicate with standard error of the mean (SEM) values. N.A. no activity at maximum concentration tested (10−4 M), N.D. not determined, N.C. not calculable.
All of these compounds were prepared according to the synthetic strategy depicted in Scheme 1. Briefly, the designed compounds were prepared by Suzuki coupling of a known intermediate 342 with known49 or commercially available brominated heterocycles.50 These syntheses employed tetrakis-(triphenylphosphine)palladium(0) as a catalyst and resulted in the corresponding desired product in moderate to good yields.
Scheme 1.
Synthesis of Compounds for Exploring the RHS Heterocyclic Moietya
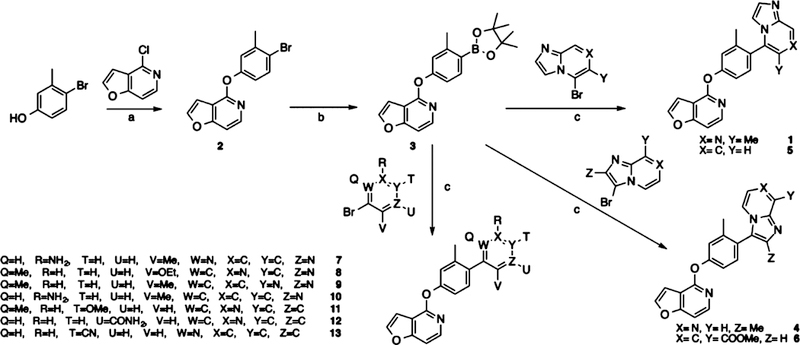
aReagents and conditions: (a) Cs2CO3, DMSO, 125 °C, 48 h; (b) Pd(dppf)Cl2, KOAc, B2(pin)2, dioxane, 80 °C, 18 h; (c) Pd(PPh3)4, K2CO3, dioxane, H2O2, 120 °C, 25 min.
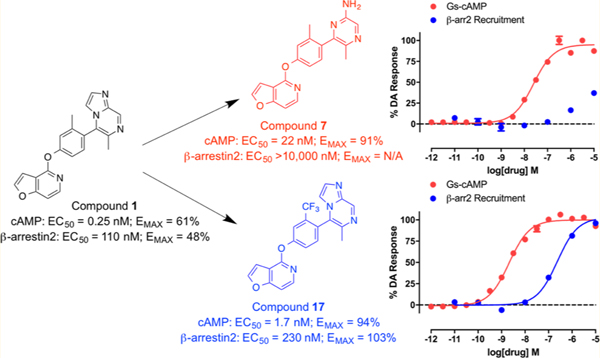
The synthesized compounds were evaluated for potency and efficacy in stimulating D1R-mediated cAMP production as measured in a D1R-GS GloSensor assay, and in activating D1R-mediated β-arrestin2 recruitment via a bioluminescence resonance energy transfer (BRET) platform-based assay, with both assays using dopamine (Figure 2A) and dihydrexidine (Figure 2B) as positive full agonist controls.51–53 Of note, the D1R-GS GloSensor assay involves a signal amplification mechanism, meaning that an EMAX may be achieved at less than full receptor occupancy, while the β-arrestin2 recruitment assay measures an unamplified, stoichiometric protein−protein interaction, meaning that an EMAX is achieved at full receptor occupancy. We began our SFSR study with compound 1, which was recently disclosed following an extensive HTS and optimization campaign.42,43 We identified 1 to be a potent partial agonist in the GloSensor assay (EC50 = 0.25 nM, EMAX = 61%) with some partial agonist activity in the β-arrestin2 recruitment BRET assay (EC50 = 110 nM, EMAX = 48%) (bias factor for cAMP accumulation: 7.7) (Figure 2C).
Figure 2.
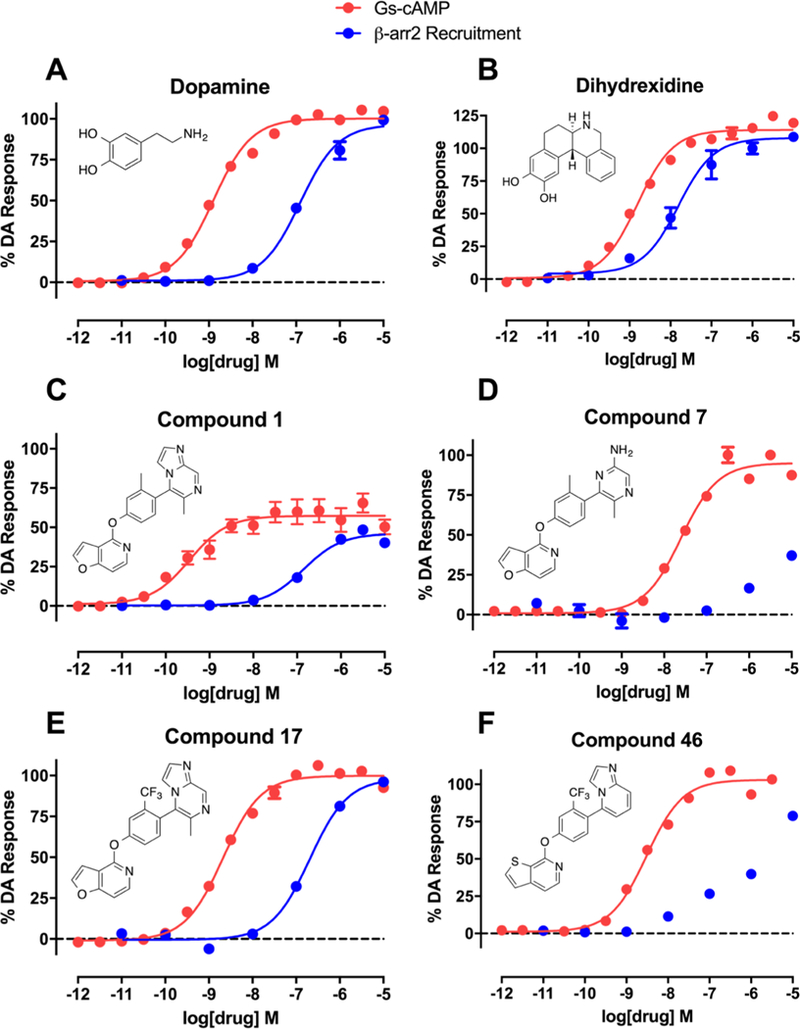
Full concentration−response curves for representative compounds and control compounds measured using the Glosensor assay for GS pathway activation (GS-cAMP) and the BRET assay for β-arrestin2 (β-arr2) recruitment as a percentage of the maximal dopamine (DA) response. Dopamine (A) is a potent full agonist of both signaling pathways, while dihydrexidine (B), a known D1/D5 selective full agonist, displays a slight β-arrestin bias (bias factor = 7.5). Compound 1 (C) is a potent GS-biased partial agonist in both pathways, while compound 7 (D) displays relatively enhanced GS bias and full agonism in the GS pathway with minimal β-arr2 recruitment. Compound 17 (E) is a potent full agonist of both signaling pathways and possesses a functional selectivity profile similar to dopamine. Compound 46 (F) also displays relatively enhanced GS bias, with potent full agonism in the GS pathway and minimal β-arr2 recruitment. Data points represent the mean concentrations ± SEM.
Through manipulation of compound 1’s RHS with diverse heterocyclic entities, we found a wide variation among compounds in terms of their activities and functional selectivity profiles. Compound 4 was designed to feature the same imidazopyrazinyl moiety as 1 but with a different methyl substitution position and a different point of attachment to the middle phenyl ring of the scaffold. Compound 4 possessed 23-fold reduced potency in the GS-cAMP accumulation assay (EC50 = 5.8 nM; EMAX = 63%) and 22-fold reduction in potency for β-arrestin2 recruitment (EC50 = 2600 nM; EMAX = 89%) (bias factor for cAMP accumulation: 4.1) compared to 1. Compound 5 featured an unsubstituted imidazopyridinyl group for the RHS and was found to maintain a similarly potent partial agonism toward β-arrestin2 recruitment as 1 (EC50 = 180 nM; EMAX = 63%). While potency in the GS-cAMP accumulation assay was approximately 10-fold weaker than 1, 5 was determined to be a full agonist of the GS pathway with an EC50 and EMAX similar to dopamine (EC50 = 2.7 nM; EMAX = 95%) (bias factor for cAMP accumulation: 1.3). The RHS of 6 featured a similar imidazopyridinyl moiety as 5, with alterations to the position of attachment and a methyl ester substitution on the ring’s 8-position. These changes resulted in a dramatically reduced potency and efficacy toward GS-cAMP accumulation (EC50 = 1000 nM; EMAX = 44%) and no detectable β-arrestin2 recruitment activity.
Switching the bicyclic imidazopyrazinyl moiety of 1 to a monocyclic aminopyrazinyl group (7) increased the efficacy of the ligand in GS-cAMP accumulation, however, with sacrificed potency (EC50 = 22 nM; EMAX = 91%). This modification also diminished the β-arrestin2 recruitment activity, resulting in a compound that is almost completely biased compound for GS compared to 1 (Figure 2D). Compared to 7 a disubstituted pyrimidinyl RHS (8) and a substituted pyridazinyl RHS (9) showed similar and improved potency (8: EC50 = 12 nM; 9: EC50 = 1 nM), respectively, but decreased efficacy in the GS-cAMP accumulation assay (8: EMAX = 67%; 9: EMAX = 61%). Similar to 7, 8 and 9 were not active in the β-arrestin2 recruitment assay. Compound 10 is a close analogue of 7, featuring a pyridinyl ring rather than a pyrazinyl ring. Compound 10 possessed similar potency in the GS pathway, but as a partial agonist compared to 7 (EC50 = 13 nM; EMAX = 67%). Notably, 10 was more active than 7 in the β-arrestin2 recruitment assay; however, it was still only a weak recruiter of β-arrestin2 overall (EC50 = 1100 nM; EMAX = 64%) (bias factor for cAMP accumulation: 1.2). Compounds 11, 12, and 13 featured different substituted pyridinyl RHS moieties and displayed different degrees of potency (11: EC50 = 77 nM; 12: EC50 = 1300 nM; 13: EC50 > 10 000 nM) as partial agonists in the GS-cAMP accumulation assay, while all being inactive in the β-arrestin2 recruitment assay. This suggests that the positions of the nitrogen in the pyridine and the ring substitutions can have a particularly important impact on both cAMP production and β-arrestin2 recruitment activity.
SFSR of the Middle Phenyl Ring.
To determine the effects of the middle phenyl ring on functional selectivity, we designed and synthesized a set of compounds with differentially substituted phenyl rings using the synthetic route outlined in Scheme 2. Various commercially available bromo phenols were substituted onto the same pyridofuran LHS group of 1, using previously described methods.42 The bromo group of the resulting intermediates was subsequently converted to a boronic ester before Suzuki coupling with the bromo imidazopyrazine RHS of 1 to afford the corresponding desired products in moderate to good yields.
Scheme 2.
Synthesis of Compounds for Exploring the Middle Phenyl Ring Moietya
aReagents and conditions: (a) Cs2CO3, DMSO, 125 °C, 48 h; (b) Pd(dppf)Cl2, KOAc, B2(pin)2, dioxane, 80 °C, 18 h; (c) Pd(PPh3)4, K2CO3, dioxane, H2O2, 120 °C, 25 min.
A few interesting SFSR trends emerged from this series of analogues, as shown in Table 2. First, changing the position of the methyl group from ortho to meta relative to the RHS, as in 14, dramatically reduced potency in the GS-cAMP accumulation assay (EC50 = 610 nM; EMAX = 62%) and completely eliminated β-arrestin2 recruitment activity. A dimethyl ring substitution at both the ortho and meta positions were introduced in compound 15 and resulted in an intermediate activity profile. In the GS-cAMP accumulation assay, 15 was found to be more potent than 14, but less potent than 1 (EC50 = 55 nM; EMAX = 54%). Compound 15 was also inactive in β-arrestin2 recruitment, similar to 14. Replacing the methyl group of 1 with an amino group resulted in compound 16, which was characterized as a potent, full agonist of the GS signaling pathway (EC50 = 1.29 nM; EMAX = 104%), while also displaying good potency and efficacy in recruiting β-arrestin2 (EC50 = 209 nM; EMAX = 81%) (bias factor for cAMP accumulation: 2.7). In compound 17, this substitution instead featured a trifluoromethyl group. This single change resulted in potent, full agonism of both the G protein (EC50 = 1.73 nM; EMAX = 94%) and β-arrestin2 (EC50 = 230 nM; EMAX = 103%) assays (bias factor for cAMP accumulation: 1.6), comparable to dopamine (Figure 2E). The enhanced efficacy of 16 and 17 in stimulating the G protein pathway may in part be due to the amino and trifluoromethyl groups being slightly bulkier and more electronegative, possibly leading to an increased energy barrier preventing torsion in this biaryl ring system.43 When this trifluoromethyl substitution was moved over one position to be meta to the RHS (compound 18), the resulting ligand was completely devoid of activity in both the G protein and β-arrestin2 recruitment assays. Interestingly, however, difluoro substitutions at both of the positions meta to the RHS, as in compound 19, eliminated β-arrestin2 recruitment activity but preserved partial agonist activity in promoting GS-cAMP accumulation (EC50 = 143 nM; EMAX = 66%).
Table 2.
SFSR of the Middle Phenyl Ring Moiety of Compound 1a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | Middle Phenyl Ring | EC50(nM) | pEC50 ± SEM | Emax ± SEM(%) | EC50(nM) | pEC50 ± SEM | Emax ± SEM(%) | Bias for cAMP accumulation |
| 1 |  |
0.25 | 9.61 ± 0.18 | 61 ±3 | 110 | 6.94 ± 0.16 | 48 ±3 | 7.7 |
| 14 |  |
610 | 6.22 ± 0.05 | 62 ±2 | N.A. | N.A. | N.A. | N.C. |
| 15 | 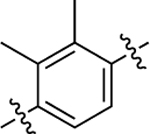 |
55 | 7.26 ± 0.11 | 54 ± 2 | N.A. | N.A. | N.A. | N.C. |
| 16 | 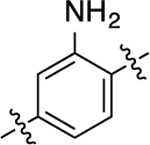 |
1.3 | 8.89 ± 0.04 | 104 ± 1 | 210 | 6.68 ± 0.10 | 81 ± 3 | 2.7 |
| 17 | 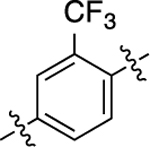 |
1.7 | 8.76 ± 0.05 | 94 ± 1 | 230 | 6.64 ± 0.06 | 103 ± 2 | 1.6 |
| 18 | 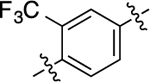 |
N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | N.C. |
| 19 | 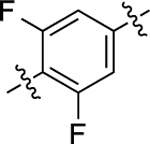 |
140 | 6.85 ± 0.04 | 66 ± 1 | N.A. | N.A. | N.A. | N.C. |
EC50 and EMAX values are the mean of at least three independent experiments performed in triplicate with standard error of the mean (SEM) values. N.A. no activity at maximum concentration tested (10−4 M), N.D. not determined, N.C. not calculable.
The above findings from our SFSR studies of the middle phenyl ring suggest that the substituent on the phenyl ring positioned ortho, but not meta, to the RHS is important for the observed potency and efficacy in the GS signaling pathway.
SFSR of the Linking Heteroatom.
A series of ligands were generated to explore the role of the linking heteroatom in D1R activity and functional selectivity. This series was synthesized using the protocol outlined in Scheme 3 and essentially involved replacing the linking oxygen atom with a variety of secondary and tertiary amine analogues. The commercially available bromo aniline was substituted onto the same pyridofuran group, using similar conditions above. The bromo group of the resulting intermediate was then replaced with a boronic ester before coupling with the same bromo imidazopyrazine RHS to afford compound 20, which was then alkylated to yield compounds 21−25 in moderate to good yields.
Scheme 3.
Synthesis of Compounds for Exploring the Linking Heteroatom Moietya
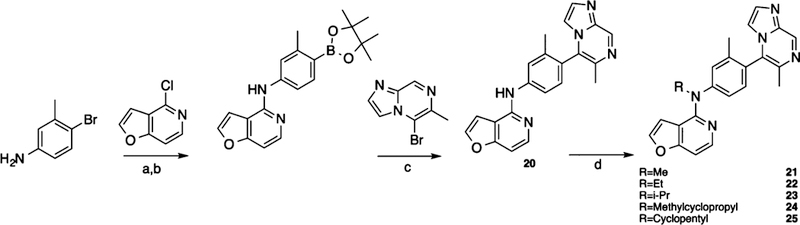
aReagents and conditions: (a) Cs2CO3, DMSO, 125 °C, 48 h; (b) Pd(dppf)Cl2, KOAc, B2(pin)2, dioxane, 80 °C, 18 h; (c) Pd(PPh3)4, K2CO3, dioxane, H2O2, 120 °C, 25 min; (d) NaH, DMF, 1 h, 80 °C, then 25−0 °C, R-I, 2 h.
Compound 20, which featured an unsubstituted secondary amino group linking the middle phenyl ring and LHS moieties, was more G protein-biased than 1 as it became only minimally active in the β-arrestin2 recruitment assay but displayed moderately potent, full agonism in the GS-cAMP accumulation assay (EC50 = 16 nM; EMAX = 98%). We next sought to explore the immediate space around this amino group. We prepared several analogues (21−25) featuring tertiary amines alkylated with side groups of varying size as the linking heteroatom moiety. The addition of a methyl group to the secondary amine to produce 21 reduced the compound’s potency and efficacy in activating the GS pathway (EC50 = 88 nM; EMAX = 79%). Methylating this amine did not alter potency for β-arrestin2 as both 20 and 21 were completely inactive in the β-arrestin2 recruitment assay. Increasing the size of the alkyl group revealed a size tolerance limit to retain potency toward GS-cAMP accumulation. An ethyl group at this position (22) possessed an intermediate potency in the GS-cAMP accumulation assay (EC50 = 36 nM; EMAX = 80%). Any larger alkyl group at this position, such as isopropyl (23) (EC50 = 100 nM; EMAX = 67%), methylcyclopropyl (24) (EC50 = 320 nM; EMAX = 78%), or cyclopentyl (25) (EC50 = 1100 nM; EMAX = 68%), significantly reduced potency and moderately reduced efficacy toward GS activity. Compared to the ether linker in 1, the amino group linkers in 20−25 dramatically reduced potency toward stimulating β-arrestin2 recruitment, rendering the compounds inactive for β-arrestin2 recruitment.
Our SFSR studies indicate that replacing the linking oxygen with unsubstituted and various substituted amino groups is not well-tolerated, as these changes reduced this scaffold’s potency toward G protein activation while eliminating ability to recruit β-arrestin2 (Table 3). This suggests that perhaps this region is spatially confined within the D1R binding pocket, or that an electronic interaction with the heteroatom at this position is important for the binding modality of this scaffold, with an oxygen atom being the preferred entity over both a secondary and tertiary amine.
Table 3.
SFSR of the Linking Heteroatom Moiety of Compound 1a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | Linking Heteroatom | EC50(nM) | pEC50 ± SEM | Emax ± SEM(%) | EC50(nM) | pEC50 ± SEM | Emax ± SEM(%) | Bias for cAMP accumulation |
| 1 |  |
0.25 | 9.61 ± 0.18 | 61 ± 3 | 114 | 6.94 ± 0.16 | 48 ± 3 | 7.7 |
| 20 | 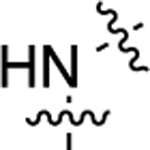 |
16 | 7.81 ± 0.04 | 98 ± 1 | >10,000 | <5.00 | N.D. | N.C. |
| 21 | 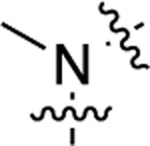 |
88 | 7.05 ± 0.03 | 79 ± 1 | N.A. | N.A. | N.A. | N.C. |
| 22 | 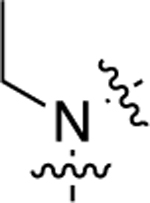 |
36 | 7.45 ± 0.05 | 80 ± 1 | N.A. | N.A. | N.A. | N.C. |
| 23 | 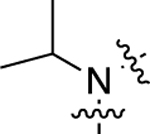 |
100 | 6.98 ± 0.09 | 67 ± 3 | N.A. | N.A. | N.A. | N.C. |
| 24 | 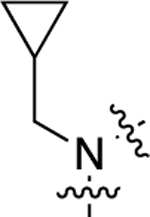 |
320 | 6.50 ± 0.03 | 78 ± 1 | N.A. | N.A. | N.A. | N.C. |
| 25 | 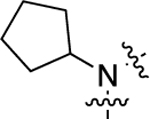 |
1,100 | 5.94 ± 0.07 | 68 ± 3 | >10,000 | <5.00 | N.D. | N.C. |
EC50 and EMAX values are the mean of at least three independent experiments performed in triplicate with standard error of the mean (SEM) values. N.A. no activity at maximum concentration tested (10−4 M), N.D. not determined, N.C. not calculable.
SFSR of the LHS Heterocycle.
A fourth set of compounds was synthesized to explore the SFSR of the LHS portion of the non-catechol scaffold (Table 4). This compound set preserved the RHS, middle phenyl ring, and linking heteroatom moieties from compound 1, while installing diverse heterocyclic entities on the LHS of the scaffold. These compounds were all synthesized from commercially available starting materials according to the synthetic routes outlined in Scheme 4. Various commercially available chlorinated heterocycles were reacted with 4-bromo-3-methylphenol, using previously described methods.42 The bromo group of the resulting intermediates was converted to a boronic ester before Suzuki coupling with the bromo imidazopyrazine RHS of 1 to afford the desired products in moderate to good yields.
Table 4.
SFSR of the LHS Moiety of Compound 1a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | LHS | EC50(nM) | pEC50 ± SEM | Emax ± SEM(%) | EC50(nM) | pEC50 ± SEM | Emax ± SEM(%) | Bias for cAMP accumulation |
| 1 | 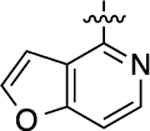 |
0.25 | 9.61 ± 0.18 | 61 ±3 | 110 | 6.94 ± 0.16 | 48 ± 3 | 7.7 |
| 26 | 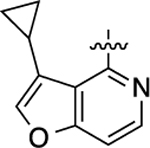 |
62 | 7.21 ± 0.08 | 67 ± 2 | N.A. | N.A. | N.A. | N.C. |
| 27 | 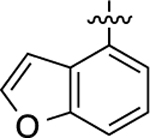 |
79 | 7.10 ± 0.05 | 69 ± 1 | N.A. | N.A. | N.A. | N.C. |
| 28 | 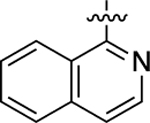 |
15 | 7.84 ± 0.06 | 82 ± 2 | >10,000 | <5.00 | N.D. | N.C. |
| 29 | 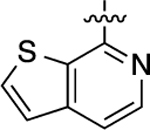 |
0.23 | 9.64 ± 0.05 | 100 ± 1 | 22 | 7.67 ± 0.10 | 63 ± 2 | 1.9 |
| 30 | 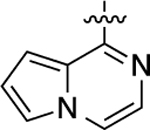 |
0.87 | 9.06 ± 0.09 | 83 ± 2 | 690 | 6.16 ± 0.19 | 59 ± 4 | 15 |
| 31 | 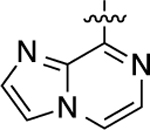 |
260 | 6.59 ± 0.03 | 85 ± 1 | N.A. | N.A. | N.A. | N.C. |
| 32 | 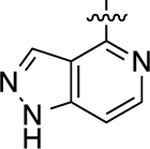 |
1.8 | 8.76 ± 0.04 | 104 ± 1 | >10,000 | <5.00 | N.D. | N.C. |
| 33 | 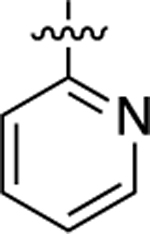 |
120 | 6.91 ± 0.06 | 83 ± 2 | >10,000 | <5.00 | N.D. | N.C. |
| 34 | 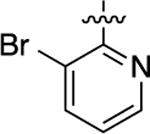 |
>10,000 | <5.00 | N.D. | N.A. | N.A. | N.A. | N.C. |
| 35 | 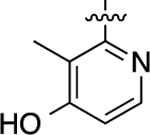 |
160 | 6.79 ± 0.11 | 34 ±1 | N.A. | N.A. | N.A. | N.C. |
| 36 | 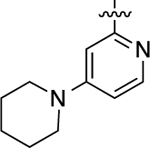 |
640 | 6.19 ± 0.07 | 26 ± 1 | N.A. | N.A. | N.A. | N.C. |
EC50 and EMAX values are the mean of at least three independent experiments performed in triplicate with standard error of the mean (SEM) values. N.A. no activity at maximum concentration tested (10−4 M), N.D. not determined, N.C. not calculable.
Scheme 4.
Synthesis of Compounds for Exploring the LHS Heterocyclic Moietya
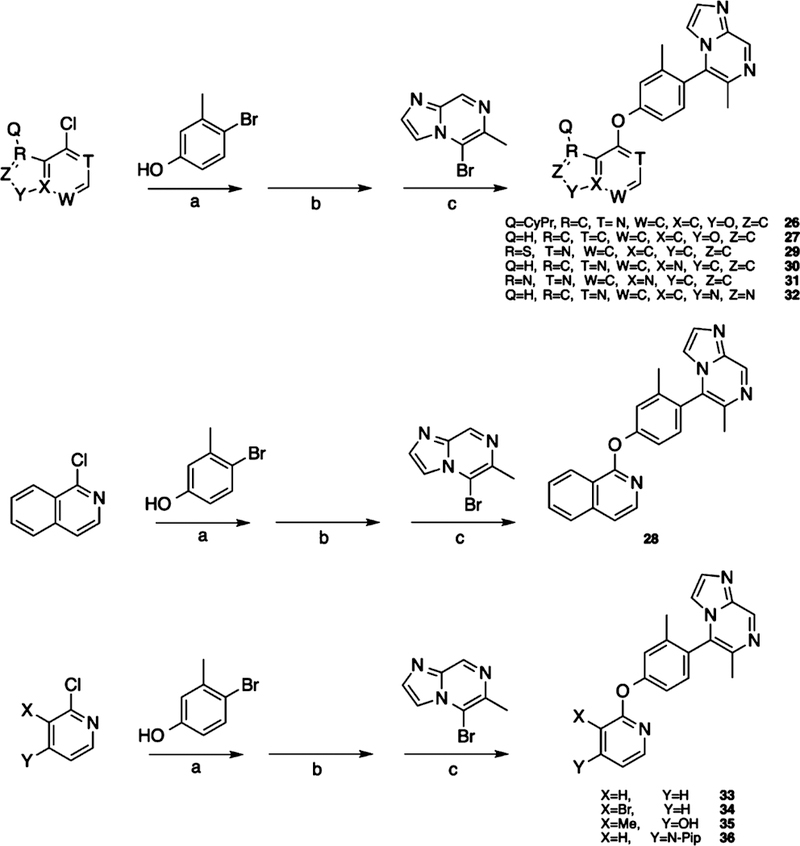
aReagents and conditions: (a) Cs2CO3, DMSO, 125 °C, 48 h; (b) Pd(dppf)Cl2, KOAc, B2(pin)2, dioxane, 80 °C, 18 h; (c) Pd(PPh3)4, K2CO3, dioxane, H2O2, 120 °C 25 min.
Cyclopropyl group substitution at the 3-position on the furopyridinyl LHS (26) dramatically reduced potency for GS-cAMP production (EC50 = 62 nM; EMAX = 67%) and diminished β-arrestin2 recruitment activity as well. The roles of the LHS pyridine nitrogen was examined with benzofuro compound 27. Omission of the nitrogen in 27 resulted in a reduction in potency for GS-cAMP accumulation (EC50 = 79 nM; EMAX = 69%) and inactivity for β-arrestin2 recruitment. Switching the furopyridinyl group of 1 to an isoquinolinyl moiety of 28 was also inactive for β-arrestin2 recruitment but retained more potency for GS-cAMP accumulation assay (EC50 = 14 nM; EMAX = 82%). Heteroatom placement within the bicyclic aromatic LHS ring was explored with the next set of compounds. A thienopyridinyl moiety was introduced in 29, which was found to be a potent full agonist of the GS pathway (EC50 = 0.23 nM; EMAX = 100%) and a potent partial agonist for β-arrestin2 recruitment (EC50 = 22 nM; EMAX = 63%) (bias factor for cAMP accumulation: 1.9). When this entity was replaced with a pyrrolopyrazinyl moiety (30), the activity in the GS-cAMP accumulation assay was retained (EC50 = 0.87 nM; EMAX = 83%), while β-arrestin2 recruitment activity dropped (EC50 = 690 nM; EMAX = 59%) (bias factor for cAMP accumulation: 15). However, the addition of a third nitrogen to create an imidazopyrazinyl moiety in this position (31) was not well-tolerated as potency for the GS pathway was dramatically reduced (EC50 = 260 nM; EMAX = 85%) and β-arrestin2 recruitment was undetectable. Rearrangement of the three nitrogen atoms in this ring to generate a pyrazolopyridinyl on the LHS (32) almost fully restored GS-cAMP accumulation activity (EC50 = 1.8 nM; EMAX = 104%). β-arrestin2 recruitment activity, however, was not recovered, making 32 a highly GS-biased D1R agonist. Our last set of compounds in this series sought to briefly explore a few pyridinyl groups as the LHS. The unsubstituted pyridinyl group led to a moderately potent ligand 33 with near full agonism in the GS-cAMP accumulation assay (EC50 = 120 nM; EMAX = 83%) and very weak activity in the β-arrestin2 recruitment assay. The addition of a bromine substituent at the 3-position (34) rendered the scaffold completely inactive in both pathways. When a disubstituted 3-methyl-4-hydroxy analogue (35) that mimicked an opened ring version of 1 was examined, only weak partial agonist activity was observed in the GS-cAMP accumulation assay (EC50 = 160 nM; EMAX = 34%). Finally, a similar effect on GS-cAMP accumulation activity (EC50 = 640 nM; EMAX = 26%) was observed with a piperidinyl substituted pyridinyl analogue (36). All of these pyridinyl analogues were also inactive in β-arrestin2 recruitment assays.
Hybrid SFSR Exploration.
During our SFSR campaign of the RHS portion of this non-catechol scaffold, we discovered compound 5, a close analogue of 1 that featured an unsubstituted imidazopyridine moiety and displayed enhanced efficacy and comparable potency in both downstream pathways relative to its parent compound. Given these robust activities, we wanted to explore whether our initial SFSR trends with 1 were consistent with 5 and whether this scaffold could also be altered to display considerably different functional selectivity profiles (Table 5).
Table 5.
SFSR of the Middle Phenyl Ring Moiety of Compound 5a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | Middle Phenyl Ring | EC50(nM) | pEC50 ± SEM | Emax ± SEM(%) | EC50(nM) | pEC50 ± SEM | Emax ± SEM(%) | Bias for cAMP accumulation |
| 5 | 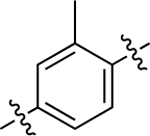 |
2.7 | 8.57 ± 0.03 | 95 ± 1 | 180 | 6.75 ± 0.09 | 63 ± 3 | 1.3 |
| 37 | 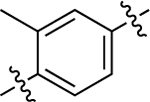 |
530 | 6.27 ± 0.09 | 29 ± 1 | N.A. | N.A. | N.A. | N.C. |
| 38 | 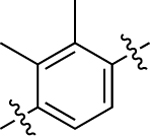 |
110 | 6.96 ± 0.13 | 30 ± 1 | N.A. | N.A. | N.A. | N.C. |
| 39 | 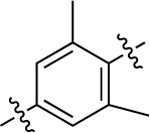 |
N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | N.C. |
| 40 | 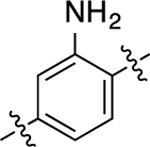 |
10 | 8.00 ± 0.05 | 91 ± 1 | 790 | 6.10 ± 0.11 | 54 + 3 | 1.7 |
| 41 | 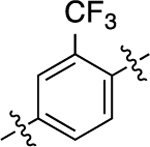 |
2.3 | 8.64 ± 0.03 | 94 ± 1 | 270 | 6.57 ± 0.12 | 93 ± 5 | 1.5 |
| 42 | 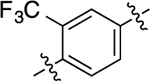 |
N.A. | N.A. | N.A. | N.A. | N.A. | N.A. | N.C. |
| 43 | 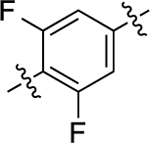 |
190 | 6.71 ± 0.04 | 57 ± 1 | >10,000 | <5.00 | N.D. | N.C. |
EC50 and EMAX values are the mean of at least three independent experiments performed in triplicate with standard error of the mean (SEM) values. N.A. no activity at maximum concentration tested (10−4 M), N.D. not determined, N.C. not calculable.
First, a set of analogues of 5 were generated by modifying the methylation pattern on the middle phenyl ring of the scaffold using the same synthetic route outlined in Scheme 2. Various commercially available bromo phenols were substituted onto the same pyridofuran LHS group of 5, using previously described methods.42 The bromo group of the resulting intermediates was subsequently converted to a boronic ester before Suzuki coupling with the bromo imidazopyridine RHS of 5 to afford the desired products in moderate to good yields.
Similar to before, migration of the methyl substituent on the middle phenyl ring to the meta position relative to the RHS (37) severely dampened GS activity (EC50 = 530 nM; EMAX = 29%) and eliminated β-arrestin2 recruitment activity. A more mild reduction in G protein activity (EC50 = 110 nM; EMAX = 30%) was observed for the substituted dimethyl analogue, 38, while β-arrestin2 recruitment was eliminated. Interestingly, this scaffold was completely inactive when both methyl groups were moved ortho to the RHS, as in 39.
When the methyl substituent on 1 was converted to an amino group, the resulting compound 16 possessed high potency and efficacy in both signaling pathways; however, when this same conversion was implemented starting with 5, the resulting analogue (40) was a GS-biased compound with full agonistic activity toward GS-cAMP accumulation (EC50 = 10 nM; EMAX = 91%) and relatively weak β-arrestin2 recruitment activity (EC50 = 790 nM; EMAX = 54%) (bias factor for cAMP accumulation: 1.7). Conversely, converting the methyl substituent on the middle phenyl ring of 5 into a trifluoromethyl group (compound 41) closely recapitulated the functional selectivity profile of the analogue 17 in both pathways (bias factor for cAMP accumulation: 1.5). When this trifluoromethyl substitution was moved meta to the RHS (compound 42), the resulting ligand was inactive in both G protein and β-arrestin2 recruitment assays. Difluoro substitutions at both of the positions meta to the RHS (compound 43) showed moderate partial agonist activity in the GS-cAMP accumulation assay with an EC50 = 190 nM and EMAX = 57%, and minimal β-arrestin2 recruitment.
Because the goal of this SFSR study was to explore the structural determinants of the non-catechol scaffold’s potency and efficacy in both the GS and β-arrestin2 pathways at D1R, we wanted to use one of the more balanced ligands we developed with potent full agonism in both pathways to further explore this topic. We decided to use 41 for this purpose because it featured slight structural modifications on the RHS and middle phenyl moieties that led to potent full agonism of both the GS and β-arrestin2 pathways. The compound set derived from 41 preserved the RHS, middle phenyl ring, and linking heteroatom moieties from compound 41, while making diverse modifications to the heterocyclic entities on the LHS portion of the scaffold (Table 6). Compounds in this set were all synthesized from commercially available starting materials using the synthetic routes similar to the ones outlined in Scheme 4. Various commercially available chlorinated heterocycles were reacted with 4-bromo-3-trifluoromethylphenol, using previously described methods.42 The bromo group of the resulting intermediate was converted to a boronic ester before Suzuki coupling with the bromo imidazopyridine RHS of 41 to afford the desired products in moderate to good yields.
Table 6.
SFSR of the LHS Moiety of Compound 41a
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Cmpd | LHS | EC50(nM) | pEC50 ± SEM | Emax ± SEM(%) | EC50(nM) | pEC50 ± SEM | Emax ± SEM(%) | Bias for cAMP accumulation |
| 41 | 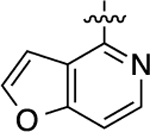 |
2.3 | 8.64 ± 0.03 | 94 ± 1 | 270 | 6.57 ± 0.12 | 93 ± 5 | 1.5 |
| 44 | 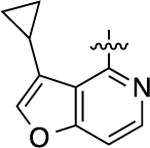 |
770 | 6.11 ± 0.02 | 83 ± 1 | N.A. | N.A. | N.A. | N.C. |
| 45 | 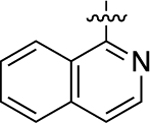 |
67 | 7.18 ± 0.07 | 61 ± 2 | N.A. | N.A. | N.A. | N.C. |
| 46 | 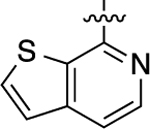 |
2.2 | 8.661 0.06 | 93 ± 2 | >10,000 | <5.00 | N.D. | N.C. |
| 47 | 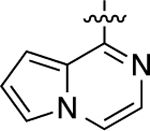 |
7.8 | 8.11 ± 0.07 | 78 ± 2 | N.A. | N.A. | N.A. | N.C. |
| 48 | 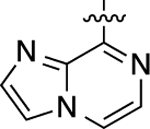 |
180 | 6.74 ± 0.06 | 54 ± 1 | N.A. | N.A. | N.A. | N.C. |
| 49 |  |
270 | 6.57 ± 0.05 | 65 ± 1 | N.A. | N.A. | N.A. | N.C. |
| 50 |  |
N.A. | N.A. | N.A. | >10,000 | <5.00 | N.D. | N.C. |
EC50 and EMAX values are the mean of at least three independent experiments performed in triplicate with standard error of the mean (SEM) values. N.A. no activity at maximum concentration tested (10−4 M), N.D. not determined, N.C. not calculable.
Substitution on the 3-position of the LHS pyridofuran moiety with a cyclopropyl group (44) abolished β-arrestin2 recruitment and dramatically hindered GS pathway activation (EC50 = 770 nM; EMAX = 83%), suggesting spatial constraints in this binding region. G protein activity was only mildly reduced (EC50 = 67 nM; EMAX = 61%) and β-arrestin2 recruitment still eliminated for compound 45 in which the furan moiety was replaced with a phenyl group on the LHS bicycle, suggesting that spatial or electronic properties on this portion of the LHS may be important for binding.
When heteroatom substitutions and rearrangements were made within the LHS core of 41, the scaffold tended to become more GS-biased. When a thienopyridine analogue was installed on the LHS (46), GS-cAMP accumulation activity showed mild attenuation (EC50 = 2.2 nM; EMAX = 93%), while β-arrestin2 recruitment activity was greatly weakened (Figure 2F). A similar profile was also observed for the pyrrolopyrazine-containing analogue 47. Compound 47 retained full agonism toward GS-cAMP accumulation with a slight reduction in potency (EC50 = 7.8 nM; EMAX = 78%), while β-arrestin2 recruitment activity was completely abrogated. Only moderate partial agonist activity was observed in the GS-cAMP accumulation assay for 48, which featured an imidazopyrazine moiety on the LHS (EC50 = 180 nM; EMAX = 54%). The pyridine-containing analogue 49 displayed modest partial agonist activity in the GS pathway (EC50 = 270 nM; EMAX = 65%) and no activity in the β-arrestin2 pathway. Finally, the substituted pyridine analogue 50 was determined to be inactive in both the GS and β-arrestin2 pathways. Together, these findings suggest that the electronic and spatial properties of 41’s LHS moiety are critical for maintaining its potent β-arrestin2 recruitment activity, as many rearrangements and substitutions disproportionately impacted β-arrestin2 recruitment over GS-cAMP accumulation activity.
In light of the fact that our SFSR campaign resulted in numerous compounds with varying functional selectivity profiles, an open question remains as to what degree of bias measured in vitro is sufficient to observe significant change in the biological activity of this ligand class at D1R in vivo. An excellent example of this is G protein-biased agonism at the MOR, which has shown the potential for improving therapeutic on-target effects for anti-nociception with reduced constipation and respiratory depression effects.20,22,26–28 Among the most clinically advanced MOR G protein-biased agonists includes TRV130, a potent G protein-biased ligand that recently completed phase 3 clinical studies for the treatment of acute pain. In in vitro studies, TRV130 demonstrated nanomolar activation of G protein signaling (pEC50 = 8.1; EMAX = 84%) and diminished activity in recruiting β-arrestin (pEC50 = 7.3; EMAX = 15%).20 Several other examples of MOR G protein-biased agonists show a correlation between their in vitro profile and reduced side effects.23 While the novel biological effects of TRV130 and other MOR G protein-biased agonists are attributed to their G protein bias at MOR, it is unclear about the degree of bias threshold necessary to produce these significant effects in vivo, especially for other GPCRs. Future studies using sets of tool compounds with a variety of bias profiles, both toward either G protein or β-arrestin, could help provide further insight into the degree of in vitro bias necessary for therapeutic effects.
Binding Affinity and Selectivity.
A selection of analogue compounds with interesting SFSR were selected for assessing their selectivity across a broad panel of over 30 class A aminergic GPCRs, ion channels, and neurotransmitter transporters using a validated radioligand binding affinity assay.54 We determined the selectivity and binding affinity of 4 ligands with diverse functional selectivity profiles generated through our SFSR studies on this non-catechol scaffold. The results, detailed in Table 7, demonstrate the remarkable selectivity of this scaffold for the D1-like class of dopamine receptors, D1R and D5R. This trend was evident across all the ligands tested, regardless of whether they only displayed GS bias (1, 46) or potent full agonism in both pathways (17, 41).
Table 7.
Radioligand Binding Affinities of Compounds 1, 17, 41, and 46 at Select GPCRs, Ion Channels, and Transportersa
| receptor | binding affinity Ki (nM) | |||
|---|---|---|---|---|
| compound 1 | compound 17 | compound 41 | compound 46 | |
| D1 | 110 | 140 | 150 | 130 |
| D2 | 8300 | 9600 | 7600 | 8700 |
| D3 | 7900 | 8100 | >10 000 | 6200 |
| D4 | 4500 | 6900 | 6900 | 4900 |
| D5 | 38 | 82 | 47 | 27 |
| 5-HT1A | 1200 | 6900 | 2500 | 3700 |
| 5-HT1B | 9300 | 8800 | 9300 | 9900 |
| 5-HT1D | 9300 | 8100 | 8900 | 4100 |
| 5-HT1E | 9800 | 8900 | 9900 | 7800 |
| 5-HT2A | 4900 | 8700 | 4300 | 3400 |
| 5-HT2B | 4100 | 6700 | 830 | 1100 |
| 5-HT3 | 7900 | 9100 | 9300 | 7600 |
| 5-HT4 | 9100 | >10 000 | >10 000 | 3700 |
| 5-HT5A | >10 000 | 9500 | 7400 | 6200 |
| 5-HT6 | 8100 | 7900 | 6900 | 7200 |
| 5-HT7A | 7800 | 8100 | 4100 | 3800 |
| alpha1A | 3400 | 8700 | 7000 | 2900 |
| alpha1B | 9200 | 8800 | 7400 | 4800 |
| alpha2A | 3400 | 6800 | 5100 | 2600 |
| beta1 | 7600 | 9600 | 8500 | 8500 |
| beta2 | 9300 | >10 000 | 8600 | 5900 |
| MOR | 8200 | 9400 | 4400 | 2100 |
| KOR | 3100 | 7200 | 1500 | 3200 |
| M1 | 8800 | 8500 | 9000 | 6600 |
| M2 | 7700 | 7300 | 8100 | 1700 |
| M3 | >10 000 | 9200 | >10 000 | 2700 |
| sigma1 | 8200 | 8000 | 6900 | 8800 |
| H1 | >10 000 | >10 000 | 9800 | 2600 |
| H2 | 8000 | 8200 | 6700 | 2500 |
| DAT | 8300 | >10 000 | 9100 | 4500 |
| SERT | >10 000 | >10 000 | 9700 | >10 000 |
Ki values represent the average of at least three triplicate experiments. SEM <±20%.
Mouse Pharmacokinetic Studies and Blood−Brain Barrier Penetrance.
Given the favorable in vivo profile of our starting compound 1, as reported by Gray et al.,42 we sought to examine whether or not the modifications we introduced into this scaffold during our SFSR campaign altered in vivo pharmacokinetic properties and blood−brain barrier (BBB) penetrance of this scaffold. We selected compound 40 for these studies because it represented a structure to which sufficient chemical modifications were made resulting in further diminished β-arrestin2 recruitment potency compared to 1 with potency in the GS pathway mostly retained. Compound 40 was administered to mice at 50 mg/kg via intraperitoneal injection and drug concentrations in the plasma and brain were assessed at four time points. These results, obtained in biological triplicates, clearly indicate that this scaffold retains high exposure in plasma and excellent BBB penetrance. At this dose, compound 40 reached a peak concentration of approximately 100 μM in the brain and plasma. The exposure levels of this drug were found to persist in both the plasma and the brain even up to 4 h after injection (Figure 3). Further, even with a high plasma and brain exposure of 40, it was well tolerated and no clinical signs were observed in the tested animals. During the course of this study, the mean brain-to-plasma ratio typically ranged around 1.2−1.6, indicating excellent BBB penetration. Taken together, these data suggest that 40 and other ligands in this series may serve as valuable chemical tools for in vivo investigations of functional selectivity and GS protein signaling at D1R.
Figure 3.
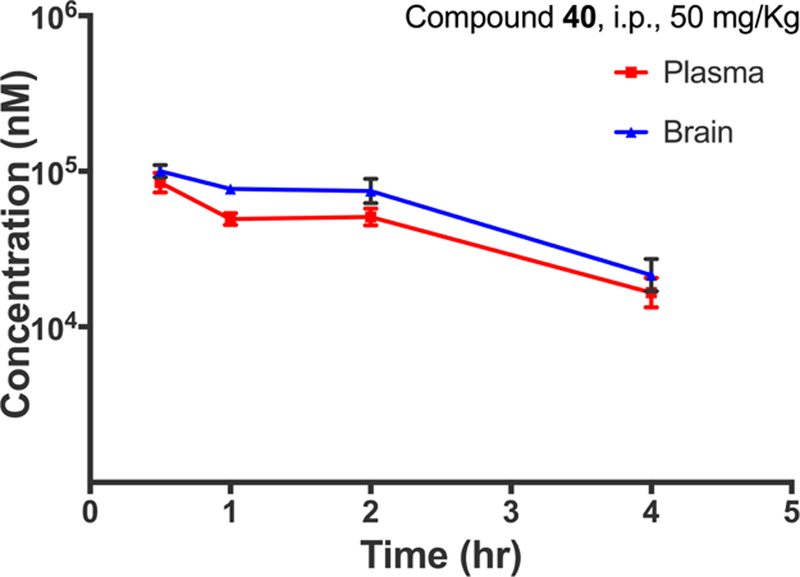
Mouse pharmacokinetic profile of compound 40 in the plasma and brain. Compound 40 was injected into the peritoneum at 50 mg/kg, and concentrations were quantitatively assessed at 0.5, 1, 2, and 4 h after administration. Experiments were carried out in biological triplicates. Data points represent the mean concentrations ± SEM.
CONCLUSIONS
In summary, we conducted a comprehensive SFSR campaign on four regions of compound 1, a non-catechol D1R agonist scaffold. By design, synthesis, and biological characterization of approximately 50 novel derivatives, we found a number of interesting SFSR trends for this scaffold. Our SFSR studies culminated in the discovery of compounds with high degrees of structural similarity but differences in their abilities to activate the G protein and β-arrestin2 signaling pathways. Some compounds, such as 16, 17, and 41, are potent full agonists of β-arrestin2 recruitment, which represents a striking result for this initially GS-biased scaffold. Conversely, other compounds, such as 7, 32, 46, and 47, displayed enhanced GS bias profiles compared to 1. Several of these compounds exhibited remarkable selectivity for the D1-like class of dopamine receptors, D1R and D5R, over a broad panel of aminergic GPCRs, ion channels, and transporters. Further characterization revealed that, despite making certain structural alterations to the scaffold, this scaffold can still possess high exposure levels in the plasma and excellent BBB permeability, as demonstrated with 40. These ligands could therefore be potential chemical tools for dissecting the downstream effects of biased signaling at D1R. Furthermore, the results described here are valuable for the research community to develop the next generation of D1R biased ligands, and biased ligands for other GPCRs.
EXPERIMENTAL SECTION
Chemistry General Procedures.
All reagents were commercial grade and were used without additional purification. All dry solvents were purchased from Sigma-Aldrich and were assured to be of anhydrous quality. A Discover SP microwave system with an Explorer 12 Hybrid Autosampler by CEM (Buckingham, UK) was used for procedures that required microwave heating. All microwave reactions were carried out at 125 °C for 25 min using 250 W of power and a pressure less than 300 psi. EMD Millipore 210−270 μm 60-F254 silica gel plates were used for analytical thin-layer chromatography, with visualization using 254 μm UV light. A Teledyne (Thousand Oaks, CA) ISCO CombiFlash Rf+ system was utilized for flash column chromatography along with normal phase RediSep Rf silica columns. This system was equipped with a UV detector and a fraction collector apparatus. All final compounds were purified using a preparative high-performance liquid chromatography (HPLC) system. Preparative HPLC was performed using an Agilent Prep 1200 series with a Phenomenex Luna 750 mm × 30 mm, 5 μm, C18 column. Samples were injected into the column at room temperature. The solvent flow rate was set to 40 mL/min, and the UV detector was set to 254 nm. A linear gradient was implemented with 10% [or 50% MeOH (A) in H20] (with 0.1% TFA) (B) to 100% MeOH (A). The purities of all target compounds were established using HPLC. All biologically evaluated compounds had >95% purity after purification via the HPLC methods described below. All compounds were characterized using a liquid chromatography−mass spectrometer (LC−MS). Key compounds were further examined by liquid chromatography−high resolution mass spectrometry (LC−HRMS) as follows. HPLC spectra were obtained for all compounds using an Agilent 1200 series system with a DAD detector. A 2.1 mm × 150 mm Zorbax 300SB-C18 5 μm column was used for chromatography. Solvent A included water containing 0.1% formic acid and solvent B included acetonitrile containing 0.1% formic acid. The flow rate was set at 0.4 mL/min and the gradient program was as follows: 1% B (0−1 min), 1−99% B (1−4 min), and 99% B (4−8 min). These spectra were used for the HPLC retention times reported below for each compound. HRMS data was obtained for each compound. Samples were ionized by electrospray ionization (ESI) in positive mode. A G1969A high-resolution API-TOF mass spectrometer by Agilent Technologies, attached to the 1200 HPLC system described above, was used to conduct the HRMS analysis reported below for each compound. Each compound was also characterized using nuclear magnetic resonance (NMR) spectroscopy. A Bruker (Billerica, MA) DRX-600 spectrometer was used to obtain NMR spectra, with shifts given in parts per million (ppm, δ) relative to residual solvent peaks (CD3OD, 1H: 3.31 ppm; CDCl3, 1H: 7.26 ppm). Data from 1H NMR spectra are provided in the form of: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, app = apparent), coupling constant, and integration.
4-(3-Methyl-4-(2-methylimidazo[1,2-a]pyrazin-3-yl)phenoxy)-furo[3,2-c]pyridine (4).
To an oven dried microwavable tube were added a stir bar, tetrakis(triphenylphosphine)palladium(0) (30 mg, 0.026 mmol), potassium carbonate (36 mg, 0.26 mmol), 3-bromo-2-methylimidazo[1,2-a]pyrazine (30 mg, 0.14 mmol), and intermediate 4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)-furo[3,2-c]pyridine (3) (46 mg, 0.14 mmol) in dioxane (1 mL), and water (0.2 mL). The mixture was subjected to microwave heating for 25 min as described in the General Chemistry Procedures section. The resulting mixture was filtered through Celite. The filter was then washed with ethyl acetate. The organic filtrate was then diluted with water and extracted three times with ethyl acetate. The combined organic layers were then washed with brine and saturated NaHCO3, dried over anhydrous MgSO4, and concentrated in vacuo. The resulting residue was taken up in 2 mL of methanol and purified by HPLC under the conditions described above to give compound 4 as a beige residue (20.3 mg, 44%). 1H NMR (600 MHz, CD3OD): δ 9.37 (d, J = 1.3 Hz, 1H), 8.33−8.28 (m, 2H), 8.01 (d, J = 5.9 Hz, 1H), 7.96 (d, J = 2.3 Hz, 1H), 7.52 (d, J = 8.3 Hz, 1H), 7.44 (dd, J = 5.9, 1.0 Hz, 1H), 7.41 (d, J = 2.5 Hz, 1H), 7.31 (dd, J = 8.3, 2.5 Hz, 1H), 7.06−7.04 (m, 1H), 2.57 (s, 3H), 2.17 (s, 3H). HPLC 99% pure, tR = 4.471 min. MS (ESI) m/z: 357.2 [M + H]+.
4-(4-(Imidazo[1,2-a]pyridin-5-yl)-3-methylphenoxy)furo[3,2-c]-pyridine (5).
Compound 5 was prepared as a light yellow oil using the same procedure as preparing 4 starting with compound 3 and 5-bromoimidazo[1,2-a]pyridine, yield 94%. 1H NMR (600 MHz, CD3OD): δ 8.15−8.08 (m, 2H), 8.07−7.98 (m, 2H), 7.95 (d, J = 2.2 Hz, 1H), 7.87 (d, J = 2.2 Hz, 1H), 7.54 (dd, J = 23.7, 7.7 Hz, 2H), 7.43 (d, J = 5.9 Hz, 1H), 7.38 (d, J = 2.3 Hz, 1H), 7.31 (dd, J = 8.3, 2.4 Hz, 1H), 7.08−7.05 (m, 1H), 2.18 (s, 3H). HPLC 99% pure, tR = 4.079 min. HRMS m/z: [M + H]+ calculated for C21 H16 N3 O2+, 342.1237; found, 342.1259.
Methyl-3-(4-(furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-imidazo[1,2-a]pyridine-8-carboxylate (6).
Compound 6 was prepared using the same procedure as preparing 4 starting with compound 3 and methyl 3-bromoimidazo[1,2-a]pyridine-8-carboxylate. The title compound was obtained as a beige residue, yield 60%. 1H NMR (600 MHz, CD3OD): δ 8.26 (dd, J = 6.8, 1.3 Hz, 1H), 8.13 (dd, J = 7.1, 1.3 Hz, 1H), 8.00 (d, J = 5.9 Hz, 1H), 7.93 (d, J = 2.3 Hz, 1H), 7.74 (s, 1H), 7.48 (d, J = 8.3 Hz, 1H), 7.41 (dt, J = 5.8, 0.6 Hz, 1H), 7.31 (d, J = 2.5 Hz, 1H), 7.22 (dd, J = 8.3, 2.5 Hz, 1H), 7.10 (t, J = 7.0 Hz, 1H), 7.01−7.00 (m, 1H), 4.05 (s, 3H), 2.16 (s, 3H). HPLC 99% pure, tR = 4.210 min. MS (ESI) m/z: 400.5 [M + H]+.
6-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-5-methylpyrazin-2-amine (7).
Compound 7 was prepared using the same procedure as preparing 4 starting with compound 3 and 6-bromo-5-methylpyr- azin-2-amine. The title compound was obtained as a tan oil, yield 52%. 1H NMR (600 MHz, CDCl3): δ 8.39 (s, 1H), 8.27 (d, J = 6.3 Hz, 1H), 7.67 (d, J = 2.3 Hz, 1H), 7.47−7.44 (m, 1H), 7.30 (d, J = 8.3 Hz, 1H), 7.27−7.26 (m, 1H), 7.21 (dd, J = 8.4, 2.5 Hz, 1H), 6.54−6.38 (m, 1H), 2.35 (s, 3H), 2.22 (s, 3H). HPLC 99% pure, tR = 4.394 min. HRMS m/z: [M + H]+ calculated for C19 H17 N4 O2+, 333.1346; found, 333.1356.
4-(4-(4-Ethoxy-6-methylpyrimidin-5-yl)-3-methylphenoxy)furo-[3,2-c]pyridine (8).
Compound 8 was prepared as a clear oil using the same procedure as preparing 4 starting with compound 3 and 5-bromo-4-ethoxy-6-methylpyrimidine, yield 66%. 1H NMR (600 MHz, CD3OD): δ 9.09−9.07 (m, 1H), 8.02 (d, J = 5.9 Hz, 1H), 7.93 (d, J = 2.3 Hz, 1H), 7.44 (dd, J = 6.0, 1.0 Hz, 1H), 7.27−7.26 (m, 1H), 7.25 (s, 1H), 7.19 (dd, J = 8.3, 2.5 Hz, 1H), 6.91−6.88 (m, 1H), 4.67 (ddq, J = 52.6, 10.8, 7.0 Hz, 2H), 2.43 (s, 3H), 2.15 (s, 3H), 1.38 (t, J = 7.1 Hz, 3H). HPLC 99% pure, tR = 4.946 min. MS (ESI) m/z: 362.2 [M + H]+.
4-(4-(3,5-Dimethylpyridazin-4-yl)-3-methylphenoxy)furo[3,2-c]-pyridine (9).
Compound 9 was prepared using the same procedure as preparing 4 starting with compound 3 and 4-chloro-3,5-dimethylpyridazine. The title compound was obtained as a golden oil, yield 55%. 1H NMR (600 MHz, CD3OD): δ 9.04 (s, 1H), 7.99 (d, J = 5.9 Hz, 1H), 7.90 (d, J = 2.2 Hz, 1H), 7.41−7.38 (m, 1H), 7.26 (d, J = 2.3 Hz, 1H), 7.19 (dd, J = 8.2, 2.4 Hz, 1H), 7.16 (d, J = 8.3 Hz, 1H), 6.95−6.93 (m, 1H), 2.41 (s, 3H), 2.15 (s, 3H), 2.03 (s, 3H). HPLC 99% pure, tR = 4.381 min. MS (ESI) m/z: 332.2 [M + H]+.
5-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)-6-methylpyridin-3-amine (10).
Compound 10 was prepared as a tan oil using the same procedure as preparing 4 starting with compound 3 and 5-bromo-6-methylpyridin-3-amine, yield 59%. 1H NMR (600 MHz, CD3OD): δ 7.99 (d, J = 5.8 Hz, 1H), 7.92 (dd, J = 4.2, 2.5 Hz, 2H), 7.55 (d, J = 2.7 Hz, 1H), 7.41 (dd, J = 5.9, 1.0 Hz, 1H), 7.30−7.23 (m, 2H), 7.18 (dd, J = 8.3, 2.5 Hz, 1H), 6.97 (q, J = 1.2 Hz, 1H), 2.37 (s, 3H), 2.17 (s, 3H). HPLC 99% pure, tR = 4.117 min. MS (ESI) m/z: 332.1 [M + H]+.
4-(4-(6-Methoxy-2-methylpyridin-3-yl)-3-methylphenoxy)furo-[3,2-c]pyridine (11).
Compound 11 was prepared as a clear oil using the same procedure as preparing 4 starting with compound 3 and 3-bromo-6-methoxy-2-methylpyridine, yield 44%. 1H NMR (600 MHz, CD3OD): δ 7.98 (d, J = 5.9 Hz, 1H), 7.89 (d, J = 2.2 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.38 (dd, J = 5.9, 1.0 Hz, 1H), 7.18−7.15 (m, 2H), 7.08 (dd, J = 8.2, 2.5 Hz, 1H), 6.89 (dd, J = 2.5, 0.9 Hz, 1H), 6.72 (d, J = 8.3 Hz, 1H), 3.96 (s, 3H), 2.26 (s, 3H), 2.10 (s, 3H). HPLC 99% pure, tR = 5.393 min. MS (ESI) m/z: 347.2 [M + H]+.
5-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)nicotinamide (12).
Compound 12 was prepared as a clear oil using the same procedure as preparing 4 starting with compound 3 and 5-bromonicotinamide, yield 34%. 1H NMR (600 MHz, CD3OD): δ 9.15 (d, J = 2.0 Hz, 1H), 8.89 (d, J = 2.0 Hz, 1H), 8.61 (d, J = 1.9 Hz, 1H), 8.01 (d, J = 5.9 Hz, 1H), 7.92 (d, J = 2.2 Hz, 1H), 7.43 (dd, J = 7.7, 2.5 Hz, 2H), 7.27 (d, J = 2.5 Hz, 1H), 7.21 (dd, J = 8.3, 2.5 Hz, 1H), 6.91−6.89 (m, 1H), 2.36 (s, 3H). HPLC 99% pure, tR = 4.244 min. MS (ESI) m/z: 346.2 [M + H]+.
6-(4-(Furo[3,2-c]pyridin-4-yloxy)-2-methylphenyl)nicotinonitrile (13).
Compound 13 was prepared using the same procedure as preparing 4 starting with compound 3 and 6-bromonicotinonitrile. The title compound was obtained as an off-white residue, yield 15%. 1H NMR (600 MHz, CDCl3): δ 9.00 (s, 1H), 8.05 (t, J = 7.7 Hz, 2H), 7.68 (s, 1H), 7.61 (d, J = 8.2 Hz, 1H), 7.52 (d, J = 8.1 Hz, 1H), 7.26 (d, J = 6.0 Hz, 1H), 7.20 (d, J = 9.4 Hz, 2H), 6.94 (s, 1H), 2.45 (s, 3H). HPLC 99% pure, tR = 328.1 min. MS (ESI) m/z: 4.925 [M + H]+.
4-(2-Methyl-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)phenoxy)-furo[3,2-c]pyridine (14).
Starting from 4-chlorofuro[3,2-c]pyridine (123 mg, 0.80 mmol) and 4-bromo-2-methylphenol (150 mg, 0.80 mmol), the intermediate 4-(4-bromo-2-methylphenoxy)furo[3,2-c]-pyridine was prepared (80 mg, 0.26 mmol, 33%) in an analogous fashion with identical reaction conditions as compound 2, described by Gray et al.42 The entirety of 4-(4-bromo-2-methylphenoxy)furo[3,2-c]pyridine was then converted to the boronic ester intermediate 4-(2-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo-[3,2-c]pyridine (63 mg, 0.18 mmol, 68%) with the same conditions described by Gray, et al. for the synthesis of compound 3. Compound 14 was prepared using the same procedure as preparing compound 4 starting with 4-(2-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine (30 mg, 0.085 mmol). The title compound was obtained as a beige residue, yield 79%. 1H NMR (600 MHz, CD3OD): δ 9.31 (s, 1H), 8.14 (d, J = 1.7 Hz, 1H), 8.01 (d, J = 1.7 Hz, 1H), 7.98−7.96 (m, 2H), 7.61 (d, J = 2.1 Hz, 1H), 7.52 (dd, J = 8.2, 2.2 Hz, 1H), 7.46 (d, J = 8.2 Hz, 1H), 7.43−7.41 (m, 1H), 7.13−7.11 (m, 1H), 2.58 (s, 3H), 2.31 (s, 3H). HPLC 99% pure, tR = 5.052 min. MS (ESI) m/z: 357.4 [M + H]+.
4-(2,3-Dimethyl-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)-phenoxy)furo[3,2-c]pyridine (15).
Starting from 4-chlorofuro[3,2-c]pyridine and 4-bromo-2,3-dimethylphenol, compound 15 was prepared as a white solid according to the same procedures and synthetic route as 14. The title compound was obtained as a brown residue, yield 99%. 1H NMR (600 MHz, CD3OD): δ 9.38 (s, 1H), 8.18 (t, J = 1.4 Hz, 1H), 7.98−7.94 (m, 2H), 7.90 (d, J = 1.7 Hz, 1H), 7.41 (dd, J = 6.0, 1.0 Hz, 1H), 7.35 (d, J = 8.3 Hz, 1H), 7.29 (d, J = 8.3 Hz, 1H), 7.12−7.10 (m, 1H), 2.51 (s, 3H), 2.26 (s, 3H), 2.10 (s, 3H). HPLC 99% pure, tR = 4.515 min. MS (ESI) m/z: 371.3 [M + H]+.
5-(Furo[3,2-c]pyridin-4-yloxy)-2-(6-methylimidazo[1,2-a]-pyrazin-5-yl)aniline (16).
Starting from 4-chlorofuro[3,2-c]pyridine and 3-amino-4-bromophenol, compound 16 was prepared as a white solid according to the same procedures and synthetic route as 14. The title compound was obtained as a beige residue, yield 43%. 1H NMR (600 MHz, CD3OD): δ 9.37 (s, 1H), 8.21 (s, 1H), 8.03 (d, J = 5.9 Hz, 1H), 7.99 (d, J = 1.8 Hz, 1H), 7.95 (d, J = 2.2 Hz, 1H), 7.44 (dd, J = 6.0, 1.0 Hz, 1H), 7.28 (d, J = 8.4 Hz, 1H), 7.02 (d, J = 2.2 Hz, 1H), 6.80 (d, J = 2.3 Hz, 1H), 6.67 (dd, J = 8.4, 2.3 Hz, 1H), 2.61 (s, 3H). HPLC 99% pure, tR = 4.081 min. HRMS m/z: [M + H]+ calculated for C20H16N5O2+, 358.1299; found, 358.1303.
4-(4-(6-Methylimidazo[1,2-a]pyrazin-5-yl)-3-(trifluoromethyl)-phenoxy)furo[3,2-c]pyridine (17).
Starting from 4-chlorofuro[3,2-c]pyridine and 4-bromo-3-(trifluoromethyl)phenol, compound 17 was prepared as a white solid according to the same procedures and synthetic route as 14. The title compound was obtained as a clear oil, yield 42%. 1H NMR (600 MHz, CD3OD): δ 9.31 (t, J = 1.4 Hz, 1H), 8.07 (t, J = 1.6 Hz, 1H), 8.05 (d, J = 5.8 Hz, 1H), 8.00−7.97 (m, 2H), 7.85−7.81 (m, 1H), 7.76−7.73 (m, 2H), 7.52 (d, J = 7.5 Hz, 1H), 7.15 (q, J = 1.2 Hz, 1H), 2.43 (s, 3H). HPLC 99% pure, tR = 4.514 min. HRMS m/z: [M+ H]+ calculated for C21H14F3N4O2+, 411.1063; found, 411.1065.
4-(4-(6-Methylimidazo[1,2-a]pyrazin-5-yl)-2-(trifluoromethyl)-phenoxy)furo[3,2-c]pyridine (18).
Starting from 4-chlorofuro[3,2-c]pyridine and 4-bromo-2-(trifluoromethyl)phenol, compound 18 was prepared as a white solid according to the same procedures and synthetic route as 14. The title compound was obtained as a clear oil, yield 85%. 1H NMR (600 MHz, CD3OD): δ 9.28 (d, J = 1.7 Hz, 1H), 8.43 (d, J = 2.3 Hz, 1H), 8.28 (d, J = 2.1 Hz, 1H), 8.22 (d, J = 2.3 Hz, 1H), 8.10 (dd, J = 8.5, 2.2 Hz, 1H), 8.07 (dd, J = 2.9, 1.5 Hz, 1H), 7.96 (d, J = 8.4 Hz, 1H), 7.86 (s, 1H), 7.81 (d, J = 8.9 Hz, 1H), 7.08 (d, J = 7.9 Hz, 1H), 2.53 (s, 3H). HPLC 99% pure, tR = 3.646 min. MS (ESI) m/z: 411.2 [M + H]+.
4-(2,6-Difluoro-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)-phenoxy)furo[3,2-c]pyridine (19).
Starting from 4-chlorofuro[3,2-c]pyridine and 4-bromo-2,6-difluorophenol, compound 19 was prepared as a white solid according to the same procedures and synthetic route as 14. The title compound was obtained as a tan residue, yield 59%. 1H NMR (600 MHz, CD3OD): δ 9.28 (s, 1H), 8.09 (d, J = 1.4 Hz, 1H), 8.00 (d, J = 2.2 Hz, 1H), 7.98 (d, J = 1.6 Hz, 1H), 7.67−7.65 (m, 1H), 7.59−7.56 (m, 1H), 7.52 (d, J = 7.5 Hz, 1H), 7.48−7.46 (m, 1H), 7.20−7.18 (m, 1H), 2.57 (s, 3H). HPLC 99% pure, tR = 4.624 min. MS (ESI) m/z: 379.3 [M + H]+.
N-(3-Methyl-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)phenyl)furo-[3,2-c]pyridin-4-amine (20).
Starting from 4-chlorofuro[3,2-c]-pyridine (1.66 g, 10.8 mmol) and 4-bromo-3-methylaniline (2.0 g, 10.8 mmol), the intermediate N-(4-bromo-3-methylphenyl)furo[3,2-c]pyridin-4-amine was prepared (851 mg, 2.81 mmol, 26%) in an analogous fashion with identical reaction conditions as compound 2, described by Gray et al. N-(4-bromo-3-methylphenyl)furo[3,2-c]-pyridin-4-amine (97 mg, 0.32 mmol) was then converted to the boronic ester intermediate N-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)furo[3,2-c]pyridin-4-amine (106 mg, 0.30 mmol, 94%) with the same conditions described by Gray, et al. for the synthesis of compound 3. Compound 20 was prepared using the same procedure as preparing compound 4 starting with N-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenyl)furo[3,2-c]-pyridin-4-amine (52 mg, 0.148 mmol). The title compound was obtained as a light yellow oil, yield 99%. 1H NMR (600 MHz, CD3OD): δ 9.25 (d, J = 2.7 Hz, 1H), 8.17−8.15 (m, 1H), 8.04−8.02 (m, 1H), 7.89 (d, J = 6.9 Hz, 1H), 7.71 (s, 1H), 7.63−7.61 (m, 2H), 7.52 (s, 1H), 7.48 (d, J = 7.1 Hz, 1H), 7.34 (d, J = 2.4 Hz, 1H), 2.46 (d, J = 2.4 Hz, 3H), 2.18 (d, J = 2.5 Hz, 3H). HPLC 99% pure, tR = 4.159 min. MS (ESI) m/z: 356.2 [M + H]+.
N-Methyl-N-(3-methyl-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)-phenyl)furo[3,2-c]pyridin-4-amine (21).
To an oven-dried flask was added a stir bar, compound 20 (20 mg, 0.056 mmol), sodium hydride (2.0 mg, 0.084 mmol), and anhydrous N,N-dimethylformamide (0.5 mL). The reaction was allowed to stir under N2 for 1 h. The mixture was then cooled to 0 °C and methyl iodide (8.8 mg, 0.062 mmol) was added dropwise. The reaction was then stirred for 2 h, allowing the temperature to slowly rise to room temperature. The reaction was quenched with saturated sodium bicarbonate, extracted three times with ethyl acetate, dried over anhydrous sodium sulfate, concentrated in vacuo, and purified by HPLC to give compound 21 as a clear oil. (7.8 mg, 43%). 1H NMR (600 MHz, CD3OD): δ 9.16 (s, 1H), 8.01 (d, J = 7.0 Hz, 1H), 7.94 (d, J = 2.3 Hz, 2H), 7.74 (d, J = 2.2 Hz, 1H), 7.67 (d, J = 8.1 Hz, 1H), 7.64 (dd, J = 8.1, 2.2 Hz, 1H), 7.50 (s, 1H), 7.48−7.46 (m, 1H), 5.89−5.86 (m, 1H), 3.86 (s, 3H), 2.44 (s, 3H), 2.16 (s, 3H). HPLC 99% pure, tR = 3.481 min. MS (ESI) m/z: 370.3 [M + H]+.
N-Ethyl-N-(3-methyl-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)-phenyl)furo[3,2-c]pyridin-4-amine (22).
Using the same procedure as 21, an oven-dried flask was charged with a stir bar, compound 20, sodium hydride, and anhydrous N,N-dimethylformamide. The reaction was allowed to stir under N2 for 1 h. The mixture was then cooled to 0 °C and ethyl iodide was added dropwise. The reaction was then stirred for 2 h, allowing the temperature to slowly rise to room temperature. The reaction was quenched with saturated sodium bicarbonate, extracted three times with ethyl acetate, dried over anhydrous sodium sulfate, concentrated in vacuo, and purified by HPLC to give compound 22 as a clear oil. (Yield: 29%). 1H NMR (600 MHz, CD3OD): δ 9.03 (s, 1H), 8.13−8.10 (m, 1H), 7.83 (s, 1H), 7.61−7.58 (m, 1H), 7.43−7.38 (m, 2H), 7.36 (s, 1H), 7.29 (d, J = 8.2 Hz, 1H), 7.11 (d, J = 5.9 Hz, 1H), 5.74 (s, 1H), 4.27−4.21 (m, 2H), 2.41 (dd, J = 3.5, 1.7 Hz, 3H), 2.05 (d, J = 2.8 Hz, 3H), 1.36 (ddd, J = 7.0, 4.5, 2.7 Hz, 3H). HPLC 99% pure, tR = 4.160 min. MS (ESI) m/z: 384.2 [M + H]+.
N-Isopropyl-N-(3-methyl-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)phenyl)furo[3,2-c]pyridin-4-amine (23).
Using the same procedure as 21, an oven-dried flask was charged with a stir bar, compound 20, sodium hydride, and anhydrous N,N-dimethylformamide. The reaction was allowed to stir under N2 for 1 h. The mixture was then cooled to 0 °C and isopropyl iodide was added dropwise. The reaction was then stirred for 2 h, allowing the temperature to slowly rise to room temperature. The reaction was quenched with saturated sodium bicarbonate, extracted three times with ethyl acetate, dried over anhydrous sodium sulfate, concentrated in vacuo, and purified by HPLC to give compound 23 as a light yellow oil. (Yield: 36%). 1H NMR (600 MHz, CD3OD): δ 9.06 (s, 1H), 8.06 (dd, J = 6.6, 2.2 Hz, 1H), 7.85 (s, 1H), 7.51 (d, J = 8.1 Hz, 1H), 7.48 (t, J = 2.6 Hz, 1H), 7.40 (s, 1H), 7.35 (s, 2H), 7.00 (d, J = 5.9 Hz, 1H), 5.33 (dd, J = 13.4, 6.8 Hz, 1H), 5.29 (d, J = 3.0 Hz, 1H), 2.42 (d, J = 2.7 Hz, 3H), 2.09 (d, J = 2.8 Hz, 3H), 1.33 (d, J = 2.2 Hz, 3H), 1.31 (s, 3H). HPLC 99% pure, tR = 4.173 min. MS (ESI) m/z: 398.2 [M + H]+.
N-(Cyclopropylmethyl)-N-(3-methyl-4-(6-methylimidazo[1,2-a]-pyrazin-5-yl)phenyl)furo[3,2-c]pyridin-4-amine (24).
Using the same procedure as 21, an oven-dried flask was charged with a stir bar, compound 20, sodium hydride, and anhydrous N,N-dimethylformamide. The reaction was allowed to stir under N2 for 1 h. The mixture was then cooled to 0 °C and (iodomethyl)cyclopropane was added dropwise. The reaction was then stirred for 2 h, allowing the temperature to slowly rise to room temperature. The reaction was quenched with saturated sodium bicarbonate, extracted three times with ethyl acetate, dried over anhydrous sodium sulfate, concentrated in vacuo, and purified by HPLC to give compound 24 as a light yellow oil. (Yield: 35%). 1H NMR (600 MHz, CD3OD): δ 9.03 (s, 1H), 8.11 (dd, J = 5.5, 1.9 Hz, 1H), 7.83 (s, 1H), 7.60−7.57 (m, 1H), 7.43−7.38 (m, 2H), 7.38−7.32 (m, 2H), 7.10 (d, J = 5.9 Hz, 1H), 5.74 (s, 1H), 4.10−4.04 (m, 2H), 2.40 (d, J = 1.9 Hz, 3H), 2.07−2.03 (m, 3H), 1.31 (s, 1H), 0.52−0.47 (m, 2H), 0.22 (d, J = 4.9 Hz, 2H). HPLC 99% pure, tR = 4.217 min. MS (ESI) m/z: 410.2 [M + H]+.
N-Cyclopentyl-N-(3-methyl-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)phenyl)furo[3,2-c]pyridin-4-amine (25).
Using the same procedure as 21, an oven-dried flask was charged with a stir bar, compound 20, sodium hydride, and anhydrous N,N-dimethylformamide. The reaction was allowed to stir under N2 for 1 h. The mixture was then cooled to 0 °C and iodocyclopentane was added dropwise. The reaction was then stirred for 2 h, allowing the temperature to slowly rise to room temperature. The reaction was quenched with saturated sodium bicarbonate, extracted three times with ethyl acetate, dried over anhydrous sodium sulfate, concentrated in vacuo, and purified by HPLC to give compound 25 as a light yellow oil. (Yield: 17%). 1H NMR (600 MHz, CD3OD): δ 9.05 (s, 1H), 8.09−8.06 (m, 1H), 7.84 (s, 1H), 7.50 (s, 2H), 7.38 (s, 1H), 7.33 (d, J = 10.1 Hz, 2H), 7.03 (s, 1H), 5.36 (s, 1H), 2.42−2.40 (m, 3H), 2.08 (d, J = 2.5 Hz, 3H), 1.71 (s, 5H), 1.63 (d, J = 12.0 Hz, 4H). HPLC 99% pure, tR = 4.458 min. MS (ESI) m/z: 424.2 [M + H]+.
3-Cyclopropyl-4-(3-methyl-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)phenoxy)furo[3,2-c]pyridine (26).
To a flame dried microwavable vessel were added a stir bar, tetrakis(triphenylphosphine)palladium(0) (81 mg, 0.07 mmol), potassium carbonate (99 mg, 0.72 mmol), 3-bromo-4-chlorofuro[3,2-c]pyridine (100 mg, 0.43 mmol), and cyclopropylboronic acid (31 mg, 0.36 mmol) in dioxane (2 mL) and water (0.3 mL). The mixture was subjected to microwave heating for 25 minutes as described in the General Chemistry Procedures section. The resulting mixture was filtered through Celite. The filter was then washed with ethyl acetate. The organic filtrate was then diluted with water and extracted three times with ethyl acetate. The combined organic layers were then washed with brine and saturated NaHCO3, dried over anhydrous MgSO4, and concentrated in vacuo. The resulting crude product, 4-chloro-3-cyclopropylfuro[3,2-c]pyridine (177 mg, 0.91 mmol), was obtained as a brown residue and used directly in the next step. The intermediate 4-(4-bromo-3-methylphenoxy)-3-cyclopropylfuro[3,2-c]pyridine (28 mg, 0.08 mmol) was prepared using 4-bromo-3-methylphenol (94 mg, 0.50 mmol) and the crude 4-chloro-3-cyclopropylfuro[3,2-c]pyridine (88 mg, 0.45 mmol) obtained from the previous step in an analogous fashion with identical reaction conditions as compound 2, described by Gray et al. 4-(4-bromo-3-methylphenoxy)-3-cyclopropylfuro[3,2-c]pyridine (28 mg, 0.08 mmol) was then converted to the boronic ester intermediate 3-cyclopropyl-4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)-furo[3,2-c]pyridine (11 mg, 0.03 mmol, 35%) with the same conditions described by Gray, et al. for the synthesis of compound 3. Compound 26 was prepared using the same procedure as preparing compound 4 starting with 3-cyclopropyl-4-(3-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo[3,2-c]pyridine (11 mg, 0.03 mmol). The title compound was obtained as a clear oil, yield 58%. 1H NMR (600 MHz, CD3OD): δ 9.23 (s, 1H), 8.05−7.96 (m, 2H), 7.70 (s, 1H), 7.62 (s, 1H), 7.48 (d, J = 8.5 Hz, 1H), 7.42−7.29 (m, 3H), 2.46 (d, J = 2.3 Hz, 3H), 2.11 (d, J = 3.1 Hz, 3H), 1.01 (d, J = 8.2 Hz, 2H), 0.93 (s, 1H), 0.79 (s, 2H). HPLC 99% pure, tR = 4.827 min. MS (ESI) m/z: 397.2 [M + H]+.
5-(4-(Benzofuran-4-yloxy)-2-methylphenyl)-6-methylimidazo-[1,2-a]pyrazine (27).
Starting from 4-chlorobenzofuran and 4-bromo-3-methylphenol, compound 27 was prepared as a white solid according to the same procedures and synthetic route as 26. The title compound was obtained as a beige residue, yield 69%. 1H NMR (600 MHz, CDCl3): δ 9.49 (s, 1H), 7.95 (s, 1H), 7.65 (d, J = 2.1 Hz, 1H), 7.44 (d, J = 8.3 Hz, 1H), 7.35 (t, J = 8.2 Hz, 1H), 7.24 (d, J = 7.6 Hz, 2H), 7.13 (d, J = 2.5 Hz, 1H), 7.09−7.05 (m, 1H), 7.00 (d, J = 7.9 Hz, 1H), 6.73 (s, 1H), 2.47 (s, 3H), 2.03 (s, 3H). HPLC 99% pure, tR = 4.860 min. MS (ESI) m/z: 356.1 [M + H]+.
1-(3-Methyl-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)phenoxy)-isoquinoline (28).
Starting from 1-chloroisoquinoline and 4-bromo-3-methylphenol, compound 28 was prepared as a white solid according to the same procedures and synthetic route as 26. The title compound was obtained as a yellow oil, yield 95%. 1H NMR (600 MHz, CD3OD): δ 9.30 (s, 1H), 8.50 (d, J = 8.4 Hz, 1H), 8.11 (s, 1H), 7.98 (d, J = 8.0 Hz, 1H), 7.95 (d, J = 5.4 Hz, 1H), 7.88 (t, J = 7.6 Hz, 1H), 7.81 (s, 1H), 7.79−7.75 (m, 1H), 7.56 (d, J = 6.1 Hz, 1H), 7.52 (d, J = 7.8 Hz, 1H), 7.47 (s, 1H), 7.39 (d, J = 8.2 Hz, 1H), 2.50 (d, J = 2.2 Hz, 3H), 2.14 (s, 3H). HPLC 99% pure, tR = 4.813 min. MS (ESI) m/z: 367.2 [M + H]+.
7-(3-Methyl-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)phenoxy)-thieno[2,3-c]pyridine (29).
Starting from 7-chlorothieno[2,3-c]-pyridine and 4-bromo-3-methylphenol, compound 29 was prepared as a white solid according to the same procedures and synthetic route as 26. The title compound was obtained as a clear oil, yield 99%. 1H NMR (600 MHz, CD3OD): δ 9.33 (s, 1H), 8.13 (d, J = 2.1 Hz, 1H), 8.07− 8.00 (m, 2H), 7.79 (s, 1H), 7.66 (dd, J = 5.6, 3.1 Hz, 1H), 7.59 (dd, J = 6.0, 2.4 Hz, 1H), 7.51 (dd, J = 8.4, 2.6 Hz, 1H), 7.45 (d, J = 2.8 Hz, 1H), 7.37 (d, J = 8.0 Hz, 1H), 2.50 (d, J = 2.8 Hz, 3H), 2.14 (d, J = 2.8 Hz, 3H). HPLC 99% pure, tR = 4.679 min. MS (ESI) m/z: 373.1 [M + H]+.
6-Methyl-5-(2-methyl-4-(pyrrolo[1,2-a]pyrazin-1-yloxy)phenyl)-imidazo[1,2-a]pyrazine (30).
Starting from 1-chloropyrrolo[1,2-a]-pyrazine and 4-bromo-3-methylphenol, compound 30 was prepared as a white solid according to the same procedures and synthetic route as 26. The title compound was obtained as a light brown residue, yield 99%. 1H NMR (600 MHz, CDCl3): δ 9.58 (d, J = 2.6 Hz, 1H), 8.01 (s, 1H), 7.73−7.67 (m, 1H), 7.57−7.55 (m, 1H), 7.53 (d, J = 7.6 Hz, 1H), 7.45 (s, 1H), 7.39 (dd, J = 11.1, 6.5 Hz, 2H), 7.16 (dd, J = 5.0, 2.5 Hz, 1H), 7.10 (s, 1H), 6.94 (d, J = 3.0 Hz, 1H), 2.51 (d, J = 2.7 Hz, 3H), 2.11 (d, J = 2.7 Hz, 3H). HPLC 99% pure, tR = 4.512 min. MS (ESI) m/z: 356.2 [M + H]+.
5-(4-(Imidazo[1,2-a]pyrazin-8-yloxy)-2-methylphenyl)-6-methylimidazo[1,2-a]pyrazine (31).
Starting from 8-chloroimidazo-[1,2-a]pyrazine and 4-bromo-3-methylphenol, compound 31 was prepared as a white solid according to the same procedures and synthetic route as 26. The title compound was obtained as a light brown residue, yield 96%. 1H NMR (600 MHz, CDCl3): δ 9.57 (d, J = 2.7 Hz, 1H), 8.05−7.96 (m, 3H), 7.85 (dd, J = 2.9, 1.4 Hz, 1H), 7.59 (dd, J = 4.6, 2.6 Hz, 1H), 7.55−7.49 (m, 2H), 7.43−7.39 (m, 1H), 7.33 (s, 1H), 2.52 (d, J = 2.7 Hz, 3H), 2.13 (d, J = 2.7 Hz, 3H). HPLC 99% pure, tR = 4.202 min. MS (ESI) m/z: 357.1 [M + H]+.
4-(3-Methyl-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)phenoxy)-1H-pyrazolo[4,3-c]pyridine (32).
Starting from 4-chloro-1H-pyrazolo-[4,3-c]pyridine and 4-bromo-3-methylphenol, compound 32 was prepared as a white solid according to the same procedures and synthetic route as 26. The title compound was obtained as a clear oil, yield 10%. 1H NMR (600 MHz, CD3OD): δ 9.21 (s, 1H), 8.00 (s, 1H), 7.96 (s, 1H), 7.71 (d, J = 6.4 Hz, 1H), 7.49 (d, J = 8.1 Hz, 1H), 7.44 (s, 1H), 7.36 (d, J = 8.4 Hz, 1H), 7.06 (s, 1H), 7.01 (s, 1H), 6.92 (s, 1H), 2.47−2.45 (m, 3H), 2.12 (s, 3H). HPLC 99% pure, tR = 4.527 min. MS (ESI) m/z: 356.2 [M + H]+.
6-Methyl-5-(2-methyl-4-(pyridin-2-yloxy)phenyl)imidazo[1,2-a]-pyrazine (33).
Starting from 2-chloropyridine and 4-bromo-3-methylphenol, compound 33 was prepared as a white solid according to the same procedures and synthetic route as 26. The title compound was obtained as a clear oil, yield 31%. 1H NMR (600 MHz, CD3OD): δ 9.21 (s, 1H), 8.21 (s, 1H), 8.01 (s, 1H), 7.94 (t, J = 7.8 Hz, 1H), 7.64 (s, 1H), 7.45 (d, J = 8.2 Hz, 1H), 7.32 (s, 1H), 7.23 (d, J = 8.8 Hz, 2H), 7.16 (d, J = 8.5 Hz, 1H), 2.44 (d, J = 3.1 Hz, 3H), 2.10 (d, J = 2.8 Hz, 3H). HPLC 99% pure, tR = 4.398 min. MS (ESI) m/z: 317.1 [M + H]+.
5-(4-((3-bromopyridin-2-yl)oxy)-2-methylphenyl)-6-methylimidazo[1,2-a]pyrazine (34).
Starting from 3-bromo-2-chlor-opyridine and 4-bromo-3-methylphenol, compound 34 was prepared as a white solid according to the same procedures and synthetic route as 26. The title compound was obtained as a beige residue, yield 23%. 1H NMR (600 MHz, CD3OD): δ 9.21 (s, 1H), 8.42 (d, J = 4.5 Hz, 1H), 8.31 (s, 1H), 8.11 (d, J = 7.4 Hz, 1H), 8.04 (s, 1H), 7.86 (s, 1H), 7.76 (s, 1H), 7.02 (s, 1H), 6.82 (d, J = 8.6 Hz, 1H), 2.54−2.51 (m, 3H), 2.35 (d, J = 3.1 Hz, 3H). HPLC 99% pure, tR = 4.736 min. MS (ESI) m/z: 395.1 [M + H]+.
3-Methyl-2-(3-methyl-4-(6-methylimidazo[1,2-a]pyrazin-5-yl)-phenoxy)pyridin-4-ol (35).
Starting from 2-chloro-3-methylpyridin-4-ol and 4-bromo-3-methylphenol, compound 35 was prepared as a white solid according to the same procedures and synthetic route as 26. The title compound was obtained as a light yellow residue, yield 88%. 1H NMR (600 MHz, CD3OD): δ 9.27 (s, 1H), 8.08−8.04 (m, 1H), 7.52 (d, J = 3.2 Hz, 1H), 7.33−7.29 (m, 1H), 6.99 (d, J = 3.2 Hz, 1H), 6.91 (ddt, J = 8.3, 6.4, 2.5 Hz, 2H), 6.77 (d, J = 3.2 Hz, 1H), 6.73−6.68 (m, 1H), 2.44−2.41 (m, 3H), 2.16 (d, J = 3.2 Hz, 3H), 2.03 (d, J = 3.2 Hz, 3H). HPLC 99% pure, tR = 4.455 min. MS (ESI) m/z: 346.2 [M + H]+.
6-Methyl-5-(2-methyl-4-((4-(piperidin-1-yl)pyridin-2-yl)oxy)-phenyl)imidazo[1,2-a]pyrazine (36).
Starting from 2-chloro-4-(piper-idin-1-yl)pyridine and 4-bromo-3-methylphenol, compound 36 was prepared as a white solid according to the same procedures and synthetic route as 26. The title compound was obtained as a light brown oil, yield 91%. 1H NMR (600 MHz, CD3OD): δ 9.20 (s, 1H), 7.97 (s, 1H), 7.92 (d, J = 7.4 Hz, 1H), 7.58 (d, J = 8.4 Hz, 1H), 7.50 (d, J = 7.3 Hz, 2H), 7.41 (d, J = 8.4 Hz, 1H), 7.01 (d, J = 7.6 Hz, 1H), 6.41 (d, J = 2.7 Hz, 1H), 3.66 (d, J = 6.1 Hz, 4H), 2.43−2.39 (m, 3H), 2.14 (s, 3H), 1.79 (s, 2H), 1.72 (s, 4H). HPLC 99% pure, tR = 4.101 min. MS (ESI) m/z: 400.2 [M + H]+.
4-(4-(Imidazo[1,2-a]pyridin-5-yl)-2-methylphenoxy)furo[3,2-c]-pyridine (37).
Compound 37 was prepared starting from 5-bromoimidazo[1,2-a]pyridine and the boronic ester intermediate 4-(2-methyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)-furo[3,2-c]pyridine that was initially prepared for the synthesis of 14. This synthesis followed the same procedure as was used for the conversion of the boronic ester intermediate 3 into compound 4. Compound 37 was obtained as a light brown residue, yield 99%. 1H NMR (600 MHz, CD3OD): δ 8.25 (d, J = 2.2 Hz, 1H), 8.12−8.08 (m, 2H), 8.00−7.94 (m, 3H), 7.75 (d, J = 2.2 Hz, 1H), 7.67 (dd, J = 8.3, 2.3 Hz, 1H), 7.56−7.53 (m, 1H), 7.44−7.40 (m, 2H), 7.11−7.09 (m, 1H), 2.32 (s, 3H). HPLC 99% pure, tR = 4.835 min. MS (ESI) m/z: 342.4 [M + H]+.
4-(4-(Imidazo[1,2-a]pyridin-5-yl)-2,3-dimethylphenoxy)furo[3,2-c]pyridine (38).
Compound 38 was prepared starting from 5-bromoimidazo[1,2-a]pyridine and the boronic ester intermediate 4-(2,3-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-phenoxy)furo[3,2-c]pyridine that was initially prepared for the synthesis of 15. This synthesis followed the same procedure as was described compound 4. Compound 38 was obtained as a light brown residue, yield 93%. 1H NMR (600 MHz, CD3OD): δ 8.14−8.09 (m, 2H), 8.04 (d, J = 9.0 Hz, 1H), 7.96 (t, J = 1.9 Hz, 1H), 7.94 (d, J = 5.9 Hz, 1H), 7.91 (d, J = 2.2 Hz, 1H), 7.53−7.51 (m, 1H), 7.40 (ddd, J = 8.2, 4.5, 3.1 Hz, 2H), 7.25 (d, J = 8.1 Hz, 1H), 7.11 (d, J = 2.7 Hz, 1H), 2.25 (s, 3H), 2.11 (s, 3H). HPLC 99% pure, tR = 4.351 min. MS (ESI) m/z: 356.3 [M + H]+.
4-(4-(Imidazo[1,2-a]pyridin-5-yl)-3,5-dimethylphenoxy)furo[3,2-c]pyridine (39).
Starting from 4-chlorofuro[3,2-c]pyridine (154 mg, 1.0 mmol) and 4-bromo-3,5-dimethylphenol (221 mg, 1.1 mmol), the intermediate 4-(4-bromo-3,5-dimethylphenoxy)furo[3,2-c]pyridine was prepared (195 mg, 0.61 mmol, 61%) in an analogous fashion with identical reaction conditions as compound 2, described by Gray et al. 4-(4-bromo-3,5-dimethylphenoxy)furo[3,2-c]pyridine (92 mg, 0.29 mmol) was then converted to the boronic ester intermediate 4-(3,5-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)-furo[3,2-c]pyridine (67 mg, 0.18 mmol, 63%) with the same conditions described by Gray, et al. for the synthesis of compound 3. Compound 39 was prepared using the same procedure as preparing compound 4 starting with 4-(3,5-dimethyl-4-(4,4,5,5-tetramethyl-1,3,2-dioxaboro-lan-2-yl)phenoxy)furo[3,2-c]pyridine and 5-bromoimidazo[1,2-a]-pyridine. The title compound was obtained as a clear oil, yield 33%. 1H NMR (600 MHz, CD3OD): δ 8.42 (q, J = 3.6, 2.9 Hz, 1H), 8.15 (ddt, J = 14.1, 4.6, 2.2 Hz, 2H), 8.10 (d, J = 8.9 Hz, 1H), 8.02−7.97 (m, 1H), 7.71 (dd, J = 6.0, 3.7 Hz, 2H), 7.57−7.53 (m, 1H), 7.42 (d, J = 4.4 Hz, 2H), 6.89 (dt, J = 9.5, 3.1 Hz, 1H), 2.13−2.10 (m, 6H). HPLC 99% pure, tR = 3.238 min. MS (ESI) m/z: 355.2 [M + H]+.
5-(Furo[3,2-c]pyridin-4-yloxy)-2-(imidazo[1,2-a]pyridin-5-yl)-aniline (40).
Compound 40 was prepared starting from 5-bromoimidazo[1,2-a]pyridine and intermediate 5-(furo[3,2-c]pyridin-4-yloxy)-2-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)aniline that was initially prepared for the synthesis of 16. This synthesis followed the same procedure as was used for the conversion of the boronic ester intermediate 3 into compound 4. Compound 40 was obtained as a brown oil, yield 29%. 1H NMR (600 MHz, CD3OD): δ 8.09−8.07 (m, 1H), 8.06 (d, J = 2.1 Hz, 1H), 8.02 (d, J = 5.9 Hz, 1H), 7.98−7.96 (m, 1H), 7.94 (d, J = 2.2 Hz, 1H), 7.91 (d, J = 2.2 Hz, 1H), 7.54 (dd, J = 7.2, 1.1 Hz, 1H), 7.43 (dd, J = 6.0, 1.0 Hz, 1H), 7.33 (d, J = 8.3 Hz, 1H), 7.00−6.99 (m, 1H), 6.77 (d, J = 2.3 Hz, 1H), 6.64 (d, J = 2.3 Hz, 1H). HPLC 99% pure, tR = 4.061 min. HRMS m/z: [M + H]+ calculated for C20H15N4O2+, 343.1190; found, 343.1195.
4-(4-(Imidazo[1,2-a]pyridin-5-yl)-3-(trifluoromethyl)phenoxy)-furo[3,2-c]pyridine (41).
Compound 41 was prepared starting from 5-bromoimidazo[1,2-a]pyridine and the intermediate 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-(trifluoromethyl)phenoxy)-furo[3,2-c]pyridine that was initially prepared for the synthesis of 17. This synthesis followed the same procedure as was used for the conversion of the boronic ester intermediate 3 into compound 4. Compound 41 was obtained as a clear oil, yield 89%. 1H NMR (600 MHz, CD3OD): δ 8.16−8.13 (m, 1H), 8.13−8.10 (m, 2H), 8.04−8.02 (m, 1H), 7.99 (d, J = 2.3 Hz, 1H), 7.95 (d, J = 2.3 Hz, 1H), 7.89 (d, J = 2.3 Hz, 1H), 7.83 (d, J = 8.3 Hz, 1H), 7.80 (dd, J = 8.4, 2.3 Hz, 1H), 7.59 (dd, J = 6.9, 1.4 Hz, 1H), 7.48 (dd, J = 5.9, 1.1 Hz, 1H), 7.15−7.14 (m, 1H). HPLC 99% pure, tR = 4.081 min. HRMS m/z: [M + H]+ calculated for C21H13F3N3O2+, 396.0954; found, 396.0971.
4-(4-(Imidazo[1,2-a]pyridin-5-yl)-2-(trifluoromethyl)phenoxy)-furo[3,2-c]pyridine (42).
Compound 42 was prepared starting from 5-bromoimidazo[1,2-a]pyridine and the intermediate 4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-2-(trifluoromethyl)phenoxy)-furo[3,2-c]pyridine that was initially prepared for the synthesis of 18. This synthesis followed the same procedure as was used for the conversion of the boronic ester intermediate 3 into compound 4. Compound 42 was obtained as a clear oil, yield 71%. 1H NMR (600 MHz, CD3OD): δ 8.42−8.37 (m, 2H), 8.23−8.20 (m, 2H), 8.15 (dq, J = 2.5, 1.3 Hz, 1H), 8.13 (s, 1H), 8.09−8.06 (m, 1H), 8.04 (ddd, J = 9.0, 7.7, 1.2 Hz, 1H), 7.94 (d, J = 8.5 Hz, 1H), 7.81 (d, J = 8.9 Hz, 1H), 7.65−7.62 (m, 1H), 7.02 (d, J = 7.9 Hz, 1H). HPLC 99% pure, tR = 3.256 min. MS (ESI) m/z: 395.1 [M + H]+.
4-(2,6-Difluoro-4-(imidazo[1,2-a]pyridin-5-yl)phenoxy)furo[3,2-c]pyridine (43).
Compound 43 was prepared starting from 5-bromoimidazo[1,2-a]pyridine and boronic ester intermediate 4-(2,6-difluoro-4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)phenoxy)furo-[3,2-c]pyridine that was initially prepared for the synthesis of 19. This synthesis followed the same procedure as was used for the conversion of the boronic ester intermediate 3 into compound 4. Compound 43 was obtained as a clear oil, yield 65%. 1H NMR (600 MHz, CD3OD): δ 8.29 (d, J = 2.3 Hz, 1H), 8.14−8.11 (m, 1H), 8.11−8.09 (m, 1H), 8.05 (d, J = 9.0 Hz, 1H), 7.99 (d, J = 2.3 Hz, 1H), 7.95 (d, J = 5.8 Hz, 1H), 7.69−7.64 (m, 2H), 7.61 (dd, J = 7.1, 1.2 Hz, 1H), 7.45 (dd, J = 5.9, 1.1 Hz, 1H), 7.18−7.17 (m, 1H). HPLC 99% pure, tR = 3.894 min. MS (ESI) m/z: 364.2 [M + H]+.
3- Cyclopropyl- 4 -(4-(imidazo[1,2 -a]pyridin-5-yl)-3-(trifluoromethyl)phenoxy)furo[3,2-c]pyridine (44).
Starting from 4-chloro-3-cyclopropylfuro[3,2-c]pyridine (88 mg, 0.45 mmol), which was previously prepared and described for compound 26, and 4-bromo-3-(trifluoromethyl)phenol (120 mg, 0.50 mmol), the intermediate 4-(4-bromo-3-(trifluoromethyl)phenoxy)-3-cyclopropylfuro[3,2-c]-pyridine was prepared (26 mg, 0.065 mmol, 13%) in an analogous fashion with identical reaction conditions as compound 2, described by Gray et al. 4 -(4-bromo-3-(trifluoromethyl)phenoxy)- 3-cyclopropylfuro[3,2-c]pyridine (26 mg, 0.065 mmol) was then converted to the boronic ester intermediate 3-cyclopropyl-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-(trifluoromethyl)-phenoxy)furo[3,2-c]pyridine (5.8 mg, 0.013 mmol, 20%) with the same conditions described by Gray, et al. for the synthesis of compound 3. Compound 44 was prepared using the same procedure as preparing compound 4 starting with 3-cyclopropyl-4-(4-(4,4,5,5-tetramethyl-1,3,2-dioxaborolan-2-yl)-3-(trifluoromethyl)phenoxy)furo[3,2-c]-pyridine. The title compound was obtained as a beige residue, yield 30%. 1H NMR (600 MHz, CD3OD): δ 9.09 (s, 1H), 8.01 (d, J = 5.9 Hz, 1H), 7.92 (s, 1H), 7.83 (s, 2H), 7.78 (d, J = 8.3 Hz, 1H), 7.70 (d, J = 8.5 Hz, 1H), 7.64 (s, 1H), 7.49 (s, 2H), 7.40 (d, J = 5.9 Hz, 1H), 1.01 (d, J = 8.2 Hz, 2H), 0.92 (s, 1H), 0.78 (s, 2H). HPLC 99% pure, tR = 4.931 min. MS (ESI) m/z: 436.1 [M + H]+.
1-(4-(Imidazo[1,2-a]pyridin-5-yl)-3-(trifluoromethyl)phenoxy)-isoquinoline (45).
Starting from 1-chloroisoquinoline and 4-bromo-3-(trifluoromethyl)phenol, compound 45 was prepared in an analogous fashion with identical conditions as compound 44. The title compound was obtained as a clear oil, yield 99%. 1H NMR (600 MHz, CDCl3): δ 8.46 (d, J = 8.6 Hz, 2H), 8.04 (dd, J = 5.9, 2.8 Hz, 1H), 7.91 (s, 2H), 7.85 (t, J = 7.6 Hz, 1H), 7.79 (d, J = 8.0 Hz, 1H), 7.75 (d, J = 7.8 Hz, 1H), 7.69 (t, J = 10.0 Hz, 1H), 7.60 (d, J = 8.2 Hz, 1H), 7.52 (d, J = 6.2 Hz, 2H), 7.33 (d, J = 6.0 Hz, 2H). HPLC 99% pure, tR = 4.237 min. MS (ESI) m/z: 406.2 [M + H]+.
7-(4-(Imidazo[1,2-a]pyridin-5-yl)-3-(trifluoromethyl)phenoxy)-thieno[2,3-c]pyridine (46).
Starting from 7-chlorothieno[2,3-c]-pyridine and 4-bromo-3-(trifluoromethyl)phenol, compound 46 was prepared in an analogous fashion with identical conditions as compound 44. The title compound was obtained as a light yellow oil, yield 97%. 1H NMR (600 MHz, CD3OD): δ 8.14−8.10 (m, 2H), 8.07 (d, J = 5.4 Hz, 1H), 8.03 (t, J = 5.0 Hz, 1H), 7.98 (t, J = 3.3 Hz, 1H), 7.91−7.87 (m, 1H), 7.85−7.81 (m, 2H), 7.70 (t, J = 5.6 Hz, 1H), 7.68−7.65 (m, 1H), 7.60 (q, J = 6.6, 5.8 Hz, 2H). HPLC 99% pure, tR = 4.234 min. HRMS m/z: [M + H]+ calculated for C21H12F3N3OS+, 412.0726; found, 412.0738.
5-(4-(Pyrrolo[1,2-a]pyrazin-1-yloxy)-2-(trifluoromethyl)phenyl)-imidazo[1,2-a]pyridine (47).
Starting from 1-chloropyrrolo[1,2-a]-pyrazine and 4-bromo-3-(trifluoromethyl)phenol, compound 47 was prepared in an analogous fashion with identical conditions as compound 44. The title compound was obtained as a beige residue, yield 95%. 1H NMR (600 MHz, CDCl3): δ 8.44 (d, J = 9.1 Hz, 1H), 7.93 (s, 3H), 7.79 (dd, J = 8.7, 2.7 Hz, 1H), 7.72 (d, J = 4.7 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.56 (d, J = 2.9 Hz, 1H), 7.34−7.29 (m, 2H), 7.12 (dd, J = 5.0, 2.8 Hz, 1H), 7.07 (d, J = 4.0 Hz, 1H), 6.96−6.92 (m, 1H). HPLC 99% pure, tR = 4.057 min. MS (ESI) m/z: 395.1 [M + H]+.
8-(4-(Imidazo[1,2-a]pyridin-5-yl)-3-(trifluoromethyl)phenoxy)-imidazo[1,2-a]pyrazine (48).
Starting from 8-chloroimidazo[1,2-a]pyrazine and 4-bromo-3-(trifluoromethyl)phenol, compound 48 was prepared in an analogous fashion with identical conditions as compound 44. The title compound was obtained as a beige residue, yield 89%. 1H NMR (600 MHz, CDCl3): δ 8.45 (d, J = 9.2 Hz, 1H), 8.05−8.01 (m, 2H), 7.96 (s, 1H), 7.94−7.91 (m, 2H), 7.89 (s, 1H), 7.87−7.85 (m, 1H), 7.63 (d, J = 8.4 Hz, 1H), 7.54 (d, J = 4.5 Hz, 1H), 7.35 (d, J = 7.2 Hz, 1H), 7.32−7.29 (m, 1H). HPLC 99% pure, tR = 3.431 min. MS (ESI) m/z: 396.1 [M + H]+.
5-(4-(Pyridin-2-yloxy)-2-(trifluoromethyl)phenyl)imidazo[1,2-a]-pyridine (49).
Starting from 2-chloropyridine and 4-bromo-3-(trifluoromethyl)phenol, compound 49 was prepared in an analogous fashion with identical conditions as compound 44. The title compound was obtained as a light brown oil, yield 79%. 1H NMR (600 MHz, CD3OD): δ 8.22 (d, J = 4.4 Hz, 1H), 8.10 (td, J = 15.3, 14.7, 8.6 Hz, 3H), 8.01−7.97 (m, 1H), 7.86−7.81 (m, 2H), 7.78 (d, J = 8.4 Hz, 1H), 7.69 (d, J = 7.3 Hz, 1H), 7.57 (d, J = 7.4 Hz, 1H), 7.26 (dd, J = 12.5, 7.9 Hz, 2H). HPLC 99% pure, tR = 4.134 min. MS (ESI) m/z: 356.1 [M + H]+.
5-(4-((4-(Piperidin-1-yl)pyridin-2-yl)oxy)-2-(trifluoromethyl)-phenyl)imidazo[1,2-a]pyridine (50).
Starting from 2-chloro-4-(piper-idin-1-yl)pyridine and 4-bromo-3-(trifluoromethyl)phenol, compound 50 was prepared in an analogous fashion with identical conditions as compound 44. The title compound was obtained as a brown residue, yield 96%. 1H NMR (600 MHz, CD3OD): δ 8.12 (d, J = 11.3 Hz, 3H), 7.95−7.90 (m, 2H), 7.85 (d, J = 8.4 Hz, 1H), 7.80 (s, 1H), 7.77 (d, J = 8.3 Hz, 1H), 7.56 (d, J = 6.8 Hz, 1H), 6.95 (d, J = 7.2 Hz, 1H), 6.56 (s, 1H), 3.63 (t, J = 5.4 Hz, 4H), 1.80−1.76 (m, 2H), 1.72 (s, 4H). HPLC 99% pure, tR = 4.249 min. MS (ESI) m/z: 439.2 [M + H]+.
Experimental Procedures for in Vitro Pharmacology Assays.
General Procedures.
Protocols of the cAMP biosensor and bioluminescent resonance energy transfer (BRET) assays are detailed below.
D1R GS-mediated GS-cAMP Accumulation Assay.
D1R GS-mediated GS-cAMP accumulation assays with HEK293T cells co-expressing human D1 and the cAMP biosensor GloSensor-22F (Promega) were performed. Cells were seeded (15 000 cells/40 μL/ well) into white 384 clear-bottom, tissue culture plates in DMEM containing 1% dialyzed fetal bovine serum (FBS). Next day, drug dilutions were diluted in HBSS, 20 mM N-(2-hydroxyethyl)piperazine-N′-ethanesulfonic acid (HEPES), 0.1% bovine serum albumin (BSA), 0.01% ascorbic acid, pH 7.4. Next, media was decanted from 384 well plates and 20 μL of drug buffer (HBSS, 20 mM HEPES, pH 7.4) containing GloSensor reagent was added per well and allowed to equilibrate for at least 15 min room temperature. To start the assay, cells were treated with 10 μL per well of 3× drug diluted in HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4 using a FLIPR (molecular devices). After 15 min, GS-cAMP accumulation was read on a TriLux Microbeta (PerkinElmer) plate counter. Data were normalized to maximum GS-cAMP accumulation by dopamine (100%). Data were analyzed using the sigmoidal dose−response function built into GraphPad Prism 5.0.
BRET β-Arrestin2 Assay.
To measure D1R-mediated β-arrestin2 recruitment, HEK293T cells were co-transfected in a 1:15 ratio with human D1 containing a C-terminal renilla luciferase (RLuc8), and Venus-tagged N-terminal β-arrestin2. Next day, transfected cells were plated in polylysine coated 96-well white clear bottom cell culture plates in plating media (DMEM + 1% dialyzed FBS) at a density of 25−40 000 cells in 200 μL per well and incubated overnight. Next day, media was decanted and cells were washed twice with 60 μL of drug buffer (1× HBSS, 20 mM HEPES, 0.1% BSA, 0.01% ascorbic acid, pH 7.4). Then, 60 μL of drug buffer, 30 μL of drug (3×), and 10 μL (10×) of RLuc substrate, coelenterazine h (Promega, 5 μM final concentration) 15 min was added per well before reading. Plates were read for both luminescence at 485 nm and fluorescent eYFP emission at 530 nm for 1 s per well using a Mithras LB940. The ratio of eYFP/RLuc was calculated per well, and the net BRET ratio was calculated by subtracting the eYFP/RLuc per well from the eYFP/RLuc ratio in wells without Venus-β-arrestin2 present. The net BRET ratio was plotted as a function of drug concentration using Graphpad Prism 5 (Graphpad Software Inc., San Diego, CA). Data were normalized to % dopamine stimulation and analyzed using nonlinear regression log(agonist) versus response.
Bias Calculation.
Transduction coefficients [log(τ/KA)] were calculated using the Black and Leff operational model in Graphpad Prism 5.0, where τ is agonist efficacy and KA is the equilibrium dissociation constant. Using dopamine as the full agonist reference, transduction coefficients for GS activity and β-arrestin2 recruitment were calculated and averaged across experiments. Calculation of bias factors utilized the method by Kenakin et al., where the Δlog(τ/KA) was calculated relative to the reference and the ΔΔlog(τ/KA) was calculated by subtracting the GS from β-arrestin2 transduction coefficient.55
Radioligand Binding Affinity Assay.
Assays were performed exactly as described in previous publications.54 Further information regarding the assay protocols can be found online in greater detail at https://pdspdb.unc.edu/pdspWeb/?site=assays.
Experimental Procedures for in Vivo Pharmacokinetic Studies.
Compound 40 was taken up in 5% NMP, 5% Solutol HS-15, and 90% normal saline for formulation. Nine male Swiss Albino mice were administered intraperitoneally with the formulated solution of compound 40 at 50 mg/kg dose. Blood samples (approximately 60 μL) were collected from a set of three mice at 0.5, 1.5, and 4 h under light isoflurane anesthesia. Plasma was harvested by centrifugation of blood and stored at −70 ± 10 °C until analysis. Brain samples were collected from a set of three mice at each time point at 0.5, 1.5 and 4 h immediately after blood collection. Brain samples were homogenized using ice-cooled phosphate buffer saline (pH 7.4) in a ratio of 2 (buffer):1 (brain). Homogenates were stored below −70 ± 10 °C until analysis. Total homogenate volume was three times the brain weight. The plasma and brain concentration−time data of compound 40 was used for the pharmacokinetic analysis. Pharmacokinetic analysis was performed using the NCA module of Phoenix WinNonlin (Version 7.0). Plasma and brain samples were quantified by fit-for-purpose LCMS−MS method (LLOQ: 1.01 ng/mL for plasma and 3.03 ng/g for brain).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by the grant R01NS100930 (to M.G.C., J.J. and N.U.) from the U.S. National Institutes of Health. J.J. and B.L.R. acknowledge the support by the grant U24DK116195 from the U.S. National Institutes of Health. M.L.M. acknowledges the support by the NIGMS-funded Integrated Pharmacological Sciences Training Program T32 GM062754 and the MSTP training grants at the Icahn School of Mount Sinai (NIH T32 GM007280). The authors acknowledge Drs. H. Ümit Kaniskan and Yudao Shen for their critical review of this manuscript and advice in experimental planning and manuscript preparation.
ABBREVIATIONS
- GPCR
G protein-coupled receptor
- SFSR
structure−functional selectivity relationship
- D1R
dopamine D1 receptor
- GS
stimulatory G protein
- cAMP
cyclic adenosine monophosphate
- RHS
right-hand side
- LHS
left-hand side
- BRET
bioluminescence resonance energy transfer
- D2R
dopamine D2 receptor
- D3R
dopamine D3 receptor
- D4R
dopamine D4 receptor
- D5R
dopamine D5 receptor
- 5-HT
5-hydroxytryptamine
- H1
histamine receptor 1
- H2
histamine receptor 2
- M1
muscarinic receptor 1
- M2
muscarinic receptor 2
- M3
muscarinic receptor 3
- MOR
μ opioid receptor
- KOR
kappa opioid receptor
- SERT
serotonin transporter
- DAT
dopamine transporter
- TFA
trifluoroacetic acid
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmed-chem.9b00351.
1H NMR spectra of compounds 40 and 41 (PDF)
Molecular formula strings for all compounds (CSV)
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Rask-Andersen M; Almén MS; Schiöth HB Trends in the exploitation of novel drug targets. Nat. Rev. Drug Discovery 2011, 10, 579–590. [DOI] [PubMed] [Google Scholar]
- (2).Wacker D; Stevens RC; Roth BL How Ligands Illuminate GPCR Molecular Pharmacology. Cell 2017, 170, 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Beaulieu J-M; Gainetdinov RR The physiology, signaling, and pharmacology of dopamine receptors. Pharmacol. Rev 2011, 63, 182–217. [DOI] [PubMed] [Google Scholar]
- (4).Spano PF; Govoni S; Trabucchi M Studies on the pharmacological properties of dopamine receptors in various areas of the central nervous system. Adv. Biochem. Psychopharmacol 1978, 19, 155–165. [PubMed] [Google Scholar]
- (5).Greengard P The neurobiology of dopamine signaling. Biosci. Rep 2001, 21, 247–269. [DOI] [PubMed] [Google Scholar]
- (6).Beaulieu J-M; Sotnikova TD; Marion S; Lefkowitz RJ; Gainetdinov RR; Caron MG An Akt/β-Arrestin 2/PP2A Signaling Complex Mediates Dopaminergic Neurotransmission and Behavior. Cell 2005, 122, 261–273. [DOI] [PubMed] [Google Scholar]
- (7).Urs NM; Daigle TL; Caron MG A Dopamine D1 Receptor-Dependent β-Arrestin Signaling Complex Potentially Regulates Morphine-Induced Psychomotor Activation but not Reward in Mice. Neuropsychopharmacology 2011, 36, 551–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Lefkowitz RJ; Shenoy SK Transduction of Receptor Signals by -Arrestins. Science 2005, 308, 512–517. [DOI] [PubMed] [Google Scholar]
- (9).Attramadal H; Arriza JL; Aoki C; Dawson TM; Codina J; Kwatra MM; Snyder SH; Caron MG; Lefkowitz RJ Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J. Biol. Chem 1992, 267, 17882–17890. [PubMed] [Google Scholar]
- (10).Luttrell LM; Roudabush FL; Choy EW; Miller WE; Field ME; Pierce KL; Lefkowitz RJ Activation and targeting of extracellular signal-regulated kinases by -arrestin scaffolds. Proc. Natl. Acad. Sci. U.S.A 2001, 98, 2449–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).DeFea KA; Zalevsky J; Thoma MS; Déry O; Mullins RD; Bunnett NW β-Arrestin-Dependent Endocytosis of Proteinase-Activated Receptor 2 Is Required for Intracellular Targeting of Activated Erk½. J. Cell Biol 2000, 148, 1267–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).McDonald PH; Chow C-W; Miller WE; Laporte SA; Field ME; Lin F-T; Davis RJ; Lefkowitz RJ beta -Arrestin 2: A Receptor-Regulated MAPK Scaffold for the Activation of JNK3. Science 2000, 290, 1574–1577. [DOI] [PubMed] [Google Scholar]
- (13).Tohgo A; Choy EW; Gesty-Palmer D; Pierce KL; Laporte S; Oakley RH; Caron MG; Lefkowitz RJ; Luttrell LM The Stability of the G Protein-coupled Receptor-β-Arrestin Interaction Determines the Mechanism and Functional Consequence of ERK Activation. J. Biol. Chem 2003, 278, 6258–6267. [DOI] [PubMed] [Google Scholar]
- (14).Imamura T; Huang J; Dalle S; Ugi S; Usui I; Luttrell LM; Miller WE; Lefkowitz RJ; Olefsky JM β-Arrestin-mediated Recruitment of the Src Family Kinase Yes Mediates Endothelin-1-stimulated Glucose Transport. J. Biol. Chem 2001, 276, 43663–43667. [DOI] [PubMed] [Google Scholar]
- (15).Barlic J; Andrews JD; Kelvin AA; Bosinger SE; DeVries ME; Xu L; Dobransky T; Feldman RD; Ferguson SSG; Kelvin DJ Regulation of tyrosine kinase activation and granule release through β-arrestin by CXCR1. Nat. Immunol 2000, 1, 227–233. [DOI] [PubMed] [Google Scholar]
- (16).Luttrell LM; Ferguson SSG; Daaka Y; Miller WE; Maudsley S; Della Rocca GJ; Lin F-T; Kawakatsu H; Owada K; Luttrell DK; Caron MG; Lefkowitz RJ β-Arrestin-Dependent Formation of β2Adrenergic Receptor-Src Protein Kinase Complexes. Science 1999, 283, 655–661. [DOI] [PubMed] [Google Scholar]
- (17).Yano H; Cai N-S; Xu M; Verma RK; Rea W; Hoffman AF; Shi L; Javitch JA; Bonci A; Ferre S Gs-versus Golf-dependent functional selectivity mediated by the dopamine D1 receptor. Nat. Commun 2018, 9, 486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Urs NM; Bido S; Peterson SM; Daigle TL; Bass CE; Gainetdinov RR; Bezard E; Caron MG Targeting β-arrestin2 in the treatment of l-DOPA-induced dyskinesia in Parkinson’s disease. Proc. Natl. Acad. Sci. U.S.A 2015, 112, E2517–E2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tan L; Yan W; McCorvy JD; Cheng J Biased ligands of G protein-coupled receptors (GPCRs): structure-functional selectivity relationships (SFSRs) and therapeutic potential. J. Med. Chem 2018, 61, 9841–9878. [DOI] [PubMed] [Google Scholar]
- (20).DeWire SM; Yamashita DS; Rominger DH; Liu G; Cowan CL; Graczyk TM; Chen X-T; Pitis PM; Gotchev D; Yuan C; Koblish M; Lark MW; Violin JD A G Protein-Biased Ligand at the -Opioid Receptor Is Potently Analgesic with Reduced Gastrointestinal and Respiratory Dysfunction Compared with Morphine. J. Pharmacol. Exp. Ther 2013, 344, 708–717. [DOI] [PubMed] [Google Scholar]
- (21).Violin JD; Crombie AL; Soergel DG; Lark MW Biased ligands at G-protein-coupled receptors: promise and progress. Trends Pharmacol. Sci 2014, 35, 308–316. [DOI] [PubMed] [Google Scholar]
- (22).Bohn LM; Lefkowitz RJ; Gainetdinov RR; Peppel K; Caron MG; Lin F-T Enhanced Morphine Analgesia in Mice Lacking -Arrestin 2 . Science 1999, 286, 2495–2498. [DOI] [PubMed] [Google Scholar]
- (23).Schmid CL; Kennedy NM; Ross NC; Lovell KM; Yue Z; Morgenweck J; Cameron MD; Bannister TD; Bohn LM Bias factor and therapeutic window correlate to predict safer opioid analgesics. Cell 2017, 171, 1165–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).White KL; Robinson JE; Zhu H; DiBerto JF; Polepally PR; Zjawiony JK; Nichols DE; Malanga CJ; Roth BL The G protein-biased kappa-opioid receptor agonist RB-64 is analgesic with a unique spectrum of activities in vivo. J. Pharmacol. Exp. Ther 2015, 352, 98–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Dogra S; Yadav PN Biased agonism at kappa opioid receptors: implication in pain and mood disorders. Eur. J. Pharmacol 2015, 763, 184–190. [DOI] [PubMed] [Google Scholar]
- (26).Raehal KM; Walker JK; Bohn LM Morphine Side Effects in -Arrestin 2 Knockout Mice. J. Pharmacol. Exp. Ther 2005, 314, 1195–1201. [DOI] [PubMed] [Google Scholar]
- (27).Manglik A; Lin H; Aryal DK; McCorvy JD; Dengler D; Corder G; Levit A; Kling RC; Bernat V; Hübner H; Huang X-P; Sassano MF; Giguère PM; Löber S; Da Duan D; Scherrer G; Kobilka BK; Gmeiner P; Roth BL; Shoichet BK Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Hill R; Disney A; Conibear A; Sutcliffe K; Dewey W; Husbands S; Bailey C; Kelly E; Henderson G The novel μ-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br. J. Pharmacol 2018, 175, 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Allen JA; Yost JM; Setola V; Chen X; Sassano MF; Chen M; Peterson S; Yadav PN; Huang X.-p.; Feng B; Jensen NH; Che X; Bai X; Frye SV; Wetsel WC; Caron MG; Javitch JA; Roth BL; Jin J Discovery of -Arrestin-Biased Dopamine D2 Ligands for Probing Signal Transduction Pathways Essential for Antipsychotic Efficacy. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 18488–18493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chen X; McCorvy JD; Fischer MG; Butler KV; Shen Y; Roth BL; Jin J Discovery of G protein-biased D2 dopamine receptor partial agonists. J. Med. Chem 2016, 59, 10601–10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Park SM; Chen M; Schmerberg CM; Dulman RS; Rodriguiz RM; Caron MG; Jin J; Wetsel WC Effects of β-Arrestin-Biased Dopamine D2 Receptor Ligands on Schizophrenia-Like Behavior in Hypoglutamatergic Mice. Neuropsychopharmacology 2016, 41, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wang S; Che T; Levit A; Shoichet BK; Wacker D; Roth BL Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Urs NM; Gee SM; Pack TF; McCorvy JD; Evron T; Snyder JC; Yang X; Rodriguiz RM; Borrelli E; Wetsel WC; Jin J; Roth BL; O’Donnell P; Caron MG Distinct cortical and striatal actions of a β-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc. Natl. Acad. Sci. U.S.A 2016, 113, E8178–E8186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).McCorvy JD; Butler KV; Kelly B; Rechsteiner K; Karpiak J; Betz RM; Kormos BL; Shoichet BK; Dror RO; Jin J; Roth BL Structure-inspired design of β-arrestin-biased ligands for aminergic GPCRs. Nat. Chem. Biol 2018, 14, 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Urban JD; Vargas GA; von Zastrow M; Mailman RB Aripiprazole has functionally selective actions at dopamine D2 receptor-mediated signaling pathways. Neuropsychopharmacology 2007, 32, 67–77. [DOI] [PubMed] [Google Scholar]
- (36).Hiller C; Kling RC; Heinemann FW; Meyer K; Hübner H; Gmeiner P Functionally selective dopamine D2/D3 receptor agonists comprising an enyne moiety. J. Med. Chem 2013, 56, 5130–5141. [DOI] [PubMed] [Google Scholar]
- (37).Möller D; Kling RC; Skultety M; Leuner K; Hübner H; Gmeiner P Functionally Selective Dopamine D2, D3 Receptor Partial Agonists. J. Med. Chem 2014, 57, 4861–4875. [DOI] [PubMed] [Google Scholar]
- (38).Möller D; Banerjee A; Uzuneser TC; Skultety M; Huth T; Plouffe B; Hübner H; Alzheimer C; Friedland K; Müller CP; Bouvier M; Gmeiner P Discovery of G protein-biased dopaminergics with a pyrazolo[1,5-a]pyridine substructure. J. Med. Chem 2017, 60, 2908–2929. [DOI] [PubMed] [Google Scholar]
- (39).Shonberg J; Herenbrink CK; Lpez L; Christopoulos A; Scammells PJ; Capuano B; Lane JR A structure-activity analysis of biased agonism at the dopamine D2 receptor. J. Med. Chem 2013, 56, 9199–9221. [DOI] [PubMed] [Google Scholar]
- (40).Bonifazi A; Yano H; Ellenberger MP; Muller L; Kumar V; Zou M-F; Cai NS; Guerrero AM; Woods AS; Shi L; Newman AH Novel bivalent ligands based on the sumanirole pharmacophore reveal dopamine D2 receptor (D2R) biased agonism. J. Med. Chem 2017, 60, 2890–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Weïwer M; Xu Q; Gale JP; Lewis M; Campbell AJ; Schroeder FA; Van de Bittner GC; Walk M; Amaya A; Su P; Dordevic L; Skepner A; Fei D; Dennehy K; Nguyen S; Faloon PW; Perez J; Cottrell JR; Liu F; Palmer M; Pan JQ; Hooker JM; Zhang Y-L; Scolnick E; Wagner FF; Holson EB Functionally Biased D2R Antagonists: Targeting the β-Arrestin Pathway to Improve Antipsychotic Treatment. ACS Chem. Biol 2018, 13, 1038–1047. [DOI] [PubMed] [Google Scholar]
- (42).Gray DL; Allen JA; Mente S; O’Connor RE; DeMarco GJ; Efremov I; Tierney P; Volfson D; Davoren J; Guilmette E; Salafia M; Kozak R; Ehlers MD Impaired beta-arrestin recruitment and reduced desensitization by non-catechol agonists of the D1 dopamine receptor. Nat. Commun 2018, 9, 674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Davoren JE; Nason D; Coe J; Dlugolenski K; Helal C; Harris AR; LaChapelle E; Liang S; Liu Y; O’Connor R; Orozco CC; Rai BK; Salafia M; Samas B; Xu W; Kozak R; Gray D Discovery and lead optimization of atropisomer D1 agonists with reduced desensitization. J. Med. Chem 2018, 61, 11384. [DOI] [PubMed] [Google Scholar]
- (44).Fisone G; Bezard E Molecular mechanisms of L-DOPA-induced dyskinesia. Int. Rev. Neurobiol 2011, 98, 95–122. [DOI] [PubMed] [Google Scholar]
- (45).Jenner P Molecular mechanisms of L-DOPA-induced dyskinesia. Nat. Rev. Neurosci 2008, 9, 665–677. [DOI] [PubMed] [Google Scholar]
- (46).Weed MR; Vanover KE; Woolverton WL Reinforcing effect of the D1 dopamine agonist SKF 81297 in rhesus monkeys. Psychopharmacology 1993, 113, 51–52. [DOI] [PubMed] [Google Scholar]
- (47).Dubois A; Savasta M; Curet O; Scatton B Autoradiographic distribution of the D1 agonist [3H]SKF 38393, in the rat brain and spinal cord. Comparison with the distribution of D2 dopamine receptors. Neuroscience 1986, 19, 125–137. [DOI] [PubMed] [Google Scholar]
- (48).Mottola DM; Brewster WK; Cook LL; Nichols DE; Mailman RB Dihydrexidine, a novel full efficacy D1 dopamine receptor agonist. J. Pharmacol. Exp. Ther 1992, 262, 383–393. [PubMed] [Google Scholar]
- (49).Harris AR; Nason DM; Collantes EM; Xu W; Chi Y; Wang Z; Zhang B; Zhang Q; Gray DL; Davoren JE Synthesis of 5-bromo-6-methyl imidazopyrazine, 5-bromo and 5-chloro-6-methyl imidazopyridine using electron density surface maps to guide synthetic strategy. Tetrahedron 2011, 67, 9063–9066. [Google Scholar]
- (50).Miyaura N; Suzuki A Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem. Rev 1995, 95, 2457–2483. [Google Scholar]
- (51).Hanson BJ; Wetter J; Bercher MR; Kopp L; Fuerstenau-Sharp M; Vedvik KL; Zielinski T; Doucette C; Whitney PJ; Revankar C A homogeneous fluorescent live-cell assay for measuring 7-transmembrane receptor activity and agonist functional selectivity through beta-arrestin recruitment. J. Biomol. Screening 2009, 14, 798–810. [DOI] [PubMed] [Google Scholar]
- (52).Wang T; Li Z; Cvijic ME; Krause C; Zhang L; Sum CS Measurement of Beta-Arrestin Recruitment for GPCR Targets. In Assay Guidance Manual; Sittampalam GS, Coussens NP, Brimacombe K, Grossman A, Arkin M, Auld D, Austin C, Baell J, Bejcek B, Caaveiro JMM, Chung TDY, Dahlin JL; Devanaryan V, Foley TL, Glicksman M, Hall MD, Haas JV, Inglese J, Iversen PW, Kahl SD, Kales SC, Lal-Nag M, Li Z, McGee J, McManus O, Riss T, Trask OJ Jr., Weidner JR, Wildey MJ, Xia M, Xu X, Eds.; Eli Lilly & Company: Bethesda MD, 2004. [PubMed] [Google Scholar]
- (53).Zhang R; Xie X Tools for GPCR drug discovery. Acta Pharmacol. Sin 2012, 33, 372–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Besnard J; Ruda GF; Setola V; Abecassis K; Rodriguiz RM; Huang X-P; Norval S; Sassano MF; Shin AI; Webster LA; Simeons FRC; Stojanovski L; Prat A; Seidah NG; Constam DB; Bickerton GR; Read KD; Wetsel WC; Gilbert IH; Roth BL; Hopkins AL Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Kenakin T; Watson C; Muniz-Medina V; Christopoulos A; Novick S A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci 2012, 3, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.