

Abstract
Aims:
Brain oedema is a major complication of acute toxic liver failure (ATLF). We evaluated the ability of brain affected by oedema due to ATLF, to control the ionic composition and acid-base balance of the interstitial fluid.
Methods:
ATLF was induced in 10–12 weeks old male C57Bl mice by single intraperitoneal (i.p.) injection of 100μg/g azoxymethane (AOM). Analyses were carried out in cerebral cortex of precomatous mice at ~20h after AOM administration. Brain fluid status was evaluated by measuring apparent diffusion coefficient [ADC] using NMR spectroscopy, Evans Blue extravasation, and accumulation of an intracisternally-injected fluorescent tracer. Extracellular pH ([pH]e) and ([K+]e) were measured in situ with ion-sensitive microelectrodes. Cerebral cortical microdialysates were subjected to photometric analysis of extracellular potassium ([K+]e), sodium ([Na+]e) and luminometric assay of extracellular lactate ([Lac]e). Potassium transport was measured ex vivo as 86Rb uptake to cerebral cortical slices.
Results:
Cerebral cortex of AOM mice presented decreased ADC pointing to cytotoxic oedema, increased Evans blue extravasation indicative of blood brain barrier leakage, and increased fluorescent tracer accumulation suggesting impaired interstitial fluid passage. However, ([K+]e), ([Na+]e), ([Lac]e), ([pH]e) and potassium transport in brain of AOM-treated mice was not different from control mice.
Conclusions:
In spite of deregulated fluid control, severely oedematous brain of mice with ATLF retains the ability to maintain interstitial ion homeostasis and acid-base balance. Tentatively, uncompromised brain ion homeostasis and acid-base may contribute to the relatively good prognosis of brain function recovery and spontaneous survival rate in human patients with ATLF.
Keywords: acute liver failure, azoxymethane (AOM) model, cytotoxic brain oedema, interstitial brain oedema, apparent diffusion coefficient (ADC), extracellular ions, extracellular pH, lactate
Introduction
Brain oedema is a major and often fatal complication of hepatic encephalopathy (HE) that follows acute liver failure (ALF), which develops in humans or experimental animals in consequence of viral infection or after ingestion of hepatotoxic drugs; the latter condition is further defined as acute toxic liver failure (ATLF) (Blei, 2008; Gupta et al., 2017; McCormick et al., 2003). It is generally acknowledged that ALF-induced oedema is mainly cytotoxic in nature, resulting from water movement to the intracellular (predominantly intra-astrocytic) compartment, with moderate contribution of interstitial or vasogenic oedema (Chavarria et al., 2010; Scott et al., 2013). Exposure of mice to azoxymethane (AOM) induces major behavioural, biochemical, and neurophysiological changes resembling human HE resulting from ALF, including (i) reproducible development of four stages of neurological decline, (ii) increased levels of ammonia in the blood and in the brain, (iii) severe neurological dysfunctions (Belanger et al., 2006; Matkowskyj et al., 1999; McMillin et al., 2014; Popek et al., 2017) and (iv) brain oedema (Belanger et al., 2006; Chastre et al., 2014; Nguyen et al., 2006; Rangroo Thrane et al., 2012). The principal question of the present study was whether and in what degree HE associated with brain oedema accompanying ALF affects the ion composition and pH of the interstitial space, parameters reflecting the ability of the brain to cope with pathophysiological consequences of cerebral fluid imbalance. To our knowledge, this problem has so far escaped analysis in the context of HE related to ATLF; only two studies known to us have dealt with potassium homeostasis in severe hyperammonemia, a condition not equivalent to ALF (Rangroo Thrane et al., 2013; Sugimoto et al., 1997).
In this study we first analysed the nature of brain oedema in AOM-treated mice in which neurological HE symptoms progressed to the pre-comatose stage III (Matkowskyj et al., 1999; Popek et al., 2017). The MRI analysis revealed a decrease of the apparent diffusion coefficient (ADC), reflecting cytotoxic oedema (Chavarria and Cordoba, 2015). Subsequently, we confirmed leaky blood-brain barrier indicative of a vasogenic component, and found increased retention of an intracisternally administered fluorescent marker, indicating impairment of a paravascular cerebrospinal fluid (CSF)/interstitial fluid (ISF) flow, termed glymphatic system (Iliff et al., 2012). Having established the parameters of the brain oedema, we then measured, in vivo in awake animals or in brain microdialysates, the extracellular concentrations of sodium ([Na+]e, potassium ([K+]e), lactate ([Lac]e) and extracellular pH ([pH]e). To account for the effect of ALF on cerebral potassium transport we compared the uptake of a radiolabelled potassium surrogate, 86Rb, in cerebral cortical slices derived from control and ALF-affected brains.
Materials and methods
Azoxymethane (AOM) model of ALF
Experiments were performed on 10–12 weeks old male C57Bl mice, with approval and under surveillance of the IVth Local Ethical Committee, National Drug Institute, Warsaw or of the University Committee on Animal Resources, University of Rochester Medical Center. Mice received a single intraperitoneal (i.p.) injection of 100μg/g azoxymethane (AOM) (Sigma-Aldrich). Experiments were carried out at ~20 h post AOM, when neurological status of the animals reached stage III-IV (Popek et al., 2017).
Determination of apparent diffusion coefficient (ADC)
Brains of control and AOM-treated animals were scanned with Bruker BioSpec 70/30 Advance III system working at 7T, with a transmit cylindrical radiofrequency coil (8.6 cm inner diameter) and a mouse brain dedicated receive-only array coil (2×2 elements) positioned over the animal’s head. The animals were positioned prone with the head placed in the stereotactic apparatus and anesthesia mask, and were anesthetized at around 1.5–2% isoflurane in a mixture of oxygen and air. Respiration was monitored throughout the experiment.
Structural transverse MR images covering the whole brain were acquired with T2-weighted TurboRARE (TR/TE = 6000/30ms, RARE factor = 4, spatial resolution = 78μm × 78μm × μm, 35 slices, no gaps, number of averages (NA) = 4, scan time = 25min). Spin echo diffusion-weighted images covering the whole brain were acquired (TR/TE = 7000/27ms, b-values = 0, 600, 1110 s/mm2, number of directions = 1, spatial resolution = 156μm × 156μm × 700μm, 25 slices, no gaps, scan time = 33 min). ADC maps were calculated using Bruker ParaVision 5.1 software. Atlas of each individual brain structure was obtained by automatically labelling whole brain using MRM NeAt atlas and transformation matrix (obtained in the normalization step). That automatic labelling algorithm was implemented as custom-made MATLAB script (http://www.mathworks.com/products/matlab/) exploiting modified functions provided by IBASPM software (http://www.thomaskoenig.ch/Lester/ibaspm.htm). At the top of the analysis, ADC maps were overlaid with the corresponding individual brain structure atlas and mean ADC values from each structure voxels were calculated using custom-made MATLAB scripts.
Evans blue extravasation measurement
The Evans Blue (EB) (4ml/kg in saline) was injected intravenously and allowed to circulate for 2h. Mice were then perfused transcardially under anaesthesia with heparinised saline until perfusion fluid in effluent became colourless. Tissue samples (cortex and hippocampus) were extracted and weighed, homogenized in 50% trichloracetic acid and centrifuged at 4°C. Supernatant absorbance was measured spectrophotometrically at 620nm (FLUOstar Omega microplate reader). Results were quantified using a standard curve.
Extracellular potassium ([K+]e) and pH measurements with ion-sensitive microelectrodes in awake animals
The animals were anesthetised using isoflurane (2.5% in air), a 3mm cranial window was prepared over somatosensory cortex, and dura mater was removed. The exposed scull was submerged in ACSF with 3 mM KCl. During the experiment lasting~2h, awake but restrained mice were resting on heated pads to maintain body temperature at 37°C.
In vivo recordings were obtained from somatosensory cortex (200μm below the exposed pial surface). Ion-sensitive microelectrodes (ISE) for K+, and H+ were pulled from single-barreled pipette glass, salinised and loaded with a ~300 μm column of valinomycin-based K+ ion exchange resin (Potassium Ionophore I-Cocktail B, Sigma-Aldrich), or H+ ion exchange resin (Hydrogen Ionophore I-Cocktail A, Sigma-Aldrich) and backfilled with 150 mM KCl or PBS with a pH of 7.4, respectively. A reference electrode was backfilled with 150 mM NaCl. The ISEs were calibrated before and following the K+/H+ recording using aCSF solutions with a stepwise K+ or H+ gradient. The extracellular ion concentrations data were sampled at 10 kHz and filtered from 0.1–2 kHz using an MultiClamp 700A/700B, digitized using Digidata 1322A, recorded using Clampex 9.2 and analysed using Clampfit 10.2 (Ding et al., 2016).
Microdialysis on freely moving mice
Mice were anesthetised with isoflurane (3.5% in air, and two 1.2-mm-diameter holes were drilled in the head, first at atlas coordinates AP −0.5, ML +0.5, and second at coordinates AP +2.0, ML −0.5, DV −0.5, in which skull screw and guide cannula were inserted, respectively. Guide cannula was implanted at 0.5mm depth.
Before arousal, antibiotic (Baytril, 2.5%; 0.2 ml/kg b.w.) and a painkiller (Ketonal, 2.5 mg/kg b.w.) were subcutaneously administered. Animals were placed in 21-cm-diameter cylindrical cages with ad libitum access to food and water. One day after surgery, mice were anesthetised with isoflurane (3.5% in air) and a probe was implanted. Brain was superfused with aCSF of the following composition (in mM): NaCl 130, KCl 5, CaCl2 2.5, MgSO4 1.3, KH2PO4 1.25, NaHCO3 26, and D-glucose 10, aerated with a mixture of 95% O2 and 5% CO2. After 1 h in 2.5-μl/h flow, samples were collected every 40 min (100 μl) for 4 h (six fractions). After this, AOM was i.p. injected, and after 16 h, samples were collected every 40 min. All samples were immediately frozen at −80 °C.
Photometric determination of extracellular sodium ([Na+]e), potassium ([K+]e)
Concentration of sodium and potassium ions in microdialysates from brain cortex was performed using flame photometer (PFP7, Jenway) (Kuczeriszka et al., 2013). Results were quantified using a standard curve calculated from samples of known Na+/K+ concentrations prepared in the Ringer solution.
Determination of extracellular lactate ([LAC]e)
Concentration of lactate in microdialysates from brain cortex was measured using a luminometric Lactate-Glo assay kit (Promega), according to the manufacturer protocol. The luminometry was measured with FLUOstar Omega microplate reader. Results were quantified using a standard curve.
Fluorescence tracer accumulation
To inject the tracer, the animals were cannulated into the subarachnoid CSF of the cisterna magna. The surgery was performed under ketamine/xylasine anaesthesia 24 hours prior to AOM or saline administration, to allow the animals to recover. Fluorescence tracer (BSA-conjugated Alexa 488; BSA MW ~60 kDa) was infused by peristaltic pump at 2 μM/min rate for 5 minutes (10 μl total volume) to awake animals which 20 h earlier received saline or AOM. Dissected brains were drop-fixed in 4% paraformaldehyde 30 min post tracer injection, and the influx of the CSF tracer into the brain parenchyma was evaluated by whole-slice fluorescence microscopy (Olympus; Stereo Investigator Software). First, the 100 μm coronal slices of PFA fixed brains were cut on a vibratome and mounted on microscope glass slides with ProLong Gold Antifade Mountant (Thermo Fisher Scientific). Then, the densitometric analysis of fluorescence in each slice was performed with ImageJ programme and calculated as a per cent of the total slice area (Iliff et al., 2014).
Brain samples and slice preparation
The mice were anesthetised using gaseous Isoflurane, decapitated and after removal of meninges, the cerebral cortex was dissected. To obtain brain slices for the uptake experiments, the cortices the cortices were cut into 300 μM slices, using McIlwain tissue chopper.
86Rb uptake in cerebral cortical slices
Uptake of radioactive rubidium (86Rb) used as an analogue of K+ was assayed using a modification of a procedure described in (Larsen et al. 2014). Cortical slices were placed in a K+ free buffer containing (mM): 118 NaCl, 25 NaHCO3, 1.2 NH2PO4, 2.5 CaCl2, 1.2 MgSO4, 10 glucose and 30mM choline chloride (ChCl), aerated with 95% O2 and 5% CO2 at pH 7.4, for 30 minutes at 37°C. Next, the slices were transferred to isosmotic solutions in which KCl was substituted for ChCl to change [K+]e within the range of 0.5 – 40 mM, with 1μCi/ml 86Rb (Polatom) added. The uptake experiments were performed at 37°C for 5 min, the time point being established basing on a kinetic experiment (data not shown). The assay was terminated with three brief washes in ice-cold Krebs “stop” buffer containing 30mM KCl and 50μM BaCl2. The slices were weighed and put into vials with 2ml of scintillation liquid (InstaGel). The radioactivity was determined with a Perkin Elmer scintillation counter.
Statistical analysis
Data were were expressed as the mean ± SD and analyzed with nonparametric Mann-Whitney test, using the GraphPad Prism 6 software. p value < 0.05 was regarded as statistically significant.
Results
Water diffusion imaging by magnetic resonance spectroscopy disclosed a ~ 10% decrease of ADC in AOM-treated mice (0.697 ± 0.034 and 0.626 ± 0.054 μm2/s in control and AOM-injected animals, respectively) (Fig. 1A) which is interpreted to reflect cytotoxic oedema (Chavarria and Cordoba, 2015). Brains of AOM-treated mice accumulated significantly higher amount of intracisternally injected fluorescent CSF tracer than control brains (Fig.1 B,C), which indicates impaired para-vascular CSF passage. The paravascular level of the fluorescent CSF tracer in the brain parenchyma of mice with the AOM-induced HE was higher by 67.5% (264.5% ± 97.3) compared to control animals (157.9% ± 58.7) (Fig.1 B,C).
Figure 1.
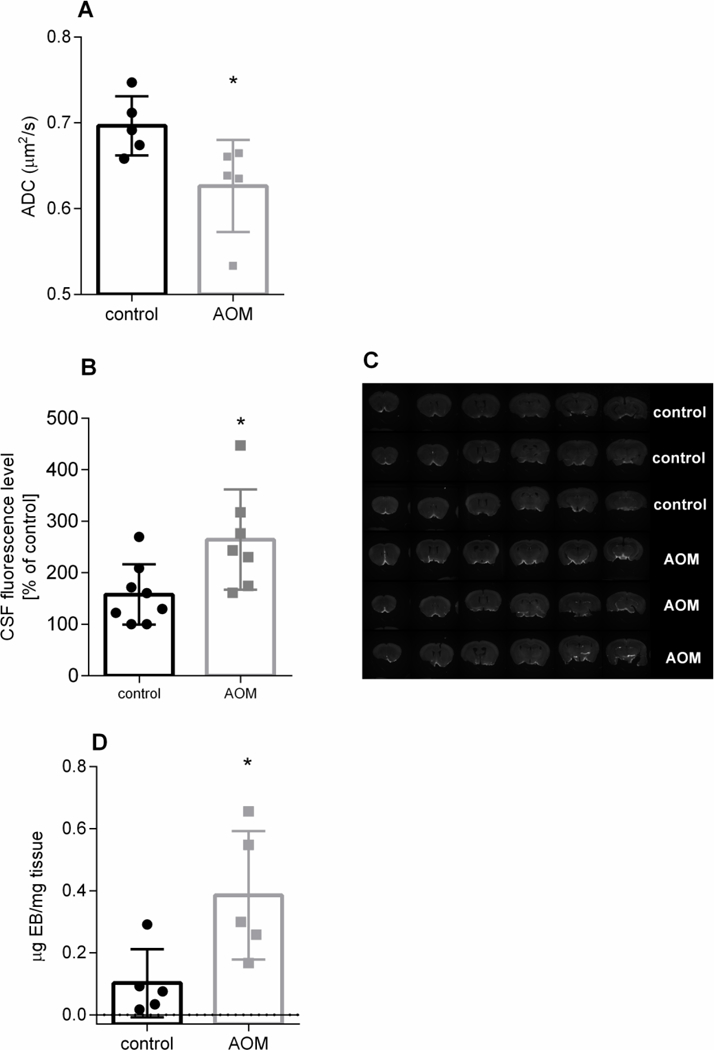
The effect of azoxymethane (AOM)-induced acute liver failure on tissue water accumulation (ADC coefficient) (A), level of intracisternally injected fluorescent CSF tracer in coronal brain slice (B; typical fluorescence image of the tracer is shown in C), and blood-brain barrier integrity (Evans Blue extravasation) (D) in mouse brain cerebral cortex. * p < 0.05 vs control for 5 (A,D) or 8–10 animals (B) in each group (Mann-Whitney test).
Massive accumulation (to ~ 380% of control) of Evans Blue in brain of AOM-treated mice was observed (Fig. 1D) (0.102 ± 0.11 and 0.386 ± 0.207 μg Evans Blue /g tissue in control and AOM-injected animals, respectively), confirming brain extravasation reported earlier in this model (Nguyen et al., 2006) and indicating possible contribution of vasogenic component to brain oedema.
Recordings of [K+]e in cerebral cortex of control and AOM-treated mice in vivo, in awake animals with ion selective microelectrodes (ISE) (Fig. 2A), or in microdialysates, assessed photometrically (Fig. 2B) revealed no differences. The average [K+]e when recorded with ISE was 3.78 ± 0.25 mM and 4.19 ± 0.95 mM in control and AOM-injected animals, respectively. When measured photometrically, the respective levels of [K+]e were 3.007 ± 0.418 mM and 2.901 ± 0.264 mM.
Figure 2.

The effect of azoxymethane (AOM)-induced acute liver failure on brain extracellular K+ (A, measured with ISE in awake animals; B, measured photometrically in microdialysates ), 86Rb uptake in mice cortical slices (C), and brain extracellular Na+ (D, measured photometrically in microdialysates ) and lactate level (E, measured in microdialysates) and pH (F, measured with ISE in awake animals). The numbers of animals in experimental groups were 12–14 (A), 9 – 14 (B, D), 5 (C), 7–12 E, 10–16 (F)
Inducing liver failure with AOM did not affect an uptake of 86Rb, a potassium analogue, by cerebral cortical slices (Michaelis-Menten plot in Fig. 2C). The Vmax and Km values were 169.4 ± 81.3 mmol/mg protein/min and 35.9 ± 29 mM for controls, and 154.3 ± 37.3 mmol/mg protein/min and 36.5 ± 15.0 mM for AOM-treated animals.
Similarly, AOM exposure did not affect [Na+]e measured in microdialysates. The values of ([Na+]e) were 146.8 ± 7.7 mM and 147 ± 6.5 mM in control and AOM-injected animals, respectively (Fig. 2D). The [Lac]e measured in microdialysates were likewise unchanged in AOM-treated mice. The values of [Lac]e were 15.53 ± 5.75 μM and 14.2 ± 8.73 μM in control and AOM-injected animals, respectively (Fig. 2E). The [pH]e recorded from the in vivo cerebral cortex with ISE, remained unaffected by AOM treatment (7.33 ± 0.07 vs. 7.29 ± 0.07) (Fig. 2F).
Discussion
Evidence from NMR analysis of human pathology strongly suggests that ALF instigates HE with cytotoxic brain oedema, the contribution of vasogenic oedema being relatively minor (Bemeur et al., 2016; Scott et al., 2013). In the AOM model and most of the other animal models of ALF caused by hepatotoxins, cytotoxic oedema has so far been documented by ultrastructural demonstration of profound astrocytic swelling (Dixit and Chang, 1990; Kato et al., 1992; Norenberg, 1977; Willard-Mack et al., 1996). The HE induced in the AOM model is accompanied by profound extravasation indicating blood brain barrier leakage (Chastre et al., 2014; Nguyen et al., 2006; this study, Fig. 1B), and net gain in brain water content which may at least in part reflect its leakage across the blood-brain barrier (Rangroo Thrane et al., 2012). The here observed significant decrease of ADC (Fig. 1A) provided objective evidence for the presence of cytotoxic oedema in the AOM model, which is consistent with observations in human ALF of varying causes (Chavarria and Cordoba et al., 2015) and in the liver devascularization rat ALF model (Chavarria et al., 2010). However, contribution of isolated ISF expansion cannot be excluded (Thrane et al., 2014).
Increased intra-brain retention of a fluorescent tracer following its intracisternal accumulation indicated disturbed CSF passage in the glymphatic system, recently described paravascular ISF/CSF exchange system (Iliff et al., 2012). However, given the complexity of mechanisms underlying the CSF flow (Hladky and Barrand, 2014), the previous data are insufficient to predict whether and in what degree the impairment of glymphatic flow of fluid contributes to interstitial water accumulation and brain oedema. The absence of extracellular (Fig. 2A) and tissue (NMR study, Popek et al., 2017) lactate accumulation, the lately identified marker of glymphatic dysfunction (Lundgaard et al., 2017) argues against the contribution of glymphatic system. Whatever the nature of this phenomenon, water accumulation in the interstitial space may per se decrease the ADC in the brain tissue (Badaut et al., 2011; Thrane et al., 2014), further aggravating brain oedema. Of note, the vasogenic component may not significantly contribute to oedema in AOM-induced HE, since the extravasation is limited to the nearest vicinity of the cerebral capillaries, with no evidence of its expansion to deeper regions of cortical parenchyma (Nguyen, 2012). Moreover, the blood-brain barrier (BBB)-leakage in the AOM model is considered to be partly a consequence of direct toxic effects of AOM or its metabolites at the barrier (Jayakumar et al., 2013).
The principal finding of the present study is that neither [K+]e and [Na+]e, which directly reflect the brain ion homeostasis, nor pHe and [Lac]e which characterize the acid-base balance are altered at the neurologically advanced stage of AOM-induced HE (Popek et al., 2017) which is associated with brain oedema. The resistance of ion homeostasis and proton-base homoeostasis in the development of HE and oedema deserves comment with regard to changes observed in the clinic cases and in other models of ALF and HE. To the best of authors’ knowledge neither [K+]e nor [Na+]e have been hitherto investigated in ALF-affected brain. An increase in [K+]e has been reported in the brains of rats with systemically induced hyperammonemia (Sugimoto et al., 1997) and in ornithine transcarbamylase – deficient mice following acute systemic or intracerebral administration of ammonium (Rangroo Thrane et al., 2013). However, the above procedures increased blood ammonia to a much higher level than that observed in AOM-induced ALF (~10 or 7-fold of the control level in hyperammonemia (Rangroo Thrane et al., 2013; Sugimoto et al., 1997) vs 2.5-fold in the AOM-induced ALF (Popek et al., 2017). Moreover, in the study described in (Rangroo Thrane et al., et al., 2013) astrocytic swelling was absent in hyperammonemic animals. The result confirms the now generally accepted view that HE associated with ALF develops in consequence of the toxic effects of ammonium in conjunction with the inflammatory response and contribution of other blood-derived toxins (Aldridge et al., 2015). Otherwise this view is corroborated by earlier findings that hyperammonemia is not equivalent to HE associated with ALF in terms of metabolic responses of the brain (Faff-Michalak and Albrecht, 1991; Faff-Michalak and Albrecht, 1993; Hilgier et al., 1991).
The absence of difference in the 86Rb uptake in brain slices derived from AOM-treated mice as compared to control mice (Fig. 2C) adds credibility to, and provides mechanistic basis for the conclusion that induction of brain oedema by ALF and severe neurological deficit in the present model may become apparent despite preserved potassium homeostasis. However, the resistance of K+ controlling cascades may not be reflected in other conditions mimicking ALF or acute hyperammonemia. Markedly reduced 86Rb fluxes were previously observed in vitro, in conditions modelling hyperammonemia. Decreased 86Rb efflux was observed in primary rat astrocytes treated with 5 mM of ammonium (Obara-Michlewska, et al., 2011), in HEK 293 cells exposed to 10 mM of ammonium (Obara-Michlewska et al., 2010). Decreased rubidium uptake was also reported for cultured mice astrocytes treated with 10 mM ammonia (Rangroo Thrane et al., 2013).
Increased brain tissue or extracellular lactate content (or lactate/pyruvate ratio) is has been recorded, and considered to reflect impaired oxidative metabolism, in brain of comatose human ALF patients (Bjerring et al., 2010; Tofteng et al., 2002), of rats (Zwingmann et al., 2003) or guinea pigs in which ALF was induced by hepatic artery ligation (HAL) (Rose et al., 2007), and in pigs with surgically resected liver (Prazak et al., 2013). Here we found that extracellular lactate does not change in the brains of mice under conditions where ALF was induced by a hepatotoxin, AOM (Fig. 2E), which is in agreement with the lack of changes of whole brain tissue lactate measured in the same stage of the AOM model in situ using NMR spectroscopy (Popek et al., 2017). The absence of change in extracellular pH, consistent with unaltered extracellular lactate, indicates that the interstitial acid/base balance remained indifferent to the insult. Studies with other chemical hepatotoxins will clarify whether the maintenance of proper acid-base balance and lactate level is the general feature characterizing ATLF.
In conclusion, the study shows that in the mouse AOM model, ATLF associated with advanced brain oedema is not accompanied by discernible impairment of interstitial ion and acid-base homeostasis. It is likely that the ability of the oedema-affected brain of ATLF mice to counteract the increase of extracellular K+ is related to its relatively intact energy metabolism as reflected by unchanged brain tissue lactate. It is well established that intra-astrocytic water accumulation responsible for cytotoxic brain oedema in HE is related to the impaired function of a heterogeneous set of ion and water channels, and metabolite-transporting molecules operating in astrocytes (Albrecht et al., 1985; Allert et al., 1998; Kala et al., 2000; Lichter-Konecki et al., 2008; Sugimoto et al., 1997). The simplest interpretation of our observations is that in ALF induced by AOM, astrocytes suffered a “half-way” loss of homeostatic function: having lost the ability to maintain their own cell volume, they are still capable of controlling the extracellular ionic and acid-base balance of the brain. Such an efficient control of brain ionostasis may not occur when subacute toxic liver failure is induced by a prolonged (few day) exposure to a hepatotoxin (vide: the rat thioacetamide model; Faff-Michalak and Albrecht, 1991; Faff-Michalak and Albrecht, 1993; Hilgier et al., 1991), or when ALF is due to liver devascularisation.
It is tempting to speculate that interstitial ion and acid-base homeostasis is likewise unaltered in human ALF cases in which hepatotoxic drug overdose is a causative factor. This would explain the relatively high incidence of spontaneous or treatment-supported neurophysiological recovery in this group of patients (Lee, 2012; Reddy et al., 2016). The present results further suggest that monitoring of extracellular concentrations of monovalent ions and pH in brain microdialysates of ALF patients could be of prognostic value and, as such, a useful addition to a battery of other brain interstitial metabolites monitored using the brain microdialysis technique (Tofteng and Larsen, 2002).
Highlights.
-
1)
Acute toxic liver failure (ATLF) causes cytotoxic oedema and blood extravasation in mouse brain
-
2)
ATLF compromises brain interstitial fluid movement suggesting glymphatic system dysfunction
-
3)
Brain extracellular K+, Na+, lactate and pH are not affected by ATLF
-
4)
ATLF-affected brain tissue retains the ability to control interstitial ion homeostasis
Acknowledgments
This work was supported by the National Science Centre (NCN) of the Republic of Poland (2013/08/M/NZ3/00869) and National Centre for Research (NCBiR) (Pol-Nor/196190/23/2013).
We thank Prof. Adam Szewczyk and Dr Zbigniew Zielinski from the Nencki Institute of Experimental Biology, Warsaw, Poland for expert advice on 86Rb flux experiments.
Abbreviation list
- ADC
apparent diffusion coefficient
- ALF
acute liver failure
- AOM
azoxymethane
- ATLF
acute toxic liver failure
- BBB
blood brain barrier
- CSF
cerebrospinal fluid
- EB
Evans Blue
- MRI
magnetic resonance imaging
- [K+]e)
extracellular potassium concentration
- [Na+]e
extracellular sodium concentration
- [Lac]e)
extracellular lactate concentration
Footnotes
Competing interests
The authors declare no competing financial interests.
Conflicts of interest
None.
REFERENCES
- Albrecht J, Wysmyk-Cybula U, Rafalowska U, 1985. Na+/K+-ATPase activity and GABA uptake in astroglial cell-enriched fractions and synaptosomes derived from rats in the early stage of experimental hepatogenic encephalopathy. Acta Neurol. Scand. 72, 317–320. [DOI] [PubMed] [Google Scholar]
- Aldridge DR, Tranah EJ, Shawcross DL, 2015. Pathogenesis of hepatic encephalopathy: role of ammonia and systemic inflammation. J. Clin. Exp. Hepatol. 5, S7–S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allert N, Koller H, Siebler M, 1998. Ammonia-induced depolarization of cultured rat cortical astrocytes. Brain Res. 782, 261–270. [DOI] [PubMed] [Google Scholar]
- Badaut J, Ashwal S, Obenaus A, 2011. Aquaporins in cerebrovascular disease: a target for treatment of brain edema? Cerebrovasc. Dis. 31, 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belanger M, Cote J, Butterworth RF, 2006. Neurobiological characterization of an azoxymethane mouse model of acute liver failure. Neurochem. Int. 48, 434–440. [DOI] [PubMed] [Google Scholar]
- Bemeur C, Cudalbu C, Dam G, Thrane AS, Cooper AJ, Rose CF, 2016. Brain edema: a valid endpoint for measuring hepatic encephalopathy? Metab Brain Dis. 31, 1249–1258. [DOI] [PubMed] [Google Scholar]
- Bjerring PN, Hauerberg J, Jorgensen L, Frederiksen HJ, Tofteng F, Hansen BA, Larsen FS, 2010. Brain hypoxanthine concentration correlates to lactate/pyruvate ratio but not intracranial pressure in patients with acute liver failure. J. Hepatol. 53, 1054–1058. [DOI] [PubMed] [Google Scholar]
- Blei AT, 2008. Brain edema in acute liver failure. Crit Care Clin. 24, 99–114, ix. [DOI] [PubMed] [Google Scholar]
- Chastre A, Belanger M, Nguyen BN, Butterworth RF, 2014. Lipopolysaccharide precipitates hepatic encephalopathy and increases blood-brain barrier permeability in mice with acute liver failure. Liver Int. 34, 353–361. [DOI] [PubMed] [Google Scholar]
- Chavarria L, Cordoba J, 2015. Magnetic resonance imaging and spectroscopy in hepatic encephalopathy. J. Clin. Exp. Hepatol. 5, S69–S74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavarria L, Oria M, Romero-Gimenez J, Alonso J, Lope-Piedrafita S, Cordoba J, 2010. Diffusion tensor imaging supports the cytotoxic origin of brain edema in a rat model of acute liver failure. Gastroenterology 138, 1566–1573. [DOI] [PubMed] [Google Scholar]
- Ding F, O’Donnell J, Xu Q, Kang N, Goldman N, Nedergaard M, 2016. Changes in the composition of brain interstitial ions control the sleep-wake cycle. Science 352, 550–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit V, Chang TM, 1990. Brain edema and the blood brain barrier in galactosamine-induced fulminant hepatic failure rats. An animal model for evaluation of liver support systems. ASAIO Trans. 36, 21–27. [PubMed] [Google Scholar]
- Faff-Michalak L, Albrecht J, 1991. Aspartate aminotransferase, malate dehydrogenase, and pyruvate carboxylase activities in rat cerebral synaptic and nonsynaptic mitochondria: effects of in vitro treatment with ammonia, hyperammonemia and hepatic encephalopathy. Metab Brain Dis. 6, 187–197. [DOI] [PubMed] [Google Scholar]
- Faff-Michalak L, Albrecht J, 1993. The two catalytic components of the 2-oxoglutarate dehydrogenase complex in rat cerebral synaptic and nonsynaptic mitochondria: comparison of the response to in vitro treatment with ammonia, hyperammonemia, and hepatic encephalopathy. Neurochem. Res. 18, 119–123. [DOI] [PubMed] [Google Scholar]
- Gupta T, Dhiman RK, Ahuja CK, Agrawal S, Chopra M, Kalra N, Duseja A, Taneja S, Khandelwal N, Chawla Y, 2017. Characterization of Cerebral Edema in Acute-on-Chronic Liver Failure. J. Clin. Exp. Hepatol. 7, 190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgier W, Haugvicova R, Albrecht J, 1991. Decreased potassium-stimulated release of [3H]D-aspartate from hippocampal slices distinguishes encephalopathy related to acute liver failure from that induced by simple hyperammonemia. Brain Res. 567, 165–168. [DOI] [PubMed] [Google Scholar]
- Hladky SB, Barrand MA, 2014. Mechanisms of fluid movement into, through and out of the brain: evaluation of the evidence. Fluids Barriers. CNS. 11, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Chen MJ, Plog BA, Zeppenfeld DM, Soltero M, Yang L, Singh I Deane R, Nedergaard M, 2014. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J. Neurosci. 34, 16180–16193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M, 2012. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including amyloid beta. Sci. Transl. Med. 4, 147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayakumar AR, Ruiz-Cordero R, Tong XY, Norenberg MD, 2013. Brain edema in acute liver failure: role of neurosteroids. Arch. Biochem. Biophys. 536, 171–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kala G, Kumarathasan R, Peng L, Leenen FH, Hertz L, 2000. Stimulation of Na+,K+-ATPase activity, increase in potassium uptake, and enhanced production of ouabain-like compounds in ammonia-treated mouse astrocytes. Neurochem. Int. 36, 203–211. [DOI] [PubMed] [Google Scholar]
- Kato M, Hughes RD, Keays RT, Williams R, 1992. Electron microscopic study of brain capillaries in cerebral edema from fulminant hepatic failure. Hepatology 15, 1060–1066. [DOI] [PubMed] [Google Scholar]
- Kuczeriszka M, Dobrowolski L, Walkowska A, Sadowski J, Kompanowska-Jezierska E, 2013. Adenosine effects on renal function in the rat: role of sodium intake and cytochrome P450. Nephron Physiol 123, 1–5. [DOI] [PubMed] [Google Scholar]
- Larsen BR, Assentoft M, Cotrina ML, Hua SZ, Nedergaard M, Kaila K, Voipio J, MacAulay N, 2014. Contributions of the Na(+)/K(+)-ATPase, NKCC1, and Kir4.1 to hippocampal K(+) clearance and volume responses. Glia 62, 608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WM, 2012. Acute liver failure. Semin. Respir. Crit Care Med. 33, 36–45. [DOI] [PubMed] [Google Scholar]
- Lichter-Konecki U, Mangin JM, Gordish-Dressman H, Hoffman EP, Gallo V, 2008. Gene expression profiling of astrocytes from hyperammonemic mice reveals altered pathways for water and potassium homeostasis in vivo. Glia 56, 365–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundgaard I, Lu ML, Yang E, Peng W, Mestre H, Hitomi E, Deane R, Nedergaard M, 2017. Glymphatic clearance controls state-dependent changes in brain lactate concentration. J. Cereb. Blood Flow Metab 37, 2112–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matkowskyj KA, Marrero JA, Carroll RE, Danilkovich AV, Green RM, Benya RV, 1999. Azoxymethane-induced fulminant hepatic failure in C57BL/6J mice: characterization of a new animal model. Am. J. Physiol 277, G455–G462. [DOI] [PubMed] [Google Scholar]
- McCormick PA, Treanor D, McCormack G, Farrell M, 2003. Early death from paracetamol (acetaminophen) induced fulminant hepatic failure without cerebral oedema. J. Hepatol. 39, 547–551. [DOI] [PubMed] [Google Scholar]
- McMillin M, Frampton G, Thompson M, Galindo C, Standeford H, Whittington E, Alpini G, DeMorrow S, 2014. Neuronal CCL2 is upregulated during hepatic encephalopathy and contributes to microglia activation and neurological decline. J. Neuroinflammation. 11, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen JH, 2012. Blood-brain barrier in acute liver failure. Neurochem. Int. 60, 676–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen JH, Yamamoto S, Steers J, Sevlever D, Lin W, Shimojima N, Castanedes-Casey M, Genco P, Golde T, Richelson E, Dickson D, McKinney M, Eckman CB, 2006. Matrix metalloproteinase-9 contributes to brain extravasation and edema in fulminant hepatic failure mice. J. Hepatol. 44, 1105–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norenberg MD, 1977. A light and electron microscopic study of experimental portal-systemic (ammonia) encephalopathy. Progression and reversal of the disorder. Lab Invest 36, 618–627. [PubMed] [Google Scholar]
- Obara-Michlewska M, Jiang H, Aschner M, Albrecht J, 2010. Gain of function of Kir4.1 channel increases cell resistance to changes of potassium fluxes and cell volume evoked by ammonia and hypoosmotic stress. Pharmacol. Rep. 62, 1237–1242. [DOI] [PubMed] [Google Scholar]
- Obara-Michlewska M, Pannicke T, Karl A, Bringmann A, Reichenbach A, Szeliga M, Hilgier W, Wrzosek A, Szewczyk A, Albrecht J, 2011. Down-regulation of Kir4.1 in the cerebral cortex of rats with liver failure and in cultured astrocytes treated with glutamine: Implications for astrocytic dysfunction in hepatic encephalopathy. J. Neurosci. Res. 89, 2018–2027. [DOI] [PubMed] [Google Scholar]
- Popek M, Bobula B, Sowa J, Hess G, Polowy R, Filipkowski RK, Frontczak-Baniewicz M, Zablocka B, Albrecht J, Zielinska M, 2017. Cortical Synaptic Transmission and Plasticity in Acute Liver Failure Are Decreased by Presynaptic Events. Mol. Neurobiol doi: 10.1007/s12035-016-0367-4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prazak J, Laszikova E, Pantoflicek T, Ryska O, Koblihova E, Ryska M, 2013. Cerebral microdialysis reflects the neuroprotective effect of fractionated plasma separation and adsorption in acute liver failure better and earlier than intracranial pressure: a controlled study in pigs. BMC. Gastroenterol. 13, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangroo T,V, Thrane AS, Chang J, Alleluia V, Nagelhus EA, Nedergaard M, 2012. Real-time analysis of microglial activation and motility in hepatic and hyperammonemic encephalopathy. Neuroscience 220, 247–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangroo T,V, Thrane AS, Wang F, Cotrina ML, Smith NA, Chen M, Xu Q, Kang N, Fujita T, Nagelhus EA, Nedergaard M, 2013. Ammonia triggers neuronal disinhibition and seizures by impairing astrocyte potassium buffering. Nat. Med. 19, 1643–1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy KR, Ellerbe C, Schilsky M, Stravitz RT, Fontana RJ, Durkalski V, Lee WM, 2016. Determinants of outcome among patients with acute liver failure listed for liver transplantation in the United States. Liver Transpl. 22, 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose C, Ytrebo LM, Davies NA, Sen S, Nedredal GI, Belanger M, Revhaug A, Jalan R, 2007. Association of reduced extracellular brain ammonia, lactate, and intracranial pressure in pigs with acute liver failure. Hepatology 46, 1883–1892. [DOI] [PubMed] [Google Scholar]
- Scott TR, Kronsten VT, Hughes RD, Shawcross DL, 2013. Pathophysiology of cerebral oedema in acute liver failure. World J. Gastroenterol. 19, 9240–9255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto H, Koehler RC, Wilson DA, Brusilow SW, Traystman RJ, 1997. Methionine sulfoximine, a glutamine synthetase inhibitor, attenuates increased extracellular potassium activity during acute hyperammonemia. J. Cereb. Blood Flow Metab 17, 44–49. [DOI] [PubMed] [Google Scholar]
- Thrane AS, Rangroo T,V, Nedergaard M, 2014. Drowning stars: reassessing the role of astrocytes in brain edema. Trends Neurosci. 37, 620–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tofteng F, Jorgensen L, Hansen BA, Ott P, Kondrup J, Larsen FS, 2002. Cerebral microdialysis in patients with fulminant hepatic failure. Hepatology 36, 1333–1340. [DOI] [PubMed] [Google Scholar]
- Tofteng F, Larsen FS, 2002. Monitoring extracellular concentrations of lactate, glutamate, and glycerol by in vivo microdialysis in the brain during liver transplantation in acute liver failure. Liver Transpl. 8, 302–305. [DOI] [PubMed] [Google Scholar]
- Willard-Mack CL, Koehler RC, Hirata T, Cork LC, Takahashi H, Traystman RJ, Brusilow SW, 1996. Inhibition of glutamine synthetase reduces ammonia-induced astrocyte swelling in rat. Neuroscience 71, 589–599. [DOI] [PubMed] [Google Scholar]
- Zwingmann C, Chatauret N, Leibfritz D, Butterworth RF, 2003. Selective increase of brain lactate synthesis in experimental acute liver failure: results of a [H-C] nuclear magnetic resonance study. Hepatology 37, 420–428. [DOI] [PubMed] [Google Scholar]