

Abstract
Vinyl carbocations have been the subject of extensive experimental and theoretical studies over the past five decades. Despite this long history in chemistry, the utility of vinyl cations in chemical synthesis has been limited, with most reactivity studies focusing on solvolysis reactions or intramolecular processes. Here we report synthetic and mechanistic studies of vinyl cations generated through silylium–weakly coordinating anion catalysis. We find that these reactive intermediates undergo mild intermolecular carbon-carbon bond–forming reactions, including carbon-hydrogen (C–H) insertion into unactivated sp3 C–H bonds and reductive Friedel-Crafts reactions with arenes. Moreover, we conducted computational studies of these alkane C–H functionalization reactions and discovered that they proceed through nonclassical, ambimodal transition structures. This reaction manifold provides a framework for the catalytic functionalization of hydrocarbons using simple ketone derivatives.
For more than a century, carbocations have played a central role in the chemical sciences, inspiring the development of broadly applied chemical reactions and a greater understanding of the fundamental properties of molecules (1). This is pointedly true for vinyl cations. Beginning with Jacobs’ proposal of a vinyl cation intermediate (2) and continuing through the investigations by Grob et al. (3), Hanack (4), Rappoport and Gal (5), Schleyer and others (6), Stang and Rappoport (7), and others (8–11), this class of reactive intermediates has been the subject of extensive theoretical and experimental studies. The structure and bonding of these cations has fascinated theorists, driving the development of computational techniques over the past five decades (12–15). Vinyl cation intermediates have also been implicated in modern transition metal-catalyzed processes (16) and in classical acid-catalyzed reactions (17). Despite this rich history and broad scientific relevance, most reactivity studies have focused on solvolysis reactions where the reactive vinyl cation is intercepted by heteroatom-containing solvent molecules (18, 19).
Recently, our group reported that putative phenyl cations (1; Fig. 1A), a related class of dicoordinated carbocations, undergo insertion into sp3 C–H bonds (20). Inspired by this result, we hypothesized that vinyl cations may display analogous C–H insertion reactivity. Challenging this hypothesis was a lack of substantial precedent for this mode of reactivity, despite the extensive experimental studies of vinyl cations over the past five decades. However, a few examples of vinyl cations engaging in C–C bond–forming reactions have been reported. Hanack and others reported the formation of fused products 2 and 3 from the solvolysis of cyclononenyl triflate 4 (Fig. 1B) (21, 22). In this reaction, and in analogous reports from Olah and Mayr (23) and Caple and others (24), this unusual C–C bond–forming process was rationalized via a carbocation rebound mechanism. Here the vinyl cation 5 is quenched via 1,5-hydride shift, and the neutralized alkene (6) attacks the newly formed alkyl carbocation. This sequence forges the ring fusion of the bicyclononane cation 7, leading to products 2 and 3. The insertion of vinyl cations into methane C–H bonds has also been observed in the gas phase (25). More recently, Metzger and others (26) and Brewer and others (27) have independently proposed the intramolecular C–H insertion of vinyl cations generated from alkynes or diazo compounds in solution.
Fig. 1. C–C bond–forming reactions of dicoordinated carbocations.

(A) Phenyl cation insertion into sp3 C–H bonds. (B) Initial proposal of carbocation rebound mechanism in ring-contraction reactions of cyclononenyl triflates. (C) Proposed catalytic cycle for reductive alkane alkylation. (D) Identification of cyclohexenyl triflate as a persistent vinyl cation precursor owing to the prohibitive barrier to 1,2-hydride shift. Calculations performed at M062X/6–311+G(d,p) level of theory; [WCA]− omitted for clarity. R, alkyl; R1, aryl, alkyl, halide, or silyl ether; R2, alkyl or aryl; cat., catalytic; TMS, trimethylsilyl; OTf, trifluoromethanesulfonate; Et, ethyl; EtOH, ethanol; ΔG, Gibbs free energy; ΔG‡, Gibbs free energy of activation.
The solvolysis studies described above relied on polar solvents to facilitate thermal ionization of vinyl species bearing halide or pseudohalide leaving groups (for example, Fig. 1B). This approach almost invariably leads to attack of solvent on the cationic carbon center, leaving reactivity beyond the SN1 manifold largely undiscovered. We drew on our phenyl cation studies to overcome this limitation by using silylium-monocarba-closo-dodecaborate salts (formed in situ through the reaction of triethylsilane with [Ph3C]+[HCB11Cl11]−) in hydrophobic solvents to generate vinyl cations. Here the nonnucleophilic and nonbasic properties of weakly coordinating anions (WCA), such as undecachlorinated monocarba-closo-dodecaborate anion (HCB11Cl11−) (28), would enhance the Lewis acidity of the cationic silicon center, allowing for mild ionization of alkene derivatives. In this scenario, silylium-mediated ionization of a vinyl triflate (8) would generate a kinetically persistent vinyl cation–WCA ion pair (9; Fig. 1C). Insertion of this reactive dicoordinated cation (9) into an alkane C–H bond would lead to formation of alkyl carbocation 10. A 1,2-hydride shift would lead to the more stable tertiary cation (11) that, upon reduction by a sacrificial silane (29), would generate the functionalized hydrocarbon product 12 and regenerate the silylium-carborane initiator. In its entirety, this process would enable the direct C–H alkylation of alkanes and arenes by simple ketone derivatives. We now report the successful execution of this mechanistic hypothesis, entailing intermolecular, reductive C–H alkylation mediated by silane and catalytic quantities of carborane salts. Moreover, we describe the unexpected discovery of an ambimodal transition state and post–transition state bifurcation in this reaction that leads to several products directly through a nonclassical mechanism (30–32). Ambimodal transition states have characteristics of two or more classical transition states, just as nonclassical ions simultaneously possess the features of two classical ions.
Vinyl cation generation from cyclohexenyl triflates
Given the known propensity of substituted cyclic vinyl cations to undergo rapid intramolecular rearrangements (33), we identified prospective cation precursors through density functional theory calculations. First, we verified that a 1,2-hydride shift in cyclohexenyl cation 9, which would generate the more stable cyclic allyl cation 13, is kinetically unfavorable (Fig. 1D). The calculated high barrier, despite the impressive exothermicity of the rearrangement, results from the geometrical distortion required to achieve overlap of the migrating H 1s orbital with the nearly orthogonal orbitals at the origin and terminus of the migration. On the basis of this computational result, we initiated our studies with cyclohexenyl triflate (8). We were pleased to find that exposure of cyclohexenyl triflate (8) to 1.5 equivalents of triethylsilane and 2 mole % (mol %) [Ph3C]+[HCB11Cl11]− in dried cyclohexane solvent at 30°C in a nitrogen-filled glove box resulted in the formation of cyclohexylcyclohexane (14; Table 1, entry 1) in 87% yield, after 90 min.
Table 1. Reductive alkylation of cyclic vinyl triflates.
Reactions performed at 0.1 M in hydrocarbon solvent.
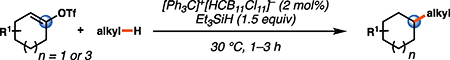 | ||||
|---|---|---|---|---|
| Entry | Substrate | Solvent | Product | Yield (%), Time (h) |
| 1 | 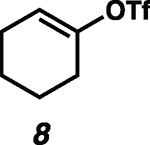 |
C6H12 | 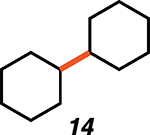 |
87*, 1.5 |
| 2 | 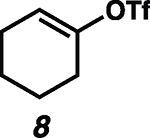 |
C7H14 | 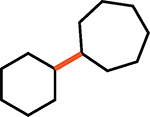 |
88*, 2 |
| 3 | 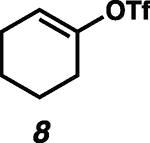 |
n-C5H12 | 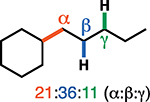 |
68*. 1.5 |
| 4 | 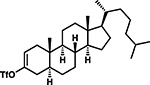 |
C6H12 | 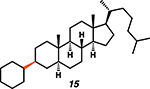 |
88†, 3 (15:1 d.r.) |
| 5 | 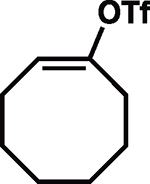 |
C6H12 | 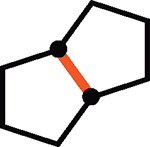 |
91*, 1 |
Yield determined by GC-FID with nonane as an internal standard.
isolated yield.
R1, alkyl.
Intrigued by the remarkably mild conditions used in this alkane alkylation reaction, we undertook a brief study of scope to further elucidate potential synthetic applications and to gain mechanistic insight. Cycloheptane and n-pentane also reacted efficiently with the cyclohexenyl cation, albeit with poor regioselectivity in the latter case (Table 1, entries 2 and 3). Although cyclohexenyl triflates bearing substituents at the carbons in the 2- or 6-position led to complex mixtures of products, presumably the result of nonproductive unimolecular decompositions (33), other positions of the cyclohexenyl ring were tolerant of substitution. For example, exposure of the enol triflate derived from 5α-cholestan-3-one (Table 1, entry 4) to our reaction conditions led to formation of the alkylated steroid 15 in 88% yield and 15:1 diastereomeric ratio (d.r.) (relative stereochemistry determined by x-ray crystallography). Analogous to the previously reported ring-contraction reactions of medium-sized cyclic vinyl triflates (21), exposure of cyclooctenyl triflate to our optimized reaction conditions led to rapid transannular C–H insertion to yield bicyclo[3.3.0]octane (Table 1, entry 5).
Evidence of a nonclassical mechanism
To probe the possibility of a hydride-abstraction, carbocation-rebound mechanism as proposed in early examples (22) (Fig. 1B), we studied the insertion reactions of unsymmetrical cyclohexenyl triflate derivatives 16 and 17 (Fig. 2A). If a stepwise rebound mechanism were operative, we would expect 4,4-dimethylcyclohexenyl triflate (16) and 5,5-dimethylcyclohexenyl triflate (17) to yield identical product distributions, as they would both proceed through alkene intermediate 18 (Fig. 2A). In the event, we found that exposure of triflate 16 to our reaction conditions led to formation of C1-cyclohexylated product 19 in 68% yield and C2-cyclohexylated product 20 in 6% yield. However, triflate 17, the 5,5-disubstituted analog, provided C1-cyclohexylated product 20 in 78% yield and C2-cyclohexylated product 19 in 6% yield. Although the C–H insertion event occurred primarily at the C1 that originally bore the triflate group, in both systems, minor products were observed that correspond to insertion at the C2 adjacent to the triflate (>10:1 C1:C2 insertion in both cases). Although these experimental results support a nonrebound, concerted C–H insertion mechanism, they do not provide mechanistic insight into the formation of C2-alkylation products. Therefore, we undertook quantum mechanical calculations and molecular dynamics (MD) simulations to probe the mechanism in more detail (movies S1 to S4).
Fig. 2. Discovery of an ambimodal transition state.

(A) Probing the possibility of a rebound mechanism. Reactions were performed under the same conditions as described in Table 1. (B) Snapshots from MD simulations representing structures taken from multiple trajectories starting from the ambimodal TS9-Cy, illustrating the most common reaction pathways, the post–transition state bifurcation, and resulting products. (C) Line drawing representations of the three-dimensional representations in (B). †Yieid determined by gas chromatography–flame ionization detector (GC-FID) with nonane as an internal standard. Calculations performed at M062X/6–311+G(d,p) level of theory. Cy, cyclohexyl.
Calculations on the reaction of the cyclohexen-1-yl (9) cation with cyclohexane gives a very early ambimodal transition structure (TS), wherein the geminai cyclohexane C–H bonds interact with the vinyl cation vacant orbital (TS9–Cy, Fig. 2B). Quasi-classical MD trajectories were initiated from normal mode–sampled structures (a Boltzmann distribution) in the region of transition structure TS9–Cy. Taken together, these represent the ambimodal transition state, a single saddle-point region that can give rise to several different products (30–32). Two typical trajectories are summarized in Fig. 2B, with several snapshots of structures illustrating the bifurcation of the reaction pathway. The common trajectory path from ambimodal TS9–Cy begins with hydride transfer from cyclohexane to the vinyl cation via DT1 (where DT stands for dynamical transient, not an intermediate or transition state) (Fig. 2B). Unlike a rebound mechanism, the cyclohexene and cyclohexyl cation moieties fail to separate; the newly formed cyclohexyl cation coordinates (DT2) with the alkene of the neutralized cyclohexene, in most trajectories, at the carbon that previously bore the cation (C1) (Fig. 2B). The resulting bridging nonclassical species (DT3) continues to different products through different trajectories (Fig. 2B). DT3 is an unsymmetrical nonclassical carbocation, with the cyclohexyl bound more strongly at C1, but still provides the three-center two-electron hypervalent bonding (very strong hyper-conjugation) to stabilize the carbocation. In most of the trajectories, DT3 collapses to the classical secondary carbocation structure, DT4, that undergoes a barrierless 1,2-hydride shift to form the tertiary cyclohexylcyclohexyl cation as a potential energy minimum at −40.9 kcal/mol (INT11A) (movies S2 and S4). In fewer trajectories, DT3 goes on to the opposite asymmetric nonclassical species DT5, favoring C2 functionalization (Fig. 2B). DT5 leads to DT6 and eventually INT11B (movies S1 and S3). Through the stabilizing effects and geometric constraints (fig. S40) (34) of these persistent nonclassical intermediates, energetically favorable hydride shifts are delayed, allowing for the experimentally observed dynamic distribution of products in our ring-substituted cyclohexen-1-yl cation substrates (Fig. 2A). The free energies of various stationary points on the surface between ambimodal TS9–Cy and final stable carbocation INT11A show that minima are very shallow and are, therefore, not equilibrated during the MD trajectories (fig. S39) (34).
Reactivity of acyclic vinyl triflates
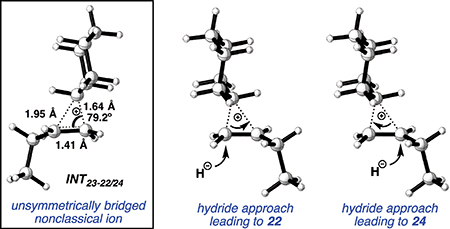
Having established the catalytic, intermolecular C–H alkylation reactions of cyclic vinyl triflates, and having come to a deeper understanding of the nonclassical reactivity of cyclic vinyl cations, we extended our investigations to the reactivities of acyclic vinyl triflates. Acyclic vinyl cations have constituted most of the previous experimental and theoretical studies of dicoordinated cations, as cyclic variants have traditionally proven difficult to generate under solvolytic conditions (35). Moreover, depending on substitution, several studies support nonclassical ground-state bonding in vinyl cations, structures reminiscent of protonated alkynes (36–38). Subjection of butenyl triflate 21 to the reaction conditions led to high-yielding reductive alkylation of cyclohexane, providing 2-cyclohexylbutane (22) in 85% yield (Table 2, entry 1). We found that the treatment of a cyclohexane solution of but-1-en-2-yl triflate (23) with 2 mol % of [Ph3C]+[HCB11Cl11]− and 1.5 equivalents of triethylsilane led to formation of a 1:1 mixture of 1-cyclohexylbutane (24) and 2-cyclohexylbutane (22) (Table 2, entry 2). Moreover, use of a 1:1 mixture of C6D12:C6H12 in this system led to exclusive formation of D12 and D0 isomers, without any detectable amount of crossover products (that is, C10H8+nD12-n or C10H20–nDn, n ≠ 0) that would be expected in a stepwise process (fig. S24) (34, 39). Use of terminal triflate 25 (Table 2, entry 3) resulted in an identical product distribution to that of entry 2, albeit requiring extended reaction times. Although primary vinyl cations are difficult to generate under solvolytic conditions (11), here the abstraction of the terminal triflate by silylium cation may provide a lower-energy transition through favorable Si–O bond formation. Moreover, a favorable pathway for a concerted triflate abstraction and hydride migration leading directly to a secondary vinyl cation has been calculated (fig. S43). Carrying out the reaction at −40°C in chloroform solvent allowed for selective formation of 1-cyclohexylbutane (24) (about 2:1) (Table 2, entry 4). These results are consistent with our view of a nonclassical intermediate (Table 2, INT23–22/24) (movie S5), where hydride delivery from silane determines product distributions. In the absence of coordinating solvent (entries 1 and 2), hyperconjugative stabilization may lead to a greater degree of bridging. As we increase the amount of chloroform solvent to attenuate hyperconjugative effects, stabilization of a more classical 2° carbocation, instead of the energetically prohibitive 1° carbocation, would lead to internal Si-H quenching and the terminal product 24. The important observation of modulated product distributions in polar solvent (entry 4) implies that the product ratios of reactions that proceed through unsymmetrical nonclassical species can be dictated by experimental factors that influence the degree of bridging.
Table 2.
Reactions of acyclic vinyl triflates and calculated nonclassical INT23–22/24 for insertion of cyclohexane into butenyl cation.
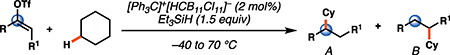 | |||||
|---|---|---|---|---|---|
| Entry | Substrate | Solvent | Temp. (°C) | Product | |
| 1* | 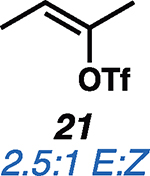 |
C6H12 | 30 | 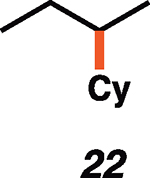 |
85∥ |
| 2† | 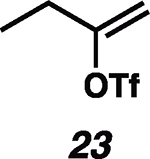 |
C6H12 | 30 | 40, 39∥ | |
| 3‡ | 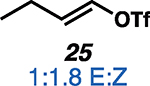 |
C6H12 | 70 | 16, 19∥ | |
| 4§ | 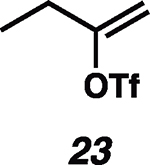 |
CHCl3/C6H12 | −40 |  |
17, 34∥ |
 | |||||
Reaction performed at 0.1 M and 30°C, with triisopropylsilane used instead of triethylsilane.
Reaction performed at 0.005 M and 30°C.
Reaction performed at 0.1 M and 70°C.
Reaction performed at 0.008 M (1:1 v:v), 5 mol % catalyst loading, and −40°C.
Yield determined by GC-FID with nonane as an internal standard.
R, alkyl or H; R1, alkyl or H.
Isotopic labeling studies
We also carried out deuterium labeling studies to further probe the mechanism. First, competition experiments with a 1:1 mixture of C6H12:C6D12 provided a 1:1 mixture of eyclohexyleyclohexane-d0 and cyclohexylcyclohexane-d12, supporting a concerted reaction pathway with C–H insertion being a diffusion-controlled event (fig. S22). The use of triethylsilane-d1 with cyclohexane and cyclo- hexenyl triflate (8) resulted in high levels of deuterium incorporation at the tertiary carbon, supporting the formation of a persistent 3° carbocation (Figs. 1C and 3A). However, this experiment does not differentiate a 1,1- from a 1,2-insertion mechanism; the 2° carbocation (10) formed via 1,1 insertion will always proceed to the more stable 3° carbocation 11 (Fig. 1C). To tease apart these different C–H insertion mechanisms, we prepared 3-phenylcyclohexenyl triflate 26 in the hope that migration of the benzylic C3 hydride of carbocation intermediate 27 would be competitive with C1-deuteride migration, allowing for direct experimental evidence of a 1,1-insertion event (Fig. 3B). Upon exposure of triflate 26 to our reaction conditions, using cyclohexane-d12 solvent and triethylsilane, we observed 67% deuterium incorporation at C2, 65% at C1, and 35% at C4 (Fig. 3C). In total, we observed 1.6 deuterides incorporated at these three carbons (C1, C2, and C4), accounting for 80% of the two cyclohexane-d12 deuterides prone to migration. Here we posit that following 1,1 insertion, C3-hydride migration produces benzylic cation 29, whereas C1-deuteride migration leads to tertiary C1 and C4 carbocations 30 and 31. All three of these species (29 to 31) can be quenched by Si–H reduction. Further confirming migration of the C3-benzylic H, using C6D12 and triethylsilane-d1, we observed 25% deuterium incorporation at C3 and 75% at C2 (32, Fig. 3D). Here D incorporation at C3 supports the formation of a benzylic carbocation that results from 1,2-hydride migration to quench a transient non-classical bridged cation (33). Moreover, use of C3-deuterated substrate 34 (Fig. 3E) with C6H12 leads to D incorporation at C2 (35, about 12%), further supporting the migration of the benzylic deuteride to quench the carbocation (36) generated upon insertion. The lower levels of C2-deuteride incorporation in product 35 can be attributed to kinetic isotope effects, as the C3 deuteride (36) will migrate slower than the C3 hydride in cation 33, whereas the C1 hydride of cation 36 will migrate faster in this system than the deuteride of transient 33 (40). The fact that less migration of the benzylic C3 hydride is observed over the tertiary C1 hydride supports the nonclassical nature of the reaction, where a discrete classical secondary cation is not formed (Fig. 3D).
Fig. 3. Deuterium labeling studies of cyclohexenyl triflate derivatives.

(A) Deuterium labeling experiment to support charge propagation step. (B) Experimental design to probe 1,1- versus 1,2-C–H insertion. (C to E) Mechanistic experiments suggesting benzylic 1,2-hydride and deuteride shifts of the resulting bridged cation. Ph, phenyl.
Reactivity of vinyl cations with arenes
Having established that vinyl triflates are competent vinyl cation precursors under silylium catalysis conditions and that these reactive intermediates undergo efficient sp3 C–H functionalization reactions, we sought to investigate their reactivity with arenes. It has been reported that cyclic vinyl triflates are poor electrophiles in Friedel-Crafts arylation reactions (41). This finding has been attributed to the poor electrophilicity of vinyl cations (15) and to the slow ionization of vinyl cation precursors in arene solvents (41). In early studies, cyclohexenyl triflates were completely unreactive toward arene nucleophiles, whereas triflates with larger ring sizes participated in Friedel-Crafts reactions at increased temperatures, albeit in poor yields (41). We posited that use of silylium-carborane salts would allow for mild ionization of cyclic vinyl triflates in nonpolar solvents, allowing for facile Friedel-Crafts arylation reactions. We were pleased to find that with four equivalents of benzene in pentane solvent (1:22 molar ratio of benzene: pentane), cyclohexenyl triflate (8) underwent smooth reductive arylation to yield phenylcyclohexane in 79% yield in 2 hours at room temperature (Fig. 4). In this reaction, the mass balance is composed of cyclohexylpentane isomers. Although we posit that triflate abstraction is rate limiting, we hypothesize that the product-determining step is the C–C bond–forming event, with the barrier for attack of the arene π-system falling nearly 20 kcal/mol below the barrier for C–H insertion (fig. S44). In addition to benzene, electron-poor haloarenes such as difluorobenzene and dichlorobenzene underwent smooth C–C bond formation to yield cyclohexylated haloarenes in synthetically useful yields. Likewise, electron-rich arenes, such as mesitylene, were competent nucleophiles. Cyclohexenyl triflates bearing substituents at the 4- or 5-position could also be arylated, including the enol triflate derived from 5α-cholestan-3-one, which yielded an arylated steroid core in 90% yield and 8:1 d.r. Various ring sizes were also competent under these reaction conditions, with cyclopentenyl triflate and cycloheptenyl triflate undergoing smooth reductive alkylation with benzene reaction partners in 64 and 71% yields, respectively. Cyclobutenyl triflate participated in this reductive Friedel-Crafts alkylation, as did aromatic alkenes. The triflate derived from α-tetralone was reductively arylated in 43% yield, and acetophenone-derived acyclic triflates were also arylated in 51 to 77% yield (Fig. 4, entries 15 and 16). Simple acyclic vinyl triflates were competent electrophiles for arylation by both electron-poor and electron-rich arenes, requiring as little as 10 equivalents of arene in chloroform solvent at −40°C (Fig. 4, entries 17 to 20).
Fig. 4. Reductive arylation of vinyl triflates.

*Reaction performed with 1.2 equivalents triethylsilane. †Triisopropylsilane used instead of triethylsilane. ‡Yield determined by GC-FID, with nonane as an internal standard. §Yield determined by nuclear magnetic resonance with an internal standard. R, alkyl, aryl, or H; R1, alkyl, halide, or H; R2, phenyl or ethyl; t-Bu, tert-butyl.
Outlook
We have shown that vinyl cations, the subject of numerous computational and experimental studies, are now accessible synthetically from simple vinyl triflates using WCA salts under mild conditions. The nonnucleophilic nature of the WCA allows these unstabilized vinyl cations to engage in C–C bond–forming reactions with alkanes and a variety of arenes, modes of reactivity that have been largely unreported despite extensive previous work. We find that the C–H insertion reactions of vinyl cations proceed through mechanisms that feature post–transition state bifurcation and nonclassical ions, mechanistic features that are common in terpene biosynthesis (32) but rarely found in synthetic methodology. These findings lay the conceptual and experimental groundwork for further discoveries in the field of alkane C–H bond functionalization using ketone derivatives and WCA catalysis.
Supplementary Material
ACKNOWLEDGMENTS
H.M.N. thanks M. Jung for reagents and advice.
Funding: Support was generously provided by the David and Lucile Packard Foundation (to H.M.N.), the Alfred P. Sloan Foundation (to H.M.N.), the National Science Foundation (CHE-1361104 to K.N.H.), and the National Natural Science Foundation of China (grant nos. 11504130, 51673164, and 21501169 to L.Z.). A.L.B. thanks the Christopher S. Foote Fellowship for funding. The authors thank the UCLA Molecular Instrumentation Center for NMR instrumentation, x-ray crystallography, and the Mass Spectrometry facility at the University of California, Irvine.
Footnotes
Competing interests: The authors declare no competing financial interests.
Data and materials availability: Crystallographic data are available free of charge from the Cambridge Crystallographic Data Centre under CCDC 1838441. Additional experimental procedures and characterization data are provided in the supplementary materials.
REFERENCES AND NOTES
- 1.Olah GA, J. Org. Chem 66, 5943–5957 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Jacobs TL, Searles S Jr., J. Am. Chem. Soc 66, 686–689 (1944). [Google Scholar]
- 3.Grob CA, Csapilla J, Cseh G, Helv. Chim. Acta 47, 1590–1602 (1964). [Google Scholar]
- 4.Hanack M, Angew. Chem. Int. Ed. Engl 17, 333–341 (1978). [Google Scholar]
- 5.Rappoport Z, Gal A, J. Am. Chem. Soc 91, 5246–5254 (1969). [Google Scholar]
- 6.Radom L, Hariharan PC, Pople JA, Schleyer PR, J. Am. Chem. Soc 95, 6531–6544 (1973). [Google Scholar]
- 7.Stang PJ, Rappoport Z, Dicoordinated Carbocations (Wiley, 1997). [Google Scholar]
- 8.Sherrod SA, Bergman RG, J. Am. Chem. Soc 91, 2115–2117 (1969). [Google Scholar]
- 9.Müller T, Juhasz M, Reed CA, Angew. Chem. Int. Ed 43, 1543–1546 (2004). [DOI] [PubMed] [Google Scholar]
- 10.Jones WM, Miller FW, J. Am. Chem. Soc 89, 1960–1962 (1967). [Google Scholar]
- 11.Hinkle RJ, McNeil AJ, Thomas QA, Andrews MN, J. Am. Chem. Soc 121, 7437–7438 (1999). [Google Scholar]
- 12.Apeloig Y, Franke W, Rappoport Z, Schwarz H, Stahl D, J. Am. Chem. Soc 103, 2770–2780 (1981). [Google Scholar]
- 13.Pople JA, Lathan WA, Hehre WJ, J. Am. Chem. Soc 93, 808–815 (1971). [Google Scholar]
- 14.Pellicciari R et al. , J. Am. Chem. Soc 118, 1–12 (1996). [Google Scholar]
- 15.Byrne PA, Kobayashi S, Würthwein EU, Ammer J, Mayr H, J. Am. Chem. Soc 139, 1499–1511 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Walkinshaw AJ, Xu W, Suero MG, Gaunt MJ, J. Am. Chem. Soc 135, 12532–12535 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Johnson WS et al. , J. Am. Chem. Soc 103, 88–98 (1981). [Google Scholar]
- 18.Hanack M, Acc. Chem. Res 3, 209–216 (1970). [Google Scholar]
- 19.Stang PJ, Rappaport Z, Hanack M, Subramanian LR, Vinyl Cations (Academic Press, 1979). [Google Scholar]
- 20.Shao B, Bagdasarian AL, Popov S, Nelson HM, Science 355, 1403–1407 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Lamparter E, Hanack M, Eur. J. Inorg. Chem 105, 3789–3793 (1972). [Google Scholar]
- 22.Hargrove RJ, Stang PJ, Tetrahedron 32, 37–41 (1976). [Google Scholar]
- 23.Olah GA, Mayr H, J. Am. Chem. Soc 98, 7333–7340 (1976). [Google Scholar]
- 24.Kanishchev MI, Shegolev AA, Smit WA, Caple R, Kelner MJ, J. Am. Chem. Soc 101, 5660–5671 (1979). [Google Scholar]
- 25.Fornarini S, Speranza M, J. Phys. Chem 91, 2154–2160 (1987). [Google Scholar]
- 26.Biermann U, Koch R, Metzger JO, Angew. Chem. Int. Ed 45, 3076–3079 (2006). [DOI] [PubMed] [Google Scholar]
- 27.Cleary SE, Hensinger MJ, Brewer M, Chem. Sci 8, 6810–6814 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Körbe S, Schreiber PJ, Michl J, Chem. Rev 106, 5208–5249 (2006). [DOI] [PubMed] [Google Scholar]
- 29.Carey FA, Tremper HS, J. Org. Chem 36, 758–761 (1971). [Google Scholar]
- 30.Ess DH et al. , Angew. Chem. Int. Ed 47, 7592–7601 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yu P, Patel A, Houk KN, J. Am. Chem. Soc 137, 13518–13523 (2015). [DOI] [PubMed] [Google Scholar]
- 32.Hong YJ, Tantillo DJ, Nat. Chem 1, 384–389 (2009). [DOI] [PubMed] [Google Scholar]
- 33.Schleyer PR et al. , J. Am. Chem. Soc 93, 1513–1516 (1971). [Google Scholar]
- 34. See supplementary materials.
- 35.Okuyama T, Takino T, Sueda T, Ochiai M, J. Am. Chem. Soc 117, 3360–3367 (1995). [Google Scholar]
- 36.Curtiss LA, Pople JA, J. Chem. Phys 88, 7405–7409 (1988). [Google Scholar]
- 37.Weber J, Yoshimine M, McLean AD, J. Chem. Phys 64, 4159–4164 (1976). [Google Scholar]
- 38.Psciuk BT, Benderskii VA, Schlegel HB, Theor. Chem. Acc 118, 75–80 (2007). [Google Scholar]
- 39.Olah GA et al. , J. Am. Chem. Soc 116, 3187–3191 (1994). [Google Scholar]
- 40.Coxon JM, Thorpe AJ, J. Org. Chem 65, 8421–8429 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Stang PJ, Anderson A, J. Am. Chem. Soc 100, 1520–1525 (1978). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.