

A solid anhydrous hydrogen fluoride equivalent was prepared by mixing HF gas with an inexpensive anion exchange resin (A26-HF, HF content 30% wt/wt). This reagent is capable of hydrofluorinating alkenes, of ring-opening aziridines to give fluoroamines, and of yielding fluorotetrahydropyrans through a fluoro-Prins reaction, all with high selectivity under mild conditions. Our ion exchange resin supported HF reagent could be easily packed into a plastic column and used in flow fluorinations.
Introduction
Fluorine’s proven worth in enhancing the pharmacokinetic and physicochemical properties of active pharmaceutical ingredients, such as improved metabolic stability and enhanced membrane permeation,1 has spurred the explosive growth of synthetic strategies for the selective incorporation of fluorine in organic molecules.2–8 Many fluorinating agents have been developed over the years with the goal of solving this fundamental problem, with varying degrees of success. The fluorinating agents reported are, for the most part, expensive or non-atom economical, they require complex reaction conditions or have a narrow range of synthetic applications.9, 10 Arguably hydrogen fluoride is the most atom-economical nucleophilic fluorination agent, but it is also corrosive, highly toxic and a reactive gas at ambient conditions.11, 12 Amines (pyridine, triethylamine) or a hydrogen bond acceptor (DMPU), have been used to stabilize gaseous HF, forming liquid solutions such as Olah’s reagent (pyridine·9HF),11 triethylamine/HF (Et3N·3HF),13 or DMPU.12HF.14 However, these stabilizers reduce the acidity of the resulting HF solution and decrease its reactivity, especially in reactions that need strong acid media. Moreover, after the reaction is completed, the excess of HF needs to be neutralized, and pyridine, triethylamine or DMPU need to be separated during the reaction work-up.
Fluorinations, like most organic reactions, are typically conducted in conventional batch reactors. In contrast to batch reactors, continuous flow and microreactor methodologies offer advantages in automation, control of the reaction conditions, heat and mass transfer, safety profile, and scalability. To the best of our knowledge, hydrofluorinations in a continuous flow reactor using HF have not been reported.15–17 Preparing a solid equivalent of anhydrous HF would enable flow hydrofluorination but, only two attempts to prepare a polymer-supported HF reagent have been reported, the most recent one over 25 years ago. Zupan and co-workers reported a copolymer of styrene and 4-vinylpyridine that complexed with HF.18–20 Olah and co-workers prepared a polymeric hydrogen fluoride reagent using poly-4-vinylpyridium as a support.21–23 Both polymer-supported HF reagents have met with limited success. In addition to the high expense of the polymers used in both methods, the pyridine presence reduces the acidity of both polymeric HF reagents, curtailing their effectiveness. We have recently reported that HF formed a stable and easily handled complex with KHSO4--an ionic hydrogen bond acceptor. Its effectiveness as a fluorinating agent was demonstrated in the hydrofluorination of alkenes.24 If we could affix HSO4− to an inexpensive ion exchange resin the bisulfate anion would not only stabilize HF through hydrogen bonding but it would also increase the acidity of the resulting ion exchange resin-supported HF complex. This complex could be used in batch hydrofluorination—with the added advantage of a simplified reaction workup (only filtration or decantation are needed)—or in continuous flow hydrofluorination. We are now pleased to report that a solid anhydrous hydrogen fluoride equivalent was prepared by mixing HF gas with an inexpensive anion exchange resin. The resulting ion exchange supported-HF reagent was used effectively in the hydrofluorination of alkenes, in the ring opening of aziridines and in the fluoro-Prins reaction as well as in the continuous flow hydrofluorination of an alkene.
Results and discussion
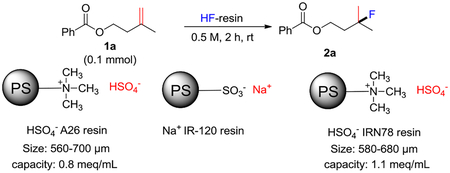
We examined several types of commercially available anionic resins: Amberlyst A-21 has a free amine functional group, whereas Amberlyst A-26 and Amberlite IRN78 possess a quaternary ammonium functional group. The preparation procedure is straightforward: the commercially available ion exchange resin was treated with a slight excess of aqueous sulfuric acid to substitute the original anion in the resin with HSO4− (Scheme 1). The modified ion exchange resin was dried and complexed with anhydrous hydrogen fluoride to furnish the ion exchange resin supported HF reagent (for detailed preparation please see the supporting information). We tested the reactivity of the ion exchange resin-supported anhydrous hydrogen fluoride reagent using the hydrofluorination of alkene 1a as our model reaction. We found that Amberlyst A-26 affixed more hydrogen fluoride per gram of dry resin and showed higher reactivity in the hydrofluorination 1a than the other two resin-HF complexes (Table 1, entry 4 vs entries 1, 2). To underscore the importance of the bisulfate in the polymeric HF complex, we tested another ion exchange resin, Amberlite IR-120 Na+, bearing an SO3−Na+ functionality. As expected, the resulting complex showed a lower reactivity than the A-26 resin HF complex because of its weaker acidity (Table 1, entry 3 vs entry 4). Solvent optimization indicated that dichloroethane (DCE) was the best (Table 1, entries 4, 7–14). Further optimization demonstrated that 15 equivalents of HF (Table 1, entries 4–6) and a reaction time of 15 h gave the best results (Table 1, entry 15).
Scheme 1.

Preparation of polymer supported HF reagent.
Table 1.
Reaction condition optimization for hydrofluorination of alkene.
| entry | resin - HF (wt/wt%) | HF equiv | solvent | yield (%)a |
|---|---|---|---|---|
| 1 | HSO4− A21 resin- HF (30%) | 10 | DCE | 19 |
| 2 | HSO4− IRN78 resin- HF (28%) | 10 | DCE | 23 |
| 3 | IR-120-Na+ resin - HF (23%) | 10 | DCE | 0 |
| 4 | HSO4− A26 resin - HF (30%) | 10 | DCE | 28 |
| 5 | HSO4− A26 resin - HF (30%) | 15 | DCE | 58 |
| 6 | HSO4− A26 resin - HF (30%) | 5 | DCE | 7 |
| 7 | HSO4− A26 resin - HF (30%) | 10 | CH3CN | 0 |
| 8 | HSO4− A26 resin - HF (30%) | 10 | DCM | 22 |
| 9 | HSO4− A26 resin - HF (30%) | 10 | Dioxane | 0 |
| 10 | HSO4− A26 resin - HF (30%) | 10 | DMF | 0 |
| 11 | HSO4− A26 resin - HF (30%) | 10 | DMSO | 0 |
| 12 | HSO4− A26 resin - HF (30%) | 10 | Et2O | 0 |
| 13 | HSO4− A26 resin - HF (30%) | 10 | EtOAc | 0 |
| 14 | HSO4− A26 resin - HF (30%) | 10 | Toluene | 51 |
| 15b | HSO4− A26 resin - HF (30%) | 15 | DCE | 87 |
NMR yield with benzotrifluoride as internal standard; b 15 hours.
Having found an optimal reaction protocol in the hydrofluorination of alkenes, we explored the substrate scope and functional group tolerance. As shown in Table 2, disubstituted and trisubstituted alkenes showed good to excellent yield. However, monosubstituted alkenes were unreactive. A wide range of functional groups such as esters (2a, 2g, 2h, 2n, 2p), ethers (2b - 2f), nitro (2c), nitrile (2d), aldehyde (2e), ketones (2f, 2k, 2m), alcohols (2l, 2o) were well tolerated. Acceptable to very good yields were obtained with different heterocyclic substrates like thiophene (2g), pyridine (2h), indazole (2i) and benzotriazole (2j). It should be noted that longer reaction times were needed for substrates containing basic moieties (2h, 2i, 2j). Since alkenes are commonly found in natural products, we screened an estrone-tethered disubstituted alkene, which furnished a good yield of the product (2k). We found that a natural product with a secondary alcohol, such as (−)-dihydrocarveol (2l), gave only a moderate yield of the product whereas nootkatone, possessing an α, β-unsaturated ketone group (2m), gave a very good chemical yield. The hydrofluorination of (−)-β-citronellol, which bears a primary alcohol functionality, gave also a very good yield of the product 2o. And we found that the cyclopropane motif remained intact after the reaction of ethyl chrysanthemate with HF (2p).
Table 2.
Hydrofluorination of alkenes.a

|
Reaction conditions: 1 (0.2 mmol), A26 Resin - HF(15 equiv HF), DCE (0.4 mL), room temperature, 15h. All yields are for the isolated product.
The synthesis of β-fluoroamines via ring-opening of aziridines with HF is, in principle, an ideal preparation method because of the low cost of reagents, mild reaction conditions and general accessibility of substrates, but the production of regioisomers and stereoisomers has muddled the efficiency of this strategy.25–28 Recently, we reported that DMPU-HF achieved regio- and stereo-control in the fluorination of aziridines.29 However, the reaction introduces DMPU, which complicates the purification step. Using our modified ion exchange resin supported HF (resin A-26 - HF), we achieved high selectivity and good functional group tolerance and avoided the introduction of impurities to the reaction mixture and simplified the workup procedure (only filtration was needed).
As shown in the Table 2, hydrofluorination of the mono alkyl-, aryl-, and benzyl-substituted N-tosylaziridines (3a - 3d) occurred at the most substituted carbon and delivered primary alkyl amines (4a - 4d) in good to excellent yields. The trisubstituted aziridine 3f was fluorinated at the more substituted carbon in acceptable yield. When 2-ethyl-3-methyl-1-tosylaziridine was the substrate, an inseparable mixture of regioisomers and diastereomers was generated (4f). A bicyclic aziridine was also tested and converted to trans-β-fluoroamine 4g in good yield and diastereoselectivity. Various N-protecting groups such as tosyl (4a - 4g), benzoyl (4h), and Cbz (carboxybenzyl) (4i) were well tolerated under the reaction conditions, affording the corresponding products in excellent yields and regioselectivity.
The Prins reaction of a homoallylic alcohol and an aldehyde in the presence of an acid is a well-established synthetic methodology for the preparation of tetrahydropyrans.30, 31 However, there are only few reports on the fluoro-Prins reaction for the synthesis of fluorinated tetrahydropyrans.32–35 Moreover, most of the reported syntheses of fluorinated tetrahydropyrans suffer from either low yields or, especially, low diastereoselectivity. With our ion exchange resin supported-HF reagent, we obtained fluorinated tetrahydropyrans with high diastereoselectivities and in good yields. The high stereoselectivity is consistent with previous studies by Rychnovsky et al.,36–38 where the cyclization proceeds through a chair transition state with a syn addition of nucleophile across the alkene. As illustrated in Table 3, both aromatic (7a - 7f) and aliphatic aldehydes (7g, 7h) gave good to excellent yields of the corresponding products.
Table 3.
Ring-opening of aziridines 3.a

|
Reaction conditions: 3 (0.2 mmol), A26 Resin – HF (5 equiv HF), DCE (0.4 mL), room temperature, 3h. All yields are for the isolated product.
Numbers in parenthesis are regioisomeric ratio determined by 19F NMR.
starting aziridine are mixture of cis- and trans-isomers.
Electron-poor aldehyde substrates (7b - 7e) gave higher yields than electron rich aldehydes (7f). Notably, a disubstituted fluorinated tetrahydropyran could be synthesized with good yield and diastereoselectivity when isoprenol was used as substrate (7i). Next, we compared the reactivity of our polymer supported HF reagent with the only commercially available polymeric hydrogen fluoride reagent--poly-4-vinylpyridium poly(hydrogen fluoride) (PVP-HF) (equations A, B and C). Our reagent showed much better efficiency in all three reactions. It should be noted that our ion exchange resin is much less costly than PVP, making it suitable for larger scale synthesis.

As mentioned above, the use of HF based reagents under flow conditions has not been reported. Our ionic exchange resin-supported HF reagent could be easily packed into a plastic tube or a column suitable for flow reactions (see supporting information). We used the hydrofluorination of alkene 1a to test the flow conditions at room temperature (Figure 1). A solution of alkene was injected into the packed column and the column was flushed with neat solvent (DCE) at 0.5 mL/h flow rate. We used an HF-scavenger at the bottom of column to prevent excess HF from enter in the eluent stream and to avoid etching the glassware. The eluent was collected and concentrated to afford the crude product in 92% NMR yield. This flow reactor could be used for large scale synthesis. In addition, if desired, the ion exchange resin could be recycled to complex with more HF and reused.
Figure 1.

Use of polymer supported HF reagent in flow reaction.

The thermal stability of the polymeric HF reagent (resin A-26 - HF) was also investigated (Figure 2). We found only a 2 wt% HF loss when an open vial containing the resin was placed in a well vented fume hood for 30 h. The stability of this reagent should facilitate its packing, transportation and storage (see supporting information).
Figure 2.

Thermostability test of polymer supported HF reagent at room temperature.
Conclusions
In summary, we have developed an inexpensive and stable ion exchange resin-supported anhydrous hydrogen fluoride reagent. The reagent showed its effectiveness in three reactions that required strong acid conditions. This reagent was also used in a flow reactor with a simplified reaction workup procedure. Further applications and possible commercialization are currently being considered in our laboratory.
Supplementary Material
Table 4.
Fluoro-Prins reaction.a

|
Reaction conditions: 5 (0.2 mmol), 6 (0.2 mmol), A26 Resin - HF(10 equiv HF), DCE (0.4 mL), room temperature, 5 h. All yields are for the isolated product.
Acknowledgements
We are grateful to the National Institutes of Health for financial support (R01GM121660). B. Xu is grateful to the National Science Foundation of China (NSFC-21672035) and the China Recruitment Program of Global Experts for financial support. B. S. Bajwa is grateful to the Camille and Henry Dreyfus Foundation and to the California State University, Fresno for a summer research scholarship.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
The authors declare no conflict of interest.
Contributor Information
Gerald B. Hammond, Email: gb.hammond@louisville.edu.
Bo Xu, Email: bo.xu@dhu.edu.cn.
Notes and references
- 1.Gillis EP, Eastman KJ, Hill MD, Donnelly DJ and Meanwell NA, J. Med. Chem, 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- 2.Uneyama K, Organofluorine chemistry, Blackwell Pub., Oxford; Ames, Iowa, 2006. [Google Scholar]
- 3.Kirsch P, Modern fluoroorganic chemistry : synthesis, reactivity, applications, Wiley-VCH, Verlag GmbH & Co. KGaA, Weinheim, Germany, Second, completely revised, and enlarged edition. edn., 2013. [Google Scholar]
- 4.Muller K, Faeh C and Diederich F, Science, 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- 5.O’Hagan D, Chem. Soc. Rev, 2008, 37, 308–319. [DOI] [PubMed] [Google Scholar]
- 6.Champagne PA, Desroches J, Hamel J-D, Vandamme M and Paquin J-F, Chem. Rev, 2015, 115, 9073–9174. [DOI] [PubMed] [Google Scholar]
- 7.Campbell MG and Ritter T, Chem. Rev, 2015, 115, 612–633. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Y, Han J, Wang J, Shibata N, Sodeoka M, Soloshonok VA, Coelho JAS and Toste FD, Chem. Rev, 2018, 118, 3887–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirk KL, Org. Process Res. Dev, 2008, 12, 305–321. [Google Scholar]
- 10.Shimizu M and Hiyama T, Angew. Chem. Int. Ed, 2005, 44, 214–231. [DOI] [PubMed] [Google Scholar]
- 11.Olah GA, Welch JT, Vankar YD, Nojima M, Kerekes I and Olah JA, J. Org. Chem, 1979, 44, 3872–3881. [Google Scholar]
- 12.Wade TN, J. Org. Chem, 1980, 45, 5328–5333. [Google Scholar]
- 13.Haufe G, J. Prakt. Chem. /Chem-Ztg, 1996, 338, 99–113. [Google Scholar]
- 14.Okoromoba OE, Han J, Hammond GB and Xu B, J. Am. Chem. Soc, 2014, 136, 14381–14384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Amii H, Nagaki A and Yoshida J, Beilstein J. Org. Chem, 2013, 9, 2793–2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McPake CB and Sandford G, Org. Process Res. Dev, 2012, 16, 844–851. [Google Scholar]
- 17.Cantillo D and Kappe CO, React. Chem. Eng, 2017, 2, 7–19. [Google Scholar]
- 18.Zupan M, šket B and Johar Y, J, Marcromol. Sci., Chem., A, 1982, 17, 759–769. [Google Scholar]
- 19.Maeda M, Abe M and Kojima M, J. Fluorine Chem, 1987, 34, 337–346. [Google Scholar]
- 20.Gregorčič A and Zupan M, J. Fluorine Chem, 1984, 24, 291–302. [Google Scholar]
- 21.Olah GA and Li X-Y, Synlett, 1990, 1990, 267–269. [Google Scholar]
- 22.Olah GA, Li X-Y, Wang Q and Surya Prakash GK, Synthesis, 1993, 1993, 693–699. [Google Scholar]
- 23.Olah GA, Prakash GKS and Reddy VP, in Ionic Liquids in Organic Synthesis, American Chemical Society, 2007, vol. 950, ch. 4, pp. 36–57. [Google Scholar]
- 24.Lu Z, Zeng X, Hammond GB and Xu B, J. Am. Chem. Soc, 2017, 139, 18202–18205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Métro T-X, Duthion B, Gomez Pardo D and Cossy J, Chem. Soc. Rev, 2010, 39, 89–102. [DOI] [PubMed] [Google Scholar]
- 26.Fan R-H, Zhou Y-G, Zhang W-X, Hou X-L and Dai L-X, J. Org. Chem, 2004, 69, 335–338. [DOI] [PubMed] [Google Scholar]
- 27.Zhang WX, Su L, Hu WG and Zhou J, Synlett, 2012, 23, 2413–2415. [Google Scholar]
- 28.van Oosten EM, Gerken M, Hazendonk P, Shank R, Houle S, Wilson AA and Vasdev N, Tetrahedron Lett, 2011, 52, 4114–4116. [Google Scholar]
- 29.Okoromoba OE, Li Z, Robertson N, Mashuta MS, Couto UR, Tormena CF, Xu B and Hammond GB, Chem. Commun, 2016, 52, 13353–13356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arundale E and Mikeska LA, Chem. Rev, 1952, 51, 505–555. [Google Scholar]
- 31.Alder RW, Harvey JN and Oakley MT, J. Am. Chem. Soc, 2002, 124, 4960–4961. [DOI] [PubMed] [Google Scholar]
- 32.Yadav JS, Subba Reddy BV, Anusha B, Subba Reddy UV and Bhadra Reddy VV, Tetrahedron Lett, 2010, 51, 2872–2874. [Google Scholar]
- 33.Bondalapati S, Reddy UC, Kundu DS and Saikia AK, J. Fluorine Chem, 2010, 131, 320–324. [Google Scholar]
- 34.Launay GG, Slawin AMZ and O’Hagan D, Beilstein J. Org. Chem, 2010, 6, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okoromoba OE, Hammond GB and Xu B, Org. Lett, 2015, 17, 3975–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jaber JJ, Mitsui K and Rychnovsky SD, J. Org. Chem, 2001, 66, 4679–4686. [DOI] [PubMed] [Google Scholar]
- 37.Jasti R, Vitale J and Rychnovsky SD, J. Am. Chem. Soc, 2004, 126, 9904–9905. [DOI] [PubMed] [Google Scholar]
- 38.Damera K, Yu B and Wang B, J. Org. Chem, 2015, 80, 5457–5463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.