

Abstract
The combination of two easily handled, highly acidic liquid HF complex reagents, DMPU-12HF and KHSO4-13HF, generated a highly acidic fluorination system that facilitated exclusive Markovnikov addition of HF to widely functionalized alkynes, including alkyne tethered drugs, or allenes to produce gem-difluorides with high atom economy, and with an easy workup.
Introduction
The value of fluorine in improving the pharmacokinetic and pharmacodynamic properties of active pharmaceutical ingredients, such as enhanced membrane permeation and superior metabolic stability1 has contributed to the explosive growth of synthetic strategies for the selective fluorination of organic molecules.2–8 More specifically, molecules with a gem-difluoride moiety have received widespread coverage in drug development. A case in point is vinflunine, a gem-difluoromethylenated derivative of a vinca alkaloid, currently used as second-line chemotherapy for advanced transitional cell carcinoma.9–11
There are three primary strategies for the synthesis of gem-difluorides from the perspective of the substrates (Scheme 1).12 The first strategy is the direct dihydrofluorination of alkynes using hydrogen fluoride reagents (Scheme 1a). This method is one of the most straightforward and atom-economical protocols. Linn13 and Plueddeman14 pioneered this approach using anhydrous hydrogen fluoride (HF), but they only investigated non-functionalized alkynes. To avoid the inconvenience of handling HF gas, Olah15–18 developed a pyridine-based HF reagent and achieved hydrofluorination with a limited scope of alkyne substrates. Later, Renoux utilized a superacid system HF-SbF5 to gem-difluorinate aminoalkynes (Scheme 1a).19 The second strategy is the gem-difluorination of carbonyl compounds with various fluorinating reagents such as gaseous SF4,20 diethylaminosulfur trifluoride (DAST),21 bis(2-methoxyethyl) aminosulfur trifluoride (Deoxo-Fluor),22 aminodifluorosulfinium tetrafluoroborate (XtalFluor)23, and 2,2-difluoro-1,3-dimethylimidazolidine (DFI) (Scheme 1b).24 A third strategy encompasses various tactics such as the indirect fluorination of carbonyl derivatives with IF or BrF3,25 or the oxidative desulfurization-fluorination of ortho-thioesters, dithiolanes or thiocarbonyl derivatives with electrophilic halonium species26, 27 (Scheme 1c). Recently, Szabó utilized a fluoro-benzoiodoxole reagent on styrene substrates to accomplish the gem-difluorination. 28 All these methods have serious limitations such as poor substrate scope, low atom-economy and safety concerns about the fluorination reagents.
Scheme 1.

Strategies for the synthesis of gem-difluorides
We have recently reported a stable and easily handled HF complex, KHSO4-13HF. The high acidity and high fluoride nucleophilicity of this reagent made possible the direct hydrofluorination of alkenes via a bi-functional activation of hydrogen fluoride.29, 30 Given the ease of its addition to the double bond, we predicted a similar behavior with other unsaturated systems such as alkynes or allenes. We are pleased to report a neat and metal-free alkyne and allene hydrofluorination protocol using an easily handled, highly acidic liquid HF complex with an unmatched substrate scope, good functional group tolerance and very simple workup—calcium carbonate neutralization followed by chromatography.
Results and discussion
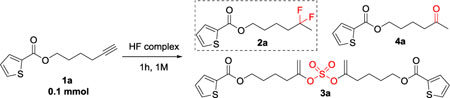
We used the hydrofluorination of alkyne 1a as our model system (Table 1). We first tested the reactivity of KHSO4-13HF (Table 1, entry 1). This reagent produced 9% of the gem-difluoride product but forming nearly equal amounts of dialkenylsulfate 3a and ketone 4a. This result showed that the HSO4− anion enhances the reactivity of the HF complex but also introduces competitive side reactions (nucleophilic attacks of HSO4− and water). For comparison, we tested other HF reagents, but no reactions took place (Table 1, entries 2, 3, 4). Using toluene as the solvent did not improve the outcome (Table 1, entry 5). We switched to HFIP since it can form a hydrogen bonding network which may further enhance the reactivity of acids.31 The reaction afforded more difluoride product but also gave an equal amount of sulfate dimer 3a and ketone 4a (Table 1, entry 6). Mixing K2SO4-14HF with different acids, such as trifluoroacetic acid (TFA) and triflic acid (TfOH), did not improve the outcome (Table 1, entries 7, 8). This result confirmed the importance of dual bifunctional activation role of KHSO4 in the system.29, 30 When we increased the reaction temperature to 50 °C, we managed to increase the conversion to product (Table 1, entry 9). To reduce the dialkenylsulfate product and augment the proportion of difluoride product, we introduced DMPU-12HF, an HF complex with high acidity developed in our laboratory, 32–35 our reason for this experiment was to increase the concentration of the fluoride source without weakening the acidity of the system. We obtained a higher proportion of difluoride compared to the sulfate byproduct (Table 1, entry 10). To underscore the importance of the KHSO4-13HF, we added KHSO4 instead of KHSO4-13HF with DMPU-12HF and we found that no reaction had occurred (Table 1, entry 11). When we conducted the reaction solventless, we obtained a higher yield of gem-difluoride product (Table 1, entry 12). We then varied the relative amounts of HF reagents and found that one equivalent of KHSO4-13HF and two equivalents of DMPU-12HF (Table 1, entries 12, 13, 14) gave the best results. When we extended the reaction time to 3 hours, we obtained optimal conversion and yield (Table 1, entry 15).
Table 1.
Reaction condition optimization for hydrofluorination of alkyne.a
 | ||||
|---|---|---|---|---|
| entry | HF complex (A-xHF) |
T (°C) |
solvent | yield (1a /2a /3a /4a)b |
| 1 | 1 equiv KHSO4-13HF | rt | DCE | 75% / 9% / 11% /5% |
| 2 | 1 equiv K2SO4-14HF | rt | DCE | 100% / 0 / 0 / 0 |
| 3 | 1 equiv Py-9HF | rt | DCE | 100% / 0 / 0 / 0 |
| 4 | 1 equiv DMPU-12HF | rt | DCE | 100% / 0 / 0 / 0 |
| 5 | 1 equiv KHSO4-13HF | rt | Toluene | 79% / 8% / 8% / 5% |
| 6 | 1 equiv KHSO4-13HF | rt | HFIP | 29% /23% /20% /28% |
| 7c | 1 equiv K2SO4-14HF | rt | DCE | 100% / 0 / 0 / 0 |
| 8d | 1 equiv K2SO4-14HF | rt | DCE | 100% / 0 / 0 / 0 |
| 9 | 1 equiv KHSO4-13HF | 50 | DCE | 62% /13%/ 16% /9% |
| 10 | 1 equiv DMPU-12HF + 1 equiv KHSO4-13HF | 50 | DCE | 48% /27% /14% /11% |
| 11e | 2 equiv DMPU-12HF | 50 | DCE | 100% / 0 / 0 / 0 |
| 12 | 2 equiv DMPU-12HF + 1 equiv KHSO4-13HF | 50 | Neat | 6% / 65% /12% /16% |
| 13 | 1 equiv DMPU-12HF + 0.5 equiv KHSO4-13HF | 50 | Neat | 46% / 39% / 6% /9% |
| 14 | 2 equiv DMPU-12HF + 0.5 equiv KHSO4-13HF | 50 | Neat | 38 / 46% /6% /10% |
| 15f | 2 equiv DMPU-12HF + 1 equiv KHSO4-13HF | 50 | Neat | 0 / 72% /12% /16% |
Reaction conditions: 1 (0.1 mmol), HF complex (1 equiv based on the complex A-xHF, equivalents of HF are x), solvent (0.1 mL), 1 hour.
NMR yields with benzotrifluoride as internal standard;
1 equiv TFA was added;
1 equiv TfOH was added;
1 equiv KHSO4 was added;
3 h reaction time;
Having established the optimal conditions for the hydrofluorination of an alkyne, we explored the substrate scope and functional group tolerance (Table 2). The distance between the location of the triple bond and the electron withdrawing substituent had a significant effect on the yield (2a-2d). This could be explained by the fact that electron withdrawing substituents reduce the electronic density of the alkyne, making it more inert to activation by protonation. Terminal and internal alkynes showed good yield (2e vs 2f). A wide range of functional groups such as esters (2a-2f, 2h-2j), ether (2g), nitro group (2e-2g, 2i), aldehyde (2h), amide (2j, 2k) and alcohol (2n) were well tolerated. The tolerance of the mesylate (2l) and tosylate groups (2m) enhanced the possibility of further functionalization. Furthermore, the hydrofluorination of a readily available iodoalkyne36–38 gave a good yield of the synthetically important β-iodo-difluoride (2i), which can be further functionalized for the synthesis of various gem-difluorides. The L-phenylalanine derivative 1j afforded the desired product 2j in 68% isolated yield.
Table 2.
Hydrofluorination of alkynes.a
 |
Reaction conditions: 1 (0.2 mmol), 1 equiv KHSO4-13HF, 2 equiv DMPU-12HF, neat, 50 °C, 3h. All yields are for the isolated product.
regioisomer ratio.
Our protocol worked satisfactorily well with substrates with a thiophene moiety (Table 2, 2a-2d). We explored other heterocycles, as illustrated in Table 3. We obtained acceptable to very good yields with different heterocyclic substrates including furan (2aa), thiazole (2ab), pyridine (2ac), pyrrole (2ad), indazole (2ae, 2af), benzotriazoles (2ag, 2ah) and indoles (2ai, 2aj, 2ak). Substrates containing basic functional groups (2ac, 2ae-2ah) needed longer reaction times. Our gem-difluorination protocol is an easy-to-use tool for the modification of drug molecules with diverse functional groups (Table 4). For example, we obtained a derivative of Probenecid (2al) in 83% yield. Probenecid is a prototypical uricosuric agent used to treat patients with renal impairment. On the other hand, a Febuxostat-tethered alkyne produced the corresponding difluoro product 2am in 71% yield. Febuxostat is used to treat hyperuricemia. This reaction tolerated an ester, nitrile, ether, and a thiazole ring present in Febuxostat. Telmisartan is used to manage hypertension; the gem-difluorination of its alkyne derivative furnished 2an in 71% yield, without affecting the benzoimidazole ring. These examples, using current drugs, demonstrate that our gem-difluorination protocol is suitable for the late-stage, protecting-group-free modification of biologically interesting molecules.
Table 3.
Hydrofluorination of alkynes with various heterocyclic substituents.a
 |
Reaction conditions: 1 (0.2 mmol), 1 equiv KHSO4-13HF, 2 equiv DMPU-12HF, neat, 50 °C, 3 h. All yields are for isolated products.
15 h reaction time.
Table 4.
Late-stage functionalization of known drugs.a
 |
Reaction conditions: 1 (0.2 mmol), 1 equiv KHSO4–13HF, 2 equiv DMPU-12HF, neat, 50 °C, 3h. All yields are for the isolated product.
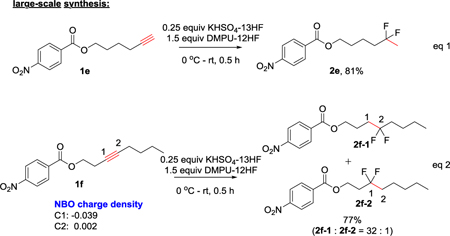
To further prove the expediency of our method, we conducted a gram scale synthesis. Both terminal (eq 1) and internal (eq 2) alkyne substrates gave good yields of products — the reaction was completed in only 30 minutes without heating and using less amount of HF reagents. Our protocol also exhibited very high regio-selectivity in the hydrofluorination of an internal alkyne (2f-1:2f-2 = 32:1, eq 2). The regio-selectivity could be easily predicted by NBO charge density via a simple DFT calculation of starting material 1f. The calculation shows the C1 is more electron rich than the C2 (eq 2). As predicted, the protonation occurred at C1 and fluorine atoms were added to the electron deficient carbon (C2 in 1f).
Finally, we investigated the gem-difluorination of allenes using our protocol (Table 5). Compared with alkyne substrates, the allene substrates were more reactive, and their reaction only needed DMPU-12HF for gem-difluorination. It should be noted that two regioisomers were obtained, with the 2,2-difluoride as the main product.
Table 5.
gem-Difluorination of allenes.a
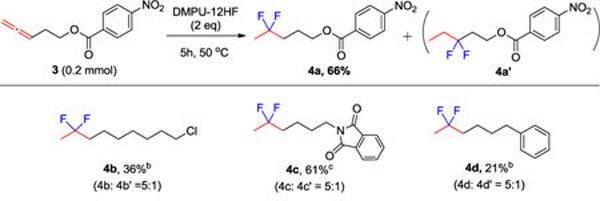 |
Reaction conditions: 3 (0.2 mmol), 2 equiv DMPU-12HF, neat, 50 °C, 5 h.
NMR yield.
isolated yield.
Conclusions
We have developed a direct regioselective gem-difluorination protocol using easily-handled hydrogen fluoride reagents. We demonstrated the effectiveness of this tactic with variously functionalized alkynes, including alkynes tethered to natural products and drug molecules. The protocol offers incomparable broad functionality tolerance, exclusive Markovnikov addition regioselectivity, high atom economy, easy workup procedure, and scalability in the synthesis of gem-difluorides from alkynes or allenes.
Supplementary Material
Acknowledgments
We are grateful to the National Institutes of Health for financial support (R01GM121660). B. Xu is thankful to the National Science Foundation of China (NSFC-21672035). B. S. Bajwa and S. Lee are grateful to the Camille and Henry Dreyfus Foundation and the California State University, Fresno for summer research scholarships.
Footnotes
Footnotes relating to the title and/or authors should appear here.
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
The authors declare no conflict of interest.
Notes and references
- 1.Gillis EP, Eastman KJ, Hill MD, Donnelly DJ and Meanwell NA, J. Med. Chem, 2015, 58, 8315–8359. [DOI] [PubMed] [Google Scholar]
- 2.Uneyama K, Organofluorine chemistry, Blackwell Pub., Oxford; Ames, Iowa, 2006. [Google Scholar]
- 3.Kirsch P, Modern fluoroorganic chemistry: synthesis, reactivity, applications, Wiley-VCH, Verlag GmbH & Co. KGaA, Weinheim, Germany, Second, completely revised, and enlarged edition. edn, 2013. [Google Scholar]
- 4.Muller K, Faeh C and Diederich F, Science, 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
- 5.O’Hagan D, Chem. Soc. Rev, 2008, 37, 308–319. [DOI] [PubMed] [Google Scholar]
- 6.Champagne PA, Desroches J, Hamel J-D, Vandamme M and Paquin J-F, Chem. Rev, 2015, 115, 9073–9174. [DOI] [PubMed] [Google Scholar]
- 7.Campbell MG and Ritter T, Chem. Rev, 2015, 115, 612–633. [DOI] [PubMed] [Google Scholar]
- 8.Zhu Y, Han J, Wang J, Shibata N, Sodeoka M, Soloshonok VA, Coelho JAS and Toste FD, Chem. Rev, 2018, 118, 3887–3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gerullis H, Wawroschek F, Köhne C-H and Ecke TH, Therapeutic Advances in Urology, 2016, 9, 28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bellmunt J, Théodore C, Demkov T, Komyakov B, Sengelov L, Daugaard G, Caty A, Carles J, Jagiello-Gruszfeld A, Karyakin O, Delgado F-M, Hurteloup P, Winquist E, Morsli N, Salhi Y, Culine S and von der Maase H, Journal of Clinical Oncology, 2009, 27, 4454–4461. [DOI] [PubMed] [Google Scholar]
- 11.Fahy J, Hellier P, Breillout F and Bailly C, Seminars in Oncology, 2008, 35, S3–S5. [DOI] [PubMed] [Google Scholar]
- 12.Tozer MJ and Herpin TF, Tetrahedron, 1996, 52, 8619–8683. [Google Scholar]
- 13.Grosse AV and Linn CB, J. Am. Chem. Soc, 1942, 64, 2289–2292. [Google Scholar]
- 14.Henne AL and Plueddeman EP, J. Am. Chem. Soc, 1943, 65, 587–589. [Google Scholar]
- 15.Olah GA, Mathew T, Goeppert A, Török B, Bucsi I, Li X-Y, Wang Q, Marinez ER, Batamack P, Aniszfeld R and Prakash GKS, J. Am. Chem. Soc, 2005, 127, 5964–5969. [DOI] [PubMed] [Google Scholar]
- 16.Olah GA, Li X-Y, Wang Q and Surya Prakash G. K., Synthesis, 1993, 1993, 693–699. [Google Scholar]
- 17.Olah GA, Prakash GKS and Reddy VP, in Ionic Liquids in Organic Synthesis, American Chemical Society, 2007, vol. 950, ch. 4, pp. 36–57. [Google Scholar]
- 18.Olah GA, Welch JT, Vankar YD, Nojima M, Kerekes I and Olah JA, J. Org. Chem, 1979, 44, 3872–3881. [Google Scholar]
- 19.Cantet A-C, Carreyre H, Gesson J-P, Jouannetaud M-P and Renoux B, J. Org. Chem, 2008, 73, 2875–2878. [DOI] [PubMed] [Google Scholar]
- 20.Hasek WR, Smith WC and Engelhardt VA, J. Am. Chem. Soc, 1960, 82, 543–551. [Google Scholar]
- 21.Middleton WJ, J. Org. Chem, 1975, 40, 574–578. [Google Scholar]
- 22.Lal GS, Pez GP, Pesaresi RJ, Prozonic FM and Cheng H, J. Org. Chem, 1999, 64, 7048–7054. [Google Scholar]
- 23.Beaulieu F, Beauregard L-P, Courchesne G, Couturier M, LaFlamme F and L’Heureux A, Org. Lett, 2009, 11, 5050–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayashi H, Sonoda H, Fukumura K and Nagata T, Chem. Commun, 2002, 0, 1618–1619. [DOI] [PubMed] [Google Scholar]
- 25.Rozen S, Acc. Chem. Res, 2005, 38, 803–812. [DOI] [PubMed] [Google Scholar]
- 26.Shimizu M and Hiyama T, Angew. Chem. Int. Ed, 2004, 44, 214–231. [DOI] [PubMed] [Google Scholar]
- 27.Kuroboshi M, Kanie K and Hiyama T, Adv. Synth. Catal, 2001, 343, 235–250. [Google Scholar]
- 28.Ilchenko NO and Szabó KJ, J. Fluorine Chem, 2017, 203, 104–109. [Google Scholar]
- 29.Lu Z, Zeng X, Hammond GB and Xu B, J. Am. Chem. Soc, 2017, 139, 18202–18205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu Z, Bajwa BS, Otome OE, Hammond GB and Xu B, Green Chemistry, 2019, DOI: 10.1039/C8GC03166E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zeng X, Liu S, Shi Z and Xu B, Org. Lett, 2016, 18, 4770–4773. [DOI] [PubMed] [Google Scholar]
- 32.Liang S, Barrios FJ, Okoromoba OE, Hetman Z, Xu B and Hammond GB, J. Fluorine Chem, 2017, 203, 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Okoromoba OE, Hammond GB and Xu B, Org. Lett, 2015, 17, 3975–3977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okoromoba OE, Li Z, Robertson N, Mashuta MS, Couto UR, Tormena CF, Xu B and Hammond GB, Chem. Commun, 2016, 52, 13353–13356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okoromoba OE, Han J, Hammond GB and Xu B, J. Am. Chem. Soc, 2014, 136, 14381–14384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hoye RC, Anderson GL, Brown SG and Schultz EE, J. Org. Chem, 2010, 75, 7400–7403. [DOI] [PubMed] [Google Scholar]
- 37.Denmark SE and Yang S-M, J. Am. Chem. Soc, 2002, 124, 2102–2103. [DOI] [PubMed] [Google Scholar]
- 38.Sun J and Kozmin SA, J. Am. Chem. Soc, 2005, 127, 13512–13513. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.