

Abstract
Fatty acids (FA) play vital biological roles as energy sources, signaling molecules and key building blocks of complex lipids in cell membranes. Modifications to FA structure and composition are associated with the onset and progression of a number of chronic diseases. Consequently, the sensitive detection and unambiguous structure elucidation of FA is integral to the advancement of biomedical sciences. Recent advances in FA analysis have taken advantage of wet chemical derivatization to enhance detection and drive unique fragmentation in tandem mass spectrometry protocols. Here, we significantly further this approach through demonstrating gas-phase charge inversion of singly deprotonated FA ions, [M − H]−, using doubly charged tris-phenanthroline alkaline earth metal complexes, [Cat(Phen)3]2+ (Cat = Mg2+, Ca2+, Sr2+, or Ba2+). Metal cationized FA, [M − H + Cat]+ are obtained after the gas-phase ion/ion reaction. Low-energy collision-induced dissociation (CID) of the [M − H + Cat]+ cations facilitates double bond localization for a variety of monounsaturated and polyunsaturated FAs. Ultimately, detailed characterization presented unambiguous distinction amongst FA double bond positional isomers, such as n-3 and n-6 isomers. The method was successfully used to identify the FA profile of corn oil, including double bond localization for unsaturated FAs present.
Graphical Abstract
The fatty acid (FA) is the most fundamental lipid class and assumes vital biological roles in cellular structure, function, and metabolism.1–3 FAs are widespread throughout nature and can appear as free circulating molecules or esterified within complex lipids such as triacyglycerols or phospholipids. While general FA composition includes an aliphatic chain and at least one carboxyl group, FAs exhibit extensive structural diversity through variations in the aliphatic chain length and variations in location, number, and geometry of double bonds. Like many biomolecules, FA structure dictates both physical properties (e.g. solubility, fluidity, thermodynamic stability) and biological function including FA metabolism. Furthermore, modification to FA composition and structure has been associated with adverse health effects, including the development and progression of chronic disease such as cardiovascular disease, diabetes, obesity and cancer.2–5 Specifically, variations in the ratio of essential n-3 and n-6 FAs has been linked with disease progression.6 Similarly, the structure of FA and consequent composition of lipid extracts is critical in the optimization of desired physical and chemical properties for applications in a range of bio-commodities (e.g., biodiesel).7,8 Thus, the ability to probe FA structure is integral to the advancement of biomedical science and biotechnology, and in turn, efforts to characterize FAs have emerged as a principal focus in lipidomics.
Mass spectrometry (MS) has been deployed extensively to characterize FA. While traditional gas chromatography (GC)-MS of fatty acid methyl esters is still the most widely used strategy for this analysis, the resulting electron ionization mass spectra cannot distinguish closely related FA structures, especially lipid isomers.9 In pursuit of detailed structure elucidation of FA, Gross and colleagues first described charge-remote fragmentation (CRF).10 In this approach FA were ionized and subjected to high energy (keV) collisions resulting in unimolecular1,4-hydrogen eliminations to produce terminally unsaturated and structurally diagnostic carboxylate product ions.11,12 Importantly, interruptions to the regular sequence of product ions could be used to identify site(s) of unsaturation or other important structural features of a FA.13–17 Primary requirements of CRF were found to be a localized charge site, which could be derived from FA cationization with a charge switching reagent (positive ion mode) or deprotonation of the carboxylic acid group (negative ion mode), and access to high collision energies. As multi-sector instruments operating in the keV regime became less prevalent access to CRF became less common, with some recent reprise on tandem time-of-flight (TOF-TOF) instruments where dilithiated FA cations, [M − H + 2Li]+, provided double bond position for a series of isomeric FAs.18
The preponderance of modern lipid analysis is undertaken using contemporary electrospray ionization-MS in conjunction with low energy collision-induced dissociation (CID). For such instruments, highly sensitive detection of FA can be achieved in negative ion mode; however CID of the resulting [M − H]− anions provides relatively little structural information with spectra dominated by water and carbon dioxide neutral loss product ions and minimal structurally informative fragments observed.12 Notably however, Yang et al. report a method exploiting these small molecule losses (i.e H2O and CO2 losses) following CID of FA [M − H]− anions in the negative ion mode to identify and quantify unsaturated FA isomers.19 Overall in order to surmount this limitation regarding FA characterization in the negative ion mode, two different derivatization strategies have been employed that can be considered in two classes. The first class targets derivatization of the functional groups within the FA chain (e.g., carbon-carbon double bonds) while the second group modify the common carboxylic acid moiety itself. Examples of the first class include reactions between unsaturated lipids and ozone20–25 or carbonyl reagents via the Paternò-Büchi reaction.26,27 These methods are now achieving wide-spread adoption but do not provide insight into other structural features of FA (e.g., chain branching, cyclization, hydroxylation etc.).
Efforts to modify the carboxylic acid functional group prior to ionization have been focused on promoting CRF at low collision energies and thereby generating rich structural details of FA. For example, Wang et al. showed that solution-based covalent derivatization of FAs via amidation of the carboxylic moiety by the amine moiety on a N-(4-aminomethylphenyl) pyridinium (AMPP) yielded an AMPP-derivatized FA. The derivatized FA can readily be ionized in the positive ion-mode. Subsequent activation of the FA-AMPP precursor provided identification and quantification of FA double bond positional isomers utilizing CRF-like spectral patterns.28,29 While powerful, these strategies rely on wet chemical derivatization prior to analysis. In contrast, other investigators have doped salts into the ESI solution to obtain metallated FA cations. For example, Hsu and Turk showed that low-energy CID of dilithiated FA cations, [M − H + 2Li]+, allowed for differentiation amongst unsaturated FA isomers in addition to double bond position identification as carbon-carbon single bond cleavage (C-C) proximal (i.e. on the carboxyl end of the aliphatic chain) and vinylic to the last double bond in the aliphatic chain produced the heaviest fragment ion (highest m/z).30,31 Low-energy activation of copper (II) adducted FAs, [M − H + CuII]+, allowed for the localization of one or two homoconjugated double bonds. In addition, CID spectra of [M − H + CuII]+ provided distinction between cis and trans FA isomers via exploitation of product ions related to the neutral loss of CO2.32 Divalent alkaline earth metals have received considerable recent attention as a means to ionize FA and drive diagnostic fragmentation of the resulting adduct ions.33,34 The body of work to-date therefore suggests an advantageous approach to FA analysis would exploit the inherent propensity to ionize these analytes as [M − H]− anions and simultaneously harness the propensity of the corresponding metallated cation, [M − H + Cat]+ to undergo CRF.
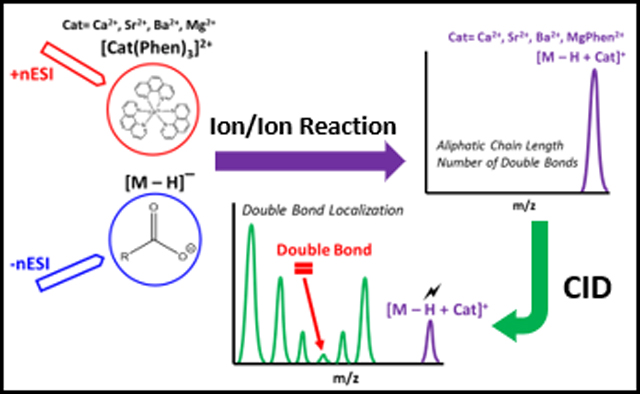
In this work, we demonstrate the gas-phase charge inversion of singly deprotonated fatty acid ions of the form [M − H]− with doubly charged tris-phenanthroline alkaline earth metal complexes, [Cat(Phen)3]2+ (Cat = Mg2+, Ca2+, Sr2+, or Ba2+). By conducting FA derivatization in the gas-phase, each reagent solution can be individually optimized with regards to solution and electrospray conditions. Additionally, gas-phase ion/ion reactions allow for enhanced control of reaction outcomes when compared to analogous transformations conducted in solution.35 Mutual storage ion/ion reaction resulted in alkaline earth metal cationization of the FA. As these complexes are inherently stable, the alkaline earth metal will not readily desorb from the carboxyl moiety upon CID providing CRF and detailed FA structural characterization.17 To prove the utility of the method, hydrolyzed corn oil was examined. The fatty acid profile of corn oil has been linked with biodiesel quality and has been recognized as an indicator of corn oil purity.7,36 The FA profile of corn oil can vary according to environmental and genetic factors, but the typical major FA constituents of corn oil include 16:0, 18:0, 18:1, 18:2, and 18:3, while minor FA components, reported at trace levels, include 14:0, 17:0, 20:0, 20:1, and 16:1.8
Experimental
Reagents.
Chloroform, HPLC-grade methanol, and HPLC-grade water were purchased from Fisher Scientific (Pittsburgh, PA). All FA standards were purchased from Cayman Chemical (Ann Arbor, MI). United States Pharmacopeia (USP) reference standard corn oil, magnesium chloride, strontium acetate, calcium acetate, barium acetate, and 1,10-phenanthroline (Phen) were purchased from Sigma-Aldrich (St. Louis, MO). FA standards were prepared in methanol to a final concentration of 0.1 mg/mL (m:v). Tris-phenanthroline alkaline earth metal complexes were prepared by mixing the 1:1(mol/mol) metal salt: Phen in methanol to final concentration of 0.1 mg/mL (m:v).37,38
Corn Oil Hydrolysis.
As corn oil consists primarily of triacylglycerols, FA were released via a TAG hydrolysis method as previously described.13 Briefly, 200 mg of corn oil was hydrolyzed at 60 °C for 1 hour with 800 μL of 98:2 (v:v) methanol: aqueous sodium hydroxide (1 M). FAs were then extracted using 2:1 (v:v) chloroform: methanol.
Nomenclature.
We adopt the nomenclature for lipids recommended by the International Union of Pure and Applied Chemistry (IUPAC).39 By convention, the first carbon (C1) in the aliphatic chain is the carboxyl carbon. The shorthand notation used denotes the number of carbon atoms followed by the number of carbon-carbon double bonds, designated after the colon, and definitive double bond position(s) indicated inside parentheses. For example, the shorthand notation 18:2 (9,12) represents an 18-carbon chain FA with 2 double bonds at the positions C9=C10 and C12=C13. Where known, the stereochemistry of carbon-carbon double bonds is assigned as Z (cis) and E (trans) (e.g., oleic acid is 18:1(9Z)), yet where unknown only the position of unsaturation is indicated (e.g., 18:1(9)).40 The n-x classification system is also used where a FA is classified according the location of the first double bond located at the xth carbon-carbon position from the methyl end of the aliphatic chain. For example, 18:3 n-3 has a double bond located at C15=C16.
Mass Spectrometry.
All experiments were performed on a Sciex QTRAP 4000 hybrid triple quadrupole/linear ion trap mass spectrometer (SCIEX, Concord, ON, Canada) that has been modified to perform ion/ion reactions.41 Alternately pulsed nano-electrospray ionization (nESI) allows for sequential injection of tris-phenanthroline alkaline earth metal complex dications, [Cat(Phen)3]2+ (Cat = Mg2+, Ca2+, Sr2+, or Ba2+), followed by singly deprotonated FA ions.42 The 1:1 (mol/mol) metal salt: Phen solution was sprayed first, followed by isolation of [Cat(Phen)3]2+. [Cat(Phen)3]2+ was then transferred to a high-pressure collision cell, q2, for storage. The FA was then ionized to an [M − H]− anion in the negative ion mode, isolated in Q1 in transient, then transferred to q2. The FA anion and [Cat(Phen)3]2+ were mutually stored in q2 for 300 milliseconds (ms). The mutual storage product ions were then subjected to 18–21 V of dipolar direct current (DDC)-CID in q2 for 25 ms.43 DDC-CID product ions were transferred to the low-pressure linear ion trap (LIT), Q3, for monoisotopic mass selection of [M − H + Cat]+, which was then back-transferred to q2 following isolation. In q2, [M − H + Cat]+ was collisionally activated for 50 ms at 200 mV via single frequency resonance excitation (q = 0.383 for unsaturated FAs, q = 0.317 for saturated FAs). Product ions generated from CID were transferred to Q3 and analyzed via mass-selective axial ejection.44
Results and Discussion
Gas-phase reaction of FA anion with [Cat(Phen)3]2+.
Gas-phase ion/ion reactions of FA [M − H]− anion with [Cat(Phen)3]2+ can charge invert FA anions to the positive ion mode. To do this experiment, [Cat(Phen)3]2+ (Cat =Mg2+, Ca2+, Sr2+, or Ba2+) was first ionized in the positive ion mode, isolated in Q1, and stored in q2 followed by the ionization of the FA in the negative ion mode, which can also be isolated in Q1, then transferred to q2. After 300 ms of mutual storage in q2, the singly deprotonated FA undergoes charge inversion generating a long-lived electrostatic complex, [M − H + Cat(Phen)2]+, produced via the replacement of a phenanthroline ligand with the FA anion. Scheme 1 shows the neutral loss of a phenanthroline ligand that occurs during the ion/ion reaction. All tris-phenanthroline alkaline earth metal complexes investigated herein produced the complex of interest (see Supporting Figure S-1). The charge inversion efficiency for the FA anion appears to be nearly unit efficient. The efficiency would be less than unit efficient if FA anion neutralization were to take place through a proton-transfer reaction with the tris-phenanthroline alkaline earth metal complex, generating a singly charged, proton deficient Cat(Phen)3 at m/z 563. As there is no m/z 563 peak in the ion/ion mass spectrum (Figure S-2), it can be concluded that single proton transfer does not occur to a significant extent.
Scheme 1.
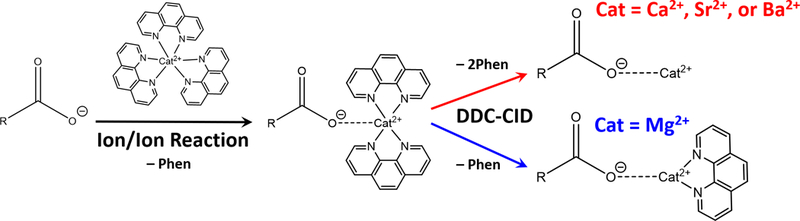
Singly deprotonated fatty acid ion, [M − H]−, reacts with [Cat(Phen)3]2+ dications in a mutual storage ion/ion reaction to produce the [M − H + Cat(Phen)2]+ complex where Cat = Ca2+, Sr2+, Ba2+, or Mg2+. Application of q2 DDC-CID results in the neutral loss of two phenanthroline ligands when Cat = Ca2+, Sr2+, or Ba2+ or one phenanthroline ligand when Cat = Mg2+ to generate the [M − H + Cat]+ complex.
Scheme 1 also outlines results of q2 DDC-CID of the [M − H + Cat(Phen)2]+ complex. Fragmentation of the complex cation results in the loss of two phenanthroline ligands and generation of a metalated FA complex, [M − H + Cat]+ (Figure S-3a-c). However, DDC-CID of the [M − H + Mg(Phen)2]+ complex resulted in the neutral loss of only one phenanthroline ligand, giving rise to [M − H + MgPhen]+ (Figure S-3d).
Gas-phase Fragmentation of Charge Inverted Saturated Fatty Acids.
Saturated FA anions can undergo charge inversion when subjected to mutual storage with [CatPhen3]2+. The resulting reaction spectrum yields the [M − H + Cat(Phen)2]+ complex, which upon DDC-CID fragments to form the [M − H + Cat]+ complex as described above. Collisional activation of metalated saturated FAs of the form [M − H + Cat]+ (Cat =MgPhen2+, Ca2+, Sr2+, or Ba2+) generates a series of CRF ions, evenly spaced 14 Da apart, corresponding to carbon-carbon (C-C) single bond cleavages along the aliphatic chain. Figure 1 shows ion-trap CID of [M − H + MgPhen]+ derived from gas-phase reaction of deprotonated octadecanoic acid (18:0) and [Mg(Phen)3]2+. The CID spectrum is dominated by a peak at m/z 221, signifying [MgPhenOH]+. [MgPhenO2]+ at m/z 236 is one of two even mass product ions observed, representing the second most abundant product ion. Herein, we did not further investigate the mechanism underlying generation of the curious product ion [MgPhenO2]+, as this product ion does not provide information relevant to FA identification. The product ion b+• (m/z 262), generated via homolytic C2-C3 cleavage, produces the only even mass, odd electron product ion derived from the aliphatic chain. Even electron, odd mass CRF ions, denoted with the prime notation (‘), begin with C3-C4 fragmentation (c’ peak at m/z 289) and continue down the aliphatic chain. Relevant m/z values are tabulated in Supporting Table S-1.
Figure 1.
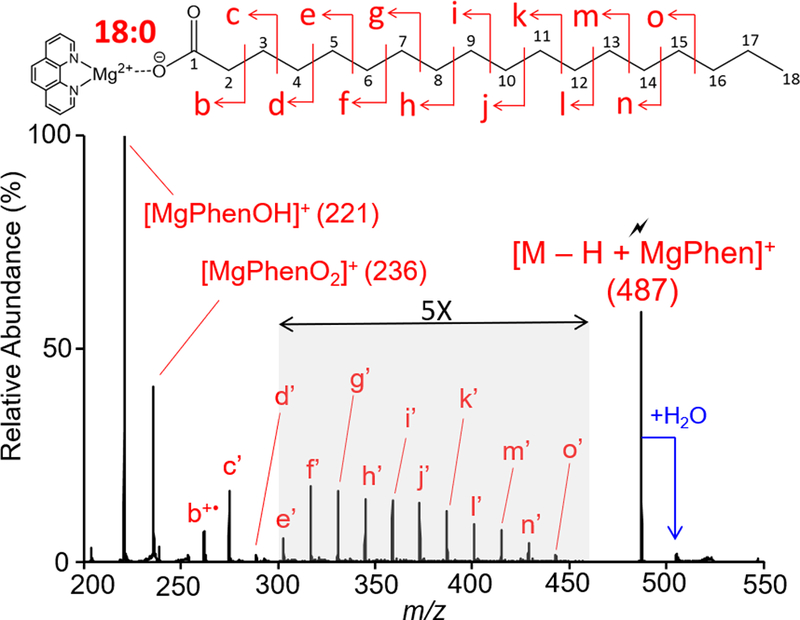
Ion-trap CID spectrum of the [M − H + MgPhen]+ complex generated via gas-phase ion/ion reaction between singly deprotonated 18:0 and [Mg(Phen)3]2+ dications. The lightning bolt ( ) signifies collisionally activated precursor ion.
) signifies collisionally activated precursor ion.
CID of [M − H + Cat]+ resulting from the ion/ion reaction between deprotonated 18:0 and [Cat(Phen)3]2+ (Cat = Ca2+, Sr2+, or Ba2+), generates CRF ions that readily adduct multiple water molecules (Figure S-4).45 Varying the q2 storage time of CRF ions resulted a direct correlation between storage time and water adduction i.e. longer storage times (up to 5000 ms) resulted in multiple water adduction. In addition, DDC-CID of the trapped ions results in sequential removal of adducted water (Figure S-5). In general, water adductions led to highly congested CID spectra when Cat = Ca2+, Sr2+, or Ba2+. However, as the size of the metal cation increases (i.e. going down the alkaline earth metal group), the relative abundance of the hydrated CRF product ion series decreases. Thus, an inverse relationship between metal cation size and water adduction of the metal cationized FA and CRF ions is observed. This trend is highlighted by the decrease in relative abundance of the doubly hydrated CRF ion series displayed in Figure S4a-c as metal cation size increased sequentially, Ca2+<Sr2+<Ba2+. The observed trend can be rationalized as an increased charge-to-size ratio of the metal cation would promote enhanced ion-dipole interactions between the metal cation and water molecules, and therefore, as the charge-to-size ratio decreases (i.e. cation size increases), the prominence of the hydrated CRF ion series decreases. Overall, the reagent cations Ca2+, Sr2+, and Ba2+ led to highly congested CID spectra complicated with water adductions. Conversely, CID spectra generated with the reagent cation MgPhen2+ were straightforward, not displaying the series of water-adducted CRF ions. Thus, the reagent cation MgPhen2+ proved to be the most useful charge inversion reagent as CID of saturated FA [M − H + MgPhen]+ ions produced the most straightforward results.
Characterization of Monounsaturated Fatty Acids.
Monounsaturated FAs ionized in the negative ion mode undergo gas-phase charge inversion via ion/ion reaction with [Cat(Phen)3]+ to produce the complex of interest, [M − H + Cat]+. The presence of a repeatable spectral pattern influenced by double bond position revealed double bond location in monounsaturated fatty acids. Double bond localization for a series of monounsaturated octadecenoic acids is demonstrated in Figure 2 using the spectral pattern highlighted with the blue curve. Observed m/z values for all product ions displayed in Figure 2 are tabulated in Table S-2.
Figure 2.
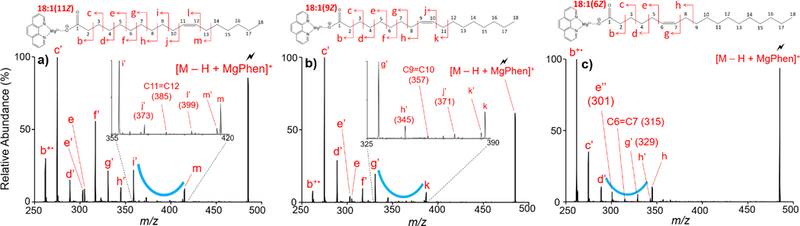
Ion-trap CID of a series of isomeric 18:1 fatty acids. Ion-trap CID spectrum of [M − H + MgPhen]+ where (a) M = 18:1(11Z), (b) M = 18:1(9Z), and (c) M = 18:1(6Z). The blue curve highlights the spectral pattern used to localize the double bond.
Figure 2(a) summarizes CID of [M − H + MgPhen]+ where M = 18:1(11Z). The spectrum shows CRF ions of the same m/z as those observed with CID of charge-inverted 18:0 (see above) until fragmentation reaches the double bond position. The C5-C6 cleavage generates a doublet, not observed with the analysis of 18:0, composed of a CRF ion (e’ peak at m/z 303) and a terminally saturated product ion (e peak at m/z 305). Formation of the saturated product ion is suggested to occur through C5-C6 fragmentation and cyclization, forming a stable six-membered ring containing the carboxyl oxygen.45
To localize the double bond in Figure 2(a), the presence of a spectral gap highlighted with the blue curve shown is used. Upon activation of the [M − H + MgPhen]+ (M = 18:1(11Z)) complex, allylic C-C bond cleavages, both proximal (i.e. on the carboxyl side) and distal (i.e. on the methyl side) to the double bond, generated high abundance product ions flanking the spectral gap. Proximal allylic cleavage, C9-C10 cleavage, produced the CRF ion i’ (m/z 359), and distal allylic cleavage, C13-C14 cleavage, produced the doublet m’ (m/z 413) and m (m/z 415). In general, C-C cleavage proximal and allylic to the double bond generates a characteristic terminally unsaturated CRF ion, whereas distal allylic C-C cleavage generates a high intensity doublet composed of a CRF ion and a terminally saturated product ion. The observed spectral gap is the result of a dramatic suppression in product ion abundance related to product ions representing the C=C double bond and C-C cleavages vinylic to the double bond. Vinylic C-C bond cleavage, both proximal and distal to the double bond, produced two CRF ions observed at very low relative abundances. The C10-C11 cleavage (peak j’ at m/z 373) and the C12-C13 cleavage (peak l’ at m/z 399) represent vinylic cleavage proximal and distal to the C11=C12 double bond respectively. Disruption of the 14 Da spacing is observed as an exceedingly small product ion representing the C11=C12 double bond (m/z 385) is spaced 12 Da from the CRF ion j’. Ultimately, perturbation of the 14 Da spacing further confirms double bond localization.
Ion-trap CID of [M − H + Cat]+ (Cat=MgPhen2+, Ca2+, Sr2+, or Ba2+), demonstrated with 11Z-octadecenoic acid (18:1(11Z)) in Figure S-6, provided double bond localization utilizing the spectral pattern described above. However, given the relative simplicity of utilizing MgPhen2+ as the reagent due to a lack of hydrated CRF ions, data presented hereafter exclusively employed MgPhen2+.
Distinction amongst FA double bond positional isomers is facilitated by direct spectral comparison. Double bond localization for a double bond positional isomer of 18:1(11Z) is first demonstrated with the analysis of 18:1(9Z). CID of the [M − H + MgPhen]+ complex (M = 18:1(9Z)) is shown in Figure 2(b). Though not entirely understood, analysis of this isomer via CID of the [M − H +MgPhen]+ complex shows a dramatic decrease in the abundance of the b+• (m/z 275) product ion relative to the CRF ion c’ (m/z 289). To localize the double bond for this isomer, the CID spectrum again displays a spectral gap flanked by allylic C-C cleavages. Allylic cleavage proximal to the double bond, C7-C8 cleavage, produced the CRF ion g’ at m/z 331. The characteristic doublet (k peak at m/z 387 and k’ peak at m/z 385) is produced via C11-C12 allylic cleavage distal to the double bond. Vinylic cleavages are once more observed at lower relative abundances compared to allylic cleavages as shown with the CRF ions h’ (m/z 345) and j’ (m/z 371). Perturbation of the 14 Da spacing was also observed, as the m/z differential between h’ and the product ion representing the C9=C10 double bond is 12 Da.
Similarly, the gas-phase charge inversion of singly deprotonated 18:1(6Z) with [Mg(Phen)3]2+ generates the [M − H + MgPhen]+ complex at m/z 485. CID of the [M − H + MgPhen]+ complex where M = 18:1(6Z) provided double bond localization and distinction from the previous octadecenoic acid isomers investigated. When the double bond is located prior to C7, as in the case of 18:1(6Z), the double bond location can still be determined, but double bond localization relies solely on the spectral gap (Figure 2(c)). An unexpected doubly unsaturated product ion (peak e’’ at m/z 301), denoted with the double prime notation (‘‘), most likely originated from hydride migration and subsequent C5-C6 fragmentation, generating a terminally conjugated diene product ion; the presence of this product ion however did not hinder double bond localization, as the spectral gap was still maintained, albeit not as dramatically as with the other octadecenoic isomers shown above. The spectral gap (blue curve) is bordered by allylic cleavage product ions, g’ (m/z 289), h (m/z 343), and h’ (m/z 345). Once again, vinylic cleavages (peaks e’’ and g’) are dramatically suppressed. The product ion corresponding to the C6=C7 double bond does not perturb the 14 Da spacing, but as this product ion is the least abundant CRF ion in the spectral gap, double bond localization is still obtained. Thus, exploitation of a spectral gap influenced by double bond position, allowed for rapid, facile localization of the double bond for monounsaturated fatty acids and distinction between double bond positional FA isomers.
Characterization of Polyunsaturated Fatty Acids.
The study of polyunsaturated fatty acids (PUFAs) was twofold, including an in-depth investigation of diunsaturated FAs and FAs with more than two degrees of unsaturation. Collisional activation of [M − H + MgPhen]+ for diunsaturated FAs provided double bond localization again through repeatable spectral patterns indicative of double bond structure (e.g. homoconjugated, conjugated), similar to those described above.
Singly deprotonated 9Z,12Z-octadecadienoic acid (18:2(9Z,12Z)), an n-6 essential fatty acid, reacted in the gas-phase with [Mg(Phen)3]2+ producing the complex of interest, [M − H + MgPhen]+ at m/z 483. Ion-trap CID of [M − H + MgPhen]+ (M = 18:2(9Z,12Z)), shown in Figure 3(a), provided bond localization through utilization of a repeatable spectral pattern. Observed m/z values for Figure 3 are tabulated in Table S-3. For the first double bond location in 18:2(9Z,12Z), a spectral gap (blue curve) like that described for monounsaturated FA analysis is observed. Allylic cleavages proximal and distal to the first double bond generated product ions g’ (m/z 331) and k’ (m/z 385), respectively, flanking the spectral gap. Cleavage vinylic to the first double bond, represented by C8-C9 (peak h’ at m/z 345) and C10-C11 (peak j at m/z 373 and peak j’ at m/z 371) fragmentation, is suppressed, as is the product ion representing the C9=C10 double bond (m/z 359). While perturbation of the 14 Da spacing is not observed, the C9=C10 double bond can still be localized through exploitation of the spectral gap pattern.
Figure 3.
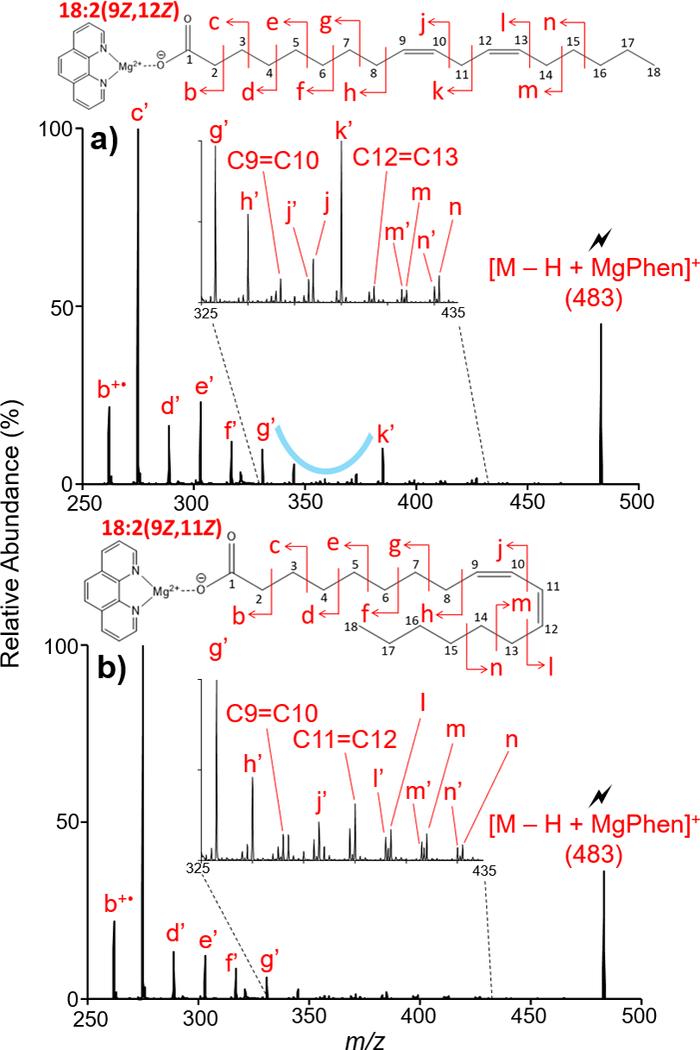
Ion-trap CID of [M − H + MgPhen]+ where (a) M =18:2(9Z, 12Z) and (b) M = 18:2(9Z, 11Z). Comparison of the two CID spectra shows marked differences between diunsaturated FAs with homoconjugated and conjugated double bonds.
To localize the second double bond in a homoconjugated diunsaturated FA, a secondary pattern is used. Once more, this is demonstrated with 18:2(9Z,12Z) analysis in Figure 3(a). However, the pattern was also observed for numerous n-6 homoconjugated fatty acids analyzed via CID of the [M − H + MgPhen]+ complex. The pattern indicative of homoconjugated double bonds consists of a series of doublets resulting from allylic and vinylic C-C cleavages distal to the double bond. Vinylic C-C cleavage distal to the double bond, formed the doublet m’ (m/z 411) and m (m/z 413). Allylic C-C cleavage distal to the second double bond, represented by C14-C15 cleavage, produced a doublet of similar product ion intensity, n’ (m/z 425) and n (m/z 427). Fragmentation of the aliphatic chain after C15 is dramatically suppressed, as shown in Figure 3(a). Therefore, through the exploitation of a repeatable spectral pattern unique to homoconjugated, diunsaturated FAs, both double bond positions could be determined.
Examination of conjugated and homoconjugated diunsaturated FA structures resulted in distinct spectral differences upon CID of the [M − H + MgPhen]+ complex. Direct spectral comparison of CID spectra of [M − H + MgPhen]+ where M = 18:2(9Z,12Z) and M = 18:2(9Z,11Z), shown in Figures 3(a) and (b) respectively, resulted in marked spectral differences, predominantly regarding C11-C12 cleavage. For 18:2(9Z,12Z), C11-C12 cleavage represented a C-C single bond cleavage, producing the CRF ion k’ (m/z 385). However, for 18:2(9Z,11Z) (Figure 3(b)), C11-C12 cleavage represented C=C double bond cleavage. Therefore, a dramatic suppression in relative abundance of the C11=C12 product ion was expected and experimentally confirmed. Recognition of this suppressed product ion thus provides distinction amongst homoconjugated and conjugated double bond systems in diunsaturated fatty acids.
Biologically-derived polyunsaturated FA are commonly homoconjugated with conjugated systems also observed under certain conditions. However, some unusual FA structures have been reported where the double bonds are neither conjugated nor homoconjugated.47 The gas-phase charge inversion of 8,14-eicosadienoic acid (20:2(8Z,14Z)) and subsequent analysis of the [M − H + MgPhen]+ complex via CID provided double bond localization. Figure 4 shows the CID spectrum of the [M − H + MgPhen]+ complex (M = 20:2(8Z,14Z)). Two spectral gaps are observed after CID of the [M − H + MgPhen]+ complex (M = 20:2(8Z,14Z)). Observed m/z values of peaks shown in Figure 4 are tabulated in Table S-4. These spectral gaps were used to localize each double bond present in 20:2(8Z,14Z). For localization of the C8=C9 double bond, slight deviations from the monounsaturated FA spectral pattern were observed, specifically concerning to C7-C8 cleavage. Typically, the peaks signifying C-C cleavages allylic to the double bond were more readily observed when compared to vinylic cleavage peaks. Yet with the analysis of 20:2(8Z,14Z), C7-C8 cleavage resulted in a more abundant CRF ion g’ (m/z 331) compared to the C6-C7 allylic cleavage CRF ion f’ (m/z 317). However, this slight deviation in spectral pattern did not hinder localization of the first double bond. Ultimately, the dramatic reduction in peak intensity observed for the product ion representing the C8=C9 double bond (m/z 343) and the disruption of the 14 Da spacing provided double bond localization for the first double bond. As for localization of the C14=C15 double bond, the position of this double bond was determined again using the observed spectral gap. Flanking the second spectral gap in Figure 4, are product ions generated via cleavages allylic to the C14=C15 double bond, denoted by the doublet p (m/z 455) and p’ (m/z 453) and the CRF ion l’ (m/z 399). Cleavages vinylic to the second double bond are suppressed, as shown via CRF ions m’ (m/z 413) and o’ (m/z 439). Disruption of the 14 Da spacing is once again observed, confirming double bond localization, as the product ion representing the C14=C15 double bond (m/z 425) is spaced 12 Da from the CRF ion m’ (m/z 413). Thus, utilizing this secondary spectral gap and the disruption of the 14 Da spacing exhibited by CRF ions, the second double bond position can be localized.
Figure 4.
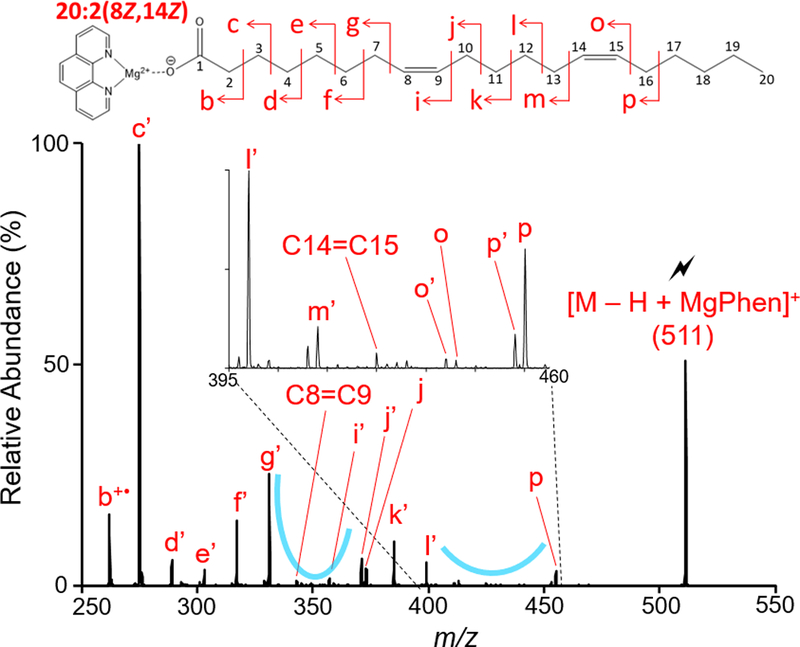
Ion-trap CID of [M − H + MgPhen]+ derived from the gas-phase ion/ion reaction of singly deprotonated 20:2(8Z, 14Z) and [Mg(Phen)3]2+.
As the degrees of unsaturation increase, overall fragmentation of the charge-inverted fatty acid [M − H +MgPhen]+ complex increases as well. Thus, localization of the double bonds proved challenging. Nevertheless, distinction amongst FA isomers, specifically n-3 and n-6 double bond positional FA isomers, is readily achieved. This is demonstrated with the comparative analysis of octadecatrienoic acid (18:3) n-3 and n-6 isomers represented by 18:3(9Z,12Z,15Z) and 18:3(6Z,9Z,12Z) respectively.
Ion-trap CID of [M − H + MgPhen]+ where M = 18:3(9Z,12Z,15Z) resulted in clear spectral differences when compared to the CID spectrum of [M − H + MgPhen]+ where M = 18:3(6Z, 9Z, 12Z), shown in Figures 5(a) and (b) respectively. Relevant m/z values are recorded in Table S-5. For both 18:3 isomers investigated, congested CID spectra suggest gas-phase double bond rearrangements, complicating double bond localization. While the localization of the double bonds is not readily visible with the CID spectrum, distinct tandem mass spectral differences allow for differentiation amongst the n-3 and n-6 18:3 isomers. CID spectral differences are predominately observed regarding fragmentation near the methyl end of the aliphatic chain, as highlighted in Figure 5 with the blue bar. Analysis of 18:3(9Z,12Z,15Z) via activation of the [M − H + MgPhen]+ complex (see Figure 5(a)) shows a doublet composed of two similarly intense peaks generated by C-C cleavage vinylic and distal to the C15=C16 double bond. The doublet, denoted by peaks n’’ (m/z 423) and n’ (m/z 425), consists of a CRF ion and a CRF ion possessing an additional degree of unsaturation, suggested to form via hydride migrations and CRF. Likewise, the CID spectrum of [M − H + MgPhen]+ where M = 18:3(6Z,9Z,12Z) shows C11-C12 cleavage produces the doublet k’’ (m/z 381) and k’ (m/z 383). Directly following the doublet, a single, intense CRF ion representing the final double bond is observed for both FA isomers, given by the peaks C15=C16 (m/z 437) and C12=C13 (m/z 395) for 18:3(9Z,12Z,15Z) and 18:3(6Z,9Z,12Z) respectively. The strong presence of this product ion reflecting the final double bond position suggests potential interaction and stabilization between the double bond and metal cation. CRF ion peaks, above the peaks representing the final double bond, are observed in decreasing relative abundance as fragmentation approaches the methyl end of the aliphatic chain. In total, despite difficulties localizing double bonds in the 18:3 isomers, clear spectral differences are observed in CID spectra obtained via activation of the described complex, providing distinction between the n-3 and n-6 isomers represented by 18:3(9Z,12Z,15Z) and 18:3(6Z,9Z,12Z).
Figure 5.
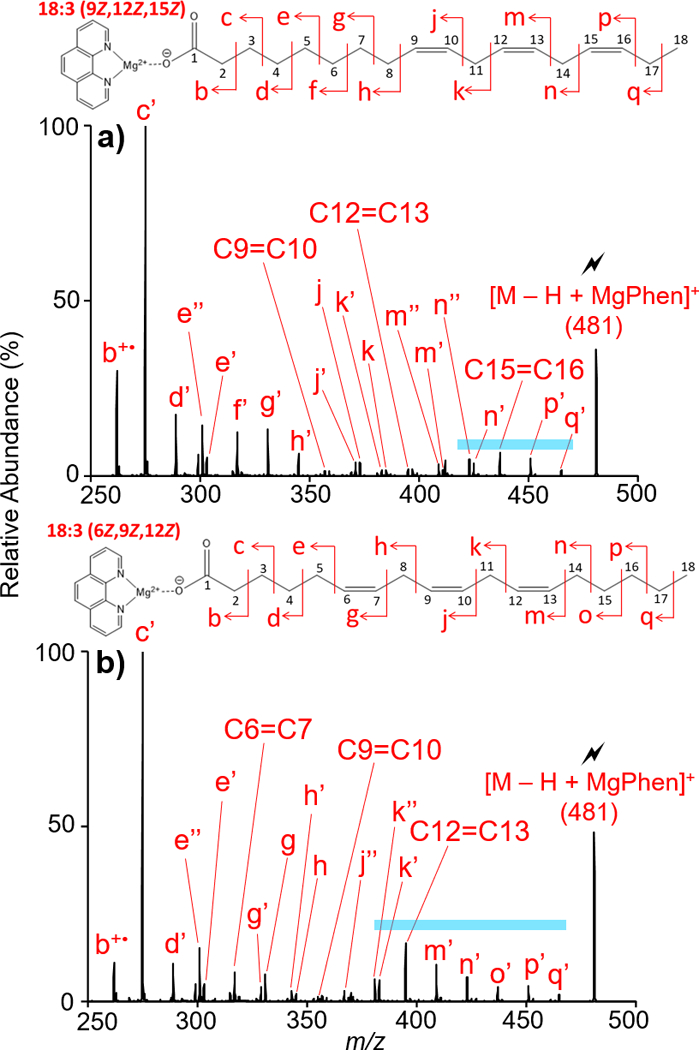
Ion-trap CID of [M − H + MgPhen]+ derived from the gas-phase ion/ion reaction of [Mg(Phen)3]2+ with singly deprotonated (a) 18:3(9Z, 12Z, 15Z) and (b) 18:3(6Z, 9Z, 12Z).
Analysis of FAs with more than three degrees of unsaturation, posed significant challenges for double bond localization. As the number of double bonds increases, fragmentation of the [M − H + MgPhen]+ complex was more abundant, generating complex CID spectra. Despite complications experienced with highly unsaturated FA standards, this problem could be circumvented if an appropriate FA standard was readily available, as tandem mass spectra were highly reproducible using the proposed method. Construction of a spectral library with hopes of applications utilizing spectral matching is under current investigation and represents the subject of future work.
Characterization of Fatty Acids in Corn Oil.
To evaluate the utility of the method, hydrolyzed corn oil was examined. Negative ion mode analysis of hydrolyzed corn oil revealed the presence of the following FA: 14:0, 15:0, 16:0, 17:0, 18:0, 16:1, 18:1, 18:2, and 18:3 (Figure S-7a). Subsequent negative ion mode analysis of a solvent blank subjected to the extraction procedure described above demonstrated the presence of FA contaminants in the hydrolyzed corn oil sample (Figure S-7b). The identified FA contaminants include: 12:0, 14:0, 15:0, 16:0, 18:0, 16:1 and 18:1. Comparison of the hydrolyzed corn oil and solvent blank nESI mass spectra shows the 18:1 FA is significantly more abundant in the hydrolyzed corn oil sample; thus, it can be reasonably concluded that the origin of the 18:1 FA can be attributed to both corn oil and sample contamination.
Application of the reported q2 mutual storage ion/ion reaction between mass selected hydrolyzed corn oil FA anions ions with [Mg(Phen)3]2+ generated the [M − H + Mg(Phen)2]+ complex. DDC-CID of the product ions resulted in the neutral loss of a phenanthroline ligand producing the [M − H + MgPhen]+ complex. Ion-trap CID of the [M − H + MgPhen]+ complex provided detailed FA characterization for the FAs in hydrolyzed corn oil.
Saturated FAs observed in hydrolyzed corn oil, though classified as contaminants due to their presence in the solvent blank, readily produced the [M − H + MgPhen]+ complex upon mutual storage with [Mg(Phen)3]2+. CID of the resulting [M − H + MgPhen]+ complex, yielded similar CID spectra for all saturated FA with product ions equally spaced 14 Da apart (Figure S-8). Numerous saturated FA contaminants were structurally confirmed via CID of the [M − H + MgPhen]+ complex.
Characterization of unsaturated FAs, including PUFAs, in corn oil via gas-phase charge inversion provided double bond localization. Here, we report an observed 16:1(6) FA. As the 16:1(6) is unique to humans and excreted naturally from the skin48, corn oil sample contamination is suggested as the source for this fatty acid (Figure S-9). The double bond in the 18:1 fatty acid observed in the corn oil sample was localized at C9=C10, classifying the FA as 18:1(9) (Figure 6(a)). Characterization of [M − H + MgPhen]+ where M = 18:2 via ion-trap CID (Figure 6(b)) confirmed the FA as 18:2(9,12), exhibiting homoconjugated double bonds at C9=C10 and C12=C13. Furthermore, double bond localization in the observed 18:3 FA leads to the identification double bonds at positions C9=C10, C12=C13, and C15=C16 (Figure 6(c)), identifying this FA as 18:3(9,12,15); an n-3 fatty acid. The dynamic range of the method is highlighted by the ability to identify double bond position in the 18:3 which is, on average, a factor of 50 lower in abundance when compared to the 18:2 and according to previous reports less than 1 percent of corn oil FA by mass.8 In total, while the FA profile of corn oil is relatively simple, the successes demonstrated here suggest the feasibility of significantly more complex sample analysis using the presented method.
Figure 6.
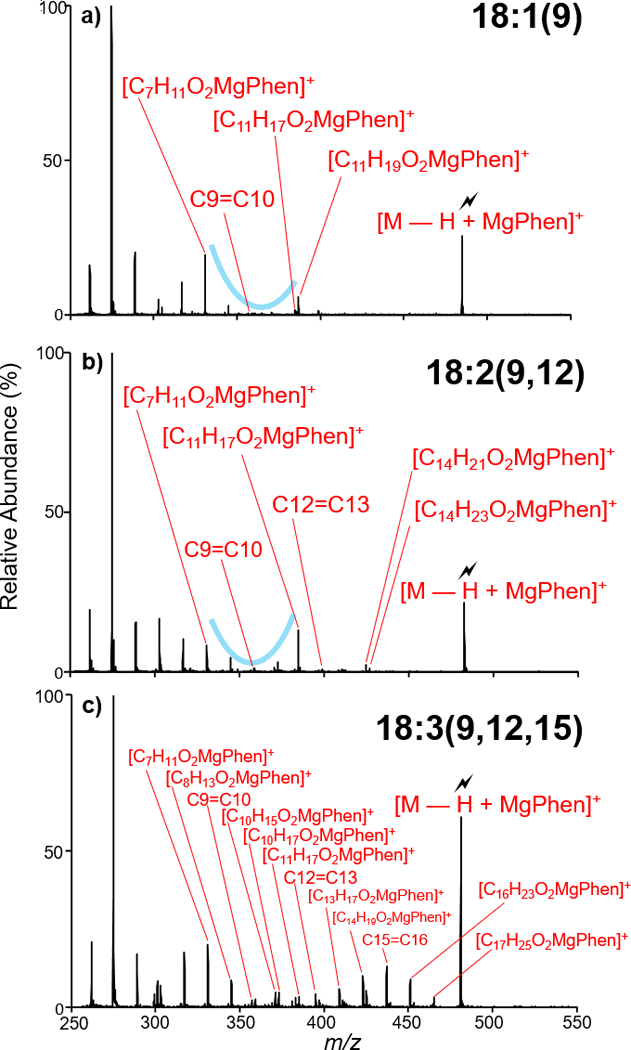
Ion-trap CID of [M − H + MgPhen]+ where M = (a) 18:1, (b) 18:2, (c) 18:3. Fatty acids were isolated from negative nESI of hydrolyzed corn oil.
Conclusions.
FAs exhibit extensive structural diversity, and double bond localization has been proven to be difficult via conventional tandem mass spectrometry. Utilization of doubly charged tris-phenanthroline alkaline earth metal complexes as charge inversion reagents, led to the metal cationization of FA anions upon a q2 mutual storage ion/ion reaction followed by DDC-CID. While all alkaline earth metals explored provided FA characterization, the mass spectral simplicity observed with [MgPhen]2+ strongly favors the usage of [Mg(Phen)3]2+ as the charge inversion reagent over [Ca(Phen)3]2+, [Sr(Phen)3]2+, and [Ba(Phen)3]2+.
Double bond localization was facile for monounsaturated and diunsaturated FAs, as a repeatable spectral gap generated via the presence of a double bond is observed, providing double bond position. Analysis of PUFAs with more than two degrees of unsaturation provided distinction amongst n-3 and n-6 fatty acid double bond positional isomers through direct spectral comparison. Complications were experienced with double bond localization in highly unsaturated FAs. However, tandem mass spectra of [M − H +MgPhen]+ were highly reproducible. Thus, there is potential for unknown FA species to be identified via spectral comparison with known FA standards. Compared to previously reported analyses of solution-based derivations of FAs, the gas-phase charge inversion method provides a rapid approach to FA detailed characterization, including double bond localization.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by the National Institutes of Health (NIH) under Grant GM R37–45372. S.J.B. acknowledged project funding through the Discovery Program (DP150101715) Australian Research Council (ARC).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Additional information discussed in the text that support the presentation of the work (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Murphy RC In Tandem Mass Spectrometry of Lipids: Molecular Analysis of Complex Lipids, 2015, pp 1–39. [Google Scholar]
- (2).Cascio G; Schiera G; Di Liegro I Curr. Diab. Rev. 2012, 8, 2–17. [DOI] [PubMed] [Google Scholar]
- (3).Kuhajda FP Nutrition 2000, 16, 202–208. [DOI] [PubMed] [Google Scholar]
- (4).Jeppesen C; Schiller K; Schulze MB Curr. Diab. Rep. 2013, 13, 279–288. [DOI] [PubMed] [Google Scholar]
- (5).Martinez-Seara H; Rog T; Pasenkiewicz-Gierula M; Vattulainen I; Karttunen M; Reigada R Biophys. J. 2008, 95, 3295–3305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Simopoulos AP Nutrients 2016, 8, 17. [Google Scholar]
- (7).Ramos MJ; Fernandez CM; Casas A; Rodriguez L; Perez A Bioresource Technol. 2009, 100, 261–268. [DOI] [PubMed] [Google Scholar]
- (8).Dunlap FG; White PJ; Pollak LM; Brumm TJ J. Am. Oil Chemists Soc. 1995, 72, 981–987. [Google Scholar]
- (9).Mitchell TW; Pham H; Thomas MC; Blanksby SJ J. Chromatogr., B: Anal. Technol. Biomedical Life Sci. 2009, 877, 2722–2735. [DOI] [PubMed] [Google Scholar]
- (10).Tomer KB; Crow FW; Gross ML J. Am. Chem. Soc. 1983, 105, 5487–5488. [Google Scholar]
- (11).Gross ML Int. J. Mass Spectrom. Ion Processes 1992, 118, 137–165. [Google Scholar]
- (12).Gross ML Int. J. Mass Spectrom. 2000, 200, 611–624. [Google Scholar]
- (13).Kerwin JL; Wiens AM; Ericsson LH J. Mass Spectrom. 1996, 31, 184–192. [DOI] [PubMed] [Google Scholar]
- (14).Jensen NJ; Gross ML Mass Spectrom. Rev. 1987, 6, 497–536. [Google Scholar]
- (15).Jensen NJ; Tomer KB; Gross ML Anal. Chem. 1985, 57, 2018–2021. [Google Scholar]
- (16).Adams J; Gross ML Anal. Chem. 1987, 59, 1576–1582. [Google Scholar]
- (17).Davoli E; Gross ML J. Am. Soc. Mass Spectrom. 1990, 1, 320–324. [DOI] [PubMed] [Google Scholar]
- (18).Trimpin S; Clemmer DE; McEwen CN J. Am. Soc. Mass Spectrom. 2007, 18, 1967–1972. [DOI] [PubMed] [Google Scholar]
- (19).Yang K; Zhao ZD; Gross RW; Han XL Anal. Chem. 2011, 83, 4243–4250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Brown SHJ; Mitchell TW; Blanksby SJ Biochim. Biophys. Acta 2011, 1811, 807–817. [DOI] [PubMed] [Google Scholar]
- (21).Poad BLJ; Pham HT; Thomas MC; Nealon JR; Campbell JL; Mitchell TW; Blanksby SJ J. Am. Soc. Mass Spectrom. 2010, 21, 1989–1999. [DOI] [PubMed] [Google Scholar]
- (22).Thomas MC; Mitchell TW; Blanksby SJ J. Am. Chem. Soc. 2006, 128, 58–59. [DOI] [PubMed] [Google Scholar]
- (23).Thomas MC; Mitchell TW; Blanksby SJ Methods Mol. Biol. 2009, 579, 413–441. [DOI] [PubMed] [Google Scholar]
- (24).Thomas MC; Mitchell TW; Harman DG; Deeley JM; Murphy RC; Blanksby SJ Anal. Chem. 2007, 79, 5013–5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Thomas MC; Mitchell TW; Harman DG; Deeley JM; Nealon JR; Blanksby SJ Anal. Chem. 2008, 80, 303–311. [DOI] [PubMed] [Google Scholar]
- (26).Ma XX; Chong L; Tian R; Shi RY; Hu TY; Ouyang Z; Xia Y Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ma XX; Xia Y Angew. Chem. Int. Ed. 2014, 53, 2592–2596. [DOI] [PubMed] [Google Scholar]
- (28).Wang M; Han RH; Han XL Anal. Chem. 2013, 85, 9312–9320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Yang K; Dilthey BG; Gross RW Anal. Chem. 2013, 85, 9742–9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Hsu FF; Turk J J. Am. Soc. Mass Spectrom. 1999, 10, 600–612. [DOI] [PubMed] [Google Scholar]
- (31).Hsu FF; Turk J J. Am. Soc. Mass Spectrom. 2008, 19, 1673–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Afonso C; Riu A; Xu Y; Fournier F; Tabet JC J. Mass Spectrom. 2005, 40, 342–349. [DOI] [PubMed] [Google Scholar]
- (33).Duncan KD; Volmer DA; Gill CG; Krogh ET J. Am. Soc. Mass Spectrom. 2016, 27, 443–450. [DOI] [PubMed] [Google Scholar]
- (34).Hale OJ; Cramer R Anal. Bioanal. Chem. 2018, 410, 1435–1444. [DOI] [PubMed] [Google Scholar]
- (35).Newton KA; Amunugama R; McLuckey SA J. Phys. Chem. 2005, 109, 3608–3616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Catharino RR; Haddad R; Cabrini LG; Cunha IBS; Sawaya A; Eberlin MN Anal. Chem. 2005, 77, 7429–7433. [DOI] [PubMed] [Google Scholar]
- (37).Barlow CK; Hodges BDM; Xia Y; O’Hair RAJ; McLuckey SA J. Am. Soc. Mass Spectrom. 2008, 19, 281–293. [DOI] [PubMed] [Google Scholar]
- (38).Crizer DM; Xia Y; McLuckey SA J. Am. Soc. Mass Spectrom. 2009, 20, 1718–1722. [DOI] [PubMed] [Google Scholar]
- (39).IUPAC-IUB Commission on Biochemical Nomenclature (1967) Eur. J. Biochem. 1977, 79, 11–21. [DOI] [PubMed] [Google Scholar]
- (40).Liebisch G; Vizcaino JA; Kofeler H; Trotzmuller M; Griffiths WJ; Schmitz G; Spener F; Wakelam MJO J. Lipid Res. 2013, 54, 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Xia Y; Wu J; McLuckey SA; Londry FA; Hager JW J. Am. Soc. Mass Spectrom. 2005, 16, 71–81. [DOI] [PubMed] [Google Scholar]
- (42).Liang XR; Xia Y; McLuckey SA Anal. Chem. 2006, 78, 3208–3212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Webb IK; Londry FA; McLuckey SA Rapid Comm. Mass Spectrom. 2011, 25, 2500–2510. [DOI] [PubMed] [Google Scholar]
- (44).Londry FA; Hager JW J. Am. Soc. Mass Spectrom. 2003, 14, 1130–1147. [DOI] [PubMed] [Google Scholar]
- (45).Liu H; Yoo HJ; Hakansson KJ Am. Soc. Mass Spectrom. 2008, 19, 799–808. [DOI] [PubMed] [Google Scholar]
- (46).Fay L; Richli U J. Chrom. 1991, 541, 89–98. [Google Scholar]
- (47).Ratnayake WMN; Chen ZY Lipids 1996, 31, S279–S282. [DOI] [PubMed] [Google Scholar]
- (48).Drake DR; Brogden KA; Dawson DV; Wertz PW J. Lipid Res. 2008, 49, 4–11. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.