

Abstract
Self-assembly of peptides is a powerful method of preparing nanostructured materials. These peptides frequently utilize charged groups as a convenient switch for controlling self-assembly in which pH or ionic strength determines the assembly state. Multidomain peptides have been previously designed with charged domains of amino acids, which create molecular frustration between electrostatic repulsion and a combination of supramolecular forces including hydrogen bonding and hydrophobic packing. This frustration is eliminated by the addition of multivalent ions or pH adjustment, resulting in a self-assembled hydrogel. However, these charged functionalities can have profound, unintended effects on the properties of the resulting material. Access to neutral self-assembled nanostructured hydrogels may allow for distinct biological properties that are not available to highly charged analogues. Here, we designed a series of peptides to determine if self-assembly could be mediated by the steric interactions created by neutral hydroxyproline (O) domains, eliminating the need for charged residues and creating a neutral peptide hydrogel. The series of peptides, On(SL)6On, was studied to determine the effect of oligo-hydroxyproline on peptide self-assembly and nanostructure. We show that peptide solubility and nanofiber length increase with a higher number of hydroxyproline residues. Within this series, O5(SL)6O5 displayed the optimal properties for self-assembly and hydrogelation. In vitro, this hydrogel supports cell viability of fibroblasts, while in vivo it is infiltrated with cells and easily degraded over time without promoting a strong inflammatory response. This neutral self-assembling peptide hydrogel shows promising properties for biomedical, cell preservation, and tissue regeneration applications.
Keywords: self-assembly, supramolecular nanofibers, neutral nonionic peptide, hydrogel
Graphical Abstract
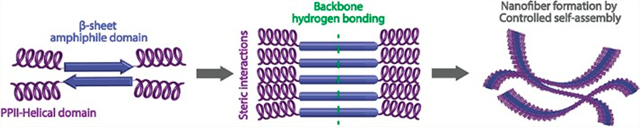
INTRODUCTION
Self-assembling peptides have been extensively studied because of their relationship to protein folding and assembly. These peptides can be designed to spontaneously self-assemble into fibrous nanomaterials, which have a chemical composition and nanostructure that is readily interfaced with living systems due to material properties that mimic the extracellular matrix.1 The resulting biointeractive nature of these supramolecular materials is useful in biomedical applications such as controlled drug and protein delivery, cell encapsulation, and tissue regeneration.2–4
Different strategies have been used to control self-assembly of peptides to form a variety of structures including nanoribbons,5 monolayers,6 nanotubes,7 and nanofibers, which utilize amphiphilic and ionic domains to govern their supramolecular structure. For instance, RADA-16 alternates hydrophobic and ionic self-complementary residues, promoting a β-sheet conformation and assembling into fibrils.8,9 Other related materials include the MAX family of peptides which contain two amphiphilic strands, alternating valine and lysine, connected by a VDPPT turn.10 The strands fold into a β-sheet structure, while the central tetrapeptide facilitates the formation of a β-hairpin structure.10 Alternative designs involve the self-assembly of fibers from hydrophobic alkyl chains covalently attached to hydrophilic peptides,11 and coiled-coil peptides with ion-complementary sticky-ends.12
Generally, self-assembling peptide design has required the incorporation of charged amino acids into the peptide sequence as a mechanism to control assembly, solubility, and material properties by stimuli such as pH and ionic strength.13–15 For example, positively charged self-assembling peptides go through conformational changes when pH is increased, neutralizing repulsive charges and resulting in either hydrogel formation or insoluble aggregates.16 These charged peptides have been widely studied as biomaterials because they are cytocompatible, have antimicrobial properties, and elicit useful biological responses.17,18 However, it is known that biological responses and cell behavior highly depend on the chemistry and charge of the interacting materials. For example, polylysine is commonly used for cell attachment and proliferation in culture techniques,19,20 but this and many other cationic polymers show an inherent concentration-dependent cytotoxicity.21,22 On the other hand, neutral polymers, such as PEG, are inert, biocompatible, and nonimmunogenic and have been used in countless studies to bestow these properties on other materials.23–25 While such neutral materials are available within the polymer design space, for peptide-based materials we are currently limited in our ability to explore biological responses to only positively or negatively charged self-assembling peptide hydrogels as strategies to control peptide self-assembly through uncharged residues are limited. Neutral, nonionic peptides tend to have poor solubility and aggregate or precipitate in aqueous solutions. This makes controlling the formation of finite supramolecular structures derived from these molecules challenging. Previously, steric interactions and size mismatch have been used to regulate the self-assembly of polymeric mushroom-shaped nanostructures.26 Herein, we incorporate this type of steric frustration as a controlling mechanism for the solubility and self-assembly of neutral peptides. The absence of charged groups allows the assembly and structure of the peptide to remain largely independent of pH and ionic strength. This work expands the scope of available selfassembling peptide nanostructured materials that may be studied for applications in biology and medicine.
Our lab has studied a class of self-assembling peptides called Multidomain Peptides (MDPs).27 These peptides are designed with an ABA motif, where the B domain alternates hydrophilic and hydrophobic amino acids while the A domain contains charged residues. The balance between charge repulsion at the ends of the peptide and stabilizing hydrogen bonding and hydrophobic packing in the core controls the extent of self-assembly. In aqueous solution, the peptides typically adopt an antiparallel β-sheet conformation and assemble into nanofibers driven by the hydrophobic effect and hydrogen bond formation between backbone amides. Fiber length and stability are modulated by pH or the addition of multivalent ions, which screen the electrostatic repulsion and shift the balance of supramolecular forces toward assembly of nanofibers. Upon achieving sufficient length these nanofibers entangle and form a viscoelastic hydrogel.27–30 Following a similar rationale, the neutral MDP design presented here is composed of the same amphiphilic core with alternating hydrophilic and hydrophobic amino acids comprised by the 12-amino acid sequence (SL)6. This domain drives intermolecular β-sheet hydrogen bonding and hydrophobic packing, which by itself yields insoluble nanofibers that aggregate and precipitate in solution. Instead of using charged termini to improve solubility and regulate selfassembly, in the current design we select hydroxyproline (here abbreviated as O) as nonionic termini. Hydroxyproline provides the necessary hydrophilicity with its hydroxyl side chain and, more importantly, cannot participate in the N—H backbone hydrogen bond donation required in the formation of β-sheets. Additionally, hydroxyproline repeats are predis-posed to form polyproline type II (PPII) helices which can provide a flexible terminus to establish molecular frustration to the growing peptide nanofibers by both steric impediment and secondary structure disruption. To study the effect of oligo-hydroxyproline on the self-assembly of nanofibers and subsequent hydrogelation, MDPs with a variable number of hydroxyproline residues, On(SL)6On, where n = 1—6, were designed and synthesized (Figure 1, Table S1).
Figure 1.
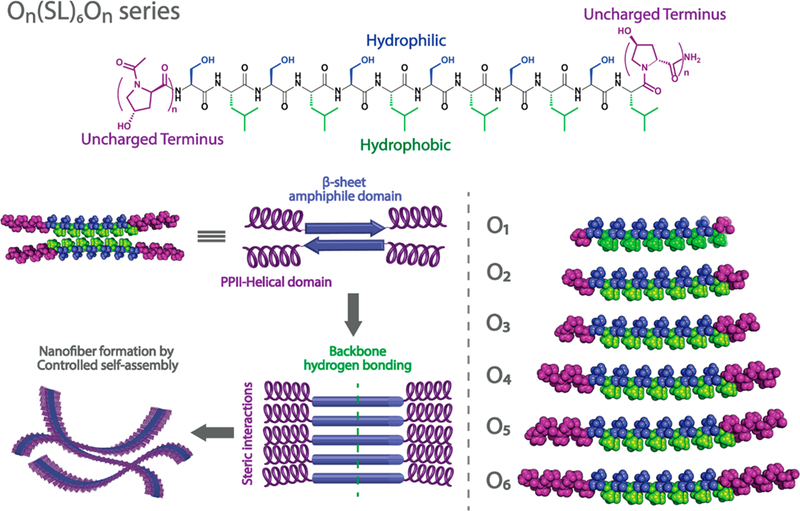
Rational design of On(SL)6On peptide series. 4-Hydroxyproline domains of variable length are incorporated into the N and C termini of MDPs to create bulky and uncharged domains. In aqueous solution, the amphiphilic domain forms an antiparallel β-sheet conformation and assembles by hydrogen bonding between peptide backbones. The uncharged domains provoke steric interactions that frustrate the infinite aggregation of the forming MDP nanofibers.
EXPERIMENTAL METHODS
Peptide Synthesis.
All peptides were synthesized using solid-phase peptide synthesis according to the methodology previously reported.28 Briefly, a semimanual synthesis was accomplish using an Apex Focus XC (Aapptec) synthesizer for mixing, solvent addition, and wash steps. Low-loading Rink Amide MBHA resin (0.32 mmol/ g) was used, and the N-termini were acetylated before cleavage with TFA and scavengers. TFA was removed by rotary evaporation and solid peptide was recovered by trituration with diethyl ether. O1 and O2 were washed with Milli-Q deionized H2O because of their poor solubility, while O3–O6 were dialyzed against Milli-Q H2O for 3–4 days in 100–500 or 1000 Da MWCO dialysis tubing (Spectra/Por, Spectrum Laboratories Inc., Rancho Dominguez, CA). Peptide solutions were adjusted to pH 7.2, sterile filtered when solubility allowed, frozen, lyophilized, and stored at –20 °C. All peptides were characterized using Autoflex MALDI-TOF MS (Bruker Instruments, Billerica, MA) to confirm purity and correct mass (Figure S1). Samples for MALDI-TOF MS characterization were prepared as follows: peptides were dissolved in TFE/H2O/MeOH (1:1:0.2) acidified with 0.05% trifluoracetic acid (TFA). The peptide solution was mixed with α-CHCA matrix solution (in 50% acetonitrile) in a 1 to 1 ratio while being spotted on a ground steel MALDI target plate. Droplets were allowed to dry before the analysis.
Circular Dichroism (CD) Spectroscopy.
For secondary structure characterization, peptide solutions of 1% by weight (molar concentration is included in Table S1) were prepared in 2,2,2-trifluoroethanol (TFE, Sigma-Aldrich, St. Louis, MO). CD spectra acquisition was performed in a CD Jasco J-810 spectropolarimeter (Jasco Inc., Easton, MD) using a 0.01 mm cuvette. Data were collected at room temperature from 180 to 250 nm at a speed of 50 nm/min, with a 0.1 nm data pitch, and the spectra were averaged over five scans. To study the hydrogelation of O5, 1% by weight peptide solutions were made in 149 mM sucrose solution in 0.5× Hank’s balanced salt solution (HBSS, Life Technologies) and analyzed as previously described.
To estimate the secondary structure of O peptide series, data fitting using the least-squares linear method was performed in MATLAB (Mathworks, Natick, MA). (Hyp)6, O1(SL)6O1, and K2(SL)6K2 were used as basis spectra for polyproline type II, β-sheet, and α-helix, respectively (Figure S2). As has been demonstrated in other systems, TFE promotes the formation of α-helical structures;31 therefore, α-helix was considered as a possible structure to achieve a better estimation.
Attenuated Total Reflectance Fourier Transform Infrared (ATR-FTIR) Spectroscopy.
Peptide samples (10 μL of 1% by weight peptide solution) were dried under nitrogen flow on a Golden Gate diamond window of an ATR stage or on a glass slide. IR spectra were collected on a Jasco FT/IR-660 plus spectrometer (Jasco Inc., Easton, MD) and a Nicolet iS5 FT/IR spectrometer (Thermo Scientific, Waltham, MA) at 1 cm−1 resolution with 64 scans accumulation, and the background was subtracted, and vapor reduction was applied to minimize the contribution of water in the resulting spectra. Spectragryph–optical spectroscopy software was used for data processing and normalization.32
Transmission Electron Microscopy (TEM).
For TEM, 1% by weight peptide solutions in TFE or Milli-Q H2O were diluted to concentrations ranging from 0.1 to 0.01% by weight with Milli-Q water. All samples were prepared via negative staining on Quantifoil R1.2/1.3 holey carbon films on copper mesh grids. Each peptide was spotted and allowed to adsorb for 1 min before excess peptide was blotted. A 2 wt % phosphotungstic acid (PTA) solution in Milli-Q water was prepared at pH 7 and sterile filtered. The sample grids were negative stained for 5 min in PTA. Excess PTA was wicked off the grids before the samples were dried and imaged at 120 kV and 40K magnification using a JEOL 2010 TEM microscope (JEOL USA Inc., Peabody, MA).
Hydrogel Formation.
Peptide solutions of O5 were prepared at 1% by weight (4.2 mM) in 149 mM sucrose and 0.5× HBSS. This buffer system was chosen to be consistent with prior MDP experiments and to find optimum gelling conditions for future in vivo and in vitro work. Peptide solutions were sonicated using a Microson Ultrasonic Cell Disruptor with a 2 mm microprobe. The instrument frequency was 22.5 kHz and a power output up to 10 W (RMS) was used. Solutions were sonicated at room temperature for 1 or 10 cycles, where each cycle consisted of 10 pulses and 1 min relaxation time between cycles. For the study of hydrogelation of O5, K2, and E2 at different pH values and ionic strength, 1% by weight peptide solutions were prepared in deionized water, PBS, or HBSS. pH was adjusted using HCl or NaOH 0.1 M solutions.
Oscillatory Rheology.
Rheological properties of O5 hydrogel were analyzed by oscillatory rheology using a TA Instruments AR-G2 rheometer (TA Instruments, New Castle, DE). Hydrogels were prepared 24 h before the analysis as described above. 150 μL of 1% by weight peptide hydrogels were transferred from a cut syringe onto the rheometer stage equipped with a 12 mm stainless-steel parallel plate set to a 1000 μm gap height. The following program was used to monitor the storage modulus (G’) and loss modulus (G”) under various conditions as has been previously published.4 Strain sweep analysis was performed using an applied strain of 0.01–200% at a frequency of 1 rad/s. Frequency sweep analysis was performed under 1% strain at 0.1–100 rad/s. Shear recovery analysis was performed by subjecting the peptide sample to 1% strain for 20 min, then 200% for 1 min, and finally 1% for 20 min, allowing for disruption of the hydrogel and monitoring of G’ and G” recovery.
Scanning Electron Microscopy (SEM).
For SEM characterization, 1% by weight peptide hydrogel was prepared as previously described. Hydrogel was dehydrated with a series of ethanol from 30% to 100% and the ethanol was removed by critical point drying using an EMS 850 critical point dryer (Electron Microscopy Sciences, Hatfield, PA). Dried samples were mounted into SEM pucks with conductive carbon tape and coated with 4 nm of gold using a Denton Desk V Sputter system (Denton Vacuum, Moorestown, NJ). Samples were imaged using a JEOL 6500F Scanning Electron Microscope (JEOL, USA Inc., Peabody, MA).
Cytocompatibility and Cell Viability Studies.
All cell culture materials, not otherwise specified, were purchased from Gibco, Life Technologies. NIH-3T3 fibroblast cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2% L-glutamine (200 mM), 1% sodium pyruvate (100 mM), and 1% pen-strep solution (5000 units/mL of penicillin and 5 mg/mL of streptomycin, Sigma-Aldrich, St. Louis, MO). Cells were maintained at 37 °C and 5% CO2 with humidity and split when reaching 80–90% confluency by treatment with 0.25% trypsin-EDTA and neutralization with complete media. Cells were centrifuged for 6 min at 1600 rpm, then the cell pellet was dispersed in HBSS, counted and adjusted to the required concentration. 3D cell culture in K2 hydrogel was achieved by mixing a 2% by weight peptide stock solution in 298 mM sucrose with a cell suspension of 2 × 106 cells/ mL in HBSS. The final concentration was 1% by weight K2 hydrogel in 149 mM sucrose and 0.5× HBSS with 1 × 106 cells/mL. For O5 peptide, hydrogel was prepared by sonication using only 90% of the required 149 mM sucrose/0.5× HBSS to obtain a 1% by weight concentration, and the remaining 10% was added as a cell suspension and mixed by pipetting into the gel to obtain a final concentration of 1 × 106 cells/mL evenly distributed though the volume of the hydrogel. A volume of 70 μL of seeded hydrogels was cultured in a Lab-Tek 16-well glass chamber (0.4 mm2, Thermo Fisher, Rochester, NY) with 200 μL of medium and fed every 2 days by replacing 100 μL with fresh media. Cell viability was analyzed using a live/dead viability kit (Invitrogen, Thermo Fisher, Carlsbad, CA). At specific time points, the medium was removed, and gels were rinsed with 1× PBS, and then 100 μL of staining solution containing 2 μM Calcein AM and 4 μM ethidium homodimer-1 in DPBS was added. Cells were incubated from 15 to 30 min and imaged using a Nikon A1 Rsi fluorescent confocal microscope (Nikon Instruments, Tokyo, Japan). Three z-stack images of 100 μm deep per sample were taken at 20× using a 488 nm green channel and 561 nm red channel lasers. Images were processed with NIS Element, and live/dead cell counting was performed on Imaris 3D/4D Image Processing software (Bitplane, Concord, MA).
To evaluate the cell morphology, hydrogels were rinsed with PBS and fixed with 10% buffered formalin for 30 min. Cells were treated with 0.5% Triton X-100 for 10 min and 100 mM glycine for 10 min, followed by 10% bovine serum albumin (BSA) in HBSS block solution. Then cells were stained for filamentous actin with Alexa Fluor 488-Phalloidin and for the nuclei with ProLong Gold Antifade DAPI mounting media. Cell in the hydrogels were analyzed by confocal microscopy using a 40× water objective and processed as previously described.
In Vivo Compatibility Studies.
All experimental procedures were approved by the Rice University Institutional Animal Care and Use Committee (IACUC) and performed according to the Animal Welfare Act and NIH guidelines. Female C57BL/6 mice from 8 to 12 weeks were purchased from Charles River (Wilmington, MA). Mice were injected with 4 implants of 100 μL 1% by weight O5 hydrogel in the dorsal subcutaneous space using a 26-gauge needle. At days 3, 7, and 14, mice were euthanized, and implants were extracted, fixed with 10% neutral buffered formalin, and processed into paraffin blocks. Tissue was sectioned at 5 μm thickness and stained with hematoxylin and eosin (H&E) and Masson’s trichome under standard procedures. For fluorescent immunostaining, tissue sections were deparaffinized and hydrated, and antigens were retrieved by boiling in sodium citrate buffer (10 mM sodium citrate, 0.05% Tween 20 at pH 6.0). Tissues were permeabilized with 0.5% Triton X-100 and blocked with 1% BSA in PBS for 30 min. Tissues were incubated overnight at 4 °C with rat anti-mouse F4/80 monoclonal antibody (1:50, clone BM8, eBioscience, cat# 14–4801-82). Then, samples were incubated for 1 h with goat anti-rat AlexaFluor 568 (1:500, Invitrogen), followed by mounting with ProLong Gold Antifade reagent with DAPI. Tissue slides were analyzed by confocal fluorescent microscopy.
RESULTS AND DISCUSSION
The series of peptides On(SL)6On were prepared by solid phase peptide synthesis. From the series, O1–O4 were poorly soluble in water (<0.5 mg/mL) and resulted in the formation of a heterogeneous suspension. In contrast, O5 and O6 were found to be soluble in water at concentrations of 1 mg/mL. At 10 mg/mL, O5 forms a hydrogel while O6 is a viscous solution. Poor solubility made peptide characterization challenging, especially for O1-O4, and therefore, in many cases, the use of 2,2,2-trifluoroethanol (TFE) facilitated the sample preparation and characterization of the series. Particularly for molecular mass determination using MALDI-MS, TFE helped with the dispersion of insoluble peptide aggregates and allowed for higher quality spectra. The use of TFE as a solvent for MALDI sample preparation may be generally useful for characterizing peptides with low solubility, a common difficulty for peptides designed to self-assemble.
Secondary Structure Characterization of On(SL)6On Series.
The ATR-FTIR spectra (Figure 2a) of the dried peptide films presented a peak between 1620 and 1630 cm−1, which corresponds to the amide I band of a β-sheet conformation, and a weak peak around 1695 cm−1 indicating an antiparallel component. These results indicate that the core β-sheet structure is not significantly disturbed by the oligo-hydroxyproline termini in the dried samples. However, there does appear to be a slight decrease in β-sheet character with the addition of more hydroxyproline residues, where the intensity of amide Ib peak decreases from O1-O6 and subtle variation is observed in the amide I band wavenumber. These observations are consistent with the circular dichroism spectroscopy analyses described below.
Figure 2.
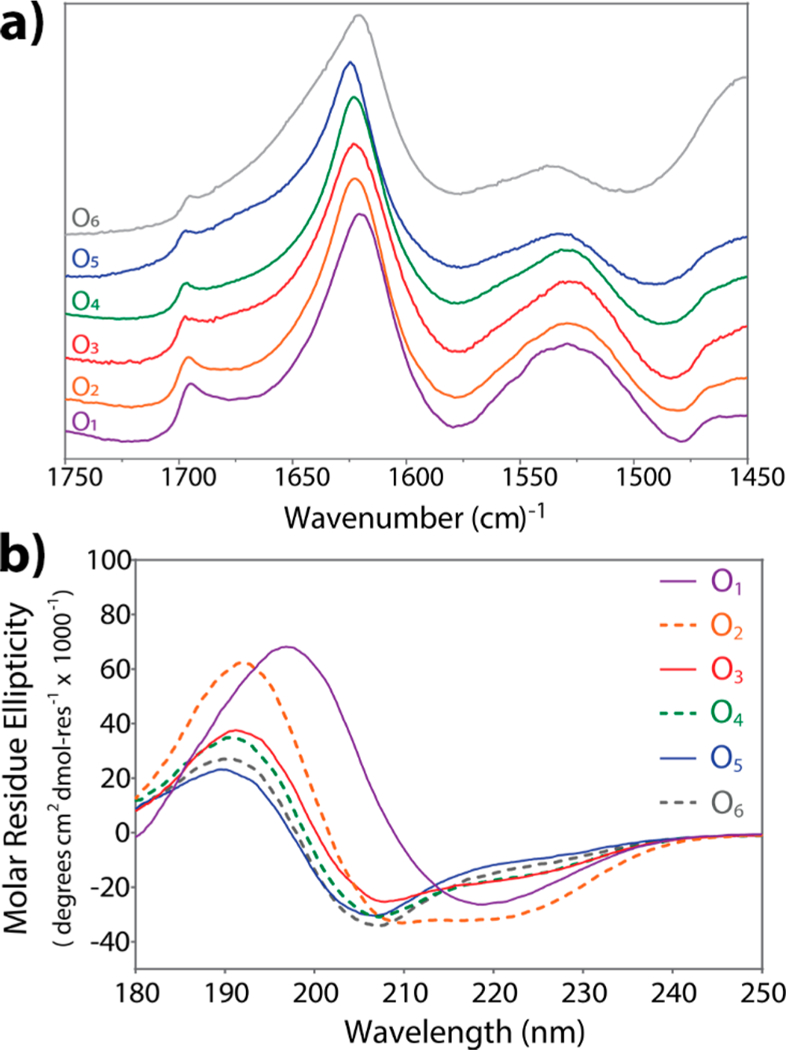
Secondary structure characterization of On(SL)6On series. (a) Normalized ATR-FTIR spectra of the amide I and amide II region of dried peptide samples. All peptides have characteristic peaks for antiparallel β-sheet structure: amide Ia between 1620–1630 cm−1 and amide Ib at 1695 cm−1. (b) Circular dichroism (CD) spectra of 1 wt % peptide solutions in 2,2,2-trifluoroethanol (TFE) at 25 °C.
Poor solubility of O1—O4 made characterization across all peptides in uniform solvent challenging; therefore, 2,2,2-trifluoroethanol (TFE) was often used as a solubilizing agent. Circular dichroism (CD) spectroscopy of peptide solutions in TFE (1% by weight, 7–4 mM) was performed (Figure 2b). CD of O1 corresponds to a canonical β-sheet structure, presenting minima at 218 nm and maxima at 197 nm. Peptides O2 through O6 display a combination of different secondary structures. However, increasing number of hydroxyproline residues resulted in a corresponding increased fraction of PPII secondary structure as determined using a least-squares linear fitting method (Tables S2 and S3). While the peptides also showed an α-helical component, this is due to the propensity of TFE to promote this conformation.33,34 Together the IR and CD suggests the secondary structure of this series of peptides is β-sheet in the amphiphilic (SL)6 core while the On termini in O2 - O6 adopt a PPII helical structure. This hydroxyproline PPII helix becomes large enough to frustrate infinite assembly and aggregation and thus, mediate solubility for O5 and O6.
Nanostructure of On(SL)6On Series.
Negative-stain TEM was used to analyze the nanostructure of the O1-O6 peptide series. The very poor water solubility of O1 and O2 resulted in dense clusters of nanofibers along with amorphous aggregates in their TEM images with only rare areas being dispersed enough to see the underlying nanofibrous structure (Figure 3). However, the dispersion of the nanofibers increases with the number of hydroxyproline residues, improving their visualization. Also, the apparent fiber length increases with greater numbers of hydroxyproline residues. While fibers from O1-O4 appear mostly short and rigid, O5 and O6 nanofibers are significantly longer and display more curvature, suggesting greater flexibility. TEM was also performed on O1-O6 which had been initially dissolved in TFE to help disperse the aggregated nanofibers and revealed similar trends to those formed only in water (Figure S4). These data suggest that the increase in hydroxyproline residues and PPII content in the peptide structure modulate the solubility and nanostructure of the fibers by increasing both the hydrophilicity and steric frustration in the supramolecular assembly. With too short of On termini, the peptides assemble into short, rigid, low solubility fibers that form dense aggregates and precipitate. In contrast, with too long of On termini, the peptide assembles into long, easily solubilized fibers, but with too few physical cross-links to support gelation.
Figure 3.
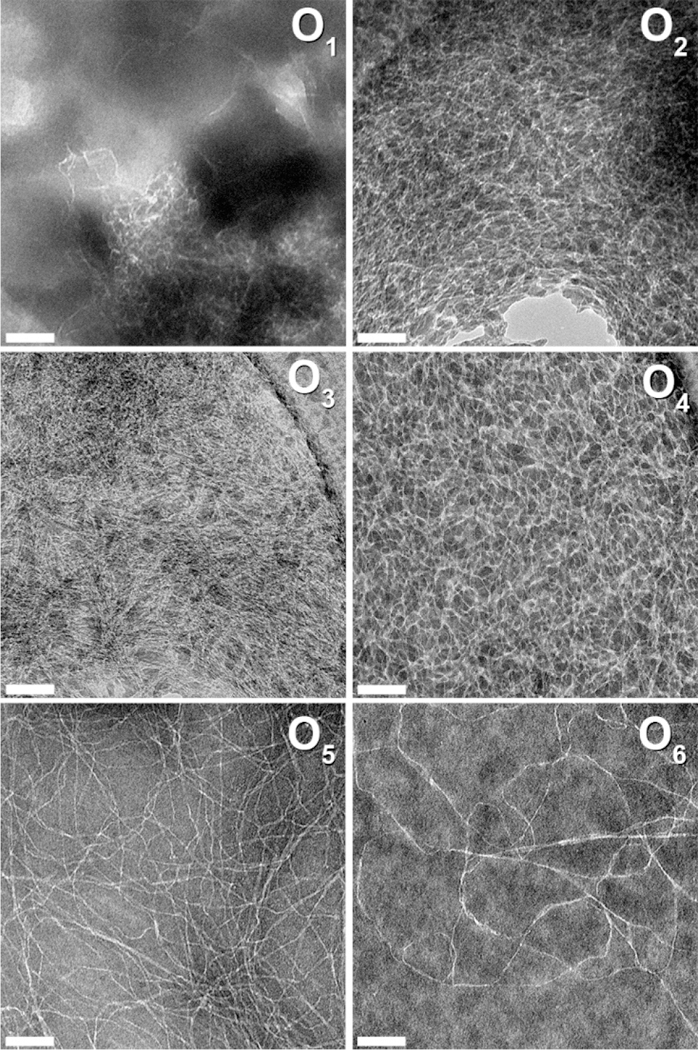
Negative-stained TEM images of On(SL)6On series in water. O1, O2, and O5 at 0.01 wt %; O3, O4, and O6 at 0.02 wt %. Scale bar = 100 nm.
Hydrogelation of O5(SL)6O5 Peptide.
Hydrogel for-mation of previously designed MDPs is achieved by the addition of multivalent ions (such as phosphate) to induce gelation at neutral pH. Under these conditions O5 forms a self-supportive hydrogel after dispersion by ultrasonication while O1-O4 remain insoluble and O6 dissolves to form a viscous solution. This suggests that only O5(SL)6O5 reaches the right balance of fiber length, rigidity and solubility to form a hydrogel. This peptide remains a hydrogel over a wide pH range (3–11) as well as in deionized water (Figure 4) and common buffers such as phosphate-buffered saline (10 mM phosphate, 137 mM NaCl) (Figure S5) and Hank’s balanced salt solution (HBSS). Similarly, the secondary structure as examined by CD has only minor changes under this range of conditions demonstrating the robustness of the neutral MDP approach. In contrast, charged MDPs such as K2(SL)6K2 and E2(SL)6E2 have a strong dependence on pH and ionic strength with gels only forming at neutral pH or at elevated ionic strength (Figures 4 and S6), and significant changes to secondary structure are observed across these conditions that correlate to the ionization or shielding of their charged residues.
Figure 4.
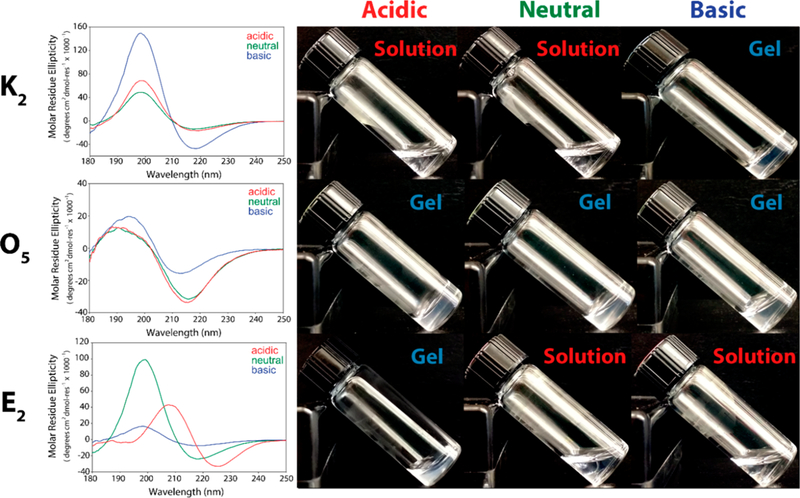
O5(SL)6O5 hydrogel properties are pH and ionic strength independent. CD spectra of the positively charged MDP, K2(SL)6K2, neutral O5(SL)6O5, and negatively charged, E2(SL)6E2, at different pH conditions in water. K2 and E2 hydrogel formation is dependent on pH, whereas O5 remains a hydrogel at acidic (pH 3), neutral (pH 7), and basic pH (pH 11).
To assess the biological potential of the O5 peptide, we tested its hydrogelation in media that supports cell culture (1% by weight in 149 mM sucrose and 0.5× HBSS) and is commonly used to test charged MDPs such as K2(SL)6K2 and E2(SL)6E2.30 Figure 5a shows the CD spectra of aqueous 1 wt % (4.2 mM) O5 peptide solution in 149 mM sucrose and 0.5× HBSS. SEM analysis of the resulting hydrogel reveals a high-density nanofiber network (Figure 5b). The viscoelastic properties of the O5 hydrogel were analyzed by oscillatory rheology, and a storage modulus (G’) of 23.3 ± 8.7 Pa and a loss modulus (G”) of 1.7 ± 0.5 Pa were found. The low G’ value compared to previously studied MDPs,28 which are typically in the range of 100–400 Pa, indicates that this material is a relatively compliant and flexible gel, which is also confirmed by the frequency sweep (Figure S7). The O5 peptide hydrogel exhibits liquid behavior when high strain is applied, while having a very rapid recovery of approximately 92% of its initial storage modulus 1 min after the strain is released (Figure 5d), making this supramolecular material easily injectable because of its shear thinning and shear recovery properties. In comparison, charged MDPs typically show a slower shear recovery of approximately 80% in the same time frame and require approximately 10 min to reach 90% recovery.28
Figure 5.
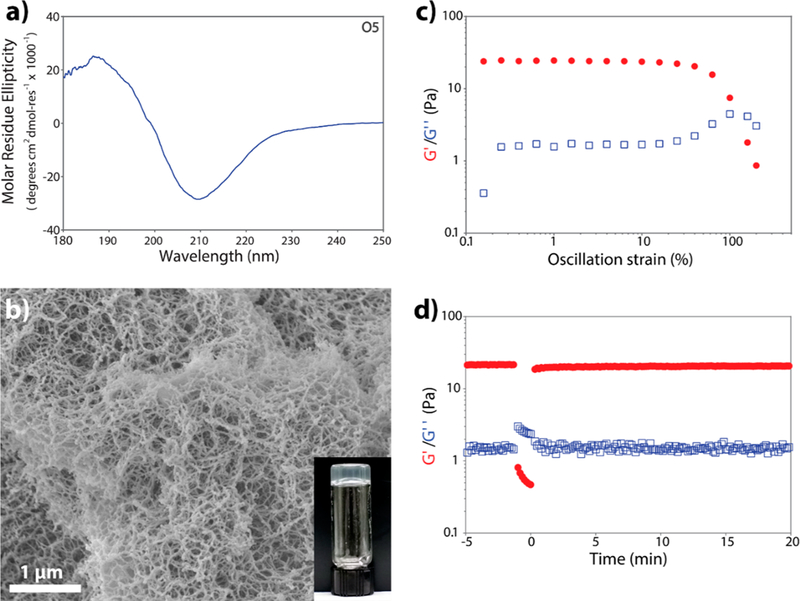
O5(SL)6O5 peptide solution forms a hydrogel upon treatment with ultrasonication. (a) Structural characterization of 1 wt % peptide solutions in 149 mM sucrose and 0.5× HBSS. (b) SEM micrograph of 1 wt % O5 hydrogel showing an entangled fibrillar structure. Scale bar = 1 μm. (c) Strain sweep and (d) shear recovery of 1 wt % O5 peptide hydrogel.
Cytocompatibility of O5 Peptide Hydrogel.
Former MDP hydrogels, particularly lysine-based peptides, have shown great promise for their use as biomaterials in part because of their cytocompatibility.30 To study the effect of using nonionic O5 flanking domains on cell compatibility, 3D culture of NIH-3T3 fibroblasts was performed in O5 peptide hydrogel and compared to culture in the positively charged K2 peptide. Cell viability was evaluated using a Calcein-AM and Ethidium homodimer-1 test (Figure 6). At day 1, approximately 20% of cells are viable in K2 hydrogel, whereas for O5 hydrogel, almost 70% survive after encapsulation (Figure 6a–d). At day 3, these levels are relatively unchanged. By day 5, fibroblasts are growing and reproducing in the positively charged K2 hydrogel, achieving a similar percentage of viable cells as O5 hydrogel. Overall, K2 is observed to be somewhat cytotoxic during the first day of cell culture, likely due to the high density of positively charged amines. However, cells recover and can attach and proliferate within the gel, and by day 5 cell density is significantly higher than day 1 and 3 (Figure 6e). On the other hand, fibroblasts encapsulated in the O5 hydrogel show good initial viability and this remains constant throughout the experiment (Figure 6d). Despite high and consistent cell viability in O5 over the course of this study, the encapsulated fibroblasts are not observed to proliferate. Cells encapsulated in O5 gel do not spread and adhere to the material like in K2, but instead they remain “balled up” as shown in Figure 6b,c. Generally, this balled-up morphology represents imminent cell death or apoptosis,35 where cells die from 12 to 24 h after initiating this process.36,37 Yet, cells in the neutral MDP preserve this morphology through day 5 (120 h) and show green fluorescence derived from Calcein AM, indicating an intact membrane and cell survival. To confirm the viable nature of these cells, they were harvested from the O5 hydrogel after 5 days and transferred to standard tissue culture plastic where the cells were found to adhere, spread and proliferate (Figure S8). All these observations suggest that fibroblasts cultured in O5 may be induced to enter a quiescent state when encapsulated within the hydrogel. The ability of O5 hydrogel to keep cells alive without proliferation is a promising result with potential applications for the use of this material in cell preservation. The differences in cell viability, morphology and proliferation are all indicators of the substantial differences induced by fibers with similar morphology but with different charge.
Figure 6.
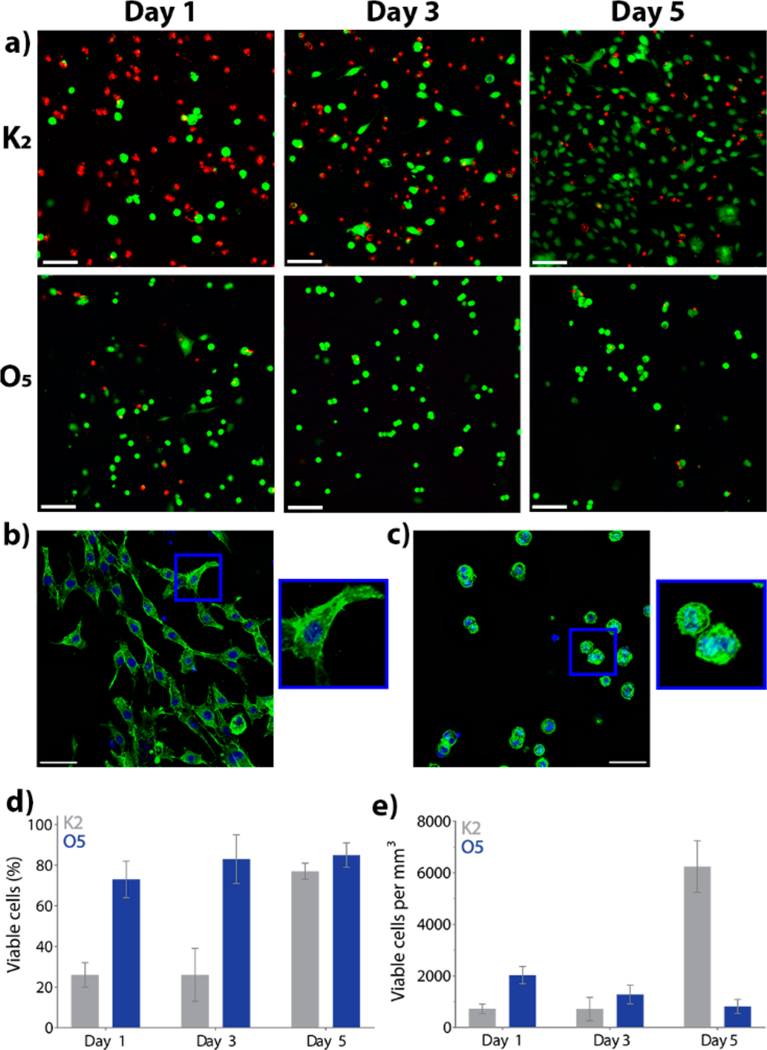
3D culture of NIH-3T3 fibroblasts in 1 wt % O5 hydrogel and the positively charged peptide hydrogel K2(SL)6K2. (a) Cell viability of NIH-3T3 fibroblasts at day 1, 3, and 5 by Calcein AM and ethidium homodimer-1 staining (viable cells in green, dead cells in red). Scale bar = 100 μm. Actin cytoskeleton staining of NIH-3T3 fibroblasts encapsulated for 5 days in (b) K2 and (c) O5. AlexaFluor 488-phalloidin (green) and DAPI (blue). Scale bar is 50 μm. (d) Percentage of viable cells present in K2 and O5 at day 1, day 3 and day 5 after seeding. (e) Viable cell density (cells/ mm3) at day 1, day 3, and day 5. Error bars represent standard deviation (n = 6).
In Vivo Host Response to O5 Peptide Hydrogel.
To investigate the application of this peptide-based material in regenerative medicine, we evaluated the host response to O5 peptide hydrogel in mice using a subcutaneous injection model. The injected hydrogel boluses were examined at 3, 7, and 14 days after injection. Figure 7 shows histological sections of the implants stained with hematoxylin and eosin and fluorescent immunostaining for F4/80+ macrophages. At day 3 (Figure 7a), F4/80+ macrophages have infiltrated the periphery of the material, while some are migrating to the core. By day 7 (Figure 7b), macrophages have completely infiltrated the implant. A decrease in the implant size suggests that the O5 hydrogel is degrading (Figure S9). By day 14 (Figure 7c), the hydrogel is almost completely degraded, with fewer macrophages observed. Additionally, natural collagen is observed as the material is remodeled into natural tissue (Figure S10). Compared to K2 hydrogel, the neutral MDP O5 elicited a weaker inflammatory response and did not promote vascularization or innervation.38 However, in contrast with other neutral polymers, such as PEG, the O5 hydrogel promotes rapid cellular infiltration, is degradable, and is not fibrously encapsulated,39 demonstrating that it promotes reduced biological responses and a decreased inflammatory response similar to the PEG materials while also maintaining the ability to interact with living systems.
Figure 7.
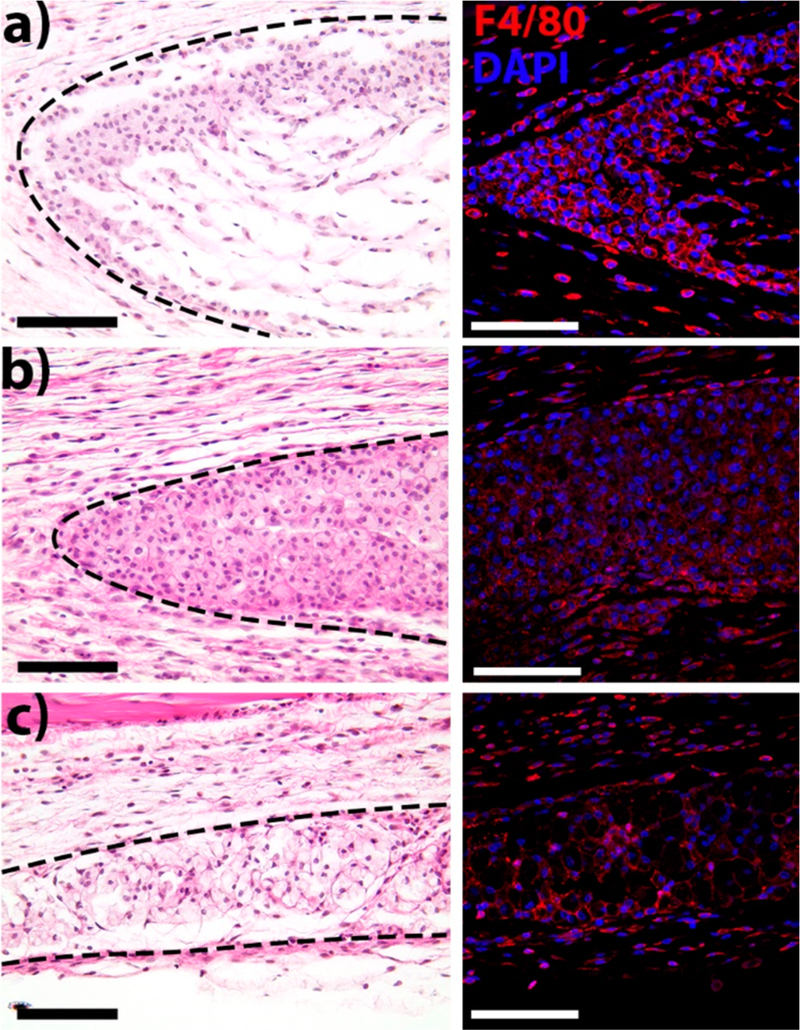
H&E and immunofluorescent staining of subcutaneous implants of O5 hydrogel. (a) Day 3, (b) day 7, and (c) day 14. After implantation, the hydrogel is infiltrated by macrophages (F4/80+ cells, red; nuclei, blue) which degrade the material and remodel the native tissue. By day 14, the implant size decreases and new collagen is observed similar to native tissue (Figures S9 and S10). Scale bar is 100 μm. Dotted line indicates borders of the hydrogel implant.
CONCLUSIONS
We have demonstrated an alternative mechanism to control the self-assembly of multidomain peptides that does not involve the use of charged amino acids. Uncharged domains of oligo-hydroxyproline at the peptide termini form bulky helical structures that create molecular frustration between the β-sheet self-assembling domain and terminal steric interactions. These steric interactions and chemical functionality from the hydroxyproline domains affect peptide solubility, aggregation, and nanofiber structure. While all peptides in the On(SL)6On peptide series (n = 1–6) formed nanofibers, only O5(SL)6O5 was able to form a hydrogel. This nanostructured hydrogel shows promising properties for biomedical applications, including rapid shear recovery and support for cell viability. It also appears to induce a quiescent state in cells over time, a property that may be desirable for cell preservation. When implanted in vivo, the hydrogel is infiltrated by macrophages without evidence of eliciting an adverse inflammatory response and the implant is degraded over time. This work establishes the creation of a neutral biocompatible hydrogel and extends the scope of peptide-based self-assembling materials to encompass positive, negative, and neutral nanofiber chemistries for diverse biomaterial designs.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from NIH NIDCR R01DE021798 and from the Welch Foundation C1557. T.L.L.-S was supported by the Mexican National Council for Science and Technology (CONACyT) Ph.D. Scholarship Program (678341). D.G.L. was supported by the NSF Graduate Research Fellowship Program under Grant 1450681. I.L. was supported by the Stauffer-Rothrock Fellowship.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsbiomater-ials.8b01348.
Peptide sequences, MALDI-TOF mass spectra data, basis spectra for CD spectroscopy data fitting, fitting results, rheological data, and tissue staining data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Lutolf MP; Hubbell JA Synthetic Biomaterials as Instructive Extracellular Microenvironments for Morphogenesis in Tissue Engineering. Nat. Biotechnol. 2005, 23 (1), 47–55. [DOI] [PubMed] [Google Scholar]
- (2).Webber MJ; Appel EA; Meijer EW; Langer R Supramolecular Biomaterials . Nat. Mater. 2016, 15 (1), 13–26. [DOI] [PubMed] [Google Scholar]
- (3).Rad-Malekshahi M; Lempsink L; Amidi M; Hennink WE; Mastrobattista E Biomedical Applications of Self-Assembling Peptides. Bioconjugate Chem. 2016, 27 (1), 3–18. [DOI] [PubMed] [Google Scholar]
- (4).Leach DG; Dharmaraj N; Piotrowski SL; Lopez-Silva TL; Lei YL; Sikora AG; Young S; Hartgerink JD STINGel: Controlled Release of a Cyclic Dinucleotide for Enhanced Cancer Immunotherapy. Biomaterials 2018, 163, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Aggeli A; Bell M; Carrick LM; Fishwick CWG; Harding R; Mawer PJ; Radford SE; Strong AE; Boden N PH as a Trigger of Peptide β-Sheet Self-Assembly and Reversible Switching between Nematic and Isotropic Phases. J. Am. Chem. Soc. 2003, 125 (32), 9619–9628. [DOI] [PubMed] [Google Scholar]
- (6).Powers ET; Yang SI; Lieber CM; Kelly JW Ordered Langmuir-Blodgett Films of Amphiphilic B-Hairpin Peptides Imaged by Atomic Force Microscopy. Angew. Chem., Int. Ed. 2002, 41, 127–130. [DOI] [PubMed] [Google Scholar]
- (7).Hartgerink JD; Granja JR; Milligan RA; Ghadiri M R Self-Assembling Peptide Nanotubes. J. Am. Chem. Soc. 1996, 118 (1), 43–50. [Google Scholar]
- (8).Holmes TC; de Lacalle S; Su X; Liu G; Rich, a; Zhang, S. Extensive Neurite Outgrowth and Active Synapse Formation on SelfAssembling Peptide Scaffolds. Proc. Natl. Acad. Sci. U. S. A. 2000, 97 (12), 6728–6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Cormier AR; Pang X; Zimmerman MI; Zhou H; Paravastu AK Molecular Structure of RADA16 - I Designer Self-Assembling Peptide Nano Fibers. ACS Nano 2013, 7 (9), 7562–7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Pochan DJ; Schneider JP; Kretsinger J; Ozbas B; Rajagopal K; Haines L Thermally Reversible Hydrogels via Intramolecular Folding and Consequent Self-Assembly of a de Novo Designed Peptide. J. Am. Chem. Soc. 2003, 125 (39), 11802–11803. [DOI] [PubMed] [Google Scholar]
- (11).Hartgerink JD; Beniash E; Stupp SI Self-Assembly and Mineralization of Peptide-Amphiphile Nanofibers. Science 2001, 294 (5547), 1684–1688. [DOI] [PubMed] [Google Scholar]
- (12).Banwell EF; Abelardo ES; Adams DJ; Birchall MA; Corrigan A; Donald AM; Kirkland M; Serpell LC; Butler MF; Woolfson DN Rational Design and Application of Responsive Alpha-Helical Peptide Hydrogels. Nat. Mater. 2009, 8 (7), 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chockalingam K; Blenner M; Banta S Design and Application of Stimulus-Responsive Peptide Systems. Protein Eng. Des. Sel. 2007, 20 (4), 155–161. [DOI] [PubMed] [Google Scholar]
- (14).Habibi N; Kamaly N; Memic A; Shafiee H Self-Assembled Peptide-Based Nanostructures: Smart Nanomaterials toward Targeted Drug Delivery. Nano Today 2016, 11 (1), 41–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Micklitsch CM; Knerr PJ; Branco MC; Nagarkar R; Pochan DJ; Schneider JP Zinc-Triggered Hydrogelation of a Self-Assembling β-Hairpin Peptide. Angew. Chem. Int. Ed. 2011, 50 (7), 1577–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Schneider JP; Pochan DJ; Ozbas B; Rajagopal K; Pakstis L; Kretsinger J Responsive Hydrogels from the Intramolecular Folding and Self-Assembly of a Designed Peptide. J. Am. Chem. Soc. 2002, 124 (50), 15030–15037. [DOI] [PubMed] [Google Scholar]
- (17).Veiga AS; Sinthuvanich C; Gaspar D; Franquelim HG; Castanho MARB; Schneider JP Arginine-Rich Self-Assembling Peptides as Potent Antibacterial Gels. Biomaterials 2012, 33 (35), 8907–8916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Nisbet DR; Williams RJ Self-Assembled Peptides: Characterisation and in Vivo Response. Biointerphases 2012, 7 (1–4), 1–14. [DOI] [PubMed] [Google Scholar]
- (19).Mazia D; Schatten G; Sale W Adhesion of Cells to Surfaces Coated with Polylysine. J. Cell Biol. 1975, 66, 198–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Blau A Cell Adhesion Promotion Strategies for Signal Transduction Enhancement in Microelectrode Array in Vitro Electrophysiology: An Introductory Overview and Critical Discussion. Curr. Opin. Colloid Interface Sci. 2013, 18 (5), 481–492. [Google Scholar]
- (21).Fischer D; Li Y; Ahlemeyer B; Krieglstein J; Kissel T In Vitro Cytotoxicity Testing of Polycations: Influence of Polymer Structure on Cell Viability and Hemolysis. Biomaterials 2003, 24, 1121–1131. [DOI] [PubMed] [Google Scholar]
- (22).Kadlecova Z; Baldi L; Hacker D; Wurm FM; Klok HA Comparative Study on the in Vitro Cytotoxicity of Linear, Dendritic, and Hyperbranched Polylysine Analogues. Biomacromolecules 2012, 13 (10), 3127–3137. [DOI] [PubMed] [Google Scholar]
- (23).Zhu J Bioactive Modification of Poly(Ethylenglykol) Hydrogels for Tissue Engineering. Biomaterials 2010, 31 (17), 4639–4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Meyers SR; Grinstaff MW Biocompatible and Bioactive Surface Modifications for Prolonged in Vivo Efficacy. Chem. Rev. 2012, 112 (3), 1615–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Li Y; Maciel D; Rodrigues J; Shi X; Tomas H Biodegradable Polymer Nanogels for Drug/Nucleic Acid Delivery. Chem. Rev. 2015, 115 (16), 8564–8608. [DOI] [PubMed] [Google Scholar]
- (26).Stupp SI; Lebonheur V; Walker K; Li LS; Huggins KE; Keser M; Amstutz A Supramolecular Materials: Self-Organized Nanostructures. Science 1997, 276 (5311), 384–389. [DOI] [PubMed] [Google Scholar]
- (27).Dong H; Paramonov SE; Aulisa L; Bakota EL; Hartgerink JD Self-Assembly of Multidomain Peptides: Balancing Molecular Frustration Controls Conformation and Nanostructure. J. Am. Chem. Soc. 2007, 129 (41), 12468–12472. [DOI] [PubMed] [Google Scholar]
- (28).Aulisa L; Dong H; Hartgerink JD Self-Assembly of Multidomain Peptides: Sequence Variation Allows Control over Cross-Linking and Viscoelasticity. Biomacromolecules 2009, 10 (9), 2694–2698. [DOI] [PubMed] [Google Scholar]
- (29).Bakota EL; Sensoy O; Ozgur B; Sayar M; Hartgerink JD Self-Assembling Multidomain Peptide Fibers with Aromatic Cores. Biomacromolecules 2013, 14 (5), 1370–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Moore AN; Hartgerink JD Self-Assembling Multidomain Peptide Nanofibers for Delivery of Bioactive Molecules and Tissue Regeneration. Acc. Chem. Res. 2017, 50 (4), 714–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Myers JK; Pace CN; Scholtz JM Trifluoroethanol Effects on Helix Propensity and Electrostatic Interactions in the Helical Peptide from Ribonuclease T1. Protein Sci. 1998, 7 (2), 383–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Menges F Spectragryph - Optical Spectroscopy Software, Version 1.2.7; 2017; http://www.effemm2.de/spectragryph/.
- (33).SOnnichsen FD; Van Eyk JE; Hodges RS; Sykes BD Effect of Trifluoroethanol on Protein Secondary Structure: An NMR and CD Study Using a Synthetic Actin Peptide. Biochemistry 1992, 31 (37), 8790–8798. [DOI] [PubMed] [Google Scholar]
- (34).Roccatano D; Colombo G; Fioroni M; Mark AE Mechanism by Which 2,2,2-Trifluoroethanol/Water Mixtures Stabilize Secondary-Structure Formation in Peptides: A Molecular Dynamics Study. Proc. Natl. Acad. Sci. U. S. A. 2002, 99 (19), 12179–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Saraste A; Pulkki K Morphologic and Biochemical Hallmarks of Apoptosis. Cardiovasc. Res. 2000, 45 (3), 528–537. [DOI] [PubMed] [Google Scholar]
- (36).Saraste A Morphologic Criteria and Detection of Apoptosis. Herz 1999, 24 (3), 189–195. [DOI] [PubMed] [Google Scholar]
- (37).Messam CA; Pittman RN Asynchrony and Commitment to Die during Apoptosis. Exp. Cell Res. 1998, 238 (2), 389–398. [DOI] [PubMed] [Google Scholar]
- (38).Moore AN; Lopez Silva TL; Carrejo NC; Origel Marmolejo CA; Li IC; Hartgerink JD Nanofibrous Peptide Hydrogel Elicits Angiogenesis and Neurogenesis without Drugs, Proteins, or Cells. Biomaterials 2018, 161, 154–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lynn AD; Kyriakides TR; Bryant SJ Characterization of the in Vitro Macrophage Response and in Vivo Host Response to Poly (Ethylene Glycol) -Based Hydrogels. J. Biomed. Mater. Res. Part A 2009, 93A (3), 941–953. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.