

Abstract
Rothmund–Thomson syndrome (RTS) is a rare autosomal recessive disorder caused by mutations in RECQL4 and has characteristic clinical features. We report two unrelated phenotypically diverse patients (cases 1 and 2) with RTS having novel variants in RECQL4 gene . Case-1 was evaluated for poor growth and recurrent fractures and skin lesions. Case-2 presented at 4 months with failure to thrive and radial ray defect and developed poikilodermatous skin lesions after infancy. Both cases were confirmed to have homozygous pathogenic variants in RECQL4 . Both patients have normal intellect and are on supportive therapy. The presence of characteristic poikiloderma lesions with specific distribution and skeletal anomalies in a patient with proportionate short stature is a clue toward the diagnosis of RTS.
Keywords: Rothmund–Thomson syndrome, RECQL4, poikiloderma
Introduction
Rothmund–Thomson syndrome (RTS; OMIM 268400) is a rare genetic disorder characterized by distinctive skin lesions of poikiloderma, skeletal abnormalities, failure to thrive and high risk for developing cancers. 1 Two-thirds of RTS cases are caused by pathogenic variants in RECQL4 on 8q24, which encodes for ATP-dependent DNA-helicase Q4 that bears homology to RecQ helicases and has a pivotal role in maintaining chromosomal integrity. Two other disorders with overlapping clinical features associated with mutation in RECQL4 gene are RAPADILINO, that is, “radial ray defect, patellar aplasia, diarrhea, little size and limb anomalies, slender nose and normal intelligence” (OMIM-266280) and Baller–Gerold syndrome (OMIM-218600). Early diagnosis needs a high index of suspicion and close follow-up is necessary. Here we report two Indian cases with RTS with novel pathogenic variants in RECQL4 .
Patient 1
This 8-year-old male child was born to a nonconsanguineous couple. He presented with poor growth and recurrent fractures of right lower and left upper limb from the age of 4 years. He had history of multiple hyperpigmented skin lesions involving his face and bilateral upper and lower limbs associated with photosensitivity. There was no history of preceding erythema with blister formation. He achieved developmental milestones at the appropriate age and was intellectually normal. He was born at 38 weeks of gestation with a low birth weight of 1.5 kg. At 8 years, his weight was 16 kg (<3rd centile), height 116 cm (3–10th centile) according to Indian Academy of Pediatrics (IAP), 2015 standards 2 and upper to lower segment ratio was 1.05. Examination revealed a dysmorphic face with pointed nose, malocclusion of teeth, and receding chin ( Fig. 1A and B ). Skin examination showed diffuse hyperpigmentation present in a reticular pattern, with superimposed discrete hypopigmented atrophic macules of 3 to 5 mm. Telangiectasia was noted over the cheeks and extensor aspect of the forearms ( Fig. 1C ), arms, legs, thighs, and buttocks. His eyebrows and scalp hair were normal. Nail and teeth examinations were also within normal limits. Dermatoscopic examination of the skin lesions over the forearm showed the presence of reticulate pigmentation associated with multiple branching telangiectasias suggestive of poikiloderma. He had restricted movement at both elbow joints. Skeletal survey revealed gross osteopenia, multiple old healed fractures in the right tibia ( Fig. 1D ), fibula, and left radius with normal skull and spine. Serum calcium, phosphate, alkaline phosphatase, vitamin D, and parathyroid hormone were normal. Targeted exome analysis was performed and libraries were sequenced to a mean 80 to 100× coverage on an Illumina sequencing platform on the Hiseq model. The sequences obtained were aligned to the human reference genome (GRCh37/hg19) using the Burrows Wheeler Aligner (BWA) program. 3 4 A homozygous two base pair deletion and one base pair insertion in exon 5 of RECQL4 (chr8:145741524_145741525; c.978_979delTCinsG) was identified that results in a frameshift and premature truncation of the protein 33 amino acids downstream to codon 326 (p.Ser326ArgfsTer33; NM_004260, NP_004251). This variant was validated by Sanger's sequencing ( Fig. 1E ). This mutation has not been reported in 1,000 genomes, gnomAD or ExAC databases. Parental testing for this variant could not be done.
Fig. 1.
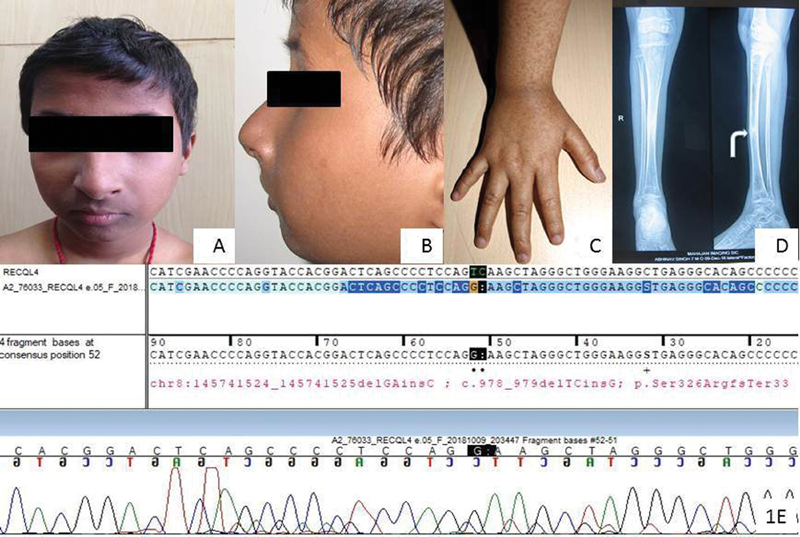
Patient-1: ( A and B ) subtle dysmorphic features including a pointed nose, receding chin, and fine eyebrows. ( C ) Hyperpigmented macules over extensor aspect of the forearm and hand. ( D ) Old healed fracture (white curved arrow) in the right tibia. ( E ) Sanger's sequencing of variant (c.978_979delTCinsG) and the variant change position is shown with a red arrow.
Patient 2
This 4-month-old female child, born to nonconsanguineous parents, presented with congenital absence of thumbs bilaterally with a short bent left forearm ( Fig. 2A ). She had a small pointed nose and smooth philtrum with a thin upper lip. She was born at full term with birth weight of 2 kg and cried immediately after birth. Her developmental milestones were normal for age. In subsequent follow-up at 18 months, her parents noticed reddening of the facial lesions on exposure to sun ( Fig. 2B and C ), failure to thrive, and recurrent diarrhea. Her weight was 5.5 kg (< 3rd centile) and height 67 cm (< 3rd centile) according to IAP 2015 standards. 2 Her height velocity was consistently less than 4 cm/year. Examination showed the presence of a small pointed nose, smooth philtrum, and thin upper lip. She had bilaterally absent thumbs with a short and bent left forearm. Skin examination revealed diffuse background hyperpigmented atrophic skin with a wrinkled surface superimposed with multiple discrete atrophic hypopigmented macules of variable size over the cheeks and extensor aspects of the forearms, sparing the chest and trunk. There was no telangiectasia, blister formation, café au lait spots, or palmoplantar hyperkeratosis. She had sparse eyebrows, particularly medially on the right, along with sparse scalp hair. Teeth and nails were normal. Her routine workup for recurrent diarrhea was normal. X-ray of the upper limb showed the presence of hypoplastic middle phalanges of the first and fourth fingers, bilateral curved ulnae, and hypoplastic radii. Her cardiac and renal evaluations were normal. Her initial workup including karyotype, neuroimaging, echocardiography, abdominal ultrasound, and testing for gastro esophageal reflux disease were normal. Chromosomal studies on peripheral blood lymphocytes were also performed using Mitomycin C to induce chromosomal breakage. The number of chromosomal breaks was counted in 100 metaphases in both this child and a control individual. This test is considered positive when the number of breaks is approximately 10 times higher in the patient compared with the control. In this patient, the results were negative. A growth hormone stimulation test using clonidine was indicated an insufficient response. In view of the dysmorphic features, absent thumbs and typical rash (poikiloderma), mutation analysis of RECQL4 was instigated. All coding exons and flanking intronic sequences of RECQL4 (Genbank NM_004260.3, NC_000008.10) were amplified by PCR and sequenced directly (Sanger's sequencing) with internal or flanking primers as described by Kitao et al. 5 RECQL4 deletion testing was performed by quantitative real-time PCR with six intragenic amplicons as described by Borozdin et al. 6 RECQL4 sequencing showed the presence of a likely pathogenic homozygous variant c.1132–2A > G in intron 5 which is not reported in 1,000 genomes and ExAC databases and is reported once in a heterozygous state (1 of 246,158 alleles) in gnomAD ( Fig. 2D ). This mutation disrupts the canonical splice acceptor site of intron 5, thereby likely resulting in use of a cryptic splice site or exon skipping with the possible consequence of mRNA degradation or formation of a truncated protein. No deletion within or including RECQL4 was detected. The parents could not be tested for this variant. Currently, the patient is 11.5 years old and has normal intellect.
Fig. 2.
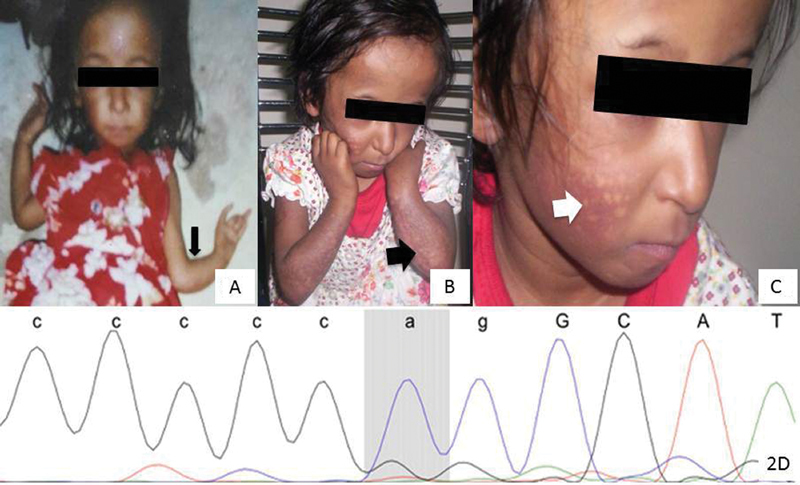
Patient 2: ( A – C ) patient 2 at 4 months ( A ) and 18 months of age ( B and C ) with a small pointed nose, smooth philtrum, and thin upper lip. Note absent thumbs and short bent left forearm (thin black arrowhead). Note the poikilodermatous skin lesions over the face (thick white arrowhead) and extensor aspects of the forearms (thick black arrowhead). ( D ) Sanger sequencing of the variant, c.1132–2A > G, IVS5–2A > G, detected in intron 5 in a homozygous state. The forward reference sequence is shown on the top line, with lower-case letters belonging to intronic and upper-case letters to exonic sequence. The peaks were obtained from sequencing of the reverse strand.
Both the children were advised to follow strict photo protection and to use a broad-spectrum sunscreen with UV (ultraviolet) A and B protection. They were also advised to have regular follow-up to monitor their growth and skin lesions to look for any change in color or texture. Annual eye examination was also suggested. The parents were given the option of prenatal testing in future pregnancies.
Discussion
RECQL4 has an important role in DNA stability, DNA replication initiation, and double strand break repair. 7 8 Homozygous/compound heterozygous mutations in RECQL4 cause three allelic disorders. RAPADILINO syndrome is characterized by skeletal and palatal defects along with gastrointestinal abnormalities. 9 Baller–Gerold syndrome includes radial ray defects, skeletal dysplasia, short stature, and craniosynostosis. The diagnostic features of RTS and RAPADILINO overlap due to common skeletal abnormalities and gastrointestinal symptoms, but RAPADILINO lacks the characteristic poikiloderma rash, though late onset of the characteristic rash of RTS can further delay the specific clinical diagnosis ( Table 1 ).
Table 1. Comparison of clinical features of the affected individuals and RECQL4 related disorders .
| Features | Patient-1 | Patient-2 | RTS | RAPADILINO syndrome | Baller–Gerold syndrome |
|---|---|---|---|---|---|
| Low birth weight | + | + | + | + | + |
| Poor growth | + | + | + | + | + |
| Facial features, palatal defects | Pointed nose, fine eyebrows, receding chin, no palatal defects | Small pointed nose, fine eyebrows, smooth philtrum, thin upper lip, no palatal defect. | Pinched nose, sparse hair, fine eyebrow, palatal defects reported in few patients. | Palatal defects present | Saddle nose, small mouth with thin vermilion border, high arched palate. |
| Radial defects | − | + | + | + | + |
| Osteopenia and fractures | + | − | + | + | − |
| Poikiloderma | + | + | + | − | + |
| Café au lait spot | − | − | − | + | |
| Craniosynostosis | − | − | − | − | + |
| Patellar abnormalities | − | − | + | + | + |
| Alopecia and sparse eye brows/ eyelashes | + | + | + | – | − |
| Eye abnormality | − | − | Cataract | Blepharophimosis | Proptosis (secondary to craniosynostosis) |
| Gastro intestinal abnormalities (recurrent diarrhea) | − | + | + | + | − |
Abbreviations: RTS, Rothmund–Thomson syndrome; RAPADILINO, radial ray defect, patellar aplasia, diarrhea, little size and limb anomalies, slender nose and normal intelligence, “−” absent, “+” present.
RTS is characterized by cutaneous rash, which presents as blisters and erythema, initially involving the face, and gradually spreads to the extremities and buttocks. With time, the rash develops into a chronic form known as poikiloderma that includes reticulated areas of depigmentation, hyperpigmentation, punctate atrophy, and telectengesia. 10 Although 90% of cases present in the first few years of life, cases have been reported with late onset poikiloderma. 11 Other features include café au lait spots, palmoplantar hyperkeratotic lesions, brittle nails, sparse and brittle scalp hair, and sparse eyebrows. Dental abnormalities including microdontia, oligodontia, delayed eruption, malocclusion, and crown malformations 12 may also be present.
Another major hallmark of RTS are various skeletal anomalies, seen in 68 to 75% of patients, including congenital radial ray defects, hypoplasia/agenesis of the patella and generalized osteopenia. 13 A study by Cao et al 14 demonstrated a positive correlation between the presence of pathogenic mutations in RECQL4 and lumber spine areal bone mineral density for age Z score ≤ − 2.0 with number of fractures. Among patients with fractures and osteopenia, 50% had nonsense mutations resulting in a truncated protein. Through mouse models, it was postulated that loss RECQL4 could affect osteoblast progenitor cells and reduce osteoid formation and hence produce weak bones. Similarly, patient-1 had a truncating mutation and presented with subtle features of RTS with low bone mineral density and a history of fractures along with skin rash.
Patient-2 also had onset of skin lesions at 1 year and was initially misclassified as Holt–Oram syndrome in the absence of skin lesions and a molecular diagnosis. The development of facial lesions led to diagnosis to RTS which changed management advice. Table 1 shows comparison of clinical features of the two patients in this study and RECQL4 related syndromes.
RECQL4 is one of the members of RecQ gene family of DNA helicases that have an important role in maintaining genomic stability and homeostasis. Loss of function mutations in RECQL4 occurs in approximately two-thirds of RTS patients. Mutations in RECQL4 lead to impaired response to oxidative stress induced DNA damage and a base excision repair defect. Mouse models have shown its differential expression in developing bones, skin, cartilage, and enterocytes which may be responsible for significant phenotypic variability of the associated genetic syndromes. 15
Mutations in RECQL4 are found in approximately 40 to 66% of RTS patients. 16 Most of the mutations in RECQL4 are truncating due to nonsense, splicing or frameshift mutations. In our study, we identified a frameshift (c.978_979delTCinsG) and splice (c.1132–2A > G) variants in two individual cases. The diagnosis of RTS in both of our cases was confirmed by homozygous variants detected in RECQL4. One of the common mutations in the Finnish population is c.1390 + 2delT (p.Ala420_Ala463del). More than 50% of these mutations are predicted to destroy the reading frame and thus disrupt the helicase domain which is essential for gene function. Truncating mutations are mostly associated with RTS and Baller–Gerold syndrome, although a few cases of RAPADILINO have been reported with truncating mutations. However, no clear genotype phenotype correlation is observed among the type of mutations and associated phenotype. 17
Conclusion
To conclude, this article describes the need for careful evaluation of the dermatological lesions and subtle facial features associated with RECQL4 related disorders. The presence of absent radii may act as a masquerader for allelic, as well as other common disorders of radial aplasia/hypoplasia such as Holt–Oram syndrome, and Thrombocytopenia Absent Radii (TAR) syndrome; however, careful follow-up in the clinical setting for the development of skin lesions and molecular evaluation is helpful for making a specific diagnosis.
Acknowledgments
We acknowledge both families for their participation and sharing their details with us.
Conflict of Interest None declared.
Note
No disclaimers. Consent for publication of their details and photographs have been taken from the parents of both the children. There is no conflict of interest among all authors.
Author Contribution
S.Y. reviewed the literature and drafted the initial version of the manuscript. N.B. helped assess the dermatological manifestations. N.G. and S.T. helped in the acquisition of data. J.K. performed molecular testing. N.G., M.K., and S.T. contributed to literature review and critically revised the manuscript. All the authors contributed to drafting of the manuscript and approved the final version of the manuscript.
References
- 1.Simon T, Kohlhase J, Wilhelm C, Kochanek M, De Carolis B, Berthold F. Multiple malignant diseases in a patient with Rothmund–Thomson syndrome with RECQL4 mutations: Case report and literature review. Am J Med Genet A. 2010;152A(06):1575–1579. doi: 10.1002/ajmg.a.33427. [DOI] [PubMed] [Google Scholar]
- 2.Khadilkar V, Yadav S, Agrawal K K et al. Revised IAP growth charts for height, weight and body mass index for 5- to 18-year-old Indian children. Indian Pediatr. 2015;52(01):47–55. doi: 10.1007/s13312-015-0566-5. [DOI] [PubMed] [Google Scholar]
- 3.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26(05):589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer L R, Zweig A S, Hinrichs A Set al. The UCSC Genome Browser database: extensions and updates 2013 Nucleic Acids Res 201341(Database issue):D64–D69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kitao S, Lindor N M, Shiratori M, Furuichi Y, Shimamoto A. Rothmund-Thomson syndrome responsible gene, RECQL4: genomic structure and products. Genomics. 1999;61(03):268–276. doi: 10.1006/geno.1999.5959. [DOI] [PubMed] [Google Scholar]
- 6.Borozdin W, Steinmann K, Albrecht B et al. Detection of heterozygous SALL1 deletions by quantitative real time PCR proves the contribution of a SALL1 dosage effect in the pathogenesis of Townes-Brocks syndrome. Hum Mutat. 2006;27(02):211–212. doi: 10.1002/humu.9396. [DOI] [PubMed] [Google Scholar]
- 7.Sangrithi M N, Bernal J A, Madine M et al. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund–Thomson syndrome. Cell. 2005;121(06):887–898. doi: 10.1016/j.cell.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 8.Petkovic M, Dietschy T, Freire R, Jiao R, Stagljar I.The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability J Cell Sci 2005118(Pt. 18):4261–4269. [DOI] [PubMed] [Google Scholar]
- 9.Kääriäinen H, Ryöppy S, Norio R. RAPADILINO syndrome with radial and patellar aplasia/hypoplasia as main manifestations. Am J Med Genet. 1989;33(03):346–351. doi: 10.1002/ajmg.1320330312. [DOI] [PubMed] [Google Scholar]
- 10.Larizza L, Roversi G, Volpi L. Rothmund–Thomson syndrome. Orphanet J Rare Dis. 2010;5:2. doi: 10.1186/1750-1172-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kumar P, Sharma P K, Gautam R K, Jain R K, Kar H K. Late-onset Rothmund–Thomson syndrome. Int J Dermatol. 2007;46(05):492–493. doi: 10.1111/j.1365-4632.2007.03248.x. [DOI] [PubMed] [Google Scholar]
- 12.Roinioti T D, Stefanopoulos P K. Short root anomaly associated with Rothmund-Thomson syndrome. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103(01):e19–e22. doi: 10.1016/j.tripleo.2006.07.021. [DOI] [PubMed] [Google Scholar]
- 13.Mehollin-Ray A R, Kozinetz C A, Schlesinger A E, Guillerman R P, Wang L L. Radiographic abnormalities in Rothmund-Thomson syndrome and genotype-phenotype correlation with RECQL4 mutation status. AJR Am J Roentgenol. 2008;191(02):W62-6. doi: 10.2214/AJR.07.3619. [DOI] [PubMed] [Google Scholar]
- 14.Cao F, Lu L, Abrams S A et al. Generalized metabolic bone disease and fracture risk in Rothmund–Thomson syndrome. Hum Mol Genet. 2017;26(16):3046–3055. doi: 10.1093/hmg/ddx178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Siitonen H A, Kopra O, Kääriäinen H et al. Molecular defect of RAPADILINO syndrome expands the phenotype spectrum of RECQL diseases. Hum Mol Genet. 2003;12(21):2837–2844. doi: 10.1093/hmg/ddg306. [DOI] [PubMed] [Google Scholar]
- 16.Kitao S, Shimamoto A, Goto M et al. Mutations in RECQL4 cause a subset of cases of Rothmund–Thomson syndrome. Nat Genet. 1999;22(01):82–84. doi: 10.1038/8788. [DOI] [PubMed] [Google Scholar]
- 17.Siitonen H A, Sotkasiira J, Biervliet M et al. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 2009;17(02):151–158. doi: 10.1038/ejhg.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]