

Abstract
We report the first case in Peru of cystic fibrosis caused by a homozygous deletion of the cystic fibrosis transmembrane conductance regulator ( CFTR ) gene. A 10-month-old child who presented with meconium ileus and pancreatic insufficiency was tested for cystic fibrosis. Both parents of the child are of Peruvian background, are nonconsanguineous, and have no personal or family history of the disease. Chromosome microarray analysis revealed a homozygous deletion of the CFTR gene on chromosome 7 (7q31.2) within a maternally derived 12.8-Mb region of loss of heterozygosity with deletion of a region that includes the CFTR gene. Parental testing confirmed this finding. This case highlights the great importance of molecular testing and the study of chromosomal rearrangements in reaching a correct diagnosis and providing proper genetic counseling to the affected families.
Keywords: cystic fibrosis, CFTR gene , chromosome microarray analysis, copy number variation, loss of heterozygosity, uniparental disomy
Introduction
Cystic fibrosis (CF) is the most common inherited childhood disease in the Caucasian population. This autosomal recessive systemic disease is caused by mutations of the gene that encodes for the CF transmembrane conductance regulator ( CFTR ), a 1,480-amino-acid protein that functions as a chloride channel, which is critical for salt homeostasis of several polarized epithelial tissues including lung, intestine, pancreas, and kidney. Patients with abnormal CFTR function suffer from impaired clearance of mucus from the respiratory tract, subsequent chronic lung infections and inflammation, and eventual respiratory failure, which is the principal cause of morbidity and mortality. Other prevalent features of CF are insufficiency of the exocrine pancreas, increased electrolytes in sweat, and male infertility. 1 2 3 4 5
More than 1,800 variants in CFTR have been identified, with variable frequencies depending on the geographic and ethnic background. 6 Six classes of mutations have been defined according to the mechanism causing disease 7 ( Table 1 ). Class I mutations in CFTR include premature termination codons, which cause truncation in the translation of the mRNA and therefore the production of little functional CFTR . These mutations are responsible for approximately 10% of CF cases worldwide, with patients usually having a phenotype of CF with exocrine pancreatic insufficiency. 8 Class IV and V mutations are habitually associated with mild disease and pancreatic sufficiency. In Peru, there is a high frequency of p.Phe508del, a class II mutation, in the population, 9 though CF in this country is severely underdiagnosed. To our knowledge, there are no other published studies investigating the incidence of microdeletions or other genetic causes of CF in Peru.
Table 1. Classes of mutations in CFTR and their consequences on the associated protein .
| Class | Consequence on the protein |
|---|---|
| I | Protein is not synthesized |
| II | Structural: folding defect |
| III | Functional: channel opening defect |
| IV | Functional: ion transport defect |
| V | Expression: decreased protein synthesis |
| VI | Stability: decreased half-life of the protein |
In this study, we describe an 18-month-old Peruvian female patient who presented with CF-associated symptoms and microcephaly. The conventional molecular screening for CF showed no amplification, and the chromosome microarray analysis (CMA) test showed a homozygous deletion of the CFTR gene. Further investigation revealed that this homozygosity arose from an interstitial maternal isodisomy containing the region deleted for CFTR , as the mother is a heterozygous carrier of the microdeletion found in the patient.
Materials and Methods
Ethical Statement
Following the Instituto Nacional de Salud del Niño regulations, informed written consent was obtained from the patient's parents.
Amplification-Refractory Mutation System/Polymerase Chain Reaction
Genomic DNA was isolated from whole blood using AxyPrep Blood Genomic DNA Miniprep Kit (Axygen; Corning Life Sciences, MA, United States). Amplification-refractory mutation system/polymerase chain reaction (ARMS-PCR) was performed to identify the most common mutations in the Peruvian population (p.Phe508del, c.489 + 1G > T, p.Asn1303Lys, p.Lys1177Serfs*15, and c.1585–1G > A), using Platinum Taq DNA Polymerase (Invitrogen; Thermo Fischer Scientific, MA, United States) and following the method described by Aquino et al. 9
Chromosome Microarray Analysis
Total DNA (250 ng) was amplified, labeled, and hybridized using GeneChip CytoScan 750K Array protocols (Affymetrix, Applied Biosystems; Thermo Fischer Scientific, MA, United States) according to the manufacturer's instructions. The array specifications include 550,000 nonpolymorphic markers and 200,436 SNP markers. CEL files obtained by scanning the arrays were analyzed using the Chromosome Analysis Suite (ChAS) software (Affymetrix) and Genome build GRCh37 (hg19). Gains and losses that affected a minimum of 25 markers and loss of heterozygosity (LOH) regions that expand over 5 Mb were initially considered.
The copy number variations (CNVs) found in the patient were compared with genomic variants in public databases, including Database of Chromosomal Imbalance and Phenotype in Humans using Ensemble Resources (DECIPHER), and The UCSC Genome Browser (hosted by the University of California, Santa Cruz). CNVs were classified into pathogenic, likely pathogenic, and of unknown clinical significance. 10 11
Results
Clinical Evaluation
The patient, a female infant aged 24 months at last assessment, is the first child of an unrelated couple of Peruvian origin (26-year-old mother and 24-year-old father). The parents are healthy and have not had any other children to this date. The proband was born by natural birth at 36 weeks' gestation due to polyhydramnios, ascites, and intestinal distention identified on antenatal ultrasound scans. Two months after the delivery, she was diagnosed with meconium ileus and was admitted into the hospital, where a colostomy was performed. With the suspected diagnosis of CF, the patient was referred to pediatrics and then to genetics. The sweat test (diagnostic cutoff for chloride being 60 mmol/L) performed at 10 weeks of age yielded a chloride of 105.9 mmol/L. She was repeatedly admitted to the hospital with pancreatic insufficiency and anemia. At the age of 5 months, she was diagnosed with microcephaly, and by the age of 7 months, she showed mild global developmental delay. At 16 months of age, she was admitted to the CF unit, with compromised pancreatic and pulmonary function.
Molecular Analyses
Molecular testing for CFTR mutations by ARMS-PCR revealed no amplification. Due to the presence of microcephaly and mild global developmental delay, a CMA test was ordered. This showed a 218-kb homozygous deletion at 7q31.2, comprising most (if not all) of the CFTR gene. This finding confirmed the CF diagnosis of the child. Since the deletion results in no mRNA or protein, the deletion is a class I CFTR mutation.
As shown in Fig. 1 , the LOH region ends proximally to the start of the deletion as the next polymorphic marker was absent. A distal polymorphic marker at position 117324588 is present and not consistent with LOH. Therefore, this 12.6-Mb LOH segment could be a longer region, 12.8 Mb, containing CFTR . If this is the case, the possibility that the homozygosity arose from a uniparental disomy (UPD) and that only one of the parents may carry the deletion existed.
Fig. 1.
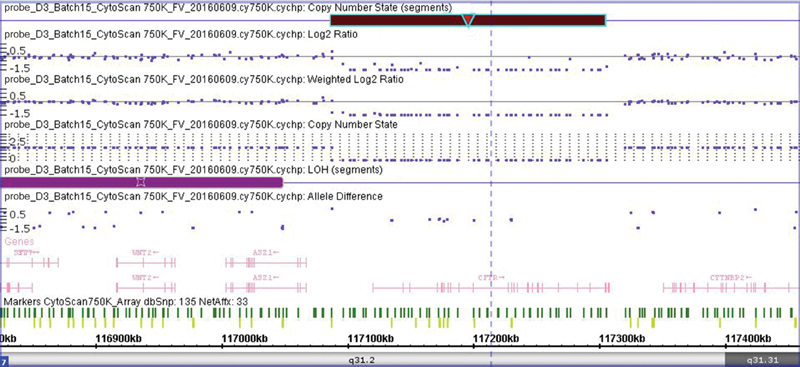
Detailed view of the homozygous microdeletion found in the patient. A 218-kb homozygous microdeletion ( dark red rectangle ) was found on chromosome 7q31.2. The purple rectangle shows a loss of heterozygosity (LOH) region. The dark green vertical lines represent nonpolymorphic markers, and the light green vertical lines represent the polymorphic (SNP) markers. The genes are shown in pink.
Following the confirmation of CF diagnosis, the carrier status of the parents was determined by molecular testing for CFTR mutations by ARMS-PCR. As shown in Fig. 2 , PCR of the paternal sample revealed bands of the same intensity and pattern as the control, showing none of the five mutations tested. PCR of maternally derived DNA revealed none of the mutations tested, but all the bands obtained showed less intensity than the bands of the control, even though the same amount of DNA was used for all samples. We suspected that the mother could be carrying only one CFTR allele; therefore, a CMA test was performed. The final results showed a 218-kb heterozygous deletion at 7q31.2, comprising most (if not all) of the CFTR gene, the same CNV that was found in the daughter ( Fig. 3 ): arr[GRCh37] 7q31.2(117087267_117305344)x0, 7q22.2q31.2(104448510_121435069) hmz mat).
Fig. 2.

Molecular testing for CFTR mutations by amplification-refractory mutation system/polymerase chain reaction (ARMS-PCR). Blood samples of the patient́s mother ( A ) and father ( B ) were tested by ARMS-PCR. The bands were obtained by electrophoresis in a 2% agarose gel. A normal control was run with the parent́s samples. In the case of the mother, a heterozygous p.Phe508del control was also included. A1: normal alleles for 621 + 1G →T (380 bp), p.Asn1303Lys (328 bp) and p.Phe508del (157 bp). A2: mutant alleles for 621 + 1G →T (380 bp), p.Asn1303Lys (328 bp) and p.Phe508del (157 bp). C: normal allele for 3659 Del C (294 bp) and mutant allele for 1717–1G →A (220 bp). D: mutant allele for 3659 Del C (294 bp) and normal allele for 1717–1G →A (220 bp).
Fig. 3.
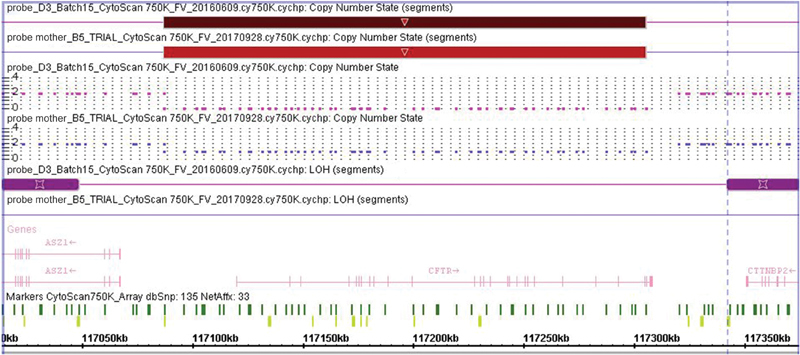
Detailed view of the microdeletions found in patient and mother. A 218-kb heterozygous microdeletion ( light red rectangle ) was found in the patient's mother. This copy number variation (CNV) is the same as the one found in the child in a homozygous state ( dark red rectangle ) on chromosome 7q31.2. The purple rectangle shows a loss of heterozygosity (LOH) region. The dark green lines are nonpolymorphic markers, and the light green lines represent the polymorphic (SNP) markers. The genes are shown in pink .
Comparing base calls of mother and daughter within the apparent region of homozygosity strengthened the hypothesis that the homozygous deletion of the patient was the consequence of segmental UPD, where a 12.8-Mb region of the maternal chromosome 7, the region lacking CFTR , was duplicated ( Supplemental Material 1 , available in online version only).
Discussion
Cystic fibrosis, a recessively inherited disease, is caused by either homozygous or compound heterozygous point mutations, small deletions or small insertions within the coding region on each CFTR allele. Some symptomatic patients carry only one detectable mutation in one CFTR allele and a large deletion on the other, but because this last event cannot be detected by standard techniques, some patients have been diagnosed as apparently homozygous for the detectable mutation. 12 Moreover, since these methods are unable to detect single or multiple exonic rearrangements, the frequency of such events is not well established.
Historically, most of the large deletions in CFTR in patients with CF were identified by coincidence, either by noting a uniparental inheritance pattern or through failure of PCR amplification. Now, these can be successfully identified by other techniques such as multiplex ligation probe amplification, semiquantitative fluorescent PCR, 13 or real-time PCR. 14 The frequency of CF alleles unidentified by conventional techniques is approximately 30% (with marked regional and ethnical variations), from which large rearrangements have been estimated to be 16 to 24% (i.e., 6% of previously unidentified alleles) according to various study cohorts. 15 16 17 18 However, it is also suggested that this range is highly underestimated.
In Peru, the sweat test is the standard test for diagnosing CF. However, there are many limitations in the access to a sweat test and immunoreactive trypsinogen measurement through newborn screening and molecular studies, since these tests are not yet implemented in a National Program and are not available in every main public hospital. Most patients who already have a molecular diagnosis were tested overseas. Therefore, CF is widely underdiagnosed and, in consequence, undertreated. Moreover, the high heterogeneity in the Latin-American population due to the different ethnic backgrounds adds to the diversity in the profile of CFTR mutations or rearrangements, 19 which is also unknown in Peru.
In this study, we reported the case of a Peruvian female patient, who is now 2 years old, with classical CF symptoms and microcephaly. Conventional ARMS-PCR showed no amplification of CFTR . CMA showed a 218-kb homozygous deletion encompassing the CFTR gene, and a segment of LOH in this region. ARMS-PCR performed in the parents suggested that the mother could possibly lack a copy of CFTR , for which a CMA was then performed, showing a heterozygous deletion of the same region that was found in the daughter. Moreover, a base call analysis suggested a segmental maternal isodisomy (UPD) in chromosome 7.
Segmental UPD is defined as UPD of a part of one chromosome (interstitial or telomeric) together with biparental inheritance of the rest of this pair of chromosomes and a normal karyotype. 20 Interstitial maternal isodisomy confined between biparentally inherited loci requires events involving errors in meiotic division and early postzygotic recombination. In the present case, a maternal meiosis II error gave rise to the formation of a trisomy 7 zygote, containing two maternal chromosomes which carried a deleted CFTR region. This event was followed by a mitotic double exchange (crossing over) between two nonuniparental chromatids, forming a paternal chromosome containing a maternally derived interstitial segment. This segment contained the maternally deleted CFTR region. Subsequently, the maternal chromatid involved in the exchange was lost such that the zygote remained with the maternal chromatid and the paternal chromosome containing the maternally derived interstitial segment with the deleted CFTR region, as described in Fig. 4 .
Fig. 4.

Possible explanation of the patient's interstitial maternal isodisomy formation. An error during maternal meiosis II may have given rise to the formation of a trisomy 7 zygote, containing the two maternal affected sister chromatids. After fertilization, a mitotic double crossing-over between two nonuniparental chromatids took place, forming a paternal chromosome containing a maternally derived interstitial segment that carried the deleted CFTR region. Subsequently, and due to trisomy rescue, the maternal chromatid involved in the exchange was lost. The red rectangle indicates the deleted CFTR region. The black circle indicates the centromeres.
To date, very few reports on UPDs in chromosome 7 (full chromosome, mosaics, segmental maternal UPD, paternal UPD) exist. 21 22 23 Most of these findings have been associated with Silver–Russel syndrome (SRS), and mostly involve regions that span all of 7q, ranging in size from the entire q arm to the distal segments of approximately 31 Mb. Some of these reports also mention findings concerning CF, but these involve point mutations in CFTR .
Knowing that to this date our patient has no clinical signs of SRS (according to Netchine–Harbison clinical scoring system 24 ) and that CMA test in peripheral blood suggested no mosaic lines, no extra chromosomal material (i.e., small supernumerary marker chromosomes), nor other chromosomal imbalances, we searched the UPD database 25 for similar reported cases. We found four cases not associated with SRS, but neither the exact location nor the size of the UPD within chromosome 7 is reported; in addition, one of the cases is a distal UPD (including the telomeric part of the chromosome).
To our knowledge, this is the first reported case of CF due to homozygous deletion of CFTR linked to an interstitial maternal isodisomy in chromosome 7 that is not associated with SRS. The fact that the patient, so far, does not show symptoms associated to SRS suggests that the region of UPD found in this study does not contain any of the genes critical for the expression of SRS phenotype. In addition, the patient's CF is controlled, and her development is progressing according to the standard development of a 2-year-old.
The chromosomal events described in the present case are extremely rare. Since only one of the parents carries the CFTR deletion, recurrence risk is predicted to be low.
This report not only describes a unique CF-causing event (homozygous complete CFTR deletion) but also highlights the great importance of the study of genetic conditions and their mechanisms of origin in genetic counseling. To be able to discover the cause of a particular disease, explain it, and predict the recurrence risk of such an event has a crucial psychosocial impact on the patient's family, who will then be able to make an informed decision regarding their future reproduction. Moreover, this particular case highlights the need for a personalized diagnosis and treatment, as the current CF drugs targeting CFTR expression, localization, and/or function will not help if there is no CFTR present. As more cases are published, more information about the frequency of these “rare events” will be known, and the correlation between genotype and phenotype will be better understood.
Footnotes
Conflict of Interest None declared.
Supplementary Material
References
- 1.Collins F S.Cystic fibrosis: molecular biology and therapeutic implications Science 1992256(5058):774–779. [DOI] [PubMed] [Google Scholar]
- 2.Dalemans W, Barbry P, Champigny Get al. Altered chloride ion channel kinetics associated with the delta F508 cystic fibrosis mutation Nature 1991354(6354):526–528. [DOI] [PubMed] [Google Scholar]
- 3.De Boeck K, Derichs N, Fajac I et al. New clinical diagnostic procedures for cystic fibrosis in Europe. J Cyst Fibros. 2011;10(02) 02:S53–S66. doi: 10.1016/S1569-1993(11)60009-X. [DOI] [PubMed] [Google Scholar]
- 4.Kerem B, Kerem E. The molecular basis for disease variability in cystic fibrosis. Eur J Hum Genet. 1996;4(02):65–73. doi: 10.1159/000472174. [DOI] [PubMed] [Google Scholar]
- 5.Lukacs G L, Chang X B, Bear C et al. The delta F508 mutation decreases the stability of cystic fibrosis transmembrane conductance regulator in the plasma membrane. Determination of functional half-lives on transfected cells. J Biol Chem. 1993;268(29):21592–21598. [PubMed] [Google Scholar]
- 6.Mall M A, Hartl D. CFTR : cystic fibrosis and beyond . Eur Respir J. 2014;44(04):1042–1054. doi: 10.1183/09031936.00228013. [DOI] [PubMed] [Google Scholar]
- 7.Rowe S M, Miller S, Sorscher E J. Cystic fibrosis. N Engl J Med. 2005;352(19):1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- 8.Wilschanski M. Class 1 CF mutations. Front Pharmacol. 2012;3:117. doi: 10.3389/fphar.2012.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Aquino R, Protzel A, Rivera J et al. Frecuencia de las mutaciones más comunes del gen CFTR en pacientes peruanos con fibrosis quística mediante la técnica ARMS-PCR . Rev Peru Med Exp Salud Publica. 2017;34(01):62–69. doi: 10.17843/rpmesp.2017.341.2767. [DOI] [PubMed] [Google Scholar]
- 10.Verma R, Babu A. New York, NY: McGraw-Hill; 1995. Human Chromosomes: Principles & Techniques. Vol. 43. 2nd ed. [Google Scholar]
- 11.Miller D T, Adam M P, Aradhya S et al. Consensus statement: chromosomal microarray is a first-tier clinical diagnostic test for individuals with developmental disabilities or congenital anomalies. Am J Hum Genet. 2010;86(05):749–764. doi: 10.1016/j.ajhg.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Neocleous V, Yiallouros P K, Tanteles G A et al. Apparent homozygosity of p.Phe508del in CFTR due to a large gene deletion of exons 4-11 . Case Rep Genet. 2014;2014:613863. doi: 10.1155/2014/613863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hantash F M, Redman J B, Starn Ket al. Novel and recurrent rearrangements in the CFTR gene: clinical and laboratory implications for cystic fibrosis screening Hum Genet 2006119(1-2):126–136. [DOI] [PubMed] [Google Scholar]
- 14.Schneider M, Joncourt F, Sanz J, von Känel T, Gallati S. Detection of exon deletions within an entire gene ( CFTR ) by relative quantification on the LightCycler . Clin Chem. 2006;52(11):2005–2012. doi: 10.1373/clinchem.2005.065136. [DOI] [PubMed] [Google Scholar]
- 15.Chevalier-Porst F, Souche G, Bozon D. Identification and characterization of three large deletions and a deletion/polymorphism in the CFTR gene . Hum Mutat. 2005;25(05):504–504. doi: 10.1002/humu.9335. [DOI] [PubMed] [Google Scholar]
- 16.Audrézet M P, Chen J M, Raguénès O et al. Genomic rearrangements in the CFTR gene: extensive allelic heterogeneity and diverse mutational mechanisms . Hum Mutat. 2004;23(04):343–357. doi: 10.1002/humu.20009. [DOI] [PubMed] [Google Scholar]
- 17.Niel F, Martin J, Dastot-Le Moal F et al. Rapid detection of CFTR gene rearrangements impacts on genetic counselling in cystic fibrosis . J Med Genet. 2004;41(11):e118–e118. doi: 10.1136/jmg.2004.022400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bombieri C, Bonizzato A, Castellani C, Assael B M, Pignatti P F. Frequency of large CFTR gene rearrangements in Italian CF patients . Eur J Hum Genet. 2005;13(05):687–689. doi: 10.1038/sj.ejhg.5201387. [DOI] [PubMed] [Google Scholar]
- 19.Pérez M M, Luna M C, Pivetta O H, Keyeux G. CFTR gene analysis in Latin American CF patients: heterogeneous origin and distribution of mutations across the continent . J Cyst Fibros. 2007;6(03):194–208. doi: 10.1016/j.jcf.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 20.Kotzot D. Complex and segmental uniparental disomy (UPD): review and lessons from rare chromosomal complements. J Med Genet. 2001;38(08):497–507. doi: 10.1136/jmg.38.8.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gerbrands L C, Haarman E G, Hankel M A, Finken M JJ. Cystic fibrosis and Silver-Russell syndrome due to a partial maternal isodisomy of chromosome 7. Clin Case Rep. 2017;5(10):1697–1700. doi: 10.1002/ccr3.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Le Caignec C, Isidor B, de Pontbriand U et al. Third case of paternal isodisomy for chromosome 7 with cystic fibrosis: a new patient presenting with normal growth. Am J Med Genet A. 2007;143A(22):2696–2699. doi: 10.1002/ajmg.a.31999. [DOI] [PubMed] [Google Scholar]
- 23.Su J, Wang J, Fan X et al. Mosaic UPD(7q)mat in a patient with silver Russell syndrome. Mol Cytogenet. 2017;10:36. doi: 10.1186/s13039-017-0337-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Azzi S, Salem J, Thibaud N et al. A prospective study validating a clinical scoring system and demonstrating phenotypical-genotypical correlations in Silver-Russell syndrome. J Med Genet. 2015;52(07):446–453. doi: 10.1136/jmedgenet-2014-102979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liehr T.Cases with uniparental disomyAvailable at:http://upd-tl.com/upd.html. Accessed December 10, 2018
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.