

Abstract
Cytochrome c oxidase deficiency is caused by mutations in any of at least 30 mitochondrial and nuclear genes involved in mitochondrial complex IV biogenesis and structure, including the recently identified PET100 gene. Here, we report two families, of which one is consanguineous, with two affected siblings each. In one family, the siblings presented with developmental delay, seizures, lactic acidosis, abnormal brain magnetic resonance imaging, and low muscle mitochondrial complex IV activity at 30%. In the other family, the two siblings, now deceased, had a history of global developmental delay, failure to thrive, muscular hypotonia, seizures, developmental regression, respiratory insufficiency, and lactic acidosis. By whole exome sequencing, a missense mutation in exon 1 of the PET100 gene (c.3G > C; [p.Met1?]) was identified in both families. A review of the clinical description and literature is discussed, highlighting the importance of this variant in the Lebanese population.
Keywords: Lebanon, mitochondria, whole exome sequencing, PET100
Introduction
Cytochrome c oxidase (COX) or complex IV is one of the four complexes that make up the mitochondrial respiratory chain. It catalyzes the last step of cellular respiration. The complex consists of 14 subunits, three of which, the core catalytic subunits, are encoded by the mitochondrial genome. 1 The rest are encoded by the nuclear genome and thus get imported into the mitochondrion. 2 3
Cytochrome c oxidase deficiency is a heterogeneous condition with clinical manifestations ranging from isolated myopathy to severe multisystem disease (OMIM 220110). Some affected individuals manifest a fatal hypertrophic cardiomyopathy resulting in neonatal death. A subset of patients manifest Leigh syndrome (OMIM 256000). Onset is usually before the age of two, but symptoms can appear later in mildly affected individuals. 4 Four types of COX deficiency are known: benign infantile mitochondrial type, French-Canadian type, infantile mitochondrial myopathy type, and Leigh syndrome. The condition is frequently fatal in childhood, but mildly affected individuals may survive into adolescence or adulthood.
Cytochrome c oxidase deficiency is caused by mutations in any of at least 30 mitochondrial and nuclear genes involved in COX biogenesis and structure. Around 20 genes involved in COX synthesis, assembly, and maturation have been reported to be frequently mutated in patients. 5 Among the latter is the assembly factor encoded by the nuclear gene PET100 , which localizes to the inner mitochondrial membrane 6 and is required for COX assembly. 7 8 9 This gene was first reported as being involved in COX IV deficiency in 10 Lebanese patients living in Australia. 8
Here, we report the identification of a missense mutation in exon 1 of the PET100 gene in two Lebanese families, and review the literature.
Materials and Methods
This study conformed to the tenets of the Declaration of Helsinki.
Patients
Family A: Two patients, male and female siblings, were born to healthy unrelated Lebanese parents ( Fig. 1A ). The girl was born at term, after an uncomplicated pregnancy. At birth, her weight was 3,180 g, her length 50 cm, and her occipitofrontal circumference (OFC) 33.5 cm. Transient high values of thyroid steroid hormone (TSH) were noted during the first 2 months of life. She was very irritable, crying for very long periods. At the age of 3 months, the parents noticed that she was unable to make eye contact, unable to focus on objects or follow movements, and was not grabbing things with her hands. At 4 months of age, the patient started to have tonic seizures, with gaze fixation and upward deviation of the eyes. The seizures started at a frequency of three times per day for 1 week, of 3 to 7 seconds duration each. The frequency increased within the following days. An electroencephalogram (EEG) showed only slowing and an irregular background during the first week. The following week, the EEG started to show several multifocal spikes during the awakening and sleeping period. Different antiepileptic drugs were tried, but without being very effective in the episodes. Instead, the seizures were aggravated upon treatment with topiramate.
Fig. 1.
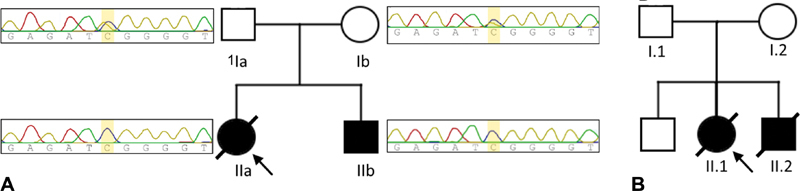
( A ) Pedigree of family A including sequence chromatograms of the mutated region (c.3G > C) in PET100, showing the variant in the heterozygous state in both parents (Ia and Ib) and the homozygous state for both siblings (IIa and IIb). ( B ) Pedigree of family B.
She exhibited high plasma lactate levels ranging from 3.94 to 5.65 mmol/L (normal range: 0.55–2.2 mmol/L); single voxel brain magnetic resonance spectroscopy revealed abnormal lactate double peaks as well as an increased choline peak in the left centrum semiovale and right corona ( Fig. 2 ). She had intermediate glucose 6-phosphate dehydrogenase deficiency at 3.3 U/g hemoglobin (normal HB range > 4.2 U/g), as well as elevated whole blood pyruvate levels (0.166 mmol/L) which were reduced to normal (0.079 mmol/L) upon follow-up (normal range: 0.056–0.1 mmol/L). Brain magnetic resonance imaging showed cortical and bilateral basal ganglia lesions ( Fig. 3 ), frontal, periventricular, and periaqueductal white matter hyperintensity as well as T2 signal involving the corpus callosum splenium and posterior limb of the internal capsules. A muscle biopsy was performed showing myofiber hypotrophy, scattered myolysis, mild mitochondrial accumulation, as well as patchy accumulation of glycogen and lipids. Muscle COX activity was reduced to 30% and complex II activity was mildly depressed at 60% relative to control. Nicotinamide adenine dinucleotide (NADH) and ATP synthase activity were normal. A mitochondrial disease was suspected; hence, in addition to her epileptic medication she received L-carnitine and coenzyme Q10 supplements.
Fig. 2.
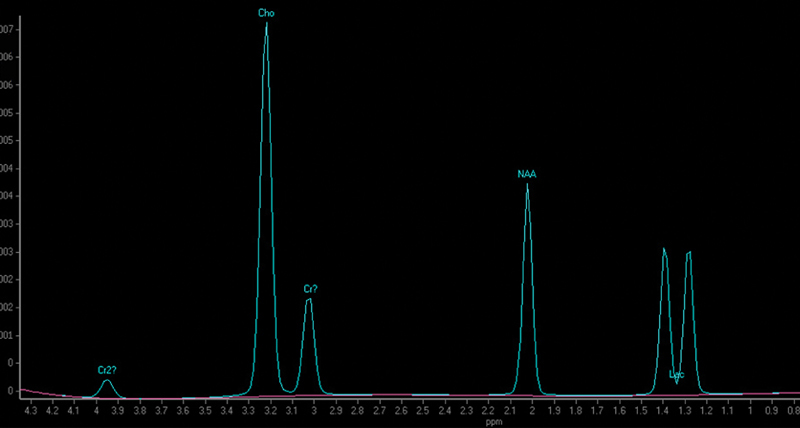
Single voxel magnetic resonance spectroscopy of patient IIa at the level of the right corona radiata showing increased choline peak and abnormal lactate double peak.
Fig. 3.
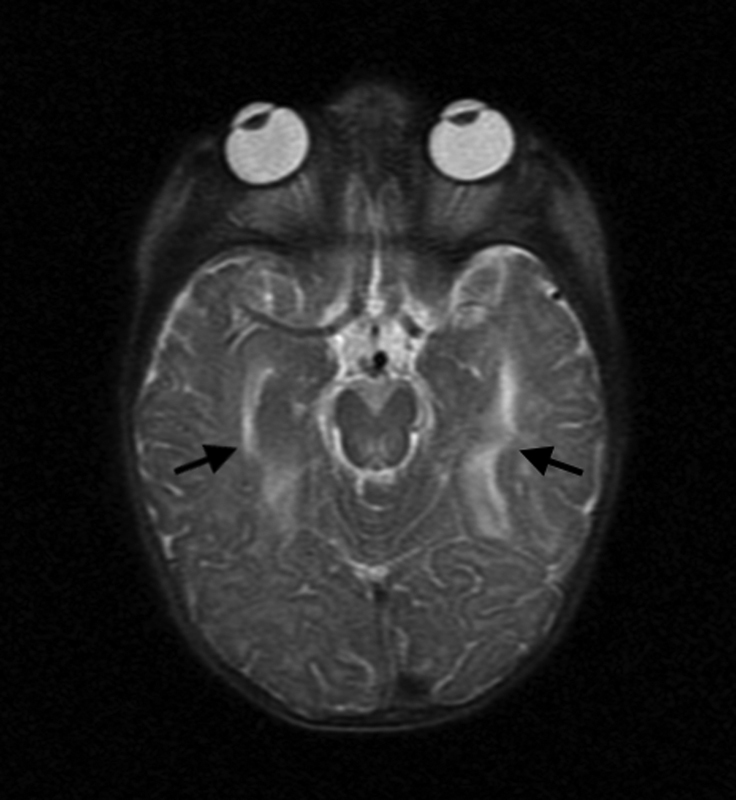
Transverse T2-weighted brain magnetic resonance imaging of patient IIa showing cortical lesions (arrows).
At the age of 8 months, her developmental milestones were delayed as she was still not able to sit, and she was very hypotonic. Despite her treatment, seizures crises were still frequent and she continued to clinically regress. She passed away at the age of 2 years and 1 month, after an episode of pulmonary infection.
A year later, the male sibling was delivered at term, following a normal pregnancy. At birth, his weight was 4,200 g and his length 51.5 cm. Clinical exam and TSH test were normal. Developmental milestones were in the normal limits till 4 months of age, when he started to become agitated and irritable. An EEG was performed and showed the same abnormalities found in his older sister. At examination at the age of 5 months, he was unable to hold his neck with head up and was not focusing on objects or making eye contact. A mitochondrial disease was suspected and the parents decided not to proceed with further laboratory or imaging analyses.
Family B: An asymptomatic first-degree consanguineous couple presented at our clinic with a history of two children, a girl and a boy, who had died at the ages of 15 and 8 months, respectively ( Fig. 1B ).
The first affected child, a girl, was born at term. Her birth weight, length, and OFC were not recorded but were considered normal by the parents. Since birth, she had a severe hypotonia and seizures and was diagnosed with hypoxic ischemic encephalopathy. A few months later, she exhibited global developmental delay, muscular hypotonia, failure to thrive, and developmental regression. A routine blood analysis showed elevated lactate levels. No further clinical investigations were performed and the baby girl died at 15 months by respiratory insufficiency following an episode labeled as septic shock.
One year later, a baby boy was born at term after an uneventful pregnancy and delivery. Birth weight, length, and OFC were not recorded but were considered normal by the parents. The neonatal tandem mass screening was normal. The child was reported to have an axial hypotonia, and showed poor eye contact. No seizures were noted. He had repetitive episodes of upper respiratory tract infections, all presenting with a severe acidosis (HCO 3 between 7 and 18 mEq/L), and anion gap of 14 mmol/L and a lactic acidosis reaching 5.6 mmol/L. Similar to his sister, he showed global developmental delay, developmental regression, muscular hypotonia, and failure to thrive.
He was seen by a clinician for the first time at the age of 6 months. Weight, length, and OFC were at the normal limits. He had a good muscle mass, normal deep tendon reflexes, clenched fists, mild axial hypotonia, and poor eye contact. Routine blood analysis, amino acids in blood, and organic acids in urine were at normal levels. He had total carnitine level of 44 µmol/L and a free carnitine at the low limit of 29 µmol/L. Parents refused further investigations.
Two weeks later, the patient was admitted to the pediatric intensive care unit following a viral infection, with high white count and high C-reactive protein and a severe lactic acidosis which led to a rapid cardiovascular failure.
Molecular Analysis
Upon obtaining informed consent and in accordance with the principles of the Declaration of Helsinki, blood sampling and DNA extraction were performed for the two siblings and their parents of family A and the parents of family B.
Family A: Whole exome sequencing (WES) was done for the affected girl as previously described using the Nextera Rapid Capture Exome (Illumina; San Diego, California, United States). 10 A targeted analysis of WES data from the patient was done using a list of nearly 1,300 genes involved in neurological, metabolic, and mitochondrial diseases selected from the Online Mendelian Inheritance in Men database and a review of the literature. The variants were analyzed and segregated using the software for variant annotation and segregation VarAFT. 11 They were filtered based on an autosomal recessive inheritance pattern: as the parents denied consanguinity, we searched for both compound heterozygous and homozygous variants in the coding sequences. In order to refine the obtained lists of candidate variants, filtering was performed by removing all variants with a frequency above 1% on the Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/ ). Additional filtering was then performed using additional frequency datasets: the Greater Middle East (GME) Variome ( http://igm.ucsd.edu/gme/ ).
Data analysis identified a homozygous missense mutation in PET100 (NM_001171155.1: c.3G > C; [p.Met1?]) resulting in a potential shift of the initiating codon and loss of the first nine amino acids. The pathogenicity of this mutation was substantiated using different bioinformatics prediction software including MutationTaster which provided a disease-causing result with 0.99 probability, and combined annotation-dependent depletion providing a Phred C-scale core of 28.2. This mutation has been previously reported as pathogenic in the homozygous state in the public variant databases dbSNP (rs587777839) and ClinVar (125441). The allele frequency of the variant reported by GnomAD is 0.001% with two heterozygous alleles belonging to the European population. By Sanger sequencing, we confirmed the presence of this mutation in the homozygous state in the affected siblings and in the heterozygous state in the parents ( Fig. 1A ).
Family B: Following the same methodology described above, a duo WES analysis was performed on the parents. The same mutation reported in family A, PET100 (NM_001171155.1: c.3G > C; [p.Met1?]), was found at the heterozygous state in both parents.
Clinical features of the patients reported here, as well as previously described individuals with mutations in PET100 , are summarized in Table 1 .
Table 1. Clinical features and cytochrome IV activity of individuals with mutated PET100 .
| Subject | Sex | Age of onset | Age at death | Clinical presentation | Brain findings | CIV activity (%) | Origin | PET100 mutation | Reference |
|---|---|---|---|---|---|---|---|---|---|
| A1 | M | 4 mo | 0.7 y | LS, lactic acidosis | Bilateral symmetrical involvement of putamen, thalamus, and basal ganglia | 17(F), 19(M), 22(L) | Lebanese | c.3G > C; p.M1? | Lim et al 8 |
| B1 | M | 1.5 mo | 0.7 y | DD, FTT, seizures, hypotonia, peripheral hypertonia, lactic acidosis | Low density white matter lesions in the caudate nuclei | 24(F) | |||
| C1 | F | 3 mo | Alive 15.5 y | LS, bilateral lamellar cataracts, lactic acidosis, elevated serum, and CSF pyruvate, low serum bicarbonate, thoracic scoliosis | Characteristic LS findings | 10(F), 21(M) | |||
| D1 | M | 6 mo | Alive 24 y | LS, dystonia, seizures, hypotonia, lactic acidosis, profound intellectual disability, microcephaly, short stature, spastic quadriplegia | High-intensity signals in the putamen, thalamus, and medulla | 27(F) | |||
| D2 | M | 4 mo | 0.6 y | LS, seizures, elevated CSF lactate, myocardial infarction | Cerebral atrophy | 22(F) | |||
| E1 | M | 4 mo | 16 y | DD, FTT, seizures, hypotonia, strabismus, spastic quadriparesis, kyphoscoliosis, dementia | Unremarkable | 29(F) | |||
| E2 | M | 9 mo | Alive 27 y | LS, seizures, hypotonia, nystagmus, elevated serum lactate | Cerebral atrophy reaching the sylvian fissures | 25(F) | |||
| F1 | M | 6 mo | Alive 9.7 y | LS, seizures, lactic acidosis, short stature, spastic quadriparesis, bulbar palsy, peripheral neuropathy, severe hearing deficit, visual impairment, mottled skin | Bilateral low attenuation signals in lentiform nuclei | 34(F) | |||
| G1 | F | 4 mo | 1 y | DD, seizures, hypotonia, lactic acidosis, elevated CSF pyruvate, hypsarrhythmia | Cortical atrophy | 12(F), 39(L) | |||
| H1 | M | 4 mo | 3.5 y | LS, seizures, hypoglycemia, lactic acidosis, elevated serum pyruvate, cortical blindness, feeding difficulties | High-intensity signals in the putamen | 51(F), 48(M), 21(L) | |||
| I1 | F | Prenatal | 55 h | IUGR, seizures, severe lactic acidosis, Hypoglycemia, generalized aminoaciduria, severe coagulopathy, elevated creatinine kinase, impaired liver function | Bilateral intraventricular cysts | <10(M), <10(F) | Pakistani | c.142C > T; p.Gln48Ter * | Oláhová et al 9 |
| IIa | F | 4 mo | 2 y | DD, seizures, lactic acidosis, transient elevated TSH, hypotonia | Basal ganglia lesions; periventricular and periaqueductal white matter hyperintensity; involvement of the corpus callosum splenium and posterior limb of the internal capsules | 30(M) | Lebanese | c.3G > C; p.Met1Ile b | This study |
| IIb | M | 4 mo | – | DD, seizures | – | – | |||
| II.1 | F | – | 15 mo | Hypotonia, seizures, DD, DR, FTT, elevated serum lactate, respiratory insufficiency | – | – | c.3G > C; p.M1? a | ||
| II.2 | M | – | 8 mo | Hypotonia, poor eye contact, DD, DR, FTT, metabolic acidosis | – | – |
Abbreviations: CSF, cerebrospinal fluid; DD, developmental delay; DR, developmental regression; (F), fibroblast; FTT, failure to thrive; IUGR, intrauterine growth retardation; (L), liver; LS, Leigh syndrome; (M), muscle.
Refers to a protein truncation mutation.
Only parents of the deceased siblings II.1 and II.2 were sequenced and both were found to be heterozygous for the c.3G > C variant.
Initiating codon mutation p.M1I (or p.Met1Ile).
Discussion
Here, we report the identification of a homozygous missense mutation in exon 1 of the PET100 gene (c.3G > C; [p.Met1?]) in a Lebanese family affected with COX deficiency. We report the same mutation in another Lebanese family with deceased children suspected to be affected with COX deficiency.
This variant was recently reported in 10 patients from six consanguineous and two nonconsanguineous Lebanese families living in Australia with COX deficiency. 8 The patients had a severe infantile-onset neuromuscular disorder, with profoundly delayed psychomotor development since early infancy, seizures, hypotonia, elevated central nervous system and plasma lactate, and brain abnormalities consistent with Leigh syndrome. We report on a similar constellation of symptoms involving developmental delay, seizures, hypotonia, lactic acidosis, and brain involvement ( Table 1 ).
Recently, a homozygous transition in PET100 resulting in a termination (NM_001171155.1: c.142C > T; [p.Gln48*]) was identified in a newborn girl, born of consanguineous British Pakistani parents, with fatal COX deficiency. 9 The patient had intrauterine growth retardation and was delivered by Cesarean section at 34 weeks of gestation. She had severe lactic and metabolic acidosis, hypoglycemia, coagulopathy, increased serum creatine kinase, and intraventricular cysts on brain ultrasound. She developed seizures at 48 hours of age and died at 55 hours of age ( Table 1 ).
PET100 likely functions as a chaperone protein in the intermediate stage of COX assembly and facilitates the interaction between several factors in the formation of the COX1-COX2 subassembly module. 12 The Lebanese PET100 mutation removes the initiation codon, potentially truncating the N-terminal transmembrane domain, which is thought to be required for protein mitochondrial localization; functional analysis performed by Lim et al showed disrupted complex IV biogenesis in mitochondria of patients with the PET100 c.3G > C; [p.Met1?] mutation, as well as in vitro restoration of complex IV formation in patient fibroblasts overexpressed with wild type PET100 . 8 Multiple Lebanese patients were reported to survive into adolescence and adulthood; these patients notably exhibited musculoskeletal features involving scoliosis and spastic quadriplegia or quadriparesis, in addition to other symptoms including intellectual disability, dementia, and ocular abnormalities. 8 The truncating mutation found in the Pakistani family was associated with the most severe phenotype, involving prenatal onset with extreme metabolic abnormalities. 9 Based on the severity, the authors suspected the mutation to result in nonsense mediated decay or the production of a protein exhibiting a dominant-negative effect. The genotype–phenotype correlation is unclear, with patients exhibiting neurologic dysfunctions with varying degrees of severity. We may also consider that phenotypes may be modified by other factors such as other genes and/or environmental elements, as well as the differing levels of mitochondrial activity in different organs; mitochondrial activity is additionally thought to be influenced by an individual's genetic makeup. 5
It is worth noting that five of the previously described Lebanese families originated from the north of Lebanon. The parents of patients in family A reported herein are not related, belong to the Maronite community, and also originate from the North of Lebanon. The parents in family B are first cousins belonging to the Sunni Muslim community, and originate from the North of Lebanon as well. We can then estimate that this founder mutation originated from the latter region. The frequency of this variant is unknown in the Lebanese population and the variant was absent from our in-house WES database of 339 individuals as well as in our recent study of 167 patients who underwent WES analysis. 13 The variable clinical description necessitates reporting recurrent variants. This study emphasizes the importance of next generation sequencing in providing a fast and accurate clinical diagnosis.
Acknowledgments
We would like to thank the family for their full cooperation throughout the study.
Footnotes
Conflict of Interest None declared.
References
- 1.Dennerlein S, Rehling P. Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance. J Cell Sci. 2015;128(05):833–837. doi: 10.1242/jcs.161729. [DOI] [PubMed] [Google Scholar]
- 2.Anderson S, Bankier A T, Barrell B Get al. Sequence and organization of the human mitochondrial genome Nature 1981290(5806):457–465. [DOI] [PubMed] [Google Scholar]
- 3.Grossman L I, Lomax M I. Nuclear genes for cytochrome c oxidase. Biochim Biophys Acta. 1997;1352(02):174–192. doi: 10.1016/s0167-4781(97)00025-0. [DOI] [PubMed] [Google Scholar]
- 4.DiMauro S, Tanji K, Schon E A. The many clinical faces of cytochrome c oxidase deficiency. Adv Exp Med Biol. 2012;748:341–357. doi: 10.1007/978-1-4614-3573-0_14. [DOI] [PubMed] [Google Scholar]
- 5.Rak M, Bénit P, Chrétien D et al. Mitochondrial cytochrome c oxidase deficiency. Clin Sci (Lond, England: 1979) 2016;130(06):393–407. doi: 10.1042/CS20150707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szklarczyk R, Wanschers B F, Nijtmans L G et al. A mutation in the FAM36A gene, the human ortholog of COX20, impairs cytochrome c oxidase assembly and is associated with ataxia and muscle hypotonia. Hum Mol Genet. 2013;22(04):656–667. doi: 10.1093/hmg/dds473. [DOI] [PubMed] [Google Scholar]
- 7.Church C, Goehring B, Forsha D, Wazny P, Poyton R O. A role for Pet100p in the assembly of yeast cytochrome c oxidase: interaction with a subassembly that accumulates in a pet100 mutant. J Biol Chem. 2005;280(03):1854–1863. doi: 10.1074/jbc.M410726200. [DOI] [PubMed] [Google Scholar]
- 8.Lim S C, Smith K R, Stroud D A et al. A founder mutation in PET100 causes isolated complex IV deficiency in Lebanese individuals with Leigh syndrome. Am J Hum Genet. 2014;94(02):209–222. doi: 10.1016/j.ajhg.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Oláhová M, Haack T B, Alston C L et al. A truncating PET100 variant causing fatal infantile lactic acidosis and isolated cytochrome c oxidase deficiency. Eur J Hum Genet. 2015;23(07):935–939. doi: 10.1038/ejhg.2014.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.El-Bazzal L, Atkinson A, Gillart A C, Obeid M, Delague V, Mégarbané A.A novel EXT2 mutation in a consanguineous family with severe developmental delay, microcephaly, seizures, feeding difficulties, and osteopenia extends the phenotypic spectrum of autosomal recessive EXT2-related syndrome (AREXT2) Eur J Med Genet 2018;(Jul):31. [DOI] [PubMed] [Google Scholar]
- 11.Desvignes J P, Bartoli M, Delague Vet al. VarAFT: a variant annotation and filtration system for human next generation sequencing data Nucleic Acids Res 201846(W1):W545–W553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vidoni S, Harbour M E, Guerrero-Castillo S et al. MR-1S interacts with PET100 and PET117 in module-based assembly of human cytochrome c oxidase. Cell Reports. 2017;18(07):1727–1738. doi: 10.1016/j.celrep.2017.01.044. [DOI] [PubMed] [Google Scholar]
- 13.Nair P, Sabbagh S, Mansour H et al. Contribution of next generation sequencing in pediatric practice in Lebanon. A study on 213 cases. Mol Genet Genomic Med. 2018;6(06):1041–1052. doi: 10.1002/mgg3.480. [DOI] [PMC free article] [PubMed] [Google Scholar]