

Abstract
Dysregulation of the Wnt signaling pathway is an underlying mechanism in multiple diseases, particularly in cancer. Until recently, identifying agents that target this pathway has been difficult and as a result, no approved drugs exist that specifically target this pathway. We reported previously that the anthelmintic drug Niclosamide inhibits the Wnt/β-catenin signaling pathway and suppresses colorectal cancer cell growth in vitro and in vivo. As part of an effort to develop agents to treat metastatic colorectal cancer, we used the SAR of Niclosamide’s inhibition of Wnt/β-catenin signaling to design a new class of Wnt/β-catenin signaling inhibitors based on a triazole motif. Similar to Niclosamide, we found that the new triazole derivatives internalized Frizzled-1 GFP receptors, inhibited Wnt/β-catenin signaling in the TOPflash assay and reduced Wnt/β-catenin target gene levels in CRC cells harboring mutations in the Wnt pathway. Moreover, we found the Wnt/β-catenin SAR trends were generally similar between the two chemical classes of inhibitors. Overall, these studies demonstrate the ability to use the SAR of the Niclosamide chemical class to design additional chemical diversity in search of Wnt/β-catenin inhibitors with sufficient properties to progress to the clinic.
Keywords: Niclosamide, Wnt signaling inhibitor, Small molecule, Triazole derivatives, β-catenin, Cancer
Graphical abstract

The Wnt signaling pathway is a fundamental growth control pathway that is essential to the regulation of many biological process and is critical to stem cell development and the proper growth and maintenance of tissues1. As a result, dysregulation of the pathway is an underlying factor in many diseases, particularly cancer, where mutations are found frequently in various components of the pathway1–3. In colorectal cancer (CRC), more than 93% of tumors had alteration in the pathway, ~80% of which were mutations in adenomatous polyposis coli (APC) or β-catenin that result in hyperactivation of Wnt signaling1, 4. Given the importance of the Wnt signaling pathway, there has been an intensive effort for more than three decades to understand the mechanistic details of Wnt signaling and to develop therapies that target the pathway1, 5.
Our understanding of the mechanistic complexities of the signaling pathway has grown considerably during this time1, 3, 6. In the canonical Wnt pathway, a key component is the cytosolic protein β-catenin, whose levels are controlled by a destruction complex consisting of Axin, APC, and two serine-theonine kinases, CK1α/δ and GSK3α/β. Signaling is activated by secreted Wnt proteins that bind the seven transmembrane receptor Frizzled and the single transmembrane receptors LRP5/6 resulting in activation of cytosolic proteins called Dishevelled (Dvl), internalization of the Frizzled receptor, and stabilization and translocation of cytosolic β-catenin proteins into the nucleus. In the nucleus, β-catenin binds and activates the transcription factor LEF/TCF to initiate the transcription of Wnt/β-catenin target genes. In CRC, most of the mutations observed in the pathway result in an increase in the level of β-catenin protein and activation of the pathway. Thus, agents that decrease the level of β-catenin could be useful therapeutic agents. Overall, the Wnt signaling pathway consists of many intracellular protein-protein interactions for which traditional drug discovery approaches have been difficult5, 7. As a result, no drugs have been approved that specifically target the pathway8.
Recent efforts, however, have provided opportunities to inhibit the pathway1, 9–18. Our contribution to this field was the discovery that the anthelmintic drug Niclosamide (Figure 1), inhibits Wnt/β-catenin signaling19, a discovery that has since been confirmed by others20–22. Mechanistically, we found that Niclosamide promoted Frizzled internalization, downregulated Dvl and β-catenin protein levels and inhibited colorectal cancer cell growth in vitro and in vivo19, 23. Initial SAR studies indicated that Niclosamide inhibition of Wnt signaling appeared unique among structurally-related anthelmintic agents, and that the potency and Wnt/β-catenin functional response was dependent on small changes in the chemical structure of Niclosamide24.
Figure 1.

Chemical structures of Niclosamide, Niclosamide analogs, and the triazole class of Wnt/β-catenin inhibitors
Niclosamide was approved by the FDA in 1982 for use in humans to treat tapeworm infections25 and has been identified as an essential medicine by the World Health Organization26. Niclosamide is a salicylic acid derivative that belongs to the salicylanilide class of anthelmintic agents. Similar to other members of the salicylanilide class, Niclosamide has been used widely as an anthelmintic agent in livestock27. Its anthelmintic activity reported involves uncoupling of oxidative phosphorylation27–30. Niclosamide has protonophore activity and contains an aryl β-hydroxy carbonyl pharmacophore that is present in a large number of natural products and drugs that have a broad range of biological activity31. Thus, it is perhaps not surprising that since Niclosamide’s discovery more than 50 years ago, additional biological activity has been discovered that clearly indicate Niclosamide is a multi-functional drug31.
Niclosamide’s multi-functional activity has generated considerable interest in its use to treat cancer, rheumatoid arthritis, diabetes, and bacterial and viral infections31–38. In addition to our clinical trial in colorectal cancer (), there are now four additional clinical trials of Niclosamide listed in ClinicalTrials.gov in cancer (, , ), and in rheumatoid arthritis (). Particular relevant to cancer therapy, Niclosamide inhibits the growth of cancer cells from multiple tumor types and is active against drug resistant cancers23, 31, 39–41. In addition to its ability to inhibit the Wnt signaling pathway, Niclosamide inhibits other key oncogenic signaling pathways such as mTOR, NF-kB, Notch and STAT-3 and has effects on metabolism, including activation of AMPK31. Niclosamide is poorly absorbed upon oral administration and is cleared rapidly from systemic distribution23, 42, 43. Although Niclosamide’s pharmacokinetic properties are appropriate for its anthelmintic indication, its pharmacokinetic and solubility properties can limit its use to treat diseases where systemic exposure is required.
Given the need to identify drugs that inhibit Wnt/β-catenin signaling and the limitations of Niclosamide’s properties, we sought to expand the chemical diversity of inhibitors available to us via a classical “scaffold-hopping” strategy44 while interrogating the SAR of Niclosamide inhibition of Wnt/β-catenin signaling. In our previous SAR studies of Niclosamide, we found that a trifluoromethyl group was a suitable replacement of the nitro group and resulted in an inhibitor of Wnt/β-catenin signaling (2) (Figure 1) with similar potency to Niclosamide9, 45. However, upon insertion of one carbon atom between the anilide ring and the amide NH group of 2, the resulting salicylic amide derivative 3 had much poorer inhibitory activity45. To better understand if loss of activity was due to an increase in the steric demands of extending the inhibitor’s structure by 1 atom and changing the position of the anilide ring or if the loss of activity resulted from the loss of conjugation of the N-H bond into the aromatic ring and a decrease in the N-H hydrogen-bond donating properties, we designed a series of triazole compounds in which the salicylic scaffold of Niclosamide was simultaneously converted into a triazole scaffold (Figure 1). This change inserts one carbon atom between the anilide ring and the amide NH group and changes the anilide ring position in a similar fashion to the less-active derivative 3 while allowing the N-H bond to maintain conjugation with the aromatic ring.
Based on SAR data indicating Niclosamide derivatives without the 2’-chloro substituent in the anilide inhibited Wnt/β-catenin signaling45 and the ready availability of the commercially available starting materials in our lab, we first prepared triazole 4 in three steps using published procedures (Figure 2)46.
Figure 2.

Synthesis of triazole derivatives.
Upon testing this compound in the Fzd1-GFP internalization and the TOPflash Wnt3A-stimulated β-catenin gene transcription assay9, 24, 45, we were delighted to find it induced a robust punctate pattern in the Fzd1-GFP assay indicative of internalization of the Fzd1 receptor and inhibited Wnt3A-stimulated β-catenin gene transcription with an IC50 of 1.41 ± 0.34 μM (Table 1 and Figure S1 in supplemental information). To enable better comparison of structure and inhibitory activity, we also prepared salicylamide 10, and tested it and the imide intermediate 11 for Wnt/β-catenin inhibitory activity. Consistent with previous results obtained with the trifluoromethyl-substituted salicylamide derivative 3, the nitro-substituted salicylamide derivative 10 was considerably less active in both assays than the similarly substituted triazole 4. The imide intermediate 11 however, had a different profile. Whereas its activity in the Fzd1-GFP assay was similar to DMSO control, it increased Wnt3A-stimulated signaling in the TOPFlash assay. The reasons for this result are not understood at this time. Based on the data obtained with triazole 4, we postulate that conjugation of the N-H group and/or its hydrogen bonding donating ability was the important SAR feature for inhibitory activity. We then prepared a set of triazole derivatives that varied the nitro substituent at the 4-position of the pendent aryl ring to understand if the SAR at this position, a key position in the Niclosamide series, tracked into the new series. Similar to the SAR studies conducted in the Niclosamide series, we selected substituents with a range of electron-donating and electron-withdrawing properties. Consistent with results obtained in the Niclosamide chemotype, derivatives substituted at the para position with an electron-donating methoxyl substituent47 (6), an electron-withdrawing fluorine atom (7), or a hydrogen atom at this position (8) showed a trend toward less activity in both the Fzd1-GFP and the TOPFlash assay although the magnitude of the difference was not as great. Similarly, derivatives with electron-withdrawing nitro or trifluoromethyl substituents were more active in both assays24, 45. In the Niclosamide series, we noted earlier that a 2’-chloro substituent increased potency when there was a nitro group at the 4’-position24, 45. To test whether the addition of a corresponding 2’chloro-substituent in the aryl ring of the triazole would produce a similar increase in potency, we prepared triazole 9. Unfortunately, a similar increase in potency in the triazole series (4 vs 9) was not observed by the addition of the chloro-substituent. Overall, however, the trend in inhibitory activity of the triazole derivatives appeared consistent with the inhibitory activity of similarly substituted Niclosamide derivatives24, 45.
Table 1.
SAR of Wnt/β-catenin signaling.
| Compound Number | Structure | FZd1-GFP Internalizatio at 12.5 μMa | n Inhibition Wnt/β-catenin transcription TopFlash IC50 (μM) ± SEb |
|---|---|---|---|
| 1 | 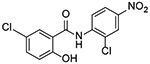 |
5 | 0.34 ± 0.07 |
| 4 | 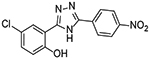 |
4 | 1.41 ± 0.34 |
| 5 | 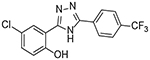 |
2 | 2.88 ± 0.84 |
| 6 | 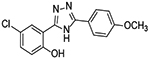 |
0 | 60% inhibition at 10 μM, 14% inhibition at 5 μM |
| 7 | 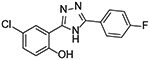 |
1 | 1.91 ± 0.60 |
| 8 | 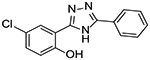 |
0 | 4.02 ± 0.98 |
| 9 | 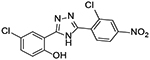 |
5 | 1.84 ± 0.60 |
| 10 | 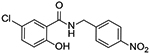 |
0 | 43% inhibition at 10μM, 3% inhibition at 5 μM |
| 11 | 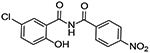 |
0 | increased signal, 153 % of control at 10μM |
Fzd1-GFP assay: Internalization of Frizzled1-GFP stably expressed in U2OS cells was determined by confocal microscopy. Cells were treated with compounds for 6 hours, fixed, and scored visually by the amount of punctate observed versus DMSO control. Punctate similar to control = 0, trace amount of punctate greater than control = 1, moderate = 3, strong = 5. Images in Supplemental Information.
Inhibition of Wnt3A-stimulated Wnt/β-catenin transcription was determined by TOPFlash assay using HEK293 cells stably expressing a TOPFlash luciferase reporter and Renilla luciferase reporter control. Cells were stimulated with Wnt3A-conditioned medium in the presence of DMSO or compounds from 0.04 to 10μM for 8 hrs. The TOPFlash reporter activity of Wnt3A conditioned media with DMSO treatment was set as 100%. Data were fit using GraphPad Prism (mean ± SEM, n ≥3). Percent inhibition at the two highest concentrations tested is reported for weak inhibitors that do not give a full dose response over the inhibitor concentration assayed.
To further confirm the Wnt/β-catenin inhibitory activity of this new chemical series, compounds with significant activity in the FZD1-GFP internalization and the Wnt3A-TOPflash assays were evaluated by immunoblot in two CRC cell lines with aberrant Wnt signaling (HCT-116, β-catenin mutant; SW480, APC mutant) for their ability to reduce the levels of Wnt/β-catenin target gene proteins (Figure 3). Consistent with the results of the Frizzled1-GFP and the Wnt3A-stimulated TOPflash assays, Niclosamide (1), and triazole derivatives 4, 5 and 9 each reduced the levels of the Wnt/β-catenin target genes: β-catenin, Axin2, c-Myc, Survivin and Cyclin D1 relative to controls in both cell lines, thereby demonstrating their ability to inhibit Wnt/β-catenin signaling in CRC cells with aberrant Wnt pathway signaling. In general, Niclosamide derivatives with IC50 values in the Wnt3A-stimulated TOPflash assay in the single digit micromolar or less range evaluated to date also typically produce a robust punctate pattern in the FZD1-GFP internalization assay. However during the course of our studies, we have identified a few compounds that do not produce a robust response in the FZD1-GFP internalization assay yet inhibit Wnt/β-catenin signaling with IC50s of 1–4 μM in the TOPflash assay9, 24, 45. Herein, compound 8 (Table 1) has this profile for which studies in HCT-116 cells confirmed the reduction in Wnt/β-catenin target genes (Supplemental Figure S2). The mechanistic underpinnings to account for this observation are not clear at present. Given Niclosamide has multifunctional activity, one explanation may be that these derivatives have a different mechanism or a different selectivity profile.
Figure 3. Reduction of Wnt/β-catenin target genes by Niclosamide and triazole 4, 5 and 9.

Cells were treated with DMSO or compounds (5 μM) in DMSO for 18 hours, the cytosolic fraction and the whole cell lysates were probed with antibodies to the proteins indicated. β-actin was used as a loading control. A. Western blot images B. Quantification of the protein levels of Western blots in A normalized to the β-Actin loading control and graphed as a percentage of protein level in the DMSO treated control cells.
Overall, these SAR studies demonstrate the ability to extend the SAR findings of the Niclosamide chemotype into a novel triazole class of Wnt/β-catenin inhibitors. Additional studies are underway to further define the Wnt/β-catenin inhibitory SAR to improve potency, the activity against pathways modulated by Niclosamide, and the pharmacokinetic properties of the series to support their evaluation in CRC tumor models and models of other diseases for which Niclosamide has significant biological activity31.
Supplementary Material
Acknowledgements
This work was funded in part by 5 R01 CA172570 (WC), BC123280 (WC), and Clinical Oncology Research Center Development Grant 5K12-CA100639–08 (RAM). Wei Chen is a V foundation Scholar and an American Cancer Society Research Scholar. NMR instrumentation in the Duke NMR Spectroscopy Center was funded by the NIH, NSF, North Carolina Biotechnology Center and Duke University. The authors gratefully acknowledge this support and the support of Professor Eric Toone and David Gooden of the Duke Small Molecule Synthesis Facility.
Abbreviations:
- APC
Adenomatous Polyposis Coli
- CRC
colorectal cancer
- Dvl
Dishevelled
- LEF/TCF
Lymphoid enhancer factor/T cell factor
- Fzd1
Frizzled1
- GFP
green fluorescent protein
- SAR
structure-activity relationships
References and notes
- 1.Nusse R; Clevers H Cell 2017, 169, 985. [DOI] [PubMed] [Google Scholar]
- 2.Zhan T; Rindtorff N; Boutros M Oncogene 2017, 36, 1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Clevers H; Nusse R Cell 2012, 149, 1192. [DOI] [PubMed] [Google Scholar]
- 4.Cancer Genome Atlas Network. Nature 2012, 487, 330.22810696 [Google Scholar]
- 5.Barker N; Clevers H Nature Reviews Drug Discovery 2006, 5, 997. [DOI] [PubMed] [Google Scholar]
- 6.Chen W; ten Berge D; Brown J; Ahn S; Hu LA; Miller WE; Caron MG; Barak LS; Nusse R; Lefkowitz RJ Science 2003, 301, 1391. [DOI] [PubMed] [Google Scholar]
- 7.Meireles LMC; Mustata G Curr. Top. Med. Chem 2011, 11, 248. [DOI] [PubMed] [Google Scholar]
- 8.Sebio A; Kahn M; Lenz H-J Expert Opinion on Therapeutic Targets 2014, 18, 611. [DOI] [PubMed] [Google Scholar]
- 9.Mook RA Jr.; Ren XR; Wang J; Piao H; Barak LS; Kim Lyerly H; Chen W Bioorg Med Chem 2017, 25, 1804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tabatabai R; Linhares Y; Bolos D; Mita M; Mita A Target Oncol 2017, 12, 623. [DOI] [PubMed] [Google Scholar]
- 11.Tran FH; Zheng JJ Protein Sci 2017, 26, 650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang K; Wang X; Zhang H; Wang Z; Nan G; Li Y; Zhang F; Mohammed MK; Haydon RC; Luu HH; Bi Y; He TC Lab Invest 2016, 96, 116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lum L; Chen C Curr Med Chem 2015, 22, 4091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y; Oliver PG; Lu W; Pathak V; Sridharan S; Augelli-Szafran CE; Buchsbaum DJ; Suto MJ Cancer Lett 2017, 389, 41. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Li B; Orton D; Neitzel LR; Astudillo L; Shen C; Long J; Chen X; Kirkbride KC; Doundoulakis T; Guerra ML; Zaias J; Fei DL; Rodriguez-Blanco J; Thorne C; Wang Z; Jin K; Nguyen DM; Sands LR; Marchetti F; Abreu MT; Cobb MH; Capobianco AJ; Lee E; Robbins DJ Science Signaling 2017, 10, published online June 27, 2017. (doi: 10.1126/scisignal.aak9916). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.You L; Zhang C; Yarravarapu N; Morlock L; Wang X; Zhang L; Williams NS; Lum L; Chen C Bioorg Med Chem Lett 2016, 26, 5891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shono T; Ishikawa N; Toume K; Arai MA; Ahmed F; Sadhu SK; Ishibashi M Bioorg Med Chem Lett 2015, 25, 2735. [DOI] [PubMed] [Google Scholar]
- 18.Lee JH; Kim MA; Park S; Cho SH; Yun E; O YS; Kim J; Goo JI; Yun MY; Choi Y; Oh S; Song GY Bioorg Med Chem Lett 2016, 26, 3529. [DOI] [PubMed] [Google Scholar]
- 19.Chen MY; Wang JB; Lu JY; Bond MC; Ren XR; Lyerly HK; Barak LS; Chen W Biochemistry 2009, 48, 10267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sack U; Walther W; Scudiero D; Selby M; Kobelt D; Lemm M; Fichtner I; Schlag Peter M; Shoemaker Robert H; Stein U Journal of the National Cancer Institute 2011, 103, 1018. [DOI] [PubMed] [Google Scholar]
- 21.Lu W; Lin C; Roberts MJ; Waud WR; Piazza GA; Li Y Plos One 2011, 6, e29290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arend RC; Londono-Joshi AI; Samant RS; Li Y; Conner M; Hidalgo B; Alvarez RD; Landen CN; Straughn JM; Buchsbaum DJ Gynecologic Oncology 2014, 134, 112. [DOI] [PubMed] [Google Scholar]
- 23.Osada T; Chen MY; Yang XY; Spasojevic I; Vandeusen JB; Hsu D; Clary BM; Clay TM; Chen W; Morse MA; Lyerly HK Cancer Research 2011, 71, 4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mook RA Jr.; Chen M; Lu J; Barak LS; Lyerly HK; Chen W Bioorg Med Chem Lett 2013, 23, 2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pearson RD; Hewlett EL Annals of Internal Medicine 1985, 102, 550. [DOI] [PubMed] [Google Scholar]
- 26.WHO, Ed. The Selection and Use of Essential Medicines World Health Organization; Geneva, 2007. [Google Scholar]
- 27.Swan GE J. S. Afr. Vet. Assoc 1999, 70, 61. [DOI] [PubMed] [Google Scholar]
- 28.Weinbach EC; Garbus J Nature 1969, 221, 1016. [DOI] [PubMed] [Google Scholar]
- 29.Williamson RL; Metcalf RL Science 1967, 158, 1694. [DOI] [PubMed] [Google Scholar]
- 30.Frayha GJ; Smyth JD; Gobert JG; Savel J General Pharmacology 1997, 28, 273. [DOI] [PubMed] [Google Scholar]
- 31.Chen W; Mook RA Jr.; Premont RT; Wang J Cellular Signalling 2017, published online April 4, 2017 ( 10.1016/j.cellsig.2017.04.001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tao H; Zhang Y; Zeng X; Shulman GI; Jin S Nature Medicine 2014, 20, 1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rajamuthiah R; Fuchs BB; Conery AL; Kim W; Jayamani E; Kwon B; Ausubel FM; Mylonakis E Plos One 2015, 10, e0124595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gooyit M; Janda KD Sci Rep 2016, 6, 33642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu M; Lee EM; Wen Z; Cheng Y; Huang WK; Qian X; Tcw J; Kouznetsova J; Ogden SC; Hammack C; Jacob F; Nguyen HN; Itkin M; Hanna C; Shinn P; Allen C; Michael SG; Simeonov A; Huang W; Christian KM; Goate A; Brennand KJ; Huang R; Xia M; Ming GL; Zheng W; Song H; Tang H Nature Medicine 2016, 22, 1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang YM; Lu JW; Lin CC; Chin YF; Wu TY; Lin LI; Lai ZZ; Kuo SC; Ho YJ Antiviral Research 2016, 135, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang M; Zeng S; Qiu Q; Xiao Y; Shi M; Zou Y; Yang X; Xu H; Liang L Int. Immunopharmacol 2016, 31, 45. [DOI] [PubMed] [Google Scholar]
- 38.Liang L; Huang M; Xiao Y; Zen S; Lao M; Zou Y; Shi M; Yang X; Xu H Inflammation Res. 2015, 64, 225. [DOI] [PubMed] [Google Scholar]
- 39.Li Y; Li PK; Roberts MJ; Arend RC; Samant RS; Buchsbaum DJ Cancer Lett 2014, 349, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu C; Lou W; Zhu Y; Nadiminty N; Schwartz CT; Evans CP; Gao AC Clinical Cancer Research 2014, 20, 3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li R; Hu Z; Sun SY; Chen ZG; Owonikoko TK; Sica GL; Ramalingam SS; Curran WJ; Khuri FR; Deng X Mol Cancer Ther 2013, 12, 2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Andrews P, Thyssen J, Lorke D Pharmac. Ther 1983, 19, 245. [DOI] [PubMed] [Google Scholar]
- 43.Chang Y-W; Yeh T-K; Lin K-T; Chen W-C; Yao H-T; Lan S-J; Wu Y-S; Hsieh H-P; Chen C-M; Chen C-T Yaowu Shipin Fenxi 2006, 14, 329. [Google Scholar]
- 44.Langdon SR; Ertl P; Brown N Molecular Informatics 2010, 29, 366. [DOI] [PubMed] [Google Scholar]
- 45.Mook RA Jr.; Wang J; Ren XR; Chen M; Spasojevic I; Barak LS; Lyerly HK; Chen W Bioorg Med Chem 2015, 23, 5829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Catarzi D; Cecchi L; Colotta V; Filacchioni G; Varano F; Martini C; Giousti L; Lucacchini A J Med Chem 1995, 38, 2196. [DOI] [PubMed] [Google Scholar]
- 47.Thomas G Medicinal Chemistry: An Introduction; West Sussex, England: John Wiley & Sons, 2007, p. 116. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.