

Abstract
Encoding the slow skeletal muscle isoform of Myosin Binding Protein-C, MYBPC1 is associated with autosomal dominant and recessive forms of arthrogryposis. We describe a novel association for MYBPC1 in four patients from three independent families with skeletal muscle weakness, myogenic tremors, and hypotonia with gradual clinical improvement. The patients carried one of two de novo heterozygous variants in MYBPC1, with the p.Leu263Arg variant seen in three individuals and the p.Leu259Pro variant in one individual. Both variants are absent from controls, well conserved across vertebrate species, predicted to be damaging, and located in the M-motif. Protein modeling studies suggested that the p.Leu263Arg variant affects the stability of the M-motif, while the p.Leu259Pro variant alters its structure. In vitro biochemical and kinetic studies demonstrated that the p.Leu263Arg variant results in decreased binding of the M-motif to myosin, which likely impairs the formation of actomyosin cross-bridges during muscle contraction. Collectively, our data substantiate that damaging variants in MYBPC1 are associated with a new form of an early-onset myopathy with tremor, which is a defining and consistent characteristic in all affected individuals, with no contractures. Recognition of this expanded myopathic phenotype can enable identification of individuals with MYBPC1 variants without arthrogryposis.
Keywords: MYBPC1, tremor, hypotonia, myopathy, Myosin Binding Protein-C, arthrogryposis
INTRODUCTION
Myosin Binding Protein-C (MyBP-C) comprises a family of accessory proteins in the muscle sarcomere that aid in the regulation of actomyosin cross bridges and the stabilization of thick filaments in striated muscles. The MyBP-C family consists of three isoforms, including slow skeletal (sMyBP-C), fast skeletal (fMyBP-C) and cardiac (cMyBP-C) proteins encoded by MYBPC1 (MIM# 160794), MYBPC2 (MIM# 160793) and MYBPC3 (MIM# 600958), respectively (Ackermann et al., 2010; Flashman et al., 2004; Oakley et al., 2007; Weber et al., 1993). MYBPC1 encodes a modular protein consisting of seven immunoglobulin (Ig) and three fibronectin-III (Fn-III) repeats, numbered C1-C10, interspersed with unique sequences, including a Pro/Ala-rich motif and a MyBP-C specific motif, termed the M-motif, which flank Ig domain C1 (Fig. 1A) (Ackermann et al., 2013) . MYBPC1 is unique among the MYBPC genes as it is heavily spliced, giving rise to at least 14 known isoforms in humans encoding proteins between 126 and 131.5 kDa. The sMyBP-C proteins are co-expressed in variable amounts and combinations in both fast and slow twitch skeletal muscles where they may coexist with fMyBP-C (Geist et al., 2016).
Figure 1:
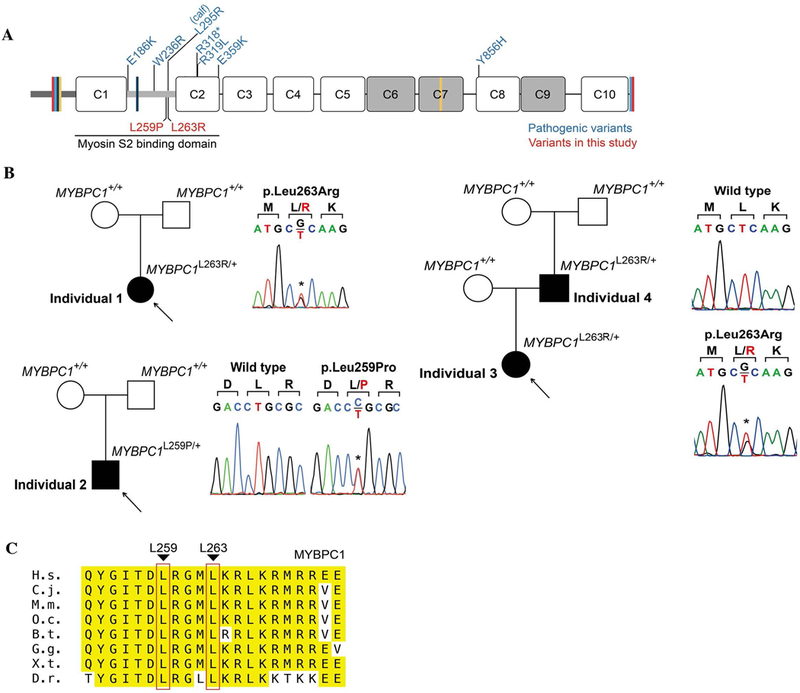
Pathogenic variants in MYBPC1. (A) Schematic representation of the domain structure of sMyBP-C encoded by MYBPC1 and depiction of known (in blue) and novel (in red) pathogenic variants; Ig and FN-III domains are shown as white and grey rectangles, the Pro/Ala rich motif and the M-motif are illustrated as dark and light grey, thick, horizontal lines, and spliced sequences are depicted as colored, perpendicular lines. (B) Pedigrees and Sanger traces of the three families containing the novel pathogenic MYBPC1 variants. Individuals 1, 3 and 4 carry the p.Leu263Arg variant, which appeared de novo in individuals 1 and 4 belonging in two different families, and was inherited by individual 3 (daughter; proband) from individual 4 (father), while individual 2 belonging to a third unrelated family carries the de novo p.Leu259Pro variant. (C) Evolutionary conservation of L259 and L263 across orthologs from vertebrate species; conserved residues surrounding L259 and L263 within the M-motif are highlighted in yellow, while L259 and L263 are marked with red boxes; H.s. denotes Homo sapiens; M.m., Mus musculus; O.c., Oryctolagus cuniculus; B.t., Bos taurus; G.g., Gallus gallus; X.t., Xenopus tropicalis; D.r., Danio rerio.
MyBP-C proteins are found in the thick filament of the sarcomere within the C-zone of the A-band in 7–9 transverse stripes, where they bind to both myosin and actin (Craig et al., 1976). Their COOH-terminus is anchored to light meromyosin (LMM) while their NH2-terminus binds to the S2 subfragment of myosin and actin to regulate muscle contraction. While the interaction of MyBP-C proteins with LMM is constant, their association with myosin S2 and actin is dynamic and in the case of cMyBP-C, and possibly sMyBP-C, regulated through phosphorylation (Ackermann et al., 2011; Moss et al., 2015). While over 500 MYBPC3 mutations have been associated with cases of hypertrophic or dilated cardiomyopathy (L. Wang et al., 2018), MYBPC1 has only recently been causally linked to the development of different forms of distal arthrogryposis (DA), which presents with congenital contractures. Two autosomal dominant variants, p.Trp236Arg and p.Tyr856His, have been linked to DA type 1B (MIM# 614335), while two additional autosomal dominant variants, p.Glu359Lys and p.Pro319Leu, have been associated with DA type II (Gurnett et al., 2010; Li et al., 2015). Moreover, two recessive variants, p.Arg318* and p.Glu186Lys, have been connected to Lethal Congenital Contracture Syndrome 4 (LCCS4: MIM# 614915) and an Arthrogryposis Multiplex Congenita presentation, respectively (Ekhilevitch et al., 2016; Markus et al., 2012). Lastly, a calf containing the de novo p.Leu295Arg variant was described to have congenital muscle tremor, weakness, contractures, and inability to fully extend its limbs, with subsequent spontaneous improvement of motor strength (Wiedemar et al., 2015). Interestingly, five of the six human MYBPC1 variants and the calf variant are located in the NH2-terminus of the molecule suggesting that it may be a hot spot for deleterious variants (Fig. 1A). Indeed, knocking down mybpc1 or introducing MYBPC1W236R and MYBPC1Y856H in zebrafish displayed human DA features, such as impaired motor excitation, disorganized myofibrils, reduced sarcomere numbers and severe body curvature (Ha et al., 2013).
Thus far, MYBPC1-linked autosomal dominant DA in humans has been associated with non-progressive congenital contractures accompanied with no overt postnatal muscle weakness. We now describe four individuals with damaging heterozygous MYBPC1 variants who presented with skeletal muscle weakness and tremor, but lacked contractures. Our protein modeling and biochemical studies provide evidence for the harmful effects of these variants on sMyBP-C stability and structure as well as its ability to bind myosin via its NH2-terminal M-motif. Our clinical, biochemical and modeling data delineate a hitherto unrecognized expanded phenotype of a non-progressive congenital/early-onset myopathy with tremor as its defining characteristic, linked to damaging variants in MYBPC1.
MATERIALS AND METHODS
Whole exome sequencing
Individual 1 underwent trio WES as part of a research study at Duke University Medical Center, with previously published methods for sequencing, data processing, variant calling, and variant annotation (Zhu X, 2014). Individual 2 had commercial trio WES and WGS according to prior published methods (Bick et al., 2017; Retterer et al., 2016) and the data were reanalyzed through the UDN. Individuals 3 and 4 underwent WES at the Yale Center for Genome Analysis and data processing was performed as described previously (Zaidi et al., 2013). Further details on the WES metrics as well as information on other variants that were identified in these individuals are in the Appendix (Table S1).
Sanger sequencing
Both MYBPC1 (NM_002465.3) variants were validated by Sanger sequencing (Applied Biosystems, Foster City, CA) and the de novo origin of each variant was confirmed by parental segregation studies.
Generation of recombinant proteins
For the blot overlay assays, human skeletal muscle cDNA was used with the following sense (5’ATGCCAGAACCCACT3’) and antisense (5’TCAATCAAGAATTTTTGC3’) primers to amplify the NH2-terminal region of sMyBP-C containing the Pro/Ala-rich motif, Ig domain C1, and the M-motif (amino acids 1–284, XP_006719468.1). The PCR fragment was subcloned into the pGex4T-1 vector (Amersham Pharmacia, Piscataway, NJ, USA) at EcoRI/XhoI sites to generate a GST-fusion protein. The p.Leu259Pro and p.Leu263Arg mutations were individually introduced into the resulting plasmid with the following set of primers: sense (5’AATCACCGATCCGCGCGGCATGC3’) and antisense (5’CCATACTGGAAGGCGATCTTCTC3’), and sense (5’GCGCGGCATGCGCAAGCGACTCA3’) and antisense (5’AGATCGGTGATTCCATACTGGAAGGCG3’), respectively.
For the circular dichroism experiments, the following set of sense (5’ACTGGAATTCATGCAGGAGGAGGAGCCC) and antisense (5’ACTGCTCGAGTCACTTCTTCTCCTCCCT’) primers was used to amplify the M-motif region. The PCR fragment was subcloned into the pET30a+ vector (Millipore Sigma, Bedford, MA, USA) at EcoRI/XhoI sites to generate a His-tagged fusion protein. The p.Leu259Pro and p.Leu263Arg mutations were introduced in the wild-type construct using the primer sets listed above. The authenticity of all constructs was verified by sequencing.
Recombinant polypeptides were expressed in BL21(DE3) cells (MilliporeSigma, Bedford, MA, USA) by induction with 1 mM IPTG overnight at 22°C and purified by affinity chromatography on Glutathione agarose (GST-proteins) and Ni-NTA-agarose (His-proteins) columns (MilliporeSigma, Bedford, MA, USA), according to the manufacturer’s instructions.
Blot overlay assays
Heavy meromyosin (HMM) and actin purified from skeletal muscle were purchased from Cytoskeleton (heavy meromyosin Cat. #: MH01-A, actin Cat. #: AKL99-A). Equivalent amounts (3 μg) of purified HMM and actin were separated by 4–12% SDS-PAGE and transferred electrophoretically to nitrocellulose. Equivalent loading and transfer were confirmed by staining the nitrocellulose membrane with Ponceau Red. Blots were incubated in buffer A (50 mM KCl, 20 mM MOPS, 4 mM MgCl2, 0.1 mM EGTA, 2 mM DTT, 3% BSA, 10 mM NaN3, 0.5% Tween-20, and 0.5% Nonidet P-40) for 8 hours at room temperature, followed by incubation with buffer A containing 0.5 μg/mL of the indicated GST-fusion protein overnight at 4 °C. Blots were washed extensively with buffer A, followed by PBS, blocked in 5% milk, and subsequently probed with antibodies to GST (1:10,000; Novagen, Billerica, MA, USA), as described in 25. Immunoreactive bands were detected with the Tropix Chemiluminescence kit (Applied Biosystems). These experiments were repeated at least three independent times, and quantification of immunoreactive bands was performed using densitometric analysis and ImageJ. Statistical significance was evaluated with t-test, p<0.01.
Circular Dichroism assays
All CD experiments were conducted in triplicate in a 1mm path length cuvette at 15 mM protein in 50 mM phosphate buffer, pH 7.0 and 100 mM NaCl. Samples were measured in triplicate at temperatures from 20 °C to 85 °C in 5 °C intervals on a Jasco- J-810 spectrometer.
Modeling of wild-type and mutant sMyBP-C
Models of the sMyBP-C M-motif were generated using Phyre2 based on the respective human cMyBP-C domain (PBD: 2LH4; 80% identical to sMyBP-C)26–28. The model was allowed to equilibrate using the computer program YASARA for ~90 ns 29,30. Substitutions were introduced to the model via the ‘swap’ command, and the resulting models were allowed to equilibrate for an additional ~100 ns. The analysis was repeated in triplicate, and models were visualized and evaluated in YASARA and PyMol.
Trypsin digestion assay
Pancreatic bovine trypsin (Sigma T9201) was added to recombinant wild-type or L259P His-tagged M-motif proteins at an enzyme to substrate ratio of 1:30 in 100 mM sodium phosphate, pH 7.0, 100 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), and 1 mM DTT, as in Wang et al (D. J. Wang et al., 2008) (Ref). The reactions were incubated at 250 C and stopped at various time points (30 sec, 1, 2, 3, 4, 5, and 10 min) by boiling for 5 min. Samples were separated on a 12% SDS-PAGE gel and stained with Coomassie Brilliant Blue-G-250 dye.
RESULTS
Clinical presentation of affected individuals
The studies were performed under prospectively reviewed and approved IRB/ethics committees. Four affected individuals in three unrelated families (Fig. 1B, Table 1 and Appendix) presented with an overlapping phenotype consisting of severe congenital hypotonia, tremors at rest, posture, and on intention in the extremities and tongue, absence of contractures, and normal cognition. Congenital hypotonia was so severe that spinal muscular atrophy was a diagnostic consideration for individuals 1–3. Remarkably, despite the severe neonatal presentation, hypotonia improved over time such that individuals 1, 3, and 4 achieved ambulation, albeit delayed.
Table 1:
Salient clinical manifestations in four individuals with heterozygous damaging MYBPC1 variants
| Feature | Individual 1 | Individual 2 | Individual 3 | Individual 4 |
|---|---|---|---|---|
| MYBPC1 variant in NM_002465.3 | c.788T>G (p. Leu263Arg) | c.776T>C (p.Leu259Pro) | c.788T>G (p. Leu263Arg) | c.788T>G (p. Leu263Arg) |
| Genomic position (hg 37) | chr12: g.102036319 T>G | chr12: g.102036307 T>C | chr12:g.102036319T>G | chr12:g.102036319T>G |
| Inheritance | de novo | de novo | Paternally inherited | de novo |
| Protein domain | M motif | M motif | M motif | M motif |
| Age | 9 years | 23 months | 9 years | 40 years (father of Individual 3) |
| Gender | Female | Male | Female | Male |
| Ethnicity | Caucasian/ Northern European | Caucasian | Korean | Korean |
| Congenital hypotonia | Profound | Profound | Severe | Mild |
| Fine tremor of extremities at birth | + | + | Noted at 2 years of age | n.a. |
| Contractures/ arthrogryposis at birth | - | - | - | - |
| Persistent fine tremor of extremities at rest/posture/intention | −/+/+ | +/+/+ | −/+/+ | −/+/+ |
| Persistent lip tremor at rest/tongue tremor with protrusion | +/+ | +/+ | +/+ | +/+ |
| Persistent hypotonia | Mild (markedly improved) | Severe (markedly improved) | Mild (improved) | Mild (improved) |
| Muscle weakness | Mild | Moderate | Moderate | Mild |
| Easy fatigability | + | + | + | + |
| Gait | Normal | Not yet ambulatory | Waddling gait | Waddling gait |
| Skeletal abnormalities | - | - | - | - |
| Cognition | Normal | Normal | Normal | Normal |
| Cardiac abnormalities | Normal echocardiogram at 7 years of age | None known | None known | None known |
| Dysmorphic features | Downslanting palpebral fissures | Hypotonic facies, tall forehead with frontal bossing, dolichocephaly | Hypotonic facies mild | Hypotonic facies marked |
| EMG | Normal at 3 months of age | Not performed | Not performed | Myopathic MUAP (motor unit action potential), but no abnormal resting potentials |
| Brain MRI | Normal | Normal | Normal, taken at 1 and 2 years of age | Normal at 34 years of age |
Individual 1 is a 9 year-old Caucasian female who was diagnosed with profound congenital hypotonia as a newborn. Tremors of the extremities and tongue were noticed at rest, along with feeding difficulties. She was discharged to hospice care due to a presumptive diagnosis of spinal muscular atrophy, but genetic testing for it was normal. Feeding difficulties resolved at 12-months of age. Gross motor development was delayed; she sat unsupported at 9-months and walked at 18-months. Cognitive development has been normal and she is pursuing advanced academic courses in school. She continues to make gains in muscle strength but has poor stamina compared to her peers. On examination at the age of 9 years, growth parameters were normal and she had down slanted palpebral fissure but no other dysmorphic features. Moreover, she had tongue tremors on protrusion, mild hypotonia, but normal motor strength and normal deep tendon reflexes. A postural tremor, which persisted on intention, was observed in all extremities (upper > lower) (Movie S1, Appendix). There was mild dysmetria, no dysdiadochokinesis, and tandem and regular gait were normal (Table 1). No contractures were seen. Prior genetic, mitochondrial, and metabolic testing had been normal, and two brain MRIs have shown nonspecific subcortical foci of white matter abnormality, with a normal cerebellum. A nerve conduction study and an electromyogram (EMG) have been normal (Table 1).
Individual 2 is a 23-month old Caucasian male who was evaluated through the Undiagnosed Diseases Network (UDN) clinical site at Duke University (https://undiagnosed.hms.harvard.edu/). At birth he had marked hypotonia, hypersomnolence, stridor, poor feeding, tachypnea, and a fine tremor in the face, trunk, hands and feet at rest and on intention. The stridor and feeding difficulties have gradually improved. Motor delays have been evident; at 8 months, he was able to roll over but not lift his head in the prone position, could sit with support and had fair head control with head bobbing, but could not bear weight on his feet. On examination at that time, growth parameters were normal, although hypotonic facies (Table 1), generalized hypotonia, and a fine tremor of his hands and feet at rest and on intention as well as tongue tremors were noted (Movie S2, Appendix). Motor strength was mildly diminished generally. Deep tendon reflexes were normal. Similar to individual 1, no contractures were present. Developmental testing revealed age-appropriate receptive language and cognition. At 23-months of age, he is able to bring himself to a sitting position and scoot on his buttocks; cognition and fine motor skills are normal, tremors are present at rest and on intention and worsen with fatigue. An eye examination and a video electroencephalography (vEEG) were normal. Prior genetic and mitochondrial testing were normal, as was brain imaging.
Individual 3 is a 9-year old Korean female who was noted to have severe hypotonia and feeding difficulties at birth. Head control was poor the first year of life, and she began rolling over at 1-year of age. On examination at 2-years of age, lingual tremor upon tongue protrusion, and postural and intentional hand tremors were noted. She also had dysarthria, and a developmental quotient was within normal limits. She began walking independently at 24-months of age with a waddling, lordotic gait due to truncal and proximal weakness of extremities, which has remained stable. At 4-years of age, she began having seizures with electrodiagnostic correlates that were fairly well controlled with anti-epileptic medication. On examination at 9 years of age, she has grown normally, has a thin and long face suggestive of myopathy, fine tremors of her extremities, and lip and tongue tremor upon tongue protrusion. She still has a waddling gait pattern with in-toeing and shoulder shrugging posture (Movie S3, Appendix), but no contractures. Brain imaging has been normal and she performs well in school (Table 1). Molecular testing of the spinal muscular atrophy gene (SMN1) was normal.
Individual 4, the father of Individual 3, is a 40-year old Korean male who had congenital hypotonia and motor developmental delays. He sat independently at 1-year of age and had delayed walking. He had a mild waddling gait pattern during childhood and adolescence, which persists today in adulthood. On examination, he has myopathic facies, intentional and postural tremor in his extremities, and lingual tremor upon tongue protrusion (Table 1) (Movie S4, Appendix). He also exhibits shoulder and pelvic girdle weakness as well as mild proximal limb weakness. Deep tendon reflexes are elicited but depressed. His cognitive function is normal and he has not had seizures. Poor stamina and tremors affecting his work productivity are ongoing concerns. His parents are asymptomatic (Fig. 1B). A brain MRI and nerve conduction study were normal, with myopathic findings on EMG.
Whole exome sequencing revealed rare MYBPC1 variants in affected individuals
Using trio whole exome sequencing (WES) analysis, Individual 1 was found to have a de novo missense variant in MYBPC1 (c.788T>G, p.Leu263Arg). This variant is located at a highly conserved site and predicted to be damaging by in silico models including a Polyphen2 score of 0.996 (Polymorphism Phenotyping v2, http://genetics.bwh.harvard.edu/pph2/) (Adzhubei et al., 2010), CADD score of 33 (Combined Annotation Dependent Depletion, https://cadd.gs.washington.edu/) (Kircher et al., 2014), and GERP++ score of 5.53 (Genomic Evolutionary Rate Profiling, http://mendel.stanford.edu/SidowLab/downloads/gerp/) (Davydov et al., 2010). The same variant was identified via WES in Individuals 3 and 4, and Sanger sequencing of the parents of Individual 4 confirmed it was a de novo alteration in the father (Individual 4). Individual 2 underwent trio WES, too, and a separate de novo missense variant in MYBPC1 (c.766C>T, p.Leu259Pro) was found at a highly conserved site (Fig. 1C) that was also predicted to be damaging (Polyphen2 = 0.999, CADD = 32, GERP++ = 5.53). Neither the p.Leu263Arg nor the p.Leu259Pro variant was present in gnomAD (https://gnomad.broadinstitute.org/) (Lek et al., 2015), wherein there is substantial coverage of both MYBPC1 residues in ~122,000 individuals. Similarly, both variants were absent in 13,039 internal control individuals at the research laboratory database at Columbia University where Individual 1 was sequenced and in 1,016 Korean control individuals. Both variants were confirmed by Sanger sequencing in all four individuals (Fig. 1B), and both have been submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). Notably, prior WES and whole genome sequencing for Individual 2 had been reported as negative at two separate commercial laboratories, and the c.766C>T, p.Leu259Pro MYBPC1 variant was eventually identified on the reanalysis of the WES and genome data performed by UDN. Upon further communication, we determined that the commercial laboratories did not report the variant because they concluded that the phenotypic fit was poor, specifically due to the lack of arthrogryposis in Individual 2.
p.Leu263Arg Results in Decreased Myosin Binding of sMyBP-C
Both the p.Leu259Pro and p.Leu263Arg variants are located within the NH2-terminal M-motif of sMyBP-C, which is included in all known isoforms contributing to myosin S2 and actin binding (Fig. 1A). We therefore examined if the presence of either variant affects binding to myosin or actin. GST-tagged recombinant proteins containing the NH2-terminal Pro/Ala-rich motif, Ig C1, and the M-motif (P/A-C1-M) were generated in the absence (wild-type) or presence of the p.Leu259Pro and p.Leu263Arg mutations (Fig. 2A). These were subsequently used in real-time kinetic analysis using a Biacore 3000 surface plasmon resonance (SPR) biosensor to monitor their binding affinity, KD, to heavy meromyosin (HMM; Fig. 2B–C). Evaluation of the obtained sensograms using BIA evaluation 3.1 software followed by fitting with the 1:1 Langmuir model yielded a KD of 16.5 nM for wild-type GST-P/A-C1-M, ~9 nM for GST-P/A-C1-M carrying the Leu259Pro mutation, and ~91 nM for GST-P/A-C1-M containing the Leu263Arg mutation (Fig. 2B–C). Thus, binding of GST-P/A-C1-M Leu259Pro to HMM was comparable to wild-type, whereas binding of GST-P/A-C1-M Leu263Arg was considerably decreased (~5.5-fold). We also attempted to perform SPR experiments to determine the binding affinity of wild-type and mutant proteins to actin; however, we were unable to obtain reliable sensograms, likely due to the sticky nature of actin and aggregation during experimentation. We therefore performed overlay binding assays in which actin was immobilized on nitrocellulose membrane and overlaid with equivalent amounts of wild-type and mutant GST-P/A-C1-M proteins (Fig. 2D). Neither mutation significantly affected binding to actin compared to wild-type protein (Fig. 2D).
Figure 2:
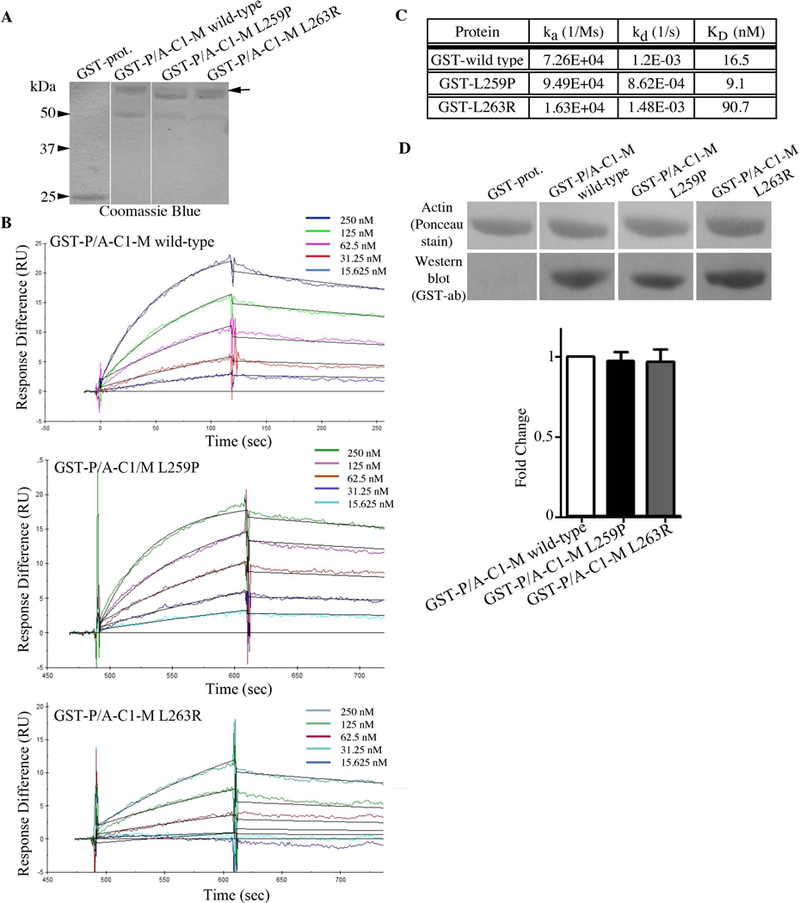
(A) Coomassie blue stained gel showing equivalent amounts (1.5 μg) of control GST, wild-type GST-P/A-C1-M, mutant GST-P/A-C1-M L259P, and mutant GST-P/A-C1-M L263R proteins used in the overlay and Biacore assays. Full length GST-P/A-C1-M recombinant proteins (~58 kDa) are denoted with an arrow; the preparations of both mutant proteins contain a very closely migrating degradation product (56–57 kDa), while a degradation product of ~50 kDa is present in all three protein preparations; non-continuous lanes are separated by a white line. (B) Real time kinetics evaluation of the interaction between wild-type or mutant GST-P/A-C1-M proteins and HMM using a Biacore 3000 SPR biosensor. (C) Kinetic measurements at equilibrium indicated that GST-P/A-C1-M L263R binding to HMM was decreased by ~5.5-fold compared to wild-type levels. (D) Equivalent amounts (3 μg) of purified actin were separated by SDS-PAGE, transferred to nitrocellulose membrane, and overlaid with 0.5 μg/mL of control GST-protein, wild-type GST-P/A-C1-M, or mutant GST-P/A-C1-M carrying the L259P or L263R mutation. Nitrocellulose membranes (Ponceau stain) and films (Western blots probed with GST-ab) were cropped to only include the area of expected signal. Densitometric quantification of at least three independent overlay/immunoblotting experiments indicated that neither mutation significantly affected binding to actin. Control GST-proteins did not bind actin.
Impact of the p.Leu259Pro and p.Leu263Arg variants on the stability and structure of the M-motif
We next examined how the presence of either variant impacts the M-motif at the molecular level. The M-motif, where both variants are located, is largely disordered except for a ~40 residue region (residues 230–270) that folds into a 3-helix bundle (Howarth et al., 2012). Circular Dichroism (CD) spectroscopy revealed that mutant M-motif containing either variant remains significantly helical, as expected. However, the presence of p.Leu259Pro reduces helicity by ~10% compared to wild-type (Fig. 3A). Molecular dynamics (MD) analysis of this variant protein suggested that this is due to the third helix, containing the L259 residue, becoming frayed upon the substitution to proline. Specifically, the introduction of a sterically constrained residue at the beginning of the helix ablates several helix-defining hydrogen bonds (Fig. 3B). This alteration, however, is local since the helix containing this variant remains amphipathic, retaining its ability to pack well into the hydrophobic interior of the 3-helix bundle (Fig. 3C). Consistent with this interpretation, the recombinant M-motif containing the Leu259Pro mutation is more susceptible to cleavage following trypsin digestion compared to wild-type protein (Fig. 3D).
Figure 3:
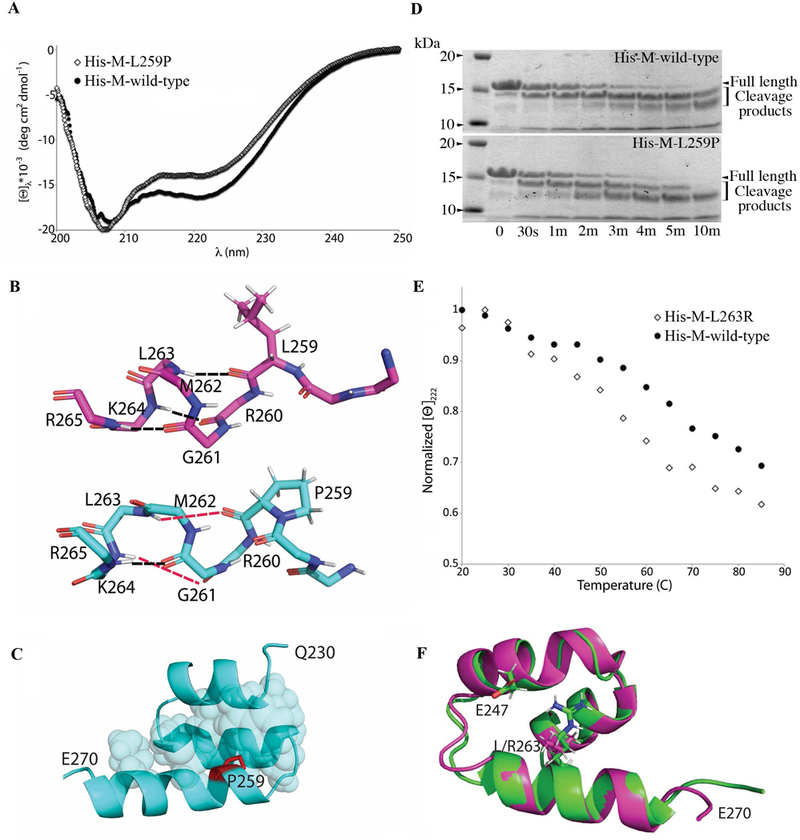
Circular dichroism (CD) experiments and computation modeling analysis of the L259P and L263R mutations. (A) Recombinant wild-type and mutant His-tagged proteins containing the M-motif that carries the L259P and L263R mutations were used in CD experiments. CD spectra at 30 °C showed that mutant M-motif containing the L259P mutation (hollow diamonds) is ~10% less helical compared to wild-type (solid circles). (B) Representative snapshot following molecular dynamics (MD) indicated that wild-type M-motif protein (top; magenta) has normal hydrogen bonds (black dashed lines) at the beginning of the M-domain helix 3 (L259-L263; R260-K264; G261-R265). On the contrary, MD of mutant M-motif protein containing the L259P variant (bottom; cyan) showed that these hydrogen bonds are no longer well-formed (red dashed lines). (C) P259 (purple) still packs well into the hydrophobic interior (cyan spheres) of the M-motif. (D) Representative SDS-PAGE gel of trypsin digestion assays for recombinant wild-type and mutant L259P M-motif proteins stained with Coomassie Brilliant Blue, showing increased cleavage products in the presence of the L259P mutation. (E) Normalized molar ellipticity at 222 nm vs. temperature showed that mutant M-motif carrying the L263R variant (hollow diamonds) is less stable than wild-type (solid circles). (F) Representative model of wild-type (purple) and mutant M-motif containing the L263R mutation (green) after a 100 ns MD equilibration. Mutant M-motif does not pack as well into the hydrophobic interior as wild-type, but does form a previously absent favorable interaction between the R263 and E248 sidechains. This decrease in stability and alterations in intramolecular hydrogen bonds may be the cause of decreased binding to myosin seen in the in vitro binding assay.
Examination of the pLeu263Arg variant via CD showed similar spectra to those of wild-type protein at room temperature ([Θ]222 of −16,500 for wild-type, and −16,200 for p.Leu263Arg at 30 °C). However, the wild-type M-motif unfolds with a midpoint around 650 while the p.Leu263Arg variant unfolds with a midpoint at 550 (Fig. 3E). This decreased stability is likely due to the insertion of a basic residue into the hydrophobic core of the M-motif. MD simulations suggested that the Arg moiety does not fully pack in the hydrophobic core as the original Leu moiety does, but instead protrudes to make an electrostatic interaction with E248 (Fig. 3F). Thus, this variant preserves the overall fold of the M-motif 3-helix bundle through the formation of favorable interactions, but it is clearly less stable than wild-type. The p.Leu263Arg variant’s decreased ability to bind myosin could therefore either be due to a slight loss of stability or a direct disruption of HMM driven by the inclusion of an additional charged residue on the M-motif surface.
DISCUSSION
The clinical, molecular and functional data in this study provide evidence that deleterious variants in MYBPC1 are associated with a previously unrecognized phenotype of a congenital myopathy with prominent tremors that are likely of myogenic origin, and with a natural history of gradual improvement in both muscle strength and tremors. Previously, MYBPC1 had been linked to severe and lethal subtypes of distal arthrogryposis (DA), in which congenital contractures of the distal extremities are the hallmark feature. Inclusive of MYBPC1, many of the underlying genes associated with the ten subtypes of DA are involved in sarcomeric structure and function (Hall, 2014). The ensuing abnormalities of muscle formation, structure and/or function due to damaging variants in these genes lead to secondarily decreased fetal movement, which in turn leads to multiple joint contractures at birth (Bamshad et al., 2009; Hall, 2014).
The deleterious variants in MYBPC1 that have been reported in association with DA syndromes include four with autosomal dominant inheritance (DA 1B and DA 2) and two with autosomal recessive inheritance [LCCS4 and a less severe form of distal arthrogryposis] (Ekhilevitch et al., 2016; Gurnett et al., 2010; Li et al., 2015; Markus et al., 2012). Of the six DA variants, two are located in the M-motif similarly to the two novel variants we report suggesting that disruption of the M-motif could lead to abnormalities in actomyosin cross-bridges and therefore muscle contractility. Our sMyBP-C protein modeling demonstrated that the p.Leu263Arg variant slightly destabilizes the M-motif fold. Additionally, the presence of a positively charged Arg moiety on the surface of the M-motif could alter direct myosin binding. This supposition is supported by our kinetic studies, which show that the p.Leu263Arg variant, which interestingly corresponds to the p.Leu295Arg variant reported in the affected calf (Figure 1) (Wiedemar et al., 2015), exhibits significantly reduced binding affinity to HMM compared to wild-type (~5.5 fold). On the other hand, the p.Leu259Pro variant affects neither target protein binding nor M-motif stability. However, it does affect the structure of the M-motif, likely due to the ablation of several helix-defining hydrogen bonds in the beginning of the 3-helix bundle. The most obvious functional outcome of this decreased helicity is some alteration of target binding that we cannot detect in vitro using protein fragments. One possible reason for this is that the M-motif may work in concert with neighboring regions in sMyBP-C to bind actin or myosin, and this mutation alters some intra-protein interactions. Alternatively, it is possible that the Leu to Pro substitution is chemically subtle enough to not significantly affect target protein binding, but instead decreases the half-life of the protein in vivo. Consistent with this, trypsin digestion indicated that recombinant M-motif containing the Leu259Pro mutation is more susceptible to cleavage than wild-type protein. The region between Arg260 and Lys268 is a top candidate for this trypsin cleavage, given 1) the abundance of Arg and Lys residues around this site, 2) the strong likelihood that this region is less well packed in the Leu259Pro variant and 3) the digested fragments are consistent in size with a cut site around residue 264. It therefore appears that the two variants act through slightly different structural or functional molecular mechanisms, yet both result is similar abnormalities in muscle function.
The clinical manifestations in our individuals are those of a congenital myopathy with lack of contractures and a clinical course of gradual improvements over time. One potential reason for the early presentation of myopathy and subsequent improved tone and motor strength may be related to the fact that sMyBP-C is the earliest of the three MyBP-C isoforms to be expressed in the embryonic period (Gautel et al., 1998; Kurasawa et al., 1999), which could lead to abnormal skeletal muscle development. The fast skeletal isoform, fMyBP-C, encoded by MYBPC2, is expressed in postnatal life (Gautel et al., 1998; Kurasawa et al., 1999), providing a possible explanation for why further progression of muscle weakness after birth is not observed in these patients. Consistent with this, with down-regulation of sMyBP-C in adult lumbricalis skeletal muscle in vivo there is ~20% upregulation of fMyBP-C (Geist et al., 2018), suggesting that it may compensate for mutant sMyBP-C in late development and adulthood therefore partially alleviating muscle weakness. Obviously, examination of the levels and distribution of fMyBP-C in patient biopsies would be highly informative in that regard.
The tremors in our individuals are likely myogenic in origin, being evident at rest and with posture, intention and fatigue. Moreover, we did not see corroborating clinical signs of cerebellar dysfunction or other signs of central nervous system involvement in these individuals (aside from epilepsy of unknown etiology in Individual 3, which could be unrelated), nor were there any structural abnormalities of the cerebellum on brain MRI or abnormalities on nerve conduction to suggest a neurogenic origin for the tremors. All affected individuals continue to exhibit mild signs of a myopathy with Individual 4 showing myopathic changes in the EMG, lending further credence to our proposition that the tremors are myogenic. These findings along with the lack of MYBPC1 expression in the brain (Atlas, version 17) also supports the myogenic origin of the tremors. The lack of contractures in our patients is intriguing; it is possible that the timing and nature of the reduced fetal movements may lead to contractures in some individuals with deleterious MYBPC1 variants and not others (Hall, 2014). The mild myopathy in our patients is also noteworthy. Given the series of pathogenic variants identified from patients with varying degrees of clinical severities, one can postulate that the functional dose of MYBPC1 can be directly associated with muscle pathology. Consistent with this, prior reports on MYBPC1-associated autosomal dominant DA syndromes specifically comment on the lack of muscle weakness in the affected patients who though presented with (relatively mild) congenital contractures (Gurnett et al., 2010; Li et al., 2015). On the contrary, more deleterious loss of function variants in MYBPC1in a homozygous state likely result in no protein expression, causing lethality or severe muscular weakness with prominent contractures possibly due to in utero fetal akinesia (Ekhilevitch et al., 2016; Markus et al., 2012).
A notable observation in Individual 2 was that the two clinical laboratories that performed the trio WES and WGS did not report the p.Leu259Pro variant, not even as a variant of unknown significance. In our conversations with the laboratories, both divulged that the variant had been called by their variant calling software, but had not been prioritized since the phenotypic fit was deemed poor, due to the lack of arthrogryposis in the phenotype provided by the ordering clinician. This was despite the bioinformatics analysis demonstrating that the p.Leu259Pro variant was de novo, rare, and predicted to be damaging in a known disease-associated gene. Therefore, the possibility that there was an expanded phenotype associated with MYBPC1 that did not involve arthrogryposis went unrecognized (Shashi et al., 2018). While such expanded phenotypes are becoming more obvious with exome and genome sequencing, this also illustrates a potential limitation, with laboratories not prioritizing a bioinformatically compelling variant when it does not match the traditional phenotypic features associated with that gene, leading to missed diagnoses.
In conclusion, we provide clinical and biochemical evidence for an expanded spectrum of muscle weakness, tremors and hypotonia, without arthrogryposis, associated with damaging heterozygous variants in the M-motif of MYBPC1. Similar to the known autosomal dominant DA syndromes associated with MYBPC1, there is no progression of the myopathy in these individuals and cognition is normal. Our report would enable recognition of individuals with a primary myopathic presentation characterized by a myogenic tremor to be associated with damaging heterozygous variants in MYBPC1.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the patients and their families for their participation.
Funding Sources: This work was supported by NIH (U01HG007672 to VS, Training Program in Muscle Biology, T32 AR007592–17 to J.G. and R21AR072981 to A.K.K.), the Muscular Dystrophy Association (Research Grant 313579 to A.K.K.), Ministry of Health and Welfare (HI16C1986 to M.C), and NSF (REU CHE-1062629 and RUI MCB-1607024) awards to N.T. W.).
Footnotes
CONFLICT OF INTEREST STATEMENT
None of the authors has a conflict of interest.
REFERENCES
- Ackermann MA, & Kontrogianni-Konstantopoulos A (2010). Myosin binding protein-C slow: an intricate subfamily of proteins. J Biomed Biotechnol, 2010, 652065. doi: 10.1155/2010/652065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackermann MA, & Kontrogianni-Konstantopoulos A (2011). Myosin binding protein-C slow is a novel substrate for protein kinase A (PKA) and C (PKC) in skeletal muscle. J Proteome Res, 10(10), 4547–4555. doi: 10.1021/pr200355w [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackermann MA, & Kontrogianni-Konstantopoulos A (2013). Myosin binding protein-C slow: a multifaceted family of proteins with a complex expression profile in fast and slow twitch skeletal muscles. Front Physiol, 4, 391. doi: 10.3389/fphys.2013.00391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, … Sunyaev SR (2010). A method and server for predicting damaging missense mutations. Nat Methods, 7(4), 248–249. doi: 10.1038/nmeth0410-248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlas, H. P. (version 17). https://www.proteinatlas.org/.
- Bamshad M, Van Heest AE, & Pleasure D (2009). Arthrogryposis: a review and update. Journal of Bone and Joint Surgery, 91 Suppl 4, 40–46. doi: 10.2106/jbjs.I.00281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bick D, Fraser PC, Gutzeit MF, Harris JM, Hambuch TM, Helbling DC, … Dimmock DP (2017). Successful Application of Whole Genome Sequencing in a Medical Genetics Clinic. J Pediatr Genet, 6(2), 61–76. doi: 10.1055/s-0036-1593968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig R, & Offer G (1976). The location of C-protein in rabbit skeletal muscle. Proc R Soc Lond B Biol Sci, 192(1109), 451–461. Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/4802 [DOI] [PubMed] [Google Scholar]
- Davydov EV, Goode DL, Sirota M, Cooper GM, Sidow A, & Batzoglou S (2010). Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP++. PLOS Computational Biology, 6(12), e1001025. doi: 10.1371/journal.pcbi.1001025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekhilevitch N, Kurolap A, Oz-Levi D, Mory A, Hershkovitz T, Ast G, … Baris HN (2016). Expanding the MYBPC1 phenotypic spectrum: a novel homozygous mutation causes arthrogryposis multiplex congenita. Clinical Genetics, 90(1), 84–89. doi: 10.1111/cge.12707 [DOI] [PubMed] [Google Scholar]
- Flashman E, Redwood C, Moolman-Smook J, & Watkins H (2004). Cardiac myosin binding protein C: its role in physiology and disease. Circulation Research, 94(10), 1279–1289. doi: 10.1161/01.Res.0000127175.21818.C2 [DOI] [PubMed] [Google Scholar]
- Gautel M, Furst DO, Cocco A, & Schiaffino S (1998). Isoform transitions of the myosin binding protein C family in developing human and mouse muscles: lack of isoform transcomplementation in cardiac muscle. Circulation Research, 82(1), 124–129. [DOI] [PubMed] [Google Scholar]
- Geist J, & Kontrogianni-Konstantopoulos A (2016). MYBPC1, an Emerging Myopathic Gene: What We Know and What We Need to Learn. Front Physiol, 7, 410. doi: 10.3389/fphys.2016.00410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geist J, Ward CW, & Kontrogianni-Konstantopoulos A (2018). Structure before function: myosin binding protein-C slow is a structural protein with regulatory properties. FASEB Journal, fj201800624R. doi: 10.1096/fj.201800624R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurnett CA, Desruisseau DM, McCall K, Choi R, Meyer ZI, Talerico M, … Dobbs MB (2010). Myosin binding protein C1: a novel gene for autosomal dominant distal arthrogryposis type 1. Human Molecular Genetics, 19(7), 1165–1173. doi: 10.1093/hmg/ddp587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha K, Buchan JG, Alvarado DM, McCall K, Vydyanath A, Luther PK, … Gurnett CA (2013). MYBPC1 mutations impair skeletal muscle function in zebrafish models of arthrogryposis. Human Molecular Genetics, 22(24), 4967–4977. doi: 10.1093/hmg/ddt344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall JG (2014). Arthrogryposis (multiple congenital contractures): diagnostic approach to etiology, classification, genetics, and general principles. Eur J Med Genet, 57(8), 464–472. doi: 10.1016/j.ejmg.2014.03.008 [DOI] [PubMed] [Google Scholar]
- Howarth JW, Ramisetti S, Nolan K, Sadayappan S, & Rosevear PR (2012). Structural insight into unique cardiac myosin-binding protein-C motif: a partially folded domain. Journal of Biological Chemistry, 287(11), 8254–8262. doi: 10.1074/jbc.M111.309591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kircher M, Witten DM, Jain P, O’Roak BJ, Cooper GM, & Shendure J (2014). A general framework for estimating the relative pathogenicity of human genetic variants. Nature Genetics, 46(3), 310–315. doi: 10.1038/ng.2892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurasawa M, Sato N, Matsuda A, Koshida S, Totsuka T, & Obinata T (1999). Differential expression of C-protein isoforms in developing and degenerating mouse striated muscles. Muscle and Nerve, 22(2), 196–207. [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski K, Minikel E, Samocha K, Banks E, Fennell T, … MacArthur D (2015). Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv. doi: 10.1101/030338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Zhong B, Han W, Zhao N, Liu W, Sui Y, … Jiang M (2015). Two novel mutations in myosin binding protein C slow causing distal arthrogryposis type 2 in two large Han Chinese families may suggest important functional role of immunoglobulin domain C2. PLoS One, 10(2), e0117158. doi: 10.1371/journal.pone.0117158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markus B, Narkis G, Landau D, Birk RZ, Cohen I, & Birk OS (2012). Autosomal recessive lethal congenital contractural syndrome type 4 (LCCS4) caused by a mutation in MYBPC1. Human Mutation, 33(10), 1435–1438. doi: 10.1002/humu.22122 [DOI] [PubMed] [Google Scholar]
- Moss RL, Fitzsimons DP, & Ralphe JC (2015). Cardiac MyBP-C regulates the rate and force of contraction in mammalian myocardium. Circulation Research, 116(1), 183–192. doi: 10.1161/circresaha.116.300561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley CE, Chamoun J, Brown LJ, & Hambly BD (2007). Myosin binding protein-C: enigmatic regulator of cardiac contraction. International Journal of Biochemistry and Cell Biology, 39(12), 2161–2166. doi: 10.1016/j.biocel.2006.12.008 [DOI] [PubMed] [Google Scholar]
- Retterer K, Juusola J, Cho MT, Vitazka P, Millan F, Gibellini F, … Bale S (2016). Clinical application of whole-exome sequencing across clinical indications. Genet Med, 18(7), 696–704. doi: 10.1038/gim.2015.148 [DOI] [PubMed] [Google Scholar]
- Shashi V, Schoch K, Spillmann R, Cope H, Tan QK, Walley N, … Goldstein DB (2018). A comprehensive iterative approach is highly effective in diagnosing individuals who are exome negative. Genet Med. doi: 10.1038/s41436-018-0044-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DJ, Brandsma M, Yin Z, Wang A, Jevnikar AM, & Ma S (2008). A novel platform for biologically active recombinant human interleukin-13 production. Plant Biotechnology Journal, 6(5), 504–515. doi:doi: 10.1111/j.1467-7652.2008.00337.x [DOI] [PubMed] [Google Scholar]
- Wang L, Geist J, Grogan A, Hu LR, & Kontrogianni-Konstantopoulos A (2018). Thick Filament Protein Network, Functions, and Disease Association. Compr Physiol, 8(2), 631–709. doi: 10.1002/cphy.c170023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber FE, Vaughan KT, Reinach FC, & Fischman DA (1993). Complete sequence of human fast-type and slow-type muscle myosin-binding-protein C (MyBP-C). Differential expression, conserved domain structure and chromosome assignment. European Journal of Biochemistry, 216(2), 661–669. [DOI] [PubMed] [Google Scholar]
- Wiedemar N, Riedi AK, Jagannathan V, Drogemuller C, & Meylan M (2015). Genetic Abnormalities in a Calf with Congenital Increased Muscular Tonus. Journal of Veterinary Internal Medicine, 29(5), 1418–1421. doi: 10.1111/jvim.13599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaidi S, Choi M, Wakimoto H, Ma L, Jiang J, Overton JD, … Lifton RP (2013). De novo mutations in histone-modifying genes in congenital heart disease. Nature, 498(7453), 220–223. doi: 10.1038/nature12141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X SP, Xie Pingxing, Ruzzo Elizabeth K., Lu Yi-Fan, Bruria Ben-Zeev Andreea Nissenkorn,Anikster Yair, Danit Oz-Levi Ryan S. Dhindsa, Hitomi Yuki, Schoch Kelly, Spillmann Rebecca C., Heimer Gali, Marek-Yagel Dina,Tzadok Michal, Han Yujun, Worley Gordon, Goldstein Jennifer, Jiang Yong-Hui, Lancet Doron, Pras Elon, Shashi Vandana, Need Anna C., and Goldstein David B.. (2014). Whole exome sequencing in undiagnosed genetic diseases: Interpreting 119 trios. Genetics in Medicine (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.