

Summary
Many microRNAs (miRNAs) exist alongside abundant miRNA isoforms (isomiRs), most of which arise from post-maturation sequence modifications, such as 3’ uridylation. However, the ways in which these sequence modifications affect miRNA function remain poorly understood. Here, using human miR-27a in cell lines as a model, we have discovered that a nonfunctional target site unable to base pair extensively with the miRNA seed sequence can regain function when an upstream adenosine is able to base-pair with a post-transcriptionally added uridine in the miR-27a tail. This Tail-U-Mediated Repression (TUMR) is abolished in cells lacking the uridylation enzymes TUT4 and TUT7, indicating that uridylation alters miRNA function by modulating target recognition. We identified a set of non-canonical targets in human cells that are specifically regulated by uridylated miR-27a. We provide evidence that TUMR expands the targets of other endogenous miRNAs. Our study reveals a function for uridylated isomiRs in regulating non-canonical miRNA targets.
Keywords: miRNA, isomiRs, uridylation, noncanonical target, miR-27a
Graphical abstract
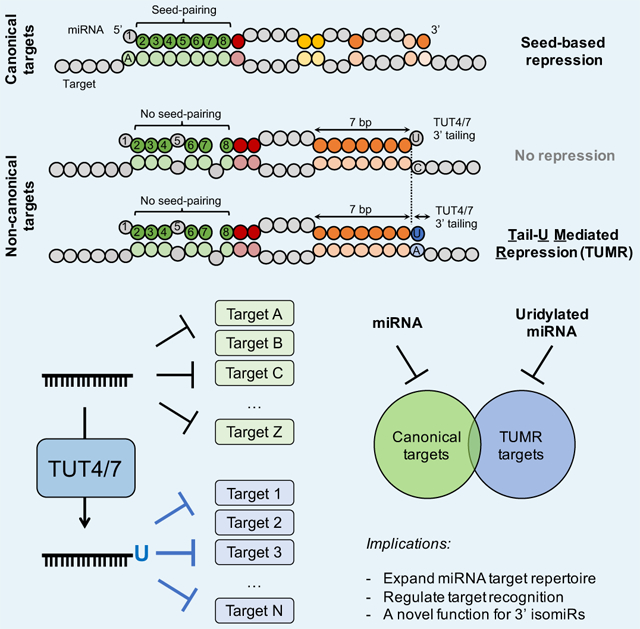
Blurb
Yang et al. demonstrate that mRNAs lacking a seed-pairing are repressed by miR-27a due to base-pairing between an upstream adenosine in the target and a non-templated U tail in miR-27a. They identify a large class of non-canonical targets regulated by uridylated miRNAs and reveal a novel function of 3' isomiRs.
Introduction
MicroRNAs (miRNAs) are a class of small non-coding RNAs with a length of ~22 nucleotides (nt). They function as key regulators of gene expression in multiple eukaryotic organisms (Bartel, 2018; Pasquinelli, 2012). More than 60% of human mRNAs contain at least one evolutionary conserved miRNA target site, and other non-conserved sites are known to be functional, indicating that most biological processes and pathways are likely under miRNA regulation (Friedman et al., 2009). MiRNA dysfunction is often associated with human disease (Lin and Gregory, 2015; Lu et al., 2008; Rupaimoole and Slack, 2017), highlighting the importance of understanding miRNA regulation in cells.
In order to function, miRNAs must both undergo a complex series of processing steps and be able to form specific base pairs with their mRNA targets. MiRNAs are typically transcribed by RNA polymerase II as primary transcripts (pri-miRNA), which are cleaved by the RNase III enzyme Drosha in nuclei, releasing a ~70 nt hairpin-like structure termed pre-miRNA. Pre-miRNA is translocated via Exportin-5 into the cytoplasm, where it is processed further by a second RNase III enzyme, Dicer, to generate a miRNA duplex (Kim, 2005; Treiber et al., 2019). Generally, one strand of the duplex (miRNA guide strand) is preferentially selected for incorporation into the Argonaute (AGO) protein, forming the RNA-induced silencing complex (RISC) (Gregory et al., 2005), while the other strand (miRNA passenger or star strand) is released and degraded (Khvorova et al., 2003; Schwarz et al., 2003). Through base-pairing, most miRNAs guide the RISC to the 3’UTR of target mRNAs, down-regulating their levels by translational repression and/or mRNA degradation (Gu and Kay, 2010; Iwakawa and Tomari, 2015; Jonas and Izaurralde, 2015). The run of 7 nucleotides from position 2 to 8 of the 5’ end of a miRNA, called the “seed”, is crucial for determining target specificity (Lewis et al., 2003). Pairing to the seed region is required, and sufficient in many cases, for an mRNA to be repressed by the complementary miRNA (Lewis et al., 2005). mRNA regulatory sites containing a perfect seed-match are considered “canonical” and those that do not are categorized as non-canonical. In non-canonical sites, the lack of perfect seed-match is often compensated by extensive pairing to other regions of a miRNA (Hausser and Zavolan, 2014; Kim et al., 2016; Shin et al., 2010).
Considerable effort has been focused on understanding how alterations in miRNA expression levels drive development and contribute to human disease. However, the functions of miRNAs can also be affected by modifications to their sequence and chemical structure. Next generation sequencing has identified a vast number of miRNA isoforms (isomiRs), most of which arise from post-transcriptional sequence modifications (Neilsen et al., 2012). Two major post-transcriptional modifications are trimming and tailing, where exoribonucleases and terminal nucleotidyltransferases remove and add non-templated nucleotides at miRNA 3’ ends, respectively. One prevalent type of tailing is uridylation, which is catalyzed by a group of terminal uridylyl transferases (TUTases) (Martin and Keller, 2007). Uridylation of pre-miRNAs modifies miRNA biogenesis (Heo et al., 2009, 2012; Thornton et al., 2012), however it is unclear how uridylation of mature miRNAs affects their function. In plants, uridylation of mature miRNA by HESO1 triggers miRNA decay (Zhao et al., 2012), whereas the connection between uridylation and miRNA turnover is not well-defined in animals. Because modifications at the 3’ end of a miRNA do not change its seed sequence, the conventional wisdom is that 3’ uridylation should not influence miRNA target recognition.
Here, we use human miR-27a as a model to investigate the functional impact of uridylation on mature miRNA. We show that a target site which is normally nonfunctional because of the absence of a seed pairing match becomes functional when an upstream adenosine in the target mRNA base-pairs to the non-templated “U”-tail of miR-27a. We identify a large class of non-canonical target sites that are regulated by uridylated miRNAs. This study reveals both a mechanism by which miRNA can recognize non-canonical targets and identifies another way in which 3’ uridylation affects RNA metabolism. We discuss the possibility that uridylation could be a general regulatory mechanism for altering miRNA function.
Results
3’ uridylated isomiRs are associated with RISC
Deep sequencing of small RNAs in HEK293T cells revealed isomiR profiles vary among different miRNAs, consistent with previous studies (Burroughs et al., 2010; Dueck et al., 2012; Westholm et al., 2012; Wyman et al., 2011; Zhou et al., 2012). For example, isomiRs are a minor portion (< 35%) of miR-148a-3p, miR-25-3p and let-7a-5p, and the dominant form (>75%) of miR-27a, miR-101-3p and miR-218-5p (Figure S1A). The sequence of these isomiRs are highly heterogeneous, with a large part of isomiRs differing through the addition of 3' non-templated nucleotide(s) (Figure S1B). miR-27a has a high percentage of tailed isomiRs, and its 3’ sequence modifications are known to be regulated: Herpesvirus saimiri (HVS) ncRNAs HSURs promote host miR-27a decay via 3’ tailing and trimming (Cazalla et al., 2010). We therefore decided to use miR-27a as a model to study the function of isomiRs.
To facilitate the detection of miR-27a and its isomiRs, we cloned the genomic sequence of miR-27a and its flanking region into a CMV (Pol II) driven expression vector and transfected it into HEK293T cells. Northern blot analysis confirmed the production of abundant miR-27a isomiRs of variable length (Figure 1A). Endogenous miR-27a was barely detectable compared to the ectopically expressed miR-27a, suggesting that the endogenous miR-27a could be neglected in subsequent analyses. Deep sequencing showed that the over-expressed miR-27a has an isomiR profile very similar to that of the endogenous miR-27a: nearly 60% of the miR-27a reads are tailed with non-templated nucleotide(s) at the 3’ end (Figure 1B); more than half of these added nucleotides are uridines (Figure 1C). This indicated that ectopically expressed miR-27a is under regulation of the cellular system that modifies the endogenous miRNAs and therefore can be used as a suitable tool to interrogate the function of uridylated isomiRs.
Figure 1. 3’ uridylated isomiRs are associated with RISC.

(A) After expressing a CMV-driven pri-miR-27a cassette in HEK293T cells, pre-miR-27a and mature miR-27a-3p were detected by Northern blotting with probe-miR-27a (Supplementary Table S1). (B) Small RNAs from HEK293T cells and HEK293T cells transfected with pri-miR-27a-expressing plasmids were extracted and subjected to deep sequencing. The average percentages of canonical miR-27a, its tailed isomiRs and trimmed isomiRs relative to the total miR-27a reads were plotted. Error bar indicates the standard deviation (n=3). (C) Nucleotide compositions of non-templated tail of miR-27a isomiRs in either HEK293T or HEK293T cells transfected with pri-miR-27a-expressing plasmids. (D) Small RNAs extracted from anti-AGO2 and anti-TNRC6C (GW182) immunoprecipitates were subjected to Northern blotting to detect miR-27a-3p. See also Figure S1.
MiRNA association with RISC is a prerequisite for target silencing. To determine whether the isomiRs are associated with RISC, we co-expressed Flag-AGO2 and miR-27a by transfecting the expression constructs into HEK293T cells. After immunoprecipitation, AGO2-associated small RNAs were subject to Northern blot. Both miR-27a and its isomiRs were recovered in the pull-down RNAs, indicating they are associated with RISC (Figure 1D). To confirm this, the same experiment was performed with the RISC component TNRC6C, a human paralog of GW182, yielding a similar result (Figure 1D). We deep sequenced the AGO2-IP samples, further validating the association of miR-27a isomiRs with AGO2 (Figure S1C). Together, those results indicate that these isomiRs are not merely degradation intermediates but are part of the functional RISC complex.
An upstream adenosine requirement for miR-27a repression
To explore the function of miR-27a isomiRs, we used a dual-luciferase reporter system in which tandem repeats of a previously validated target sequence from the PPARγ gene (Kim et al., 2010) (wild-type target) were inserted in the 3’ UTR of the Renilla (RL)-luciferase reporter gene (Figure 2A). Co-expressing miR-27a, but not a control miRNA, in HEK293T cells significantly reduced reporter activity (Figure 2A). Disrupting seed-pairing by mutating the target site sequence (seed-mutant target) abolished repression, while introduction of additional base-pairs at the 3’ region (bulged target) slightly enhanced repression (Figure 2A). Disrupting the seed-match and extending the base-pairing of the target to the miR-27a 3’ region (3’ paired target), resulted in significant repression by miR-27a (2.3 fold, p<0.0009) (Figure 2A). This result is contrary to previous studies that indicated 3' pairing by itself is not sufficient to induce miRNA-mediated repression.
Figure 2. An upstream adenosine requirement for miR-27a repression.

(A) Renilla luciferase reporters containing four tandem targets of various sequences in the 3’UTR were transfected into HEK293T cells together with either a control miRNA or miR-27a. Left, schematic representation of the potential pairing between miR-27a (top) and various target sequences (bottom). The miR-27a seed is in red. Right, dual-luciferase assays performed 48h post-transfection. Renilla luciferase activities were normalized with firefly luciferase, and the percentage of relative enzyme activity compared to the negative control (treated with the control non-target miRNA) was plotted. Error bars represent the standard deviation from four biological replicates. (B) miR-27a-mediated repression on additional reporters were measured with the dual luciferase assay. Left, schematic representation of the pairing between miR-27a (top) and target sequences (bottom). The non-templated U-tail is labeled with red circle. Yellow box indicates the adenosines upstream of the target sequence. Right, dual-luciferase assay results were plotted as described above. (C) Various renilla luciferase reporters were co-transfected with antagomirs (20 nM final concentration) of either anti-miR-27a or a non-targeting control sequence into HeLa cells. Dual-luciferase assays were performed 48h post-transfection.Results from seven biological replicates were plotted as described above. * p<0.001, ** p < 0.005, n.s. non-significant. See also Figure S2.
Examination of the 3’ paired target revealed two adenosines (“AA”) immediately upstream of the reporter target site. The 3’ paired target site was renamed "no-seed-match 7AA target site" or "7AA target" for short to indicate that the target site sequence contains (1) no perfect seed-match, (2) 7 base-pairs between the target and the 3’ region of miR-27a and (3) an upstream “AA” motif. Given that the upstream adenosines could potentially base pair with the uridines in the miR-27a tail, we wondered if this additional base pairing compensated for the lack of a perfect seed-match (Figure 2B). We mutated the “AA” motif to a “CC” (Figure 2B) and observed that the miR-27a-mediated inhibition was abolished (Figure 2B), indicating that the “AA” motif is critical for repression.
We asked whether loss of the inferred pairing between the U-tail and the “AA” motif could be compensated for by additional pairing in the miR-27a 3’ region. To this end, we generated a set of reporter constructs containing regulatory sites capable of forming 8 or 9 base pairs with the 3’ region of the miRNA. To these targets we added either two AA bases upstream of the end of the miRNA interacting region, which could potentially base-pairing with miRNA 3’ U-tail, or two CC bases, which could not. (Figures 2B and S2A). Supporting the critical role of the “AA” motif for this type of non-canonical target site, 8AA and 9AA targets but not 8CC and 9CC targets were inhibited by miR-27a (Figure 2B). Of note, the number of base-pairs between uridylated miR-27a and 7AA targets is the same as that between uridylated miR27a and 9CC targets (Figure S2A). This result suggests that the potential base-pairing between U-tail and “AA” motif plays a unique role in target recognition or miRNA function, which cannot be replaced by simply increasing base-pairing at the 3’ region of miRNA.
To rule out the possibility that repression of the 7AA target is an artifact due to miR-27a overexpression, we transfected the RL-luciferase reporters into HeLa cells, where miR-27a is endogenously expressed at high levels. A reporter containing wild-type target sites was de-repressed when endogenous miR-27a was blocked by co-transfecting a miR-27a-specific antagomir (Figures 2C and S2B). A reporter bearing 7AA target sites, but not the reporter with 7CC or 9CC target sites, was also de-repressed upon miR-27a depletion, supporting the idea that the "no-seed-match" 7AA target is repressed by endogenous miR-27a (Figure 2C). Thus, the upstream “AA” motif is required for the 3’ paired “no-seed-match” targets to be repressed by miR-27a.
Uridylated miR-27a isomiRs repress non-canonical targets
To test directly whether uridylated isomiRs are responsible for recognizing the “no-seed-match” non-canonical targets, we developed a pull-down assay to isolate functional isomiRs physically associated with the target mRNA. In brief, a set of biotinylated probes targeting the CDS region of the reporter gene were mixed with lysates of cells where miR-27a and various reporter constructs were co-expressed. IsomiRs which bind to target mRNAs were pulled down by streptavidin beads and subject to Northern blot analysis (Figure S3A). MiR-27a and its isomiRs were pulled down by a reporter containing functional target sites, whereas no signal was detected when the reporter containing nonfunctional (seed-mutant) target sites (Figure S3B). MiR-27a isomiRs were also detected in the pull-downs of 7AA, 8AA and 9AA targets but were absent in those of the corresponding CC targets (Figure 3A), demonstrating the requirement of the “AA” motif for repression. Only isomiRs with a relatively slow mobility were enriched, suggesting that the tailed isomiRs rather than the canonical miR-27a are responsible for recognizing these 3’ paired “no-seed-match” targets. Indeed, deep sequencing the pull-down RNA of 7AA target revealed that 63% of the reads are full-length miR-27a with an extended tail (Figure S3C). Among these, 83% contain an “U” at the first nucleotide position while 32% contain an “U” at both the first and the second positions of the tail (Figure 3B). This suggests that a single base pair between an uridine in the tail and an adenosine in the target may be sufficient to stabilize the association between uridylated isomiRs and the 7AA target.
Figure 3. Uridylated miR-27a isomiRs repress non-canonical targets.

(A) Various reporters were co-expressed with miR-27a in HEK293T cells. Functional isomiRs associated with the corresponding targets were pulled down using biotinylated oligonucleotides complementary to the reporter (see Figure S3A for detail) and subjected to Northern blotting with probe-miR-27a. (B) Small RNAs associated with 7AA targets were deep sequenced and mapped to the mir-27a genomic sequence (green box). All tailed isomiRs were aligned and the nucleotide composition at each position was analyzed. The percentage of each nucleotide relative to the total reads of tailed miR-27a isomiRs (y-axis) was plotted against their relative position to the end of miR-27a (x-axis). Blue circles indicate the position of the tail. (C) Synthetic miR-27a mimic or uridylated miR-27a duplex were co-transfected with various reporters into HEK293T cells. Pull downs were performed and analyzed as described above. (D) Various reporters were transfected into HeLa cells without ectopic expression of miR-27a. The endogenous miR-27a-3p isomiRs associated with the corresponding targets were analyzed by the biotin-pull-down assay. See also Figure S3.
To exclude the possibility that the miR-27a isomiRs hybridize to the 7AA targets after cells were lysed, single-stranded RNA oligos with the sequence of either miR-27a or the uridylated miR-27a (miR-27a-UU) were mixed with cell lysate directly. Neither of these RNAs were pulled down by the 7AA or 9CC target (Figure S3D), supporting the idea that the observed association between uridylated isomiRs and the target RNA occurs prior to cell lysis. Consistent with this possibility, transfection of synthetic miR-27a or miR-27a-UU duplexes, which must be loaded into the RISC complex in order to access their targets, are pulled down associated with the 7AA target but not with the 9CC target RNA (Figure 3C).
To validate that the observed phenomenon is not an artifact of miR-27a over-expression, we repeated the pull-down experiments with endogenous miR-27a by expressing either the 7AA or the 9CC target in HeLa cells, where endogenous miR-27a can be detected by Northern blot. Consistent with the results in HEK293T cells (Figure 2C), endogenous uridylated isomiRs were associated with 7AA target but not 9CC target (Figure 3D).
TUT4/TUT7-dependent repression of non-canonical targets
To further validate that uridylated isomiRs are responsible for inhibiting the 3’ paired “no-seed-match” targets, we blocked miRNA uridylation and determined its impact on repression. We generated a HEK293T double knockout (DKO) cell line lacking both TUT4 and TUT7, two enzymes which uridylate both pre-miRNAs and mature miRNAs (Heo et al., 2012; Thornton et al., 2012, 2014). Loss of TUT4 and TUT7 expression was confirmed by Western blot (Figure 4A). Deep sequencing revealed that while the percentage of ectopically expressed miR-27a carrying tails was reduced ~1.4-fold, (Figure S4A), the fraction of uridylated miR-27a dropped ~2.5-fold upon TUT4 and TUT7 depletion (from 58% in HEK293T cells to 23% in TUT4/7 DKO cells). Concomitant with the reduction in U-tails, the fraction of ectopically expressed miR-27a carrying A tails increased 2.3-fold (Figure S4B). Ectopic expression of either TUT4 or TUT7 restored the levels of U-tailed miRNAs to that of the parent HEK293T cells (Figure S4B).
Figure 4. TUT4/TUT7-dependent repression of non-canonical targets.

(A) TUT4 and TUT7 double knockout (DKO) cells were generated by CRISPR/Cas9 in HEK293T cells. Western blot was used to assess the expression of TUT4 and TUT7 proteins. Tubulin was detected and served as a loading control (B) Small RNAs associated with 7AA targets were detected by the biotin-pull-down assay performed in HEK293T cells as well as in TUT4/7 DKO cells with or without TUT4/7 rescue. (C) Various reporters were each coexpressed with miR-27a in HEK293T cells and TUT4/7 DKO cells with or without TUT4/7 rescue. Gaussia luciferase activities were first normalized with firefly luciferase activity from a co-transfected plasmid. Then the percentage of normalized Gaussia luciferase activity compared to the negative control treated with a non-targeting control miRNA was shown in the figure. The value of negative control was designated as 1. Error bars represent the standard deviation from three biological replicates. See also Figure S4.
We measured the association between miR-27a and 7AA target using the biotin pull-down experiment in either HEK293T cells or TUT4/7 DKO cells. In contrast to the wild-type cells, miR-27a and its tailed isomiRs were not bound to the 7AA target in the TUT4/7 DKO cells (Figure 4B). This association was recovered when either TUT4 or TUT7 was over-expressed in these cell lines (Figure 4B), demonstrating that TUT4/7-mediated uridylation of miR-27a was required for its association with this type of non-canonical targets.
To compare miR-27a-mediated repression in parent HEK293T cells and TUT4/7 DKO cells we used a Gaussia luciferase (GLuc) reporter system which allows secreted GLuc activity to be monitored in the medium. Target sites were inserted into the 3’UTR of GLuc and co-expressed with miR-27a or a control non-target miRNA in either wild-type or TUT4/7 DKO cells. MiR-27a-mediated repression was measured at various time points post-transfection, after normalization to the control miRNA. In both HEK293T and TUT4/7 DKO cells, the wild-type target was repressed by miR-27a while the 9CC target was not, serving as positive and negative controls, respectively (Figure 4C). Consistent with the results from the pulldown experiments (Figure 4B), miR-27a-mediated inhibition of the no-seed-match 7AA target was nearly abolished in the TUT4/7 DKO cells, and could be rescued by ectopically expressing either TUT4 or TUT7 (Figure 4C). The parallel experiment performed with the Renilla-Firefly Dual-luciferase constructs yielded a similar result (Figure S4C). Together, these findings demonstrate that the repression of the 3’ paired “no-seed-match” targets is dependent on TUT4/7-mediated uridylation. We call this type of silencing Tail-U mediated repression (TUMR).
Partial pairing of TUMR targets to uridylated isomiRs
To further characterize the requirements for TUMR, we designed a set of targets by modifying various aspects of the 7AA target site sequence (Figure 5A). Reducing the pairing to the 3’ region of miR-27a to 6bp (6AA target) abolished both association with the target and the corresponding repression (Figures 5B and 5C), indicating that a paired 3’ region with a minimal length of 7bp is required for invoking TUMR. Deep sequencing of the miR-27a isoforms that were recovered bound to the 7AA target (Figure 3A) revealed that the majority of the associated isomiRs contain a non-templated U only at the first position of the tail (Figure 3B), suggesting that a single U:A base-pair between the miR-27a tail and the target sequence is sufficient for TUMR. To test this, we generated a 7A target and compared it to the 7AA target (Figure 5A). The biotin-pull-down and dual-luciferase assays demonstrated that the 7A target was associated with, and repressed by the uridylated miR-27a isomiRs (Figures 5C and 5D), although to a lesser extent than the 7AA target, supporting the idea that a single adenosine upstream of the 3’ paired “no-seed-match” targets is sufficient to trigger TUMR.
Figure 5. Partial pairing of TUMR targets to uridylated isomiRs.

(A) Schematic representation of the pairing between miR-27a (top) and an additional set of target sequences (bottom). Non-templated Us are labeled with red circles. The yellow boxes indicate the adenosine(s) upstream of target sequence. Reporters containing these target sites in the 3’UTR were co-expressed with miR-27a. The association between miR-27a-3p isomiRs and targets was detected by biotin-pull-down assays in (B) (D) and (E) while the corresponding repressions were measured by dual-luciferase assays in (C) and (F). * p<0.001, ** p < 0.005, n.s. non-significant.
Pairing between the uridylated miR-27a and the 7AA target results in a mismatch at nucleotide 5, counting from the 5’ end of miR-27a, which precludes the seed match (nucleotide 2-8). Nonetheless, given the critical role of the seed region in target recognition, we speculated that a partial match in the seed region is required. To investigate whether mismatches at other positions of the seed region are tolerated, we generated 7AA targets with a mismatch at nucleotide 3 (7AA-M3), nucleotide 4 (7AA-M4) or nucleotide 6 (7AA-M6) (Figure 5A). The binding affinity between the uridylated miR-27a isomiRs and the target was affected by the mismatch position: 7AA-M3 did not associate with miR-27a isomiRs, 7AA-M4 had a reduced affinity, and 7AA-M6 has an enhanced affinity compared to the 7AA target (Figure 5E). Similarly, ectopic expression of miR-27a was able to repress 7AA-M4, and 7AA-M6, but not 7AA-M3 (Figure 5F). Thus, our results indicate that partial pairing in this region is required for TUMR, although the precise requirements for base pairing within the seed region remains to be defined.
Uridylated miR-27a isomiRs repress a subset of endogenous mRNAs
To identify endogenous targets of uridylated miR-27a isomiRs, we started our search by focusing on the 3’ region, given the limited understanding of the pairing requirements at the seed region. Over 4648 human genes contain at least one potential TUMR target site within their 3’UTR, with a minimum of 7 nt that could pair to the 3’ region of miR-27a plus one “A” immediately upstream. This number is far larger than the number of miR-27a canonical targets (1198 genes) predicted by TargetScan (Lewis et al., 2005). Applying a strategy similar to TargetScan, we sought to narrow our search to target genes whose pairings with the 3’ region of miR-27a are relatively conserved. Alignments of the 3’UTR sequences of all genes from 18 organisms are available through the TargetScan website. Of these, 15 organisms including human encode miR-27a of the identical sequence (Figure S5A). By requiring conservation among at least two thirds (10) of these 15 organisms as a threshold, we identified 851 genes containing at least one conserved TUMR target site (Table S2). Cross-comparing with TargetScan results revealed that 710 of the 851 genes are previously unknown miR-27a targets, not containing any canonical target sites, and expanding the potential targets of miR-27a by 59% (Figure 6A).
Figure 6. Uridylated miR-27a isomiRs repress a subset of endogenous mRNAs.

(A) Venn diagram of the canonical targets of miR-27a predicted by TargetScan (8mer, 7mer-A1, 7mer-m8) and the conserved TUMR targets. (B) Cumulative curve comparing the effect of miR-27a canonical target sites (Adj. p-value<0.0001 vs. No target) and conserved TUMR targets (adj. p-value<0.0001 vs. No target) in HEK293T wild-type cells. The number of genes analyzed in each group is in parentheses, while horizontal dash lines indicate the fraction of repressed genes (Log2(FC)<0) in each group. (C) Plot presenting the fraction of repressed genes (Log2(FC)<0) (solid blue line, left axis) and the number of genes (dashed black line, right axis) as a function of the conservation score on the x-axis. Conservation score was defined as the number of organisms among which the pairing between the target site and the miR-27a 3’ region is conserved. (D) Cumulative curve comparing the effects on miR-27a canonical target sites (Adj. p-value<0.0001 vs. No target) and conserved TUMR targets in HEK293T TUT4/7 DKO Cells. Parentheses, the number of genes analyzed in each group. The horizontal dash lines indicate the fraction of repressed genes (Log2(FC)<0) in each group. Adjusted p-values were calculated using one-way ANOVA with multiple comparisons. See also Figure S5.
To investigate whether these potential targets are regulated by miR-27a, we transfected HEK293T cells with plasmids expressing either miR-27a or a control miRNA, and compared the mRNA profiles measured by RNA-seq. We focused on those mRNA targets that both harbored the predicted TUMR target sites and were expressed in HEK293T cells (FMPK > 1) (Figure S5B). These mRNAs (n=452) were repressed in comparison to those without any predicted target sites, indicating that they are subject to miR-27a regulation (Figure 6B). Although mRNAs containing canonical miR-27a target sites (TargetScan targets) were inhibited to a greater extent compared to TUMR targets, the degree of conservation of the TUMR targets correlated with repression (Figure 6C), implying an evolutionary conserved function. mRNAs carrying both the canonical and TUMR target sites were more highly repressed, indicating that there is an additive effect between these two types of target sites (Figure 6B).
The same experiment was performed in TUT4/7 DKO cells. Consistent with a previous report that TUT4/7 are required for efficient miRNA-mediated repression (Lim et al., 2014), we observed less robust inhibition of miR-27a on canonical target genes in the DKO cells compared to that observed in wild-type cells (Figure 6D). In contrast, repression of mRNAs containing only TUMR target sites was undetectable. In addition, mRNAs bearing both type of target sites were repressed to a similar extent as mRNAs containing only canonical target sites (Figure 6D).
These results confirm that the observed repression of TUMR target genes is dependent on TUT4/7, and is specific to uridylated miR-27a. To further validate this idea, we performed a parallel TUMR target site analysis, replacing the upstream “A” with a “C”, and identified 1672 potential target genes, of which only 90 (< 6%) show conserved base pairing with the 3’ region of miR-27a (Figure S5C). In contrast to the TUMR targets, these “3’ paired plus C” targets as a group were not repressed by ectopic expression of miR-27a, compared with mRNAs lacking these sites (Figure S5D). Taken together, these results demonstrate that a set of mRNAs, that are distinct from known miR-27a targets, undergo specific repression by uridylated miR-27a isomiRs. Furthermore, reanalyzing the gene expression profile of T cells isolated from a transgenic mouse with overexpressed miR-27a (Cho et al., 2016) yielded a similar result as observed in HEK293T cells (Figure S5E), extending TUMR effects beyond cultured cells. Of note, de-repressions of neither canonical nor TUMR targets were observed when miR-27 gene was deleted, most likely due to uncharacterized genetic compensations.
TUMR expands the target range of endogenous miRNAs
To study the role of TUMR in enhancing target selection by endogenous miRNAs, we compared the isomiR profiles between HEK293T cells and the corresponding TUT4/7 DKO cells. Consistent with a recent study (Kim et al., 2019), we found that miR-148b is highly uridylated by TUT4/7 (Figure S6A). The abundance of uridylated miR-148b in HEK293T cells (Figure S6B) suggested that miR-148b has the potential to regulate a set of endogenous targets via TUMR. To evaluate this experimentally, we transfected HEK293T cells with a miR-148b-specific antagomir or an antagomir of control sequence, and compared the mRNA profiles measured by RNA-seq. The level of miR-148b and its isomiRs was reduced ~3 fold upon antagomir treatment (Figure 7A). mRNAs harboring either canonical target sites or TUMR target sites were de-repressed in comparison to those without any predicted target sites (Figure 7B). The extent of depression on canonical targets is less than expected, most likely due to compensation by miR-148a which shares the same seed sequence, but is not highly uridylated. Nevertheless, mRNAs carrying both the canonical and TUMR target sites were further de-repressed upon miR-148b depletion (Figure 7B). Parallel antagomir treatments on TUT4/7 DKO cells led to a similar reduction of miR-148b level (Figure 7A). Less robust de-repression of miR-148b targets was observed due to the fact that TUT4/7 are required for efficient mRNA degradation (Lim et al., 2014). Nonetheless, the de-repression of TUMR targets was nearly undetectable whereas a subtle de-repression of canonical targets was observed (Figure S6C). Furthermore, mRNAs bearing both type of target sites were de-repressed to a similar extent as mRNAs containing only canonical target sites (Figure S6C). These results demonstrate that a set of genes are regulated by endogenous miR-148b via TUMR in HEK293T cells.
Figure 7. TUMR expands the target range of endogenous miRNAs.

(A) Antagomirs (20 nM final concentration) of either anti-miR-148b or a control sequence were transfected into HEK293T cells or TUT4/7 DKO cells to block miR-148b function. Small RNAs were extracted and subject to Northern blotting. Endogenous miR-148b was detected by probe-miR-148b-3p. Endogenous miR-16 was detected by probe-miR-16-5p and served as loading controls. In each lane, the level of miR-148b was normalized to that of miR-16 and reported relative to control treatment in WT cells. (B) Cumulative curve comparing the effect of miR-148b reduction on mRNAs containing TargetScan sites (Adj. p-value<0.05 vs. No target), TUMR target sites (Adj. p-value<0.0001 vs. No target) and both sites (Adj. P-value<0.0001) in HEK293T wild-type cells. (C) Cumulative curve comparing the effect of miR-7 induction on mRNAs harboring TargetScan target sites (Adj. p-value<0.0001 vs. No target), TUMR target sites (adj. p-value<0.0001 vs. No target) and both sites (Adj. P-value<0.0001) in mouse brain (Kleaveland et al., 2018). (D) Analysis of miRNA target sequences identified by CLASH (Helwak et al., 2013). 79 miRNAs with more than 50 unique chimeric reads were analyzed. For each miRNA, percentages of canonical targets and TUMR targets relative to all chimeric reads of the queried miRNA were plotted. For each miRNA, the background was calculated as the chance its TUMR target sites identified in all unrelated chimeric reads. See also Figure S6.
To further evaluate the role of TUMR, we took advantage of RNA-seq results from a recent study where the endogenous level of miR-7 was perturbed in mouse brain: Knocking out long noncoding RNA Cyrano, a direct miR-7 suppressor, increased the level of miR-7, whereas knocking out two out of three pri-miR-7 paralogs resulted in a significant miR-7 reduction (Kleaveland et al., 2018). Upregulation of miR-7 led to specific repression of mRNAs containing either canonical or TUMR target sites (Figure 7C). Those mRNAs harboring both types of target sites were further repressed (Figure 7C). Correspondingly, the same sets of mRNAs were de-repressed when miR-7 levels were reduced (Figure S6D), confirming that those mRNAs with predicted TUMR target sites were subject to miR-7 regulation in mouse brain (Kleaveland et al., 2018). By performing similar analyses on published gene expression data from miR-155 knockout mice (Loeb et al., 2012), miR-205 conditional knockout mice (Lu et al., 2018) and SK-BR-3 cells depleted of the miR-200c/141 cluster (Kim et al., 2013), we demonstrated the TUMR effects of miR-155 in activated T cells (Figure S6E), TUMR effects of miR-205 in mouse mammary epithelial cells (Figure S6F) and TUMR effects of miR-141/200c in breast cancer cells (Figure S6G).
Finally, we examined miRNA-mRNA target interactome measured by the method of crosslinking, ligation, and sequencing of hybrids (CLASH) in a previous study (Helwak et al., 2013). Because an RNase treatment is included in the CLASH protocol prior to hybrid ligation, the 3’ ends of miRNAs are generally not preserved. We therefore focused on the mRNA sequences extracted from the hybrid reads. Consistent with previous analyses (Helwak et al., 2013), canonical target sites were frequently detected (11.1±0.88%) in chimeric reads of the corresponding miRNAs. Albeit less prevalent, TUMR target sites were also enriched (2.75±0.31% vs 0.49±0.03% as background, p=1.4×10−11) (Figure 7D), suggesting that TUMR could be a general mechanism for miRNA function.
Discussion
MiRNA, a master regulator of gene expression, is itself subject to regulation. Understanding how uridylation influences miRNA function has until now focused on its impact on miRNA biogenesis and turnover (Ameres and Zamore, 2013; Gebert and MacRae, 2019). Here, we show that uridylation can modulate miRNA pairing, enabling miR-27a to repress a distinct set of targets, which have the following features: (1) a minimum of 7 nt that base pair to the 3’ region of miR-27a; (2) an “A” immediately upstream of the binding site; and (3) partial base pairing in the seed region. This type of repression (TUMR) is dependent on TUT4/7 activity. Given that the expression levels of TUT4 and TUT7 are tissue-specific (Pangala et al., 2017) (Figure S7A), these findings support a model where TUT4/7-mediated uridylation regulates miRNA function by modifying its target repertoires (Figure S7B).
Although identified more than a decade ago, distinct biological functions have not been assigned to the majority of isomiRs. This is partly due to the fact that most isomiRs differ from canonical miRNAs only in their 3’ ends. Since these 3’ isomiRs share the same seed sequence with their corresponding canonical miRNAs, they are expected to function similarly. However, the profile of isomiRs is usually cell and tissue specific (McCall et al., 2017) and can be used as a biomarker for differentiating many cancers (Telonis et al., 2017), suggesting isomiRs perform specific functions (Tan et al., 2014). In addition, recent reports indicate that naturally existing isoforms of miR-26, miR-122 and miR-222 have distinct activities in regulating cytokine expression (Jones et al., 2009), facilitating virus proliferation (Yamane et al., 2017) and promoting apoptosis (Yu et al., 2017), respectively. Although our findings do not directly explain these intriguing observations, they identify a mechanism by which these isoforms could affect function. Indeed, our data suggest that other prevalent sequence modifications, such as 3’ trimming and 3’ adenylation, could also influence isomiR functions.
The seed sequence plays a critical role in determining miRNA target repertoire. Recent studies have demonstrated that pairing beyond the seed region can also contribute to target specificity (Broughton et al., 2016), explaining how miRNA family members that share the same seed sequence but have heterogeneous 3’ regions can target non-overlapping sets of genes. That TUMR requires extensive pairing in the 3’ region instead of the seed may provide further insights. Upon 3’ uridylation, each miRNA family member will be able to target distinct sets of genes via TUMR.
The seed region is exposed on the surface of the RISC and is available to base pair with targets (Schirle et al., 2014; Wang et al., 2008). This hybridization initiates at the 5’ end of the seed (position 2-4) before propagating to the rest of the seed (Chandradoss et al., 2015; Salomon et al., 2015). It is possible that the recognition of non-canonical targets follows a similar mechanism, potentially explaining why partial pairing at the seed, especially at positions 2 and 3, is required for TUMR. Unlike canonical target sites, TUMR target sites rely on extensive pairing in the 3’ region to stabilize their interaction with RISC. However, this extended basepairing is insufficient to explain why uridylated miR-27a isomiRs bind to 7AA target sites but not 9CC target sites, since both target sites have the same number of potential base-pair interactions. It is possible that the pairing between the miRNA tail and substrate is uniquely positioned within the ternary complex of Ago2/miRNA/target RNA, such that only specific additional base pairs can be accommodated. Crystal structures of uridylated miRNAs in complex with Ago and an TUMR target should provide additional insights.
Similar to other types of non-canonical targets (Helwak et al., 2013), TUMR is less robust than repression through canonical target sites (Figure 6B). It is possible that there are additional features that govern the efficacy of TUMR in addition to the requirements for 7 bp pairing in the 3’ region and an upstream “A”. Nonetheless, given the additive effect between canonical and TUMR target sites (Figure 6B), TUMR should be considered when searching for effective targets for a given miRNA. Finally, our observations may have implications beyond miRNA-mediated target repression. Several studies showed that targets of high complementarity, particularly in the miRNA 3’ region, trigger miRNA decay via 3’ trimming and tailing, a pathway termed target-directed miRNA degradation (TDMD) (Ameres et al., 2010; Baccarini et al., 2011; Buck et al., 2010; Cazalla et al., 2010; de la Mata et al., 2015; Park et al., 2017). It is possible that some of the uridylated isomiRs associated with TUMR targets are formed as a result of TDMD. Instead of being degraded, these uridylated isomiRs could base-pair with the 5’ adenosine, and enhance the association with the target transcripts. In this case, a positive feedback loop would result in stable miRNA-target hybridization and target repression. If those isomiRs were uridylated locally after association with targets, the low abundance of uridylated isomiRs by itself might not preclude their function in TUMR. Future mechanistic studies of TUMR may illuminate the reciprocal-relationship between miRNAs and their RNA targets.
STAR★Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Shuo Gu (shuo.gu@nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK293T and HeLa cells were maintained in DMEM (high glucose) (Gibco) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (Hyclone), 1X MEM Non-Essential Amino Acids (Gibco), 100 U/ml penicillin and 100 μg/ml streptomycin (Gibco) at 37°C with 5% CO2. HEK293T-TUT4/TUT7 DKO cells were generated using CRISPR/Cas9 (Shalem et al., 2014). HEK293T and HeLa cells were transfected with PolyJet™DNA Transfection Reagent (SignaGen), according to the manufacturer's’ instructions. For antagomiRs transfection, Lipofectamine RNAiMAX reagent (ThermoFisher Scientific) was used according to the manufacturer's’ instructions.
METHOD DETAILS
Northern Blotting
Total RNA was isolated from cells using Trizol (Life Technologies) and quantitated by Nanodrop. 20 μg total RNA was run on 20% (w/v) acrylamide/8M urea gels with a 32P-labeled Decade marker (Ambion), and then transferred onto Hybond-N membranes (Amersham Pharmacia Biotech). After transfer, the membrane was either UV crosslinked or EDC-mediated chemical cross-linking (Sigma)(Pall and Hamilton, 2008). Pre-hybridization was performed with PerfectHyb™ Plus Hybridization Buffer (Sigma) at 37°C for 10 min. 32P-labeled probes that reverse complement to the targeted miRNAs were hybridized with membrane overnight at 37°C. After washing with 2X SSC with 0.1% SDS buffer for 3 × 15 min at 37°C, the membrane was exposed to an Imaging Screen-K (Bio-Rad) overnight. Images were then analyzed by Typhoon Trio Imaging System (GE Healthcare).
Immunoprecipitation
One 10cm dish of HEK293T was lysed in 1mL modRIPA buffer (10mM Tris-Cl pH 7.0, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100, and 0.1% SDS) supplemented with proteinase inhibitors cocktail (Roche). Cell lysate was incubated with 50ul Anti-FLAG® M2 Magnetic Beads (Sigma) or SureBeads Protein G Magnetic Beads (Bio-Rad) plus 5ug mouse anti-c-Myc Monoclonal antibody at 4°C overnight with rotating. After wash ed for 5 times with BC150 buffer (20 mM Tris-HCl (pH 8.0), 150 mM KCl, 0.2 mM EDTA, 10% glycerol) at room temperature, the beads were lysed in 1mL Trizol (Life Technologies) for RNA extraction.
Luciferase reporter assay
For dual luciferase reporter assay, 5×104 cells were seeded into each well of a 24-well plate one day before transfection. 50 ng each of miRNA and target expression plasmids were transfected into cells. For rescue experiment, 10 ng plasmids expressing either TUT4 or TUT7 were co-transfected into TUT4/7 DKO cells. For HeLa de-repression assay, 20nM of antagomiR (Exiqon) for control or anti-miR-27a were transfected. 48h (for HEK293T cells) or 72h (for HeLa cell) post-transfection, dual-luciferase reporter assays were performed according to the manufacturer's’ protocol (Promega). The Firefly and Renilla Luciferase signals were measured by the Glomax multi-detection system (Promega). For Gaussia reporter assay, 50 ng each of miRNA and target expression plasmids with or without 50 ng TUT4 or TUT7 expression plasmid as well as 10 ng pGL3 Basic (as internal control) were co-transfected into either Wild-type or TUT4/7-DKO HEK293T cells using PolyJet. 1 mL of medium was added to each well 8h after transfection, and then 50 μL of medium from each well was collected and stored at −80°C at 12h, 24h, 36h, 48h, and 60h post-transfection for the time course analysis. The Gaussia and Firefly luciferase signals were measured by Glomax multi-detection system (Promega) according to the manufacturer's’ protocol of Pierce™ Gaussia Luciferase Glow Assay Kit (Thermo Fisher Scientific) and the Dual-Luciferase Reporter Assay program, respectively.
Biotin-pull-down assay
3×106 cells were seeded into each 10 cm dish one day before transfection. 3μg of miRNA expression vector and 2 μg of target expression vector were transfected into the cells by PolyJet. Cells were washed with PBS once and then collected by using cell scraper in 600 μL modRIPA buffer supplemented with proteinase inhibitors cocktail (Roche) and Superase-in (ThermoFisher Scientific) at 1:200. 10 pmol of biotinylated probes (Supplementary Table S1) were incubated with cell lysates in the hybridization buffer (70mM Tris-Cl pH7.0, 675mM Nacl, 5.5 mM EDTA, 1.45% SDS, 15% formamide) with proteinase inhibitors cocktail (Roche) and Superase-in (ThermoFisher Scientific), for 4h with rotating. 30 μL C1 streptavidin Dynabeads (ThermoFisher Scientific) were incubated with each sample at room temperature for 1h. After washed for 3 times with 1ml wash buffer (2x SSC, 0.5% SDS), RNAs associated with Dynabeads were extracted by Trizol.
Western Blot
Wild-type or TUT4/7-DKO HEK293T cells were lysed in modRIPA buffer with protease inhibitor cocktail (Roche). 20 μg of each protein sample was loaded into 4-20% Mini-PROTEAN® TGX Stain-Free™ Gels (Bio-Rad) and then the proteins were transferred onto a PVDF membrane using Trans-Blot Turbo Transfer System (Bio-Rad). Primary antibodies used in this study are rabbit anti-ZCCHC11 (TUT4, Proteintech), rabbit anti-ZCCHC6 (TUT7, Proteintech), and mouse anti-α-tubulin (Sigma). The signals were developed with SuperSignal West Pico Chemiluminescent Substrate (Pierce) and imaged by the Chemidoc Touch Imaging System (Bio-Rad).
Small RNA sequencing
For small RNA deep-sequencing, small RNA libraries were prepared using the NEBNext® Multiplex Small RNA Library Prep Set for Illumina® according to the manufacturer's’ protocol with minor modifications. The small RNA library quality was assessed on the Agilent 2100 Bioanalyzer (Agilent), and the quantity was determined by Qubit dsDNA HS Assay (Life Technologies). Each small RNA library was sequenced on an Illumina Miseq platform (Illumina) with MiSeq® Reagent Kit v3 kit (Illumina).
Analysis of small RNA sequencing data
The small RNA sequencing data were analyzed by in-house written scripts. Briefly, adaptors were removed, and reads were mapped using Bowtie to obtain global profiles. More detailed study of the isomiR profile was done using QuagmiR (Bofill-De Ros et al., 2018). This software uses a unique algorithm to pull specific reads and aligns them against a consensus sequence in the middle of a miRNA, allowing mismatches on the 5’ and the 3’ end to capture 5’ and 3’ isomiRs respectively.
TUMR sites prediction and conservation scoring
TUMR sites were predicted through the search on the 3’UTR sequences complementary to the last seven nucleotides of the miRNA analyzed (miR-27a-3p, miR-148b-3p, miR-7-5p, miR-155-5p, miR-205-5p, miR-141-3p, miR-200c-3p) preceded by an “A”. In the 7nt base-pairing it was allowed 1-3 G:U wobble pairs. Conservation scoring was calculated based on the preservation of equivalent sites for the same gene in a pool of species where the sequence of the miRNA analyzed is fully conserved. Accessibility was also taken into consideration during target sites scoring. Sequences for 424806 3’UTR (15 species, including human) were obtained from TargetScan downloads. Scripts of the R code used to generate the TUMR sites prediction and conservation score are available at GitHub (https://github.com/Gu-Lab-RBL-NCI/TUMR).
Global analysis of miRNA repression
HEK293T WT and DKO cells were transfected with a vector expressing either pri-miR-27a or a control pri-miRNA (pri-miR-21). Similarly, HEK293T WT and DKO cells were transfected with antagomirs against miR-148b or antagomirs of a control sequence (Exiqon). RNA was isolated and gene expression profile was obtained by RNA-seq with poly-A+ selection (Novogene and Macrogen). Raw reads were aligned to the human genome (GRCh37.75) using STAR 2.5.1. After being normalized, expression fold-change of each gene between cells transfected with miR-27a and control miRNA was calculated. To calculate the cumulative fraction plot, genes were sorted into the following groups: those only containing canonical seed-matched sites as predicted by TargetScan 7.2 (conserved 8mer, 7mer-A1 or 7mer-m8)(Agarwal et al., 2015) in their 3’UTR, those only containing conserved TUMR sites, those containing both type of sites and those contain none of those sites. Scripts of the R code used to generate the cumulative curves are available at GitHub (https://github.com/Gu-Lab-RBL-NCI/TUMR).
CLASH data analysis
Interactions between miRNA-mRNA target were analyzed from a previously published CLASH dataset (1-s2.0-S009286741300439X-mmc1) (Helwak et al., 2013). For each miRNA, we analyzed the percentage of interactions that contained either a canonical site or a TUMR sites on the mRNA segment of the chimeric read. Background frequencies for miRNA:TUMR sites were obtained by calculating the percentage of TUMR sites on the mRNA segment of the chimeric reads for any miRNA other than the one queried. Scripts of the R code used to generate the cumulative curves are available at GitHub (https://github.com/Gu-Lab-RBL-NCI/TUMR_CLASH).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using GraphPad Prism 7. The statistical tests used included two-tailed unpaired Student’s t-test and one-way ANOVA with multiple comparison correction. Time-course repression using luciferase assays were analyzed using Student’s t-test for paired samples, where comparisons for each treatment were paired across a time point. Global miRNA repression effects shown displayed as cumulative curves were assessed using Kolmogorov-Smirnov statistical test on R. Differences were considered significant if the P value was < 0.01.
DATA AND SOFTWARE AVAILABILITY
All the deep sequencing datasets can be found in GEO with the accession number GSE121327. Raw image files of Northern Blots used to prepare figure panels in the main text and supplementary information can be found at Mendeley Data:http://dx.doi.org/10.17632/pp5zz5rwvs.1
Supplementary Material
Table S1. DNA Oligonucleotides Used in This Study, Related to STAR Methods
Table S2. Predicted Targets of miR-27a, Related to Figure 6
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-c-Myc antibody | Sigma-Aldrich | Cat# M4439 |
| Anti-ZCCHC11 (TUT4) | Proteintech | Cat# 18980-1-AP |
| Anti-ZCCHC6 (TUT7) | Proteintech | Cat# 25196-1-AP |
| Mouse anti-α-tubulin | Sigma | Cat# T8203 |
| Anti-FLAG® M2 Magnetic Beads | Sigma-Aldrich | Cat# M8823 |
| Critical Commercial Assays | ||
| Dual-Luciferase® Reporter Assay System | Promega | Cat# E1980 |
| Pierce™ Gaussia Luciferase Glow Assay Kit | ThermoFisher Scientific | Cat# 16161 |
| NEBNext® Small RNA Library Prep Set for Illumina® (Multiplex Compatible) | NEB | Cat# E7330L |
| Qubit™ dsDNA HS Assay Kit | ThermoFisher Scientific | Cat# Q32854 |
| Agilent High Sensitivity DNA kit | Agilent | Cat# 5067-4626 |
| MiSeq Reagent Kit v3 (150-cycle) | Illumina | Cat# MS-102-3001 |
| Deposited Data | ||
| Raw and analyzed NGS data | This paper | GEO: GSE121327 |
| Unprocessed image files of Northern Blots | Mendeley Data | http://dx.doi.org/10.17632/pp5zz5rwvs.1 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T cells | ATCC | Cat# CRL-3216 |
| Human: HeLa cells | ATCC | Cat# CCL-2 |
| Human: HEK293T TUT4/7-DKO cells | This paper | N/A |
| Oligonucleotides | ||
| Listed in Supplementary Table S1 | This paper | Table S1 |
| Recombinant DNA | ||
| pIRESneo-FLAG/HA Ago2 | Addgene | Cat#10822 |
| psiCHECK™−2 Vector | Promega | Cat# C802A |
| pCMV-Gaussia Luc | ThermoFisher Scientific | N/A |
| Lenti-CRISPR-V2-puro | Shen lab | N/A |
| Lenti-CRISPR-V2-hygro | Shen lab | N/A |
| pGL3-Basic | Promega | Cat# E1751 |
| pIRESneo-FLAG/HA-TUT4 | This paper | N/A |
| pIRESneo-FLAG/HA-TUT7 | This paper | N/A |
| Software and Algorithms | ||
| TargetScan v7.2 | (Agarwal et al., 2015) | targetscan.org |
| QuagmiR | (Bofill-De Ros et al., 2018) | cancergenomicscloud.org |
| Bowtie2 | (Langmead and Salzberg, 2012) | bowtie-bio.sourceforge.net |
| Samtools | (Li et al., 2009) | samtools.sourceforge.net |
| STAR 2.5.1. | (Dobin et al., 2013) | cancergenomicscloud.org |
| Other | ||
| Cyrano KO mice | (Kleaveland et al., 2018) | GEO: GSE112635 |
| miR-7 DKO mice | (Kleaveland et al., 2018) | GEO: GSE112635 |
| miR-155 KO in CD4 T cells | (Loeb et al., 2012) | GEO: GSE41288 |
| miR-205 KO in mammary epithelial cells | (Lu et al., 2018) | GEO: GSE124364 |
| miR-141/200c DKO in SK-BR-3 | (Kim et al., 2013) | GEO: GSE51217 |
| Human Genome reference | GRCh37.75 | cancergenomicscloud.org |
Highlights.
Basepairing of miRNA U-tail with targets enables Tail-U-mediated Repression (TUMR)
Uridylated miR-27a isomiRs repress non-canonical targets via TUMR.
TUT4 and TUT7-mediated 3' uridylation of miRNA is required for TUMR.
3' uridylated isomiRs expand the target repertoires of the cognate miRNAs via TUMR.
Acknowledgments
We thank Dr. Sandra L. Wolin for critical reading of the manuscript and helpful discussions. We thank Dr. Jingshi Shen (University of Colorado Boulder) for the Lenti-CRISPR-V2-puro and Lenti-CRISPR-V2-hygro plasmids. We thank Richard Ma for helping with experiments. This work has been funded by the intramural research program (ZIA-BC-011566) of the National Cancer Institute, National Institutes of Health.
Footnotes
Declaration Of Interests
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agarwal V, Bell GW, Nam J-W, and Bartel DP (2015). Predicting effective microRNA target sites in mammalian mRNAs. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameres SL, and Zamore PD (2013). Diversifying microRNA sequence and function. Nat. Rev. Mol. Cell Biol. 14, 475–488. [DOI] [PubMed] [Google Scholar]
- Ameres SL, Horwich MD, Hung J-H, Xu J, Ghildiyal M, Weng Z, and Zamore PD (2010). Target RNA-directed trimming and tailing of small silencing RNAs. Science 328, 1534–1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baccarini A, Chauhan H, Gardner TJ, Jayaprakash AD, Sachidanandam R, and Brown BD (2011). Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr. Biol. 21, 369–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP (2018). Metazoan MicroRNAs. Cell 173, 20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bofill-De Ros X, Chen K, Chen S, Tesic N, Randjelovic D, Skundric N, Nesic S, Varjacic V, Williams EH, Malhotra R, et al. (2018). QuagmiR: A Cloud-based Application for IsomiR Big Data Analytics. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broughton JP, Lovci MT, Huang JL, Yeo GW, and Pasquinelli AE (2016). Pairing beyond the Seed Supports MicroRNA Targeting Specificity. Mol. Cell 64, 320–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck AH, Perot J, Chisholm MA, Kumar DS, Tuddenham L, Cognat V, Marcinowski L, Dölken L, and Pfeffer S (2010). Post-transcriptional regulation of miR-27 in murine cytomegalovirus infection. RNA 16, 307–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, Ando Y, de Hoon MJL, Tomaru Y, Nishibu T, Ukekawa R, Funakoshi T, Kurokawa T, Suzuki H, Hayashizaki Y, et al. (2010). A comprehensive survey of 3’ animal miRNA modification events and a possible role for 3’ adenylation in modulating miRNA targeting effectiveness. Genome Res. 20, 1398–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cazalla D, Yario T, and Steitz JA (2010). Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 328, 1563–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandradoss SD, Schirle NT, Szczepaniak M, MacRae IJ, and Joo C (2015). A dynamic search process underlies microrna targeting. Cell 162, 96–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Wu C-J, Yasuda T, Cruz LO, Khan AA, Lin L-L, Nguyen DT, Miller M, Lee H-M, Kuo M-L, et al. (2016). miR-23~27~24 clusters control effector T cell differentiation and function. J. Exp. Med. 213, 235–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueck A, Ziegler C, Eichner A, Berezikov E, and Meister G (2012). microRNAs associated with the different human Argonaute proteins. Nucleic Acids Res. 40, 9850–9862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK-H, Burge CB, and Bartel DP (2009). Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 19, 92–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gebert LFR, and MacRae IJ (2019). Regulation of microRNA function in animals. Nat. Rev. Mol. Cell Biol. 20, 21–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory RI, Chendrimada TP, Cooch N, and Shiekhattar R (2005). Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell 123, 631–640. [DOI] [PubMed] [Google Scholar]
- Gu S, and Kay MA (2010). How do miRNAs mediate translational repression? Silence 1, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hausser J, and Zavolan M (2014). Identification and consequences of miRNA-target interactions--beyond repression of gene expression. Nat. Rev. Genet. 15, 599–612. [DOI] [PubMed] [Google Scholar]
- Helwak A, Kudla G, Dudnakova T, and Tollervey D (2013). Mapping the human miRNA interactome by CLASH reveals frequent noncanonical binding. Cell 153, 654–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heo I, Joo C, Kim Y-K, Ha M, Yoon M-J, Cho J, Yeom K-H, Han J, and Kim VN (2009). TUT4 in concert with Lin28 suppresses microRNA biogenesis through pre-microRNA uridylation. Cell 138, 696–708. [DOI] [PubMed] [Google Scholar]
- Heo I, Ha M, Lim J, Yoon M-J, Park J-E, Kwon SC, Chang H, and Kim VN (2012). Mono-uridylation of pre-microRNA as a key step in the biogenesis of group II let-7 microRNAs. Cell 151, 521–532. [DOI] [PubMed] [Google Scholar]
- Iwakawa H-O, and Tomari Y (2015). The Functions of MicroRNAs: mRNA Decay and Translational Repression. Trends Cell Biol. 25, 651–665. [DOI] [PubMed] [Google Scholar]
- Jonas S, and Izaurralde E (2015). Towards a molecular understanding of microRNA-mediated gene silencing. Nat. Rev. Genet. 16, 421–433. [DOI] [PubMed] [Google Scholar]
- Jones MR, Quinton LJ, Blahna MT, Neilson JR, Fu S, Ivanov AR, Wolf DA, and Mizgerd JP (2009). Zcchc11-dependent uridylation of microRNA directs cytokine expression. Nat. Cell Biol. 11, 1157–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khvorova A, Reynolds A, and Jayasena SD (2003). Functional siRNAs and miRNAs exhibit strand bias. Cell 115, 209–216. [DOI] [PubMed] [Google Scholar]
- Kim VN (2005). MicroRNA biogenesis: coordinated cropping and dicing. Nat. Rev. Mol. Cell Biol. 6, 376–385. [DOI] [PubMed] [Google Scholar]
- Kim D, Sung YM, Park J, Kim S, Kim J, Park J, Ha H, Bae JY, Kim S, and Baek D (2016). General rules for functional microRNA targeting. Nat. Genet. 48, 1517–1526. [DOI] [PubMed] [Google Scholar]
- Kim H, Kim J, Kim K, Chang H, You K, and Kim VN (2019). Bias-minimized quantification of microRNA reveals widespread alternative processing and 3’ end modification. Nucleic Acids Res. 47, 2630–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SY, Kim AY, Lee HW, Son YH, Lee GY, Lee J-W, Lee YS, and Kim JB (2010). miR-27a is a negative regulator of adipocyte differentiation via suppressing PPARgamma expression. Biochem. Biophys. Res. Commun. 392, 323–328. [DOI] [PubMed] [Google Scholar]
- Kim Y-K, Wee G, Park J, Kim J, Baek D, Kim J-S, and Kim VN (2013). TALEN-based knockout library for human microRNAs. Nat. Struct. Mol. Biol. 20, 1458–1464. [DOI] [PubMed] [Google Scholar]
- Kleaveland B, Shi CY, Stefano J, and Bartel DP (2018). A network of noncoding regulatory rnas acts in the mammalian brain. Cell 174, 350–362.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih I, Jones-Rhoades MW, Bartel DP, and Burge CB (2003). Prediction of mammalian microRNA targets. Cell 115, 787–798. [DOI] [PubMed] [Google Scholar]
- Lewis BP, Burge CB, and Bartel DP (2005). Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120, 15–20. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim J, Ha M, Chang H, Kwon SC, Simanshu DK, Patel DJ, and Kim VN (2014). Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 159, 1365–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin S, and Gregory RI (2015). MicroRNA biogenesis pathways in cancer. Nat. Rev. Cancer 15, 321–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu M, Zhang Q, Deng M, Miao J, Guo Y, Gao W, and Cui Q (2008). An analysis of human microRNA and disease associations. PLoS ONE 3, e3420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Cao J, Napoli M, Xia Z, Zhao N, Creighton CJ, Li W, Chen X, Flores ER, McManus MT, et al. (2018). miR-205 Regulates Basal Cell Identity and Stem Cell Regenerative Potential During Mammary Reconstitution. Stem Cells 36, 1875–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, and Keller W (2007). RNA-specific ribonucleotidyl transferases. RNA 13, 1834–1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Mata M, Gaidatzis D, Vitanescu M, Stadler MB, Wentzel C, Scheiffele P, Filipowicz W, and Großhans H (2015). Potent degradation of neuronal miRNAs induced by highly complementary targets. EMBO Rep. 16, 500–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCall MN, Kim M-S, Adil M, Patil AH, Lu Y, Mitchell CJ, Leal-Rojas P, Xu J, Kumar M, Dawson VL, et al. (2017). Toward the human cellular microRNAome. Genome Res. 27, 1769–1781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neilsen CT, Goodall GJ, and Bracken CP (2012). IsomiRs--the overlooked repertoire in the dynamic microRNAome. Trends Genet. 28, 544–549. [DOI] [PubMed] [Google Scholar]
- Pall GS, and Hamilton AJ (2008). Improved northern blot method for enhanced detection of small RNA. Nat. Protoc. 3, 1077–1084. [DOI] [PubMed] [Google Scholar]
- Pangala SR, Enrich-Prast A, Basso LS, Peixoto RB, Bastviken D, Hornibrook ERC, Gatti LV, Marotta H, Calazans LSB, Sakuragui CM, et al. (2017). Large emissions from floodplain trees close the Amazon methane budget. Nature 552, 230–234. [DOI] [PubMed] [Google Scholar]
- Park JH, Shin S-Y, and Shin C (2017). Non-canonical targets destabilize microRNAs in human Argonautes. Nucleic Acids Res. 45, 1569–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquinelli AE (2012). MicroRNAs and their targets: recognition, regulation and an emerging reciprocal relationship. Nat. Rev. Genet. 13, 271–282. [DOI] [PubMed] [Google Scholar]
- Rupaimoole R, and Slack FJ (2017). MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat. Rev. Drug Discov. 16, 203–222. [DOI] [PubMed] [Google Scholar]
- Salomon WE, Jolly SM, Moore MJ, Zamore PD, and Serebrov V (2015). Single-Molecule Imaging Reveals that Argonaute Reshapes the Binding Properties of Its Nucleic Acid Guides. Cell 162, 84–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schirle NT, Sheu-Gruttadauria J, and MacRae IJ (2014). Structural basis for microRNA targeting. Science 346, 608–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz DS, Hutvágner G, Du T, Xu Z, Aronin N, and Zamore PD (2003). Asymmetry in the assembly of the RNAi enzyme complex. Cell 115, 199–208. [DOI] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. (2014). Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Nam J-W, Farh KK-H, Chiang HR, Shkumatava A, and Bartel DP (2010). Expanding the microRNA targeting code: functional sites with centered pairing. Mol. Cell 38, 789–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan GC, Chan E, Molnar A, Sarkar R, Alexieva D, Isa IM, Robinson S, Zhang S, Ellis P, Langford CF, et al. (2014). 5’ isomiR variation is of functional and evolutionary importance. Nucleic Acids Res. 42, 9424–9435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Telonis AG, Magee R, Loher P, Chervoneva I, Londin E, and Rigoutsos I (2017). Knowledge about the presence or absence of miRNA isoforms (isomiRs) can successfully discriminate amongst 32 TCGA cancer types. Nucleic Acids Res. 45, 2973–2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JE, Chang H-M, Piskounova E, and Gregory RI (2012). Lin28-mediated control of let-7 microRNA expression by alternative TUTases Zcchc11 (TUT4) and Zcchc6 (TUT7). RNA 18, 1875–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton JE, Du P, Jing L, Sjekloca L, Lin S, Grossi E, Sliz P, Zon LI, and Gregory RI (2014). Selective microRNA uridylation by Zcchc6 (TUT7) and Zcchc11 (TUT4). Nucleic Acids Res. 42, 11777–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Treiber T, Treiber N, and Meister G (2019). Regulation of microRNA biogenesis and its crosstalk with other cellular pathways. Nat. Rev. Mol. Cell Biol. 20, 5–20. [DOI] [PubMed] [Google Scholar]
- Wang Y, Juranek S, Li H, Sheng G, Tuschl T, and Patel DJ (2008). Structure of an argonaute silencing complex with a seed-containing guide DNA and target RNA duplex. Nature 456, 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westholm JO, Ladewig E, Okamura K, Robine N, and Lai EC (2012). Common and distinct patterns of terminal modifications to mirtrons and canonical microRNAs. RNA 18, 177–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman SK, Knouf EC, Parkin RK, Fritz BR, Lin DW, Dennis LM, Krouse MA, Webster PJ, and Tewari M (2011). Post-transcriptional generation of miRNA variants by multiple nucleotidyl transferases contributes to miRNA transcriptome complexity. Genome Res. 21, 1450–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane D, Selitsky SR, Shimakami T, Li Y, Zhou M, Honda M, Sethupathy P, and Lemon SM (2017). Differential hepatitis C virus RNA target site selection and host factor activities of naturally occurring miR-122 3’ variants. Nucleic Acids Res. 45, 4743–4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu F, Pillman KA, Neilsen CT, Toubia J, Lawrence DM, Tsykin A, Gantier MP, Callen DF, Goodall GJ, and Bracken CP (2017). Naturally existing isoforms of miR-222 have distinct functions. Nucleic Acids Res. 45, 11371–11385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Yu Y, Zhai J, Ramachandran V, Dinh TT, Meyers BC, Mo B, and Chen X (2012). The Arabidopsis nucleotidyl transferase HESO1 uridylates unmethylated small RNAs to trigger their degradation. Curr. Biol. 22, 689–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou H, Arcila ML, Li Z, Lee EJ, Henzler C, Liu J, Rana TM, and Kosik KS (2012). Deep annotation of mouse iso-miR and iso-moR variation. Nucleic Acids Res. 40, 5864–5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. DNA Oligonucleotides Used in This Study, Related to STAR Methods
Table S2. Predicted Targets of miR-27a, Related to Figure 6
Data Availability Statement
All the deep sequencing datasets can be found in GEO with the accession number GSE121327. Raw image files of Northern Blots used to prepare figure panels in the main text and supplementary information can be found at Mendeley Data:http://dx.doi.org/10.17632/pp5zz5rwvs.1