

Abstract
Insulin gene coding sequence mutations are known to cause Mutant INS-gene-induced Diabetes of Youth (MIDY), yet the cellular pathways needed to prevent misfolded proinsulin accumulation remain incompletely understood. Here we report that Akita mutant proinsulin forms detergent-insoluble aggregates that entrap wild type (WT) proinsulin in the endoplasmic reticulum (ER), thereby blocking insulin production. Two distinct quality control mechanisms operate together to combat this insult: the ER luminal chaperone Grp170 prevents proinsulin aggregation, while the ER membrane morphogenic protein RTN3 disposes of aggregates via ER-coupled autophagy (ER-phagy). We show that enhanced RTN-dependent clearance of aggregated Akita proinsulin helps to restore ER export of WT proinsulin, which can promote WT insulin production, potentially alleviating MIDY. We also find that RTN3 participates in the clearance of other mutant prohormone aggregates. Together, these results identify a series of substrates of RTN3-mediated ER-phagy, highlighting RTN3 in the disposal of pathogenic prohormone aggregates.
Graphical Abstract
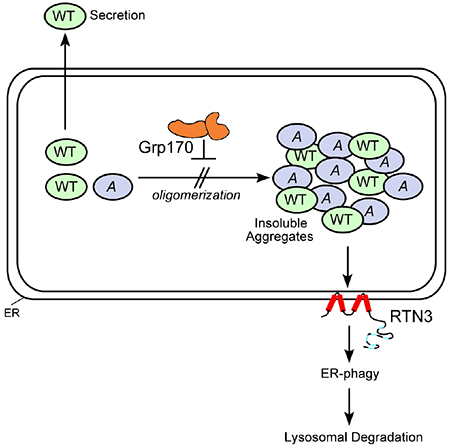
INTRODUCTION
Formation of protein aggregates is a signature of many protein misfolding diseases, including Alzheimer’s disease, Parkinson’s disease, Huntington’s disease, and diabetes (Bates, 2003; Irvine et al., 2008; Mukherjee et al., 2015). Not surprisingly, there is intense interest in learning how to stimulate cellular mechanisms that decrease the levels of potentially toxic aggregates to combat disease. In principle, these mechanisms may be comprised of pre-emptively preventing the formation of such aggregates, as well as either disaggregation or clearance of the aggregated species once they have formed. For the development of effective therapies, it may be necessary to stimulate more than one mechanism to decrease aggregate accumulation.
Proinsulin is the precursor of insulin, a hormone secreted by pancreatic β-cells that controls blood glucose levels (Steiner et al., 1967). Proinsulin biosynthesis is initiated when the precursor preproinsulin translocates into the endoplasmic reticulum (ER) (Chan et al., 1976). Upon translocation into the ER, the signal sequence of preproinsulin is removed to generate proinsulin, which contains the B chain, connecting C-peptide, and the A-chain. Folding of proinsulin ensues with concomitant generation of three conserved disulfide bonds (cysteine in the 7th residue of the B chain linked to cysteine in the 7th residue of the A-chain denoted as the B7-A7 disulfide bond, plus the B19-A20 and A6-A11 disulfide bonds). Upon proper folding, proinsulin exits the ER and is transported to the Golgi, and further, to immature secretory granules. In the granules, bioactive insulin is formed when the C-peptide is excised, with the A and B chains connected via the two interchain disulfide bonds. Exocytosis of secretory granules ultimately releases bioactive insulin to the bloodstream.
In the autosomal-dominant diabetic syndrome called Mutant INS gene–induced Diabetes of Youth (MIDY), misfolded mutant proinsulin is thought to initiate disease (Liu et al., 2010b; Stoy et al., 2007; Weiss, 2009) with association and impaired trafficking of wild-type proinsulin (Cunningham et al., 2017; Hodish et al., 2010; Liu et al., 2010a; Liu et al., 2007). Over 30 missense mutations in the human INS gene have been identified; in almost all cases, MIDY proinsulin mutants cannot fold properly and consequently fail to become bioactive insulin (Arunagiri et al., 2018). Instead, they are entrapped in the ER, dominantly interfering with the folding of wild type (WT) proinsulin which also becomes impaired in its exit from the ER, decreasing WT insulin production (Liu et al., 2010a; Liu et al., 2010b). However, whether misfolding of the mutant proinsulin leads to insoluble protein aggregation of either the mutant or WT gene product, is currently unknown. What is known is that decreased circulating insulin renders patients (and animal models) susceptible to increased blood glucose that provokes further proinsulin biosynthesis (both mutant and WT), exacerbating proinsulin misfolding in the ER and resulting in enhanced ER stress with eventual β-cell failure.
The best-characterized MIDY mutant is Akita proinsulin (herein simply called ‘Akita’), in which cysteine in the 7th position of the A chain is mutated to tyrosine, leaving cysteine in the 7th position of the B chain unpaired (Wang et al., 1999). This lone cysteine forms non-native disulfide bonds, leading to intermolecular disulfides engaging additional Akita molecules, which generate high molecular weight (MW) protein complexes (Cunningham et al., 2017; Liu et al., 2007). The accumulating presence of such complexes recruits WT proinsulin, preventing its ER exit and secretion, which leads to disease (Liu et al., 2010b). The ultimate sizes of these Akita-containing high MW complexes, and the spectrum of ER protein quality control pathways available to limit the build-up of Akita-containing high MW complexes, are questions of direct relevance to the pathogenesis and treatment of MIDY.
To date, we and others have demonstrated that a major ER quality control pathway called ER-associated degradation (ERAD) can dispose of soluble forms of Akita (Allen et al., 2004; Cunningham et al., 2017; He et al., 2015; Liu et al., 2010a). ERAD targets misfolded ER proteins for retrotranslocation to the cytosol for proteasomal destruction (Ruggiano et al., 2014; Smith et al., 2011; Tsai et al., 2002). We recently reported that the ER-resident chaperone Grp170, an atypical Hsp70 ATPase family member, facilitates ERAD of soluble Akita which enhances WT insulin secretion (Cunningham et al., 2017). However, the mechanistic basis by which Grp170 facilitates ERAD could potentially involve the targeting of Akita monomers, complexes (small or large) or even massive insoluble species.
Here we demonstrate that in the ER, Akita forms detergent-insoluble protein aggregates that entrap WT proinsulin. Strikingly, we find that two distinct ER quality control pathways are deployed to limit the levels of these aggregates. First, Grp170 acts to prevent formation of detergent-insoluble Akita aggregates which otherwise recruit and capture WT proinsulin. Second, to the extent that detergent-insoluble aggregates form despite Grp170 action, an ER-phagy pathway that relies critically upon reticulon3 (RTN3) actively removes these species. Remarkably, clearance of aggregated Akita restores proper intracellular transport of WT proinsulin. These results suggest that insoluble aggregates of Akita with WT proinsulin drives pathogenesis of MIDY, and reveal a new potential therapeutic strategy to block the disease. We also report that RTN3-dependent ER-phagy disposes of two additional, unrelated prohormone aggregates. Thus, our data identified substrates of RTN3-mediated ER-phagy, and highlight a general role of RTN3 in the disposal of pathogenic insoluble protein complexes in the ER.
RESULTS
Depletion of Grp170 promotes formation of detergent-insoluble Akita proinsulin
We previously used sucrose gradient analysis to analyze the oligomeric state of Akita (Cunningham et al., 2017). In this experiment, HEK 293T cells transiently expressing Akita-Myc (in which Myc is inserted within the C-peptide) were lysed in RIPA buffer (containing 1.0% Triton X-100 and 0.1% SDS) before sedimentation on a 10%-50% discontinuous sucrose gradient. After centrifugation, fractions were collected and subjected to reducing SDS-PAGE and immunoblotting using an antibody against Myc. As previously reported, Akita-Myc was found in all fractions (Cunningham et al., 2017). In this report, we have defined fractions 1-4 as low MW Akita species, fractions 5-8 as the mid MW species, and fractions 9-12 as high MW species (Fig. 1A).
Figure 1. Depletion of Grp170 promotes formation of detergent-insoluble Akita.
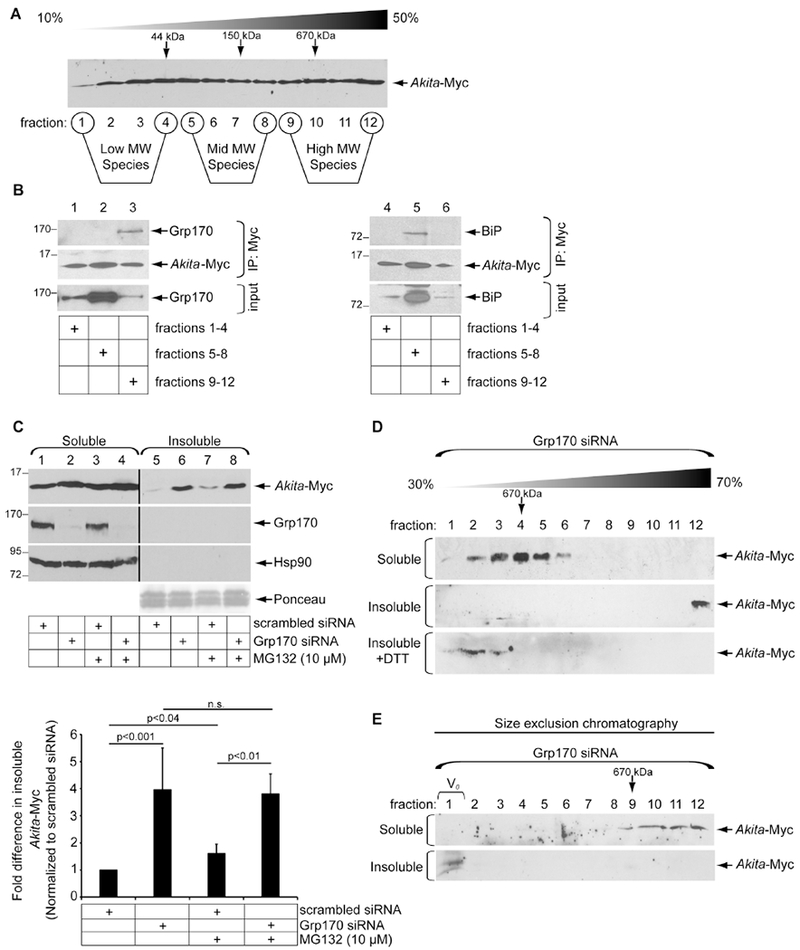
A. Whole cell extract derived from HEK 293T cells transfected with Akita-Myc was layered over a 10-50% sucrose gradient. The sample was centrifuged, and each fraction was subjected to SDS-PAGE followed by immunoblotting with an Myc antibody. B. Fractions 1-4, 5-8, and 9-12 were pooled, and Akita-Myc from the pooled fractions were immunoprecipitated. The precipitated material was subjected to SDS-PAGE and immunoblotted with the indicated antibodies (n=5). C. Cells expressing Akita-Myc and transfected with either a scrambled (control) or Grp170 siRNA were treated with or without MG132, and lysed with a RIPA buffer. After centrifugation, the supernatant represents the soluble fraction and the pellet represents the insoluble fraction; material in the insoluble fraction was resuspended in a sample buffer containing 2% SDS. Both the soluble and insoluble fractions were subjected to SDS-PAGE and immunoblotted with the indicated antibodies. Protein levels revealed by Ponceau staining serve as the loading control. The intensity of the Akita-Myc band was quantified with ImageJ (NIH). The bottom graph depicts the insoluble Akita-Myc level normalized to the level of insoluble Akita-Myc in the control. Data represent the mean ± SD (n=3); p-values are generated from student’s T-test. The black line indicates that an intervening lane from the same blot has been excised. D. The soluble (first panel) and insoluble (second panel) fractions derived from Grp170-depleted cells expressing Akita-Myc were layered over a 30-70% sucrose gradient and centrifuged as in A. Each fraction was collected, subjected to SDS-PAGE, and immunoblotted with an antibody against Myc. The insoluble fraction was also treated with DTT prior to sucrose gradient analysis (third panel) (n=3). E. Akita-Myc in the soluble and insoluble fractions was isolated by Myc-IP followed by Myc elution. Isolated Akita was subjected to FPLC using a Superose 6 Increase 3.2/300 column. Fractions were collected and subjected to SDS-PAGE and immunoblotted (n=3).
Although Grp170 binds to Akita and promotes ERAD of Akita (Cunningham et al., 2017), it is unclear which size Akita complexes this chaperone preferentially interacts with. In a discontinuous sucrose gradient, the bulk of cellular Grp170 in the steady state resides in the middle of the gradient (Fig. 1B, first set of panels). Akita in the low, mid, and high MW species were then immunoprecipitated (with anti-Myc) and subjected to SDS-PAGE followed by immunoblotting with an antibody against Grp170 (Fig. 1B, first set of panels) or the ER-resident chaperone BiP (second set of panels). Strikingly, Grp170 co-precipitated with Akita from the high MW species (Fig. 1B, lane 3) whereas pull-down of Akita from the mid MW species co-precipitated BiP (Fig. 1B, second set of panels, lane 5). These findings demonstrate that Grp170 preferentially engages the high MW Akita complexes.
What might be the functional significance of Grp170 binding to high MW Akita complexes? One possibility is that this binding prevents Akita from extending even further to form insoluble protein complexes. To determine if Grp170 prevents higher order Akita aggregation, we assessed the extent to which Akita can be extracted into the soluble phase in RIPA buffer — the molecules resistant to extraction are referred to as insoluble aggregates — but can be re-solubilized under stronger detergent conditions (1%-2% SDS). Using this method, we found that in cells expressing Akita-Myc and transfected with a control siRNA, Akita was extracted into the soluble fraction with a negligible pool of insoluble molecules (Fig. 1C, first panel, compare lane 1 to 5). By contrast, upon knockdown of Grp170 [siRNA #1, (Cunningham et al., 2017)], the Akita-Myc level in the insoluble fraction increased significantly (Fig. 1C, first panel, compare lane 6 to 5; quantified in right graph). Grp170 knockdown also modestly increased the Akita level in the soluble fraction (Fig. 1C, first panel, compare lane 2 to 1). Appearance of Akita in the insoluble phase is not due to non-specific partitioning because neither Grp170 nor cytosolic Hsp90 appeared in this fraction under any condition (Fig. 1C, second and third panels). Hence, the observation that depleting Grp170 increased the level of aggregated Akita suggests that Grp170 functions to prevent Akita aggregation.
To probe the size of Akita complexes in the soluble and insoluble fraction, we layered these fractions from Grp170-depleted cells over a heavier 30%-70% discontinuous sucrose gradient. Whereas soluble Akita migrated to the lighter fractions (2-6) as expected, insoluble Akita was found only in the densest fraction (12) (Fig. 1D, compare first to second panels). However, when Akita from the insoluble fraction was pretreated with the reducing agent dithiothreitol (DTT), it migrated to lighter fractions (2-3) (Fig. 1D, third panel), indicating that insoluble Akita consists of a massive disulfide-bonded complex. The sizes of soluble and insoluble Akita were further analyzed by size exclusion chromatography. In this approach, Akita-Myc was immunoprecipitated from the soluble or insoluble fraction, followed by release of Akita-Myc from the antibody with Myc peptide. The eluted material was then subjected to size exclusion chromatography. While soluble Akita-Myc is present in fractions corresponding to proteins approximating 660 kDa or smaller, insoluble Akita-Myc was found in the V0 void volume fraction (Fig. 1E, compare first to second panels). Because the reported exclusion size limit of the column is ≥ 40,000 kDa, the insoluble Akita represents a massive protein complex. These data are in agreement with the sucrose gradient analysis.
Given the large size of insoluble Akita, we asked to what extent proteasomal activity might play a role in clearing or preventing these insoluble aggregates in control cells (transfected with scrambled siRNA). However, our findings demonstrate that in control cells, treatment with the proteasome inhibitor MG132 increased the Akita level in the insoluble fraction to a lesser extent than in cells with Grp170 knockdown (Fig. 1C, first panel, compare lane 7 to 5; quantified in the right graph). Similarly, in cells transfected with Grp170 siRNA, the level of insoluble Akita was not enhanced by MG132 (Fig. 1C, first panel, compare lane 8 to 6; quantified in the graph below). These findings raise the possibility that a proteasome-independent pathway is used to remove aggregated Akita (see below).
Grp170 prevents formation of aggregated Akita in vitro
Our cell-based results demonstrate that Grp170 prevents accumulation of insoluble Akita aggregates. In parallel, we developed a system to directly test whether Akita-Myc released from an ER-containing fraction is predisposed to form aggregates in vitro. This simple assay is based on the idea that Akita-Myc aggregates can be pelleted by centrifugation (Fig. 2A, top). If so, addition of Grp170 could then be tested for its ability to prevent Akita-Myc aggregation (Fig. 2A, bottom).
Figure 2. Grp170 prevents formation of aggregated Akita in vitro.
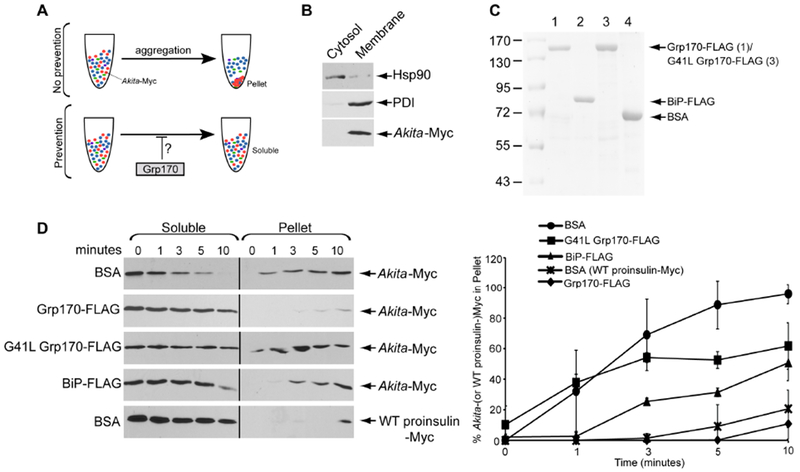
A. Schematic of the in vitro aggregation assay. (top) When Akita-Myc in a membrane extract aggregates, it forms a pellet after centrifugation. (bottom) However, if the extract containing Akita-Myc was incubated with Grp170, aggregation is prevented. Thus Akita-Myc remains soluble even after centrifugation. B. To generate Akita-Myc in the membrane extract, HEK 293T cells expressing Akita-Myc were treated with digitonin and centrifuged to generate two fractions. The supernatant fraction contains cytosol proteins (cytosol) and the pellet fraction harbors membranes including the ER (membrane). Both fractions were immunoblotted with the indicated antibodies. C. Coomassie gel of FLAG-tagged Grp170, BiP, G41L Grp170, and BSA. D. (1st – 4th panels) Membrane fraction containing Akita-Myc was incubated with the indicated protein for the indicated time. The samples were centrifuged, and the supernatant material separated from the pellet; the pellet material was resuspended in a sample buffer containing 2% SDS. Both the supernatant and pellet material were analyzed by SDS-PAGE and immunoblotted with an antibody against Myc. (5th panel) Membrane extract containing WT proinsulin-Myc was incubated with BSA and processed as above. The right graph represents the percentage of Akita (or WT proinsulin)-Myc in the pellet fraction compared to the total Akita-Myc signal at each time point from both the soluble and pellet fractions. Data represented the mean ± SD (n=3). The black line indicates that an intervening lane from the same blot has been excised.
To isolate soluble Akita-Myc, HEK 293T cells expressing Akita-Myc were treated with a low digitonin concentration (0.01%) and centrifuged to generate a supernatant that harbors cytosolic proteins and a pellet that contains the membrane fraction including the ER. When the cytosol and membrane fractions were subjected to SDS-PAGE, cytosolic Hsp90 was found predominantly in the cytosol fraction whereas the ER-resident PDI was present only in the membrane fraction (Fig. 2B). Importantly, Akita-Myc partitioned only in the membrane fraction (Fig. 2B, third panel) and the soluble pool of proteins from this fraction were released with RIPA buffer. In parallel, we immunopurified FLAG-tagged WT Grp170 (Grp170-FLAG), BiP (BiP-FLAG), and an ATPase-defective Grp170 (G41L Grp170-FLAG) from HEK 293T cells (Fig. 2C).
Remarkably, when soluble Akita-Myc was mixed with a control protein (bovine serum albumin, BSA) and incubated for various times, Akita-Myc rapidly became insoluble as detected by centrifugation: it was depleted from the soluble fraction within 10 min while increasing in the pellet (Fig. 2D upper panel; Akita-Myc in the pellet is quantified in the right graph). In contrast, when soluble WT proinsulin-Myc was incubated with BSA and processed similarly, it remained mostly soluble (Fig. 2D. fifth panel; quantified in the right graph). Strikingly, when purified Grp170-FLAG (but not the ATPase-deficient G41L Grp170-FLAG) was added, Akita-Myc solubility was preserved (Fig. 2D, compare second to first and third panels; quantified in the right graph). Incubation of soluble Akita-Myc with an equal amount of BiP-FLAG also prevented insolubility of Akita, but was less efficient than Grp170-FLAG (Fig. 2D, compare fourth to second panel; quantified in the right graph). Together, these findings support the cell-based results (Fig. 1), demonstrating that mutant Akita proinsulin has a high propensity to aggregate, and that Grp170 uses an energy-driven reaction to maintain Akita solubility.
Grp170 prevents WT proinsulin from entering detergent-insoluble aggregates
Because Akita dominantly interferes with WT proinsulin folding (Liu et al., 2007), insoluble Akita may recruit WT proinsulin entry into these insoluble aggregates. If so, Grp170’s ability to prevent formation of insoluble Akita aggregates might also prevent WT proinsulin aggregation. To test this, cells expressing superfolder GFP-tagged WT proinsulin (in which sfGFP is inserted in the C-peptide) were transfected with scrambled or Grp170 siRNA, along with either an empty vector control or Akita-Myc. Indeed, expressing Akita-Myc stimulated WT proinsulin-sfGFP to enter into the detergent-insoluble fraction (Fig. 3A, first panel, compare lane 7 to 5; quantified in the right graph), supporting the idea that Akita promotes aggregation of WT proinsulin. Knockdown of Grp170 increased the level of WT proinsulin-sfGFP in the detergent-insoluble fraction, and formation of the insoluble WT proinsulin-sfGFP was essentially additive in the presence of Akita (Fig. 3A, first panel, lanes 6-8; quantified in the right graph). Similar results were found when the experiments were repeated in the more physiologically-relevant rat pancreatic β-cell line (INS1 832/13) (Fig. 3B). Together, these data indicate that Grp170 prevents aggregation of WT proinsulin recruited by aggregated Akita. Intriguingly, the finding that Grp170 prevents WT proinsulin aggregation in the absence of Akita suggests that Grp170 may also protect against misfolding of WT proinsulin.
Figure 3. Grp170 prevents WT proinsulin from entering detergent-insoluble aggregates.
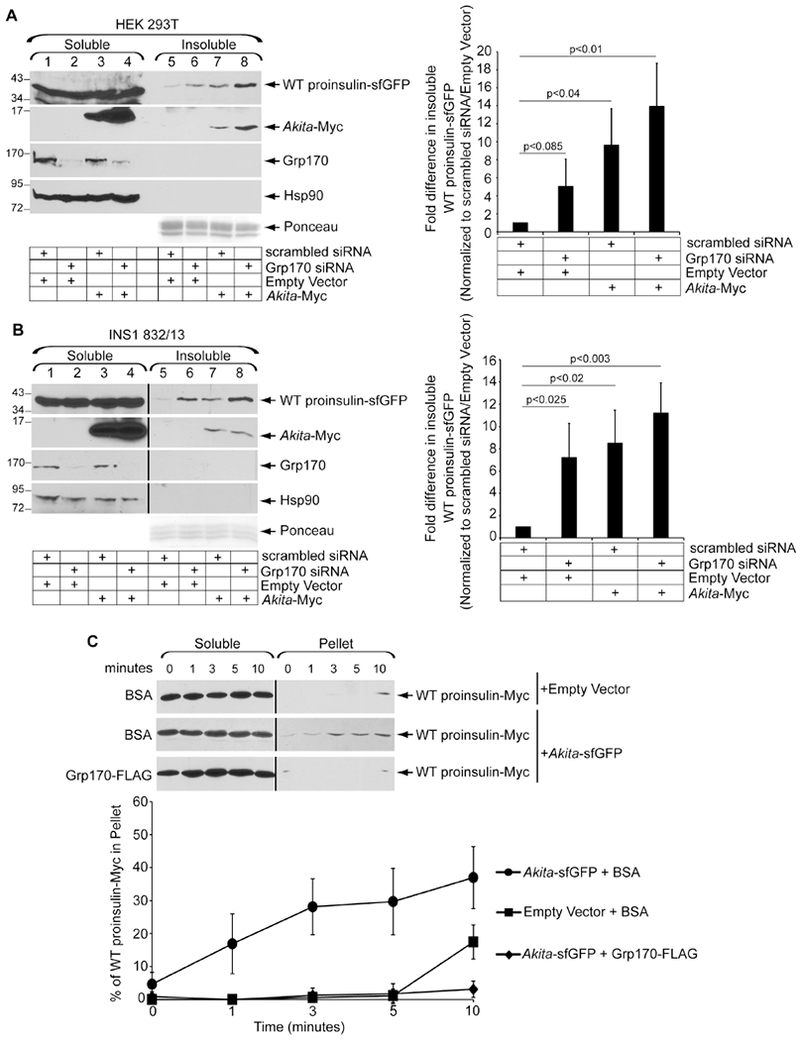
A. HEK 293T cells expressing WT proinsulin-sfGFP were transfected with scrambled or Grp170 siRNA and either Akita-Myc or an empty vector. Cells were processed to generate the soluble and insoluble fractions as in 1C. The right graph depicts the insoluble WT proinsulin-sfGFP level normalized to the level of insoluble WT proinsulin-sfGFP present in the scrambled/empty vector control. Data represent the mean ± SD (n=3); p-values are generated from student’s T-test. B. As in A, except data were generated from rat INS1 832/13 cells (n=3). The black line indicates that an intervening lane from the same blot has been excised. C. In vitro aggregation of WT proinsulin-Myc. Membrane extract derived from cells transfected with either WT proinsulin-Myc and an empty vector, or WT proinsulin-Myc and Akita-sfGFP were incubated with BSA or Grp170-FLAG. Samples were processed as in 2D. The bottom graph represents the percentage of WT proinsulin-Myc in the pellet fraction as compared to the total WT proinsulin-Myc signal at each time point from both the soluble and pellet fractions. Data represents the mean ± SD (n=3). The black line indicates that an intervening lane from the same blot has been excised.
We used the in vitro system (Fig. 2) to further examine this finding. WT proinsulin-Myc was expressed in HEK 293T cells and solubilized from an ER-enriched fraction, using the same method noted above for Akita-Myc. When soluble WT proinsulin-Myc was incubated with BSA and the sample processed as before, a low level of WT proinsulin-Myc in the pellet fraction was detected (Fig. 3C, first panel; quantified in the bottom graph). However, if soluble WT proinsulin-Myc was co-isolated from cells co-expressing sfGFP-tagged Akita (Akita-sfGFP, in which sfGFP is appended within the C-peptide), incubation with BSA now resulted in increased insolubility of WT proinsulin-Myc (Fig. 3C, second panel; quantified in the bottom graph), consistent with the idea that Akita promotes WT proinsulin aggregation. Strikingly, if purified Grp170-FLAG was added, WT proinsulin aggregation was blocked (Fig. 3C, third panel; quantified in the bottom graph), supporting the idea that Grp170 prevents WT proinsulin from recruitment into aggregates promoted by Akita.
Beclin1/ULK2/Atg5-dependent autophagy disposes of aggregated Akita
Because additional mechanisms beyond Grp170 are likely to limit accumulation of Akita aggregates (Fig. 1C), we sought to better understand the disposal pathways participating in clearance of these insoluble species. Two major ER quality control pathways are 1) ERAD and 2) ER-phagy, a degradative pathway that relies on elements of the macro-autophagy machinery in which subdomains of the ER are delivered to the lysosome for degradation (Bernales et al., 2006; Bernales et al., 2007). Hence, we compared the extent to which the key ERAD component Hrd1 (Bays et al., 2001; Bordallo et al., 1998; Hampton et al., 1996) or a critical regulator of macro-autophagy Beclin1 (Cao and Klionsky, 2007; Kang et al., 2011) play roles in clearance of insoluble Akita aggregates. Remarkably, our results demonstrated that knockdown of Beclin1 but not Hrd1 robustly increased the level of Akita-Myc in the detergent-insoluble fraction (Fig. 4A, top panel, compare lane 8 to 6; quantified in the right graph). Although Hrd1-dependent ERAD is unlikely to be responsible for removing insoluble Akita, it is possible that other E3 ligases coupled to ERAD might clear it. Nevertheless, Beclin1 knockdown caused insoluble Akita-Myc to increase to a level even higher than that observed with Grp170 knockdown (Fig. 4A, top panel, compare lane 8 to 7; quantified in the graph below). Not surprisingly, sucrose sedimentation analysis revealed that the accumulation of aggregated Akita-Myc after Beclin1 knockdown represents a massive protein complex, which is significantly larger than Akita-Myc in the soluble fraction (Fig. 4B). These results strongly suggest that macro-autophagy disposes insoluble Akita.
Figure 4. Beclin1/ULK2/Atg5-dependent autophagy disposes aggregated Akita.
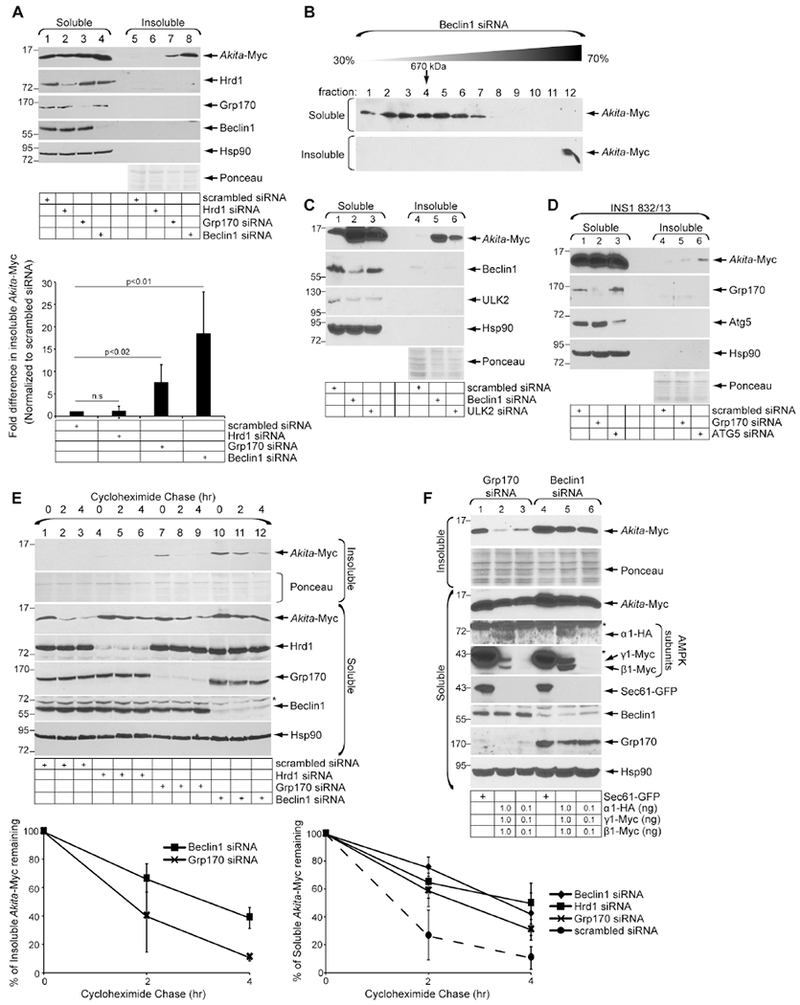
A. HEK 293T cells were transfected with Akita-Myc and the indicated siRNAs. Soluble and insoluble fractions were generated and processed as in 1C. The bottom graph represents insoluble Akita-Myc level normalized to the level of insoluble Akita-Myc in the scrambled control. Data represent the mean ± SD (n=4); p-values from student’s T-test. B. As in 1D, except the soluble (first panel) and insoluble (second panel) fractions were derived from Beclin1-depleted cells (n=3). C. As in A, except cells were transfected with the indicated siRNAs (n=3). D. INS1 832/13 cells were transfected with Akita-Myc and the indicated siRNAs. Soluble and insoluble fractions were generated and processed as in 1C (n=3). E. HEK 293T cells expressing Akita-Myc and transfected with indicated siRNAs were treated with cycloheximide for 0, 2, 4 h. Soluble and insoluble fractions were generated as in 1C. Samples were subjected to SDS-PAGE and immunoblotted with the indicated antibodies. The bottom graphs represent the percentage of insoluble (left) or soluble (right) Akita-Myc remaining. Data represents mean ±SD (n=4). Asterisks indicate non-specific bands. F. HEK 293T cells expressing Akita-Myc were co-transfected with the indicated siRNA and DNA constructs. Soluble and insoluble fractions were generated as in 1C (n=3); asterisks indicate non-specific bands.
To further establish a role of macro-autophagy in clearance of insoluble Akita, we assessed the functions of two additional autophagic components, ULK2 and Atg5. Indeed, knockdown of ULK2 robustly increased the level of insoluble Akita (Fig. 4C, first panel, compare lanes 6 to 4), and depletion of Atg5 (in the rat INS1 832/13 pancreatic β-cell line) also enhanced the level of transfected Akita-Myc in the insoluble fraction (Fig. 4D, first panel, compare lanes 6 to 4). These results further support the idea that macro-autophagy plays a critical role in clearing insoluble Akita aggregates.
We next assessed the degradation rate of Akita-Myc in the insoluble and soluble fractions under different knockdown conditions. Whereas Akita-Myc in the insoluble fraction was rapidly degraded when Grp170 was depleted, its degradation was more stabilized in Beclin1-depleted cells (Fig. 4E, top panel, compare lanes 7-9 to 10-12; quantified in the top right graph; degradation of insoluble Akita-Myc in Hrd1-depleted cells was not further analyzed due to the low level of the mutant proinsulin protein at the initial time point). These findings, demonstrating that loss of Beclin1 significantly impairs the clearance of aggregated Akita, reinforce the idea that Beclin1-dependent autophagy is critical for degrading insoluble Akita aggregates. In contrast, degradation of Akita-Myc in the soluble fraction is impaired by depletion of either Hrd1, Grp170, or Beclin1 (Fig. 4E, third panel, compare lanes 4-6, 7-9, and 10-12 to 1-3; quantified in the right bottom graph), indicating that all three proteins participate in the turnover of soluble Akita. By limiting aggregation of Akita, Grp170 retains mutant proinsulin in the soluble phase, enabling it to undergo Hrd1-mediated ERAD (He et al., 2015). However, the apparent role of Beclin1 in controlling the fate of soluble Akita might be indirect, i.e., buildup of Akita in the insoluble fraction in Beclin1-deficient cells could promote back-up into the soluble fraction.
Finally, we asked whether activation of autophagy might clear the Akita aggregates. To activate autophagy, we simultaneously overexpressed the α1, β1, and γ1 AMP Kinase (AMPK) subunits (Kim et al., 2011; Egan et al., 2011) in Grp170 or Beclin1 knockdown cells. Whereas overexpressing the AMPK subunits removed the Akita aggregates generated under Grp170 knockdown conditions (Fig. 4F, first panel, compare lanes 2-3 to lane 1), overexpressing the AMPK subunits was unable to clear the Akita aggregates in Beclin1-depleted cells (Fig. 4F, first panel, compare lanes 5-6 to lane 4). These results demonstrate that activation of autophagy via AMPK induction can clear aggregated Akita via a Beclin1-dependent pathway.
RTN3-dependent ER-phagy promotes degradation of Akita aggregates
Because Beclin1-mediated macro-autophagy removes detergent-insoluble Akita aggregates from the ER, we asked if specific ER membrane proteins involved in ER-phagy participate in this pathway. Strikingly, similar to Beclin1 knockdown, depleting RTN3 [but not CCPG1, Sec62, or FAM134B (Fumagalli et al., 2016; Grumati et al., 2017; Khaminets et al., 2015; Smith et al., 2018)] increased the level of Akita-Myc in the detergent-insoluble fraction (Fig. 5A, first panel, compare lane 10 to 7). RTN3 exists as a long (3A) form that contains an N-terminal extension protruding into the cytosol, and a short (3C) form lacking this extension; the antibody used in this experiment does not recognize RTN3C in 293T cells. Because knockdown of CCPG1 and FAM134B moderately decreased the Akita-Myc level in the soluble fraction (Fig. 5A, first panel, compare lanes 2 and 5 to 1), we asked if this could be accounted for by an inhibition of Akita-Myc translation. Accordingly, we performed 35S-amino acid pulse-labeling experiments to directly examine the relative amounts of Akita-Myc biosynthesis under the different knockdown conditions, and found no differences in the synthesis of Akita-Myc when either CCPG1 or FAM134B was knocked down (Fig. S1A). These data indicate that the modest decrease in soluble Akita-Myc in cells depleted of CCPG1 or FAM134B is unlikely due to an impairment of protein translation, raising the possibility that protein degradation of soluble Akita-Myc may account for this observation. Additionally, the small increase of Akita-Myc biosynthesis (Fig. S1A) cannot account for the significant increase in the level of insoluble Akita aggregates observed when RTN3 or Beclin1 are knocked down (Fig. 5A). Our analyses thus suggest that RTN3 exerts an important function in clearance of aggregated Akita from the ER.
Figure 5. RTN3-dependent ER-phagy promotes degradation of Akita aggregates.
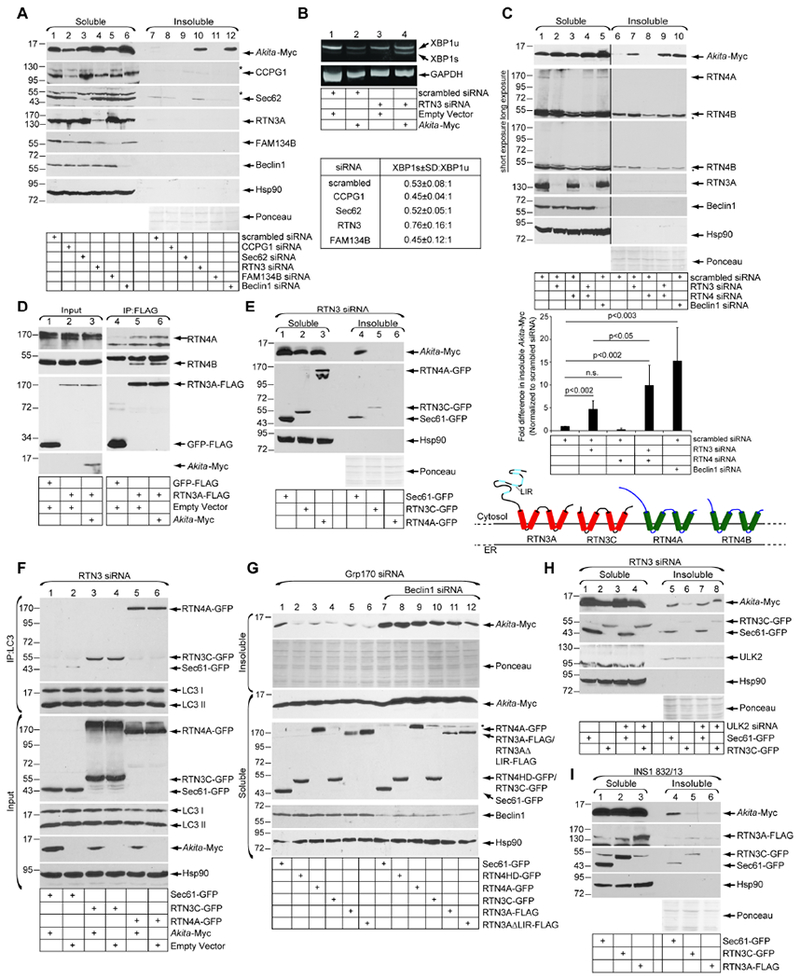
A. siRNA screen of known ER membrane proteins involved in ER-phagy. HEK 293T cells expressing Akita-Myc were transfected with the indicated siRNAs. Soluble and insoluble fractions were generated and processed as in 1C (n=3). Asterisks indicate non-specific bands. B. HEK 293T cells transfected with an empty vector or Akita-Myc were co-transfected with either scrambled or RTN3 siRNA. RNA was extracted, reverse transcribed to cDNA, and the cDNA amplified using PCR. The lower table represents quantification of the ratio of XBP1s (spliced) ±SD:XBP1u (unspliced) under the indicated knockdown conditions in cells expressing Akita-Myc (n=3). C. Cells transfected with the indicated siRNA(s) were processed as in A. The bottom graph represents insoluble Akita-Myc level normalized to the level of insoluble Akita-Myc in the scrambled control. Data represent the mean ± SD (n=5); p-values from student’s T-test. The schematic depicts the topology of RTN3A, RTN3C, RTN4A, RTN4B. The black line indicates that an intervening lane from the same blot has been excised and asterisks indicate nonspecific bands. D. HEK 293T cells transfected with the indicated constructs were chemically crosslinked with DSP. The resulting cell extract was subjected to FLAG-immunoprecipitation, with the precipitated material analyzed by SDS-PAGE and immunoblotting (n=3). E. RTN3-depleted cells expressing Akita-Myc were transfected with Sec61β -GFP, RTN3C-GFP, or RTN4A-GFP. Soluble and insoluble fractions were generated and processed as in 1C (n=4). F. HEK 293T cells were transfected with Akita-Myc and either Sec61β-GFP, RTN3C-GFP, or RTN4A-GFP under RTN3 knockdown. Endogenous LC3 was immunoprecipitated, and the precipitated material was subjected to SDS-PAGE and immunoblotted with the indicated antibodies (n=4). G. Cells expressing Akita-Myc that were depleted of either Grp170 alone or Grp170 and Beclin1 were transfected with the indicated constructs. The soluble and insoluble fractions were generated and processed as in 1C (n=3). Asterisks indicate non-specific bands. H. HEK 293T cells expressing Akita-Myc that were depleted of either RTN3 alone or RTN3 and ULK2 were transfected with either Sec61β-GFP or RTN3C-GFP. The soluble and insoluble fractions were generated and processed as in 1C (n=3). I. INS1 832/13 cells was transfected with the indicated constructs. The soluble and insoluble fractions were generated and processed as in 1C (n=3).
In addition to the increase in insoluble Akita aggregates in cells depleted of RTN3, the level of Akita-Myc in the soluble fraction also accumulated (Fig. 5A, first panel, compare lane 4 to 1), supporting that a build-up of Akita-Myc in the insoluble fraction may lead to back-up of Akita-Myc in the soluble fraction. To test if this condition triggers ER stress, we examined XBP1 splicing. In control cells, Akita-Myc expression moderately increased XBP1 splicing (Fig. 5B, first panel, compare lane 2 to 1), consistent with a previous report (Nozaki et al., 2004) and knockdown of RTN3 induced a further increase in ER stress response that was dependent upon expression of the misfolded substrate protein Akita-Myc (Fig. 5B, first panel, compare lanes 4 to 3; the ratio of spliced to unspliced XBP1 in the presence of Akita-Myc is quantified below). When the ratio of spliced to unspliced XBP1 was analyzed upon knockdown of RTN3, CCPG1, Sec62, or FAM134B in the presence of Akita-Myc, we found that this ratio was the highest when RTN3 was depleted (Fig. S1B, first panel; this ratio is indicated in Fig. 5B). These results indicate that accumulation of Akita-Myc due to impairment of RTN3-dependent ER-phagy induces marked ER stress.
We next evaluated if RTN4 might also facilitate degradation of aggregated Akita. In contrast to knockdown of RTN3, depletion of the long (4A) and short (4B) forms of RTN4 did not result in accumulation of Akita in the detergent-insoluble fraction (Fig. 5C, first panel, compare lane 8 to 6; quantified in the bottom graph), although this did modestly enhance the effect of depleting RTN3 (Fig. 5C, first panel, compare lane 9 to 7; quantified in the bottom graph). Thus, RTN3 appears to play a primary and specific role in removing Akita aggregates during ER-phagy, whereas the role of RTN4 appears to be secondary. This role may even be dependent on RTN3 as our binding analysis demonstrated that RTN3A-FLAG interacts with endogenous RTN4A and RTN4B (Fig. 5D).
We then tested whether accumulation of aggregated Akita in the detergent-insoluble fraction that results when RTN3 is knocked down can be blocked when GFP-tagged mouse RTN3C is exogenously expressed. (Due to sufficient sequence divergence, mouse RTN3C-GFP is resistant to the RTN3 siRNA designed to target human RTN3). Indeed, when compared to expression of the control ER membrane protein Sec61β-GFP, expressing RTN3C-GFP robustly decreased the level of Akita aggregates (Fig. 5E. first panel, compare lanes 5 to 4). These findings verify the integrity of the RTN3 siRNA, unambiguously demonstrating that accumulation of aggregated Akita triggered by RTN3 knockdown is not caused by unintended off-target effects. Interestingly, overexpressing RTN4A-GFP also blocked the accumulation of aggregated Akita upon RTN3 knockdown (Fig. 5E, first panel, compare lane 6 to 4), suggesting that in the absence of RTN3, exogenously expressed RTN4 can replace RTN3 activity.
Because RTN3C and RTN4A both lack a recognizable microtubule-associated protein 1A/1B-light chain 3 (LC3)-interacting region (LIR, a signature motif that couples to the autophagy machinery), we asked if these two proteins can nevertheless associate with endogenous LC3 under RTN3 knockdown. Importantly, both RTN3C-GFP and RTN4A-GFP (but not the negative control Sec61β -GFP) co-precipitated with endogenous LC3 under RTN3 knockdown, regardless of the presence of Akita-Myc (Fig. 5F, first panel, compare lanes 3-4 and 5-6 to lanes 1-2). These data demonstrate that RTN3C and RTN4A can indeed engage the autophagy machinery, suggesting that another LIR-containing protein likely mediates this interaction. Intriguingly, while RTN3C-GFP can become associated with LC3 regardless of RTN3 knockdown (Fig. S1C, first panel, compare lane 4 to 3), RTN4A-GFP can only associate with LC3 in the absence of RTN3 (Fig. S1C, first panel, compare lane 6 to 5). Although different reticulon complexes may form under different expression conditions, our data strongly suggest that both RTN3C and RTN4A can participate in the clearance of Akita aggregates through a bona fide ER-phagy pathway.
The accumulation of aggregated Akita-Myc due to knockdown of Grp170 (instead of RTN3) could similarly be decreased by overexpressing RTN4HD-GFP, RTN4A-GFP, RTN3C-GFP, RTN3A-FLAG, or RTN3AΔLIR-FLAG (Fig. 5G, first panel, compare lanes 2-6 to 1). RTN4HD-GFP is a mutant GFP-tagged RTN4 construct lacking its N-terminal cytosolic domain (aa 1-960) (Shibata et al., 2008), while RTN3AΔLIR-FLAG is a mutant FLAG-tagged RTN3A construct missing the six LIRs located in its N-terminal cytosolic domain (Grumati et al., 2017). Importantly, intact Beclin1 is required for any of these overexpressed proteins to efficiently decrease the accumulation of aggregated Akita due to knockdown of Grp170 (Fig. 5G, first panel, compare lanes 8-12 to 2-6). Likewise, intact ULK2 is required for RTN3C-GFP to remove the Akita aggregates (Figure 5H, first panel, compare lanes 7-8 to 5-6). These results further establish that RTN-dependent clearance of aggregated Akita requires an intact macro-autophagy mechanism. In the INS1 832/13 β-cell line, RTN3 also clears aggregated Akita because insoluble Akita-Myc in these cells is largely eliminated by expressing either RTN3C-GFP or RTN3A-FLAG (Fig. 5I, top panel, compare lane 5-6 to 4). Hence, in the physiologically-relevant β cells, RTN3 is deployed to remove insoluble proinsulin aggregates.
We followed the fate of aggregated Akita and reasoned that the RTN3-dependent ER-phagy pathway should deliver the Akita aggregates to the lysosome. Using HeLa cells transiently expressing Akita-Myc (and LAMP1-YFP as the lysosomal marker), confocal image analyses revealed that in control cells, Akita-Myc was expressed diffusely in the ER, and did not colocalize with LAMP1, as expected (Fig. S2, first and second panels). Strikingly, in Grp170 knockdown cells, punctate structures of Akita-Myc were seen to colocalize with enlarged LAMP1 vesicles (Fig. S2, third and fourth panels). Thus, in the absence of Grp170, Akita aggregates are formed and delivered to the lysosome. The enlarged LAMP1 vesicles likely reflect fusion of multiple autophagosomes with the lysosome. By contrast, whereas Akita-Myc punctate structures were observed in RTN3 knockdown cells, they did not colocalize with LAMP1 (Fig. S2, fifth and sixth panels), suggesting that in the RTN3-depleted cells, Akita aggregates are formed but fail to be delivered to lysosomes. Together, our findings strongly support the idea that RTN3-mediated ER-phagy couples the removal of an aggregated ER client to lysosomes for degradation.
RTN3-mediated ER-phagy clears other mutant pro-hormone aggregates
Does RTN3-dependent removal of insoluble protein aggregates operate more generally? To address this, we asked if RTN3 exerts any role during protein quality control of other mutant prohormones, such as the C28F mutation of proopiomelanocortin (C28F POMC), and the G57S mutation of pro-arginine-vasopressin (G57S Pro-AVP). Indeed, similar to Akita, knockdown of RTN3 (but not FAM134B) in HEK 293T cells increased the level of detergent-insoluble C28F POMC-FLAG (Fig. 6A, first panel, compare lane 6 to 5 and 8; quantified in graph below); this accumulation was reversed when RTN3C-GFP was expressed (Fig. 6A, first panel, compare lane 7 to 6; quantified in graph below). A comparable result was observed for G57S Pro-AVP-FLAG: while depleting RTN3 (but not FAM134B) enhanced the level of aggregated G57S Pro-AVP-FLAG (Fig. 6B, first panel, compare lane 6 to 5 and 8; quantified in graph below), expressing RTN3C-GFP decreased the accumulation of aggregates (Fig. 6B, first panel, compare lane 7 to 6; quantified in graph below). Thus, RTN3-mediated ER-phagy functions broadly to remove mutant prohormone aggregates.
Figure 6. RTN3-mediated ER-phagy clears mutant prohormone aggregates.
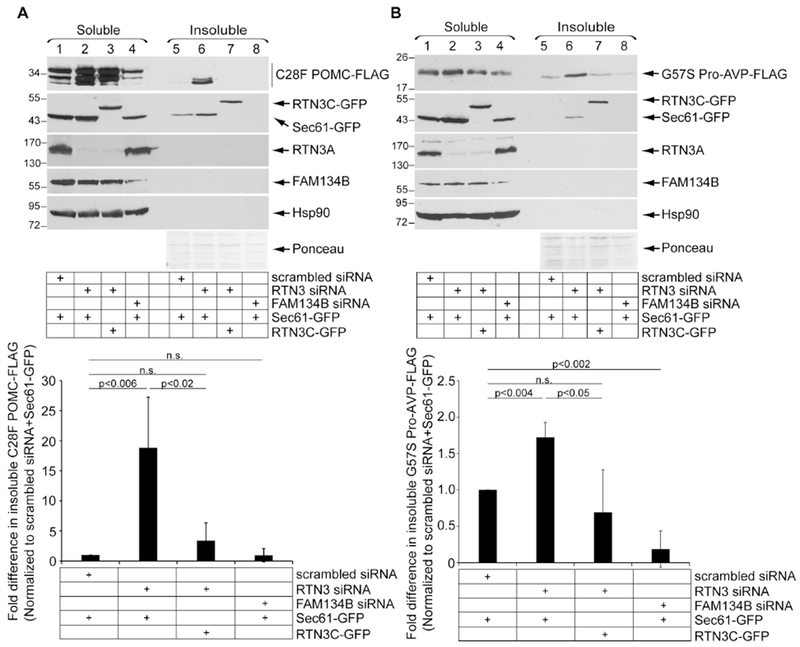
A. HEK 293T cells expressing C28F POMC-FLAG were co-transfected with the indicated siRNAs, and with either Sec61β-GFP or RTN3C-GFP. Soluble and insoluble fractions were generated as in 1C. The bottom graph represents insoluble C28F POMC-FLAG level normalized to the level of insoluble C28F POMC-FLAG in the scrambled control + Sec61β-GFP. Data represent the mean ± SD (n=3); p-values generated from student’s T-test. B. As in A, except cells were transfected with G57S Pro-AVP-FLAG. The bottom graph represents insoluble G57S Pro-AVP-FLAG level normalized to the level of insoluble G57S Pro-AVP-FLAG in the scrambled control + Sec61β-GFP (n=3).
Enhanced clearance of Akita aggregates partially rescues WT proinsulin secretion
Finally, we evaluated the functional consequence of RTN3-dependent clearance of prohormone aggregates. In the case of Akita, our findings revealed that aggregation of Akita provokes aggregation of WT proinsulin (Fig. 3), which presumably prevents WT proinsulin from exiting the ER. Indeed, in HEK 293T cells transfected with WT proinsulin-sfGFP and an empty vector, co-expressing Akita-Myc (along with the control Sec61β -GFP) decreased the level of WT proinsulin-sfGFP in the media (Fig. 7A, first panel, compare lane 2 to 1; quantified in the bottom graph). However, overexpressing increasing amounts of RTN3A-FLAG partially restored WT proinsulin-sfGFP secretion (Fig. 7A, first panel, compare lanes 3 and 4 to 2; quantified in the bottom graph). Moreover, when RTN3C-GFP, RTN4A-GFP, or RTN4HD-GFP was overexpressed (instead of RTN3A-FLAG) under the same experimental set-up as in Figure 7A, similar results were found (Fig. 7B–D, first panel, compare lane 4 to 2; quantified in the bottom graph). These findings demonstrate that clearance of Akita aggregates by overexpressing RTN3 (or RTN4) can partially restore the WT proinsulin export that is needed for insulin production.
Figure 7. Enhanced clearance of Akita aggregates partially rescues WT proinsulin secretion.
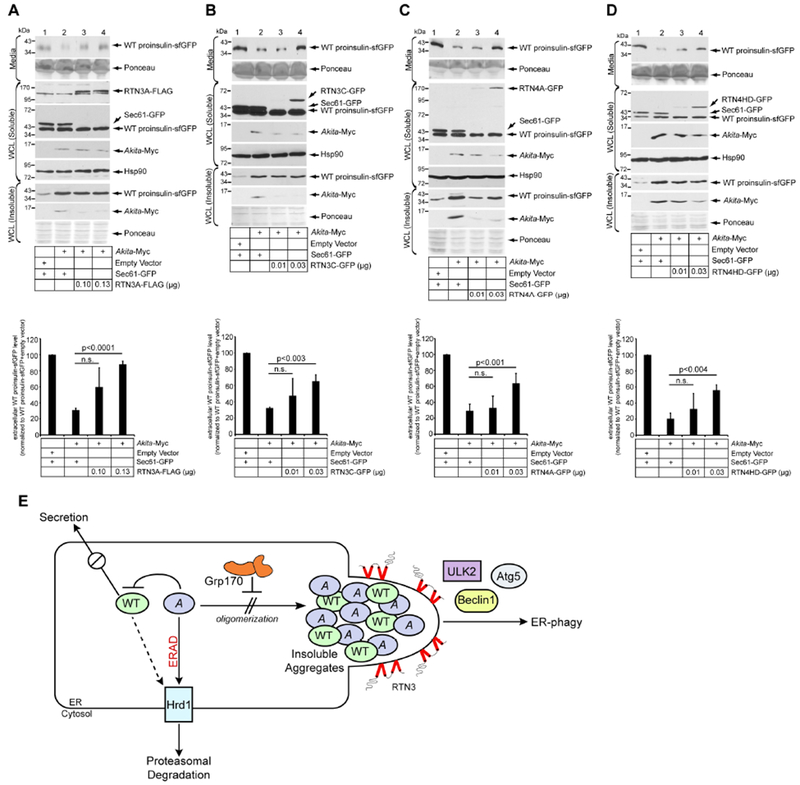
A. HEK 293T cells expressing WT proinsulin-sfGFP were transfected with the indicated construct. Media and cells were harvested. Cells were lysed and the samples subjected to SDS-PAGE followed by immunoblotting with the indicated antibodies. The bottom graph represents WT proinsulin-sfGFP level in the media normalized to the empty vector/Sec61β-GFP condition (lane 1). Data represent the mean ± SD (n=5); p-values from student’s T-test. B. As in A, except RTN3C-GFP was used (n=3). C. As in A, except RTN4A-GFP was used (n=3). D. As in A, except RTN4HD-GFP was used (n=3). E. Model depicting the deployment of a two-pronged approach to minimize the level of Akita aggregates.
DISCUSSION
Two ER quality control strategies work together to limit accumulation of misfolded proinsulin aggregates
Formation of protein aggregates is a signature feature of many human conformational diseases. Hence, identifying cellular mechanisms that minimize the accumulation of these aggregates will likely provide therapeutic approaches to alleviate such diseases. In the diabetic syndrome called MIDY, misfolding of mutant proinsulin (e.g., Akita) is thought to initiate disease progression (Liu et al., 2010b) by entrapping WT proinsulin in the ER, thereby preventing production of mature insulin (Liu et al., 2010b; Liu et al., 2007). Here, we found that in the ER, Akita forms detergent-insoluble aggregates that recruit WT proinsulin to also enter into these insoluble complexes (Fig. 7E). Importantly, our studies reveal that cells can deploy two distinct ER quality control mechanisms to minimize the buildup of aggregated Akita. First, the ER-resident Grp170 prevents aggregation of Akita (Fig. 7E). Second, RTN3-dependent ER-phagy removes any aggregates that are formed despite the action of Grp170 (Fig. 7E). Enhancing the clearance of aggregated Akita restores WT proinsulin export, suggesting that the Akita aggregates exert an important role during pathogenesis of MIDY.
Grp170 prevents Akita aggregation
Using a cell-based strategy, we found that Grp170 prevents Akita from entering into a detergent-insoluble fraction characteristic of aggregated proteins. An in vitro reconstitution approach further revealed that purified Grp170 (but not the ATPase-defective G41L Grp170) retains Akita in the soluble fraction, suggesting that Grp170 uses an energy-dependent chaperone activity to impede Akita aggregation. As Grp170 is a nucleotide-exchange factor (NEF) of BiP (Weitzmann et al., 2006) and because G41L Grp170 displays attenuated NEF activity (Inoue and Tsai, 2015), Grp170 might operate with BiP to prevent Akita aggregation. However, this seems unlikely because depletion of BiP in cells did not lead to accumulation of Akita in the detergent-insoluble fraction (data not shown), and purified BiP did not inhibit the aggregation of soluble Akita as efficiently as Grp170.
How then might Grp170 prevent Akita from aggregation? The complexes contained in detergent-insoluble Akita were found to be much larger than those in the soluble fraction, suggesting that soluble Akita progressively enlarges from the low, mid, to high MW species to eventually become detergent-insoluble aggregates. We also showed that Grp170 associates selectively with the high MW soluble species, suggesting that Grp170 is targeted to these species, interfering with further Akita extension into higher-order aggregates. Indeed, a previous study reported that Grp170 preferentially interacts with aggregation-prone amino acid sequences in a client (Behnke et al., 2016). This raises the possibility that when Akita multimerizes into the high MW species, it adopts a physical conformation in which aggregation surfaces are preferentially displayed, thereby recruiting Grp170. The idea that Grp170 prevents aggregation of Akita is also in agreement with previous studies suggesting that this chaperone can block aggregation of the model substrate luciferase under an artificial heat treatment condition (Park et al., 2003).
In MIDY, pancreatic β-cells harbor one WT and one mutant copy of the INS gene. In this setting, we found that aggregated Akita proinsulin recruits the aggregation of WT proinsulin. These findings were observed in HEK 293T cells and a pancreatic β-cell line, highlighting their physiological significance. Because aggregated WT proinsulin cannot exit the ER required for its subsequent processing, the formation of combined mutant-WT aggregates represents a critical reaction that impairs insulin production, which is central to pathogenesis of MIDY. Importantly, Grp170 suppresses aggregation of WT proinsulin recruited by Akita. This may reflect the ability of Grp170 to block aggregation of Akita, which in turn prevents WT proinsulin from aggregating. However, our cell-based studies also found that Grp170 suppresses aggregation of WT proinsulin even in the absence of Akita, suggesting that WT proinsulin itself might have some predisposition to misfold and aggregate. In such a scenario, WT proinsulin would directly benefit from the action of Grp170. Thus, prevention of WT proinsulin aggregation in MIDY is likely through the activity of Grp170 on both mutant and WT proinsulin.
We believe that upon action of Grp170, when the distribution of Akita complexes favors lower rather than higher MW species, Akita may become more competent to undergo ERAD. Based on our new results, we hypothesize that the high MW species of Akita likely exists as a dynamic protein complex that can experience extension (by further crosslinking) and shrinking (by reduction of mixed disulfide bonds). In one scenario, we envision that Grp170 might block the extension, thereby favoring a distribution towards low MW Akita species that can be disposed of by ERAD. This view is consistent with our previous report that Grp170 uses its chaperone activity to facilitate Hrd1-mediated, ERAD-dependent degradation of soluble Akita (Cunningham et al., 2017). Additionally, it is possible that Akita may use other E3 ligases coupled to ERAD for degradation, although to date, we have not obtained positive evidence for this possibility.
RTN3-dependent ER-phagy pathway clears Akita aggregates
In addition to the action of Grp170, we postulate that cells deploy additional mechanisms to minimize the accumulation of aggregated prohormones. This would provide cells with a clear advantage in maintaining proper proteostasis, as dysfunction in any one pathway can be compensated by the other. Our results demonstrate that inactivation of key autophagy components such as Beclin1 and Atg5 result in accumulation of detergent-insoluble Akita aggregates, suggesting that ER-phagy is central to the disposal of these aggregates. Moreover, the additional participation of ULK2 in clearing aggregated Akita implicates ER-phagy over other possible degradative routes involving ER-to-lysosome trafficking (Fregno et al., 2018).
Our results revealed that RTN3 promotes clearance of Akita aggregates. Specifically, we found that punctate structures containing Akita accumulate when either Grp170 or RTN3 are knocked down. However, when Grp170 is depleted, the Akita-containing puncta colocalize with lysosomes, whereas when RTN3 is depleted, Akita can no longer reach the lysosomes. These results strongly suggest lysosomal delivery of insoluble aggregates of Akita via an RTN3-dependent pathway, analogous to the mutant NPC1 and procollagen delivery to lysosomes via a FAM134B-dependent ER-phagy pathway (Forrester et al., 2019; Omari et al., 2018; Schultz et al., 2018).
Curiously, under RTN3-depleted conditions, expressing the short isoform of RTN3 (called RTN3C) or RTN4A was sufficient to eliminate insoluble Akita proinsulin. This finding is remarkable because RTN3C and RTN4A lack the LIR motif typically used to recruit core autophagy components. Nevertheless, these reticulons likely participate in complexes that do interact with LC3. This finding suggests that both the RTN3C- and RTN4A-LC3 interactions are indirect, likely mediated by another LIR-containing protein. In this context, it is intriguing to note that a previous report demonstrated that only full-length RTN3, but not RTN3C, can control the turnover of tubular ER through selective autophagy during starvation (Grumati et al., 2017). Also curious is that RTN4A can participate in a complex that binds to LC3 only in the absence of RTN3. Clearly, these are both areas of study in which more interesting work is still to come.
Functionally, the RTN-driven clearance of Akita aggregates results in improved ER export of WT proinsulin. Because Akita aggregates recruit WT proinsulin, removal of aggregated Akita would enable WT proinsulin to remain in the soluble phase so that it can proceed via anterograde intracellular transport, an essential step in production of mature insulin. These observations suggest that enhancing the activity of RTN3-mediated autophagy might offer a new therapeutic strategy to ameliorate MIDY. Moreover, given that a low level of WT proinsulin itself also aggregates, this strategy might have therapeutic value to combat the pathogenesis of other forms of diabetes.
Beyond mutant proinsulin, our findings that RTN3-dependent ER-phagy eliminates misfolded POMC and pro-AVP aggregates suggest that this ER-to-lysosome quality control pathway serves other protein clients and may function in a general mechanism to dispose of prohormone aggregates. Elucidating the structural features that enable these prohormone aggregates to exploit the function of RTN3 requires future investigations. Regardless, these data raise the possibility that therapeutic approaches targeting RTN3-mediated ER-phagy might have broader implications for a number of human conformational diseases.
STAR+METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Billy Tsai (btsai@med.umich.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
HEK 293T were cultured in Dulbecco’s modified Eagle’s medium (DMEM), supplemented with 10% fetal bovine serum in an incubator at 37°C and 5% CO2. Rat insulinoma INS1 832/13 cells were cultured in Roswell Park Memorial Institute medium (RPMI) 1640, supplemented with 10% fetal bovine serum, 10 mM HEPES (pH 7.5), 1 mM sodium pyruvate, and 50 μM β-mercaptoethanol.
METHOD DETAILS
Plasmid Transfection, Cycloheximide Chase, Immunoprecipitation, and Immunoblotting
Cells were seeded 1 day prior to transfection with 0.01-4 μg of plasmid DNA using polyethylenimine (PEI) or Lipofectamine 2000 (Invitrogen). 48 or 24 h post transfection, cells were harvested without treatment, treated with MG132 (10 μM) for 3 h, or treated with cycloheximide and chased for 0, 2, 4, and 6 h. Cells were harvested in PBS supplemented with 10 mM N-ethylmaleimide (NEM). For crosslinking immunoprecipitation experiments, cells crosslinked with DSP (per manufacturer’s protocol) were lysed in 500 μL of 1% Triton X-100 in HN buffer (50 mM HEPES pH 7.5, 150 mM NaCl) containing 10 mM NEM and 1 mM PMSF. Cells were then incubated on ice for 10 min and centrifuged. The resulting whole cell extract was incubated with M2-FLAG conjugated beads (Thermo Fisher) at 4°C for 2 h. Beads were washed with lysis buffer supplemented with NEM and PMSF, and boiled in SDS sample buffer with 100 mM dithiothreitol (DTT). For immunoblot analysis, cells were lysed and immunoblotted as previously described (Cunningham et al., 2017). Briefly, cells were lysed in 100 μL RIPA buffer supplemented with NEM and PMSF, incubated on ice for 10 min and centrifuged. The resulting extract was subjected to reducing SDS-PAGE with 100 mM DTT, transferred to nitrocellulose, and incubated with primary and secondary antibodies prior to exposure using ECL.
Small interfering RNA knockdown
Small interfering RNA (siRNA) was transfected into cells using RNAiMAX (Invitrogen), and cells were chased or harvested 48 h after treatment.
Detergent-insolubility Assay
Cells were lysed in 100 μL of RIPA supplemented with 10 mM NEM and 1 mM PMSF, and incubated on ice for 10 min and centrifuged. The extracted material represents the soluble fraction, while material in the pellet (which was extracted by 2% SDS sample buffer) represents the insoluble fraction. Soluble and insoluble samples were subjected to SDS-PAGE and immunoblotting as described above.
Sucrose Gradient Fractionation Assay
This assay was previously described (Cunningham et al., 2017). Briefly, cells were lysed in RIPA buffer to generate a whole cell extract. The extract is cleared using an ultracentrifuge at 50,000 rpm for 20 min. The resulting cleared extract is layered on top of a 10-50% or 30-70% discontinuous sucrose gradient and centrifuged on a Beckman SW50.1 rotor at 29,000 rpm for 24 h at 4°C. After centrifugation, 12-50 μL fractions were collected and analyzed by SDS-PAGE followed by immunoblotting.
Immunoprecipitation using Pooled Sucrose Fractions
Fractions were pooled as follows: fractions 1-4, 5-8, and 9-12. 50% sucrose and HN buffer were added to the three pools to normalize the sucrose concentration. The pooled samples were incubated with anti-Myc antibody (1:200) at 4°C for 24 h, followed by 2 h of incubation with A/G-agarose beads (Pierce) at 4óC. The precipitated material was subjected to SDS-PAGE followed by immunoblotting.
Fast Protein Liquid Chromatography (FPLC) on isolated Akita-Myc
Akita-Myc in the soluble and insoluble fractions was immunoprecipitated using a Myc antibody (1:300) at 4°C for 24 h, followed by 2 h of incubation with protein A/G-agarose beads (Pierce) at 4°C; note that the insoluble fraction was diluted to a final concentration 0.1% SDS before immunoprecipitation. The precipitated material was subjected to elution using a Myc peptide. Elution was performed sequentially in a volume of 50 μL three times for 20, 20, and 30 minutes, with Myc peptide concentrations of 10 μg/mL, 10 μg/mL, and 100μg/mL, respectively. Eluted Akita-Myc was subjected to FPLC on a Superose 6 Increase 3.2/300 column. Fractions were collected and pooled per groups of 2 (i.e. fractions 1-2 are indicated as fraction 1, fractions 3-4 as fraction 2).
In vitro Aggregation Assay
HEK 293T cells transfected with Akita-Myc were semipermeabilized in 300 μL of HCN buffer (50 mM HEPES pH 7.5, 150 mM NaCl, 2 mM CaCl2), supplemented with 0.01% digitonin, 10 mM NEM and 1 mM PMSF. Cells were incubated on ice for 10 min, and centrifuged at 16,100 rpm for 10 min. This generates a supernatant fraction that contains cytosolic proteins and a pellet fraction that contains membranes including the ER; Akita-Myc in the pellet fraction was subsequently solubilized by RIPA buffer containing 10 mM NEM and 1 mM PMSF. Purification of recombinant Grp170-FLAG, BiP-FLAG, and G41L Grp170-FLAG were prepared as previously described (Inoue et al., MBoC 2016). Recombinant FLAG-tagged proteins were stored at −80°C in 0.1% Triton X-100 in HN buffer. During the reaction, each tube contains a membrane extract harboring soluble Akita-Myc, 100 μM ATP, 10 mM MgCl2, 1 mM KCl, and either 0.3 mg/mL of BSA or the indicated purified protein (Grp170-FLAG, G41L Grp170-FLAG, or BiP-FLAG). Each tube was then incubated in a thermomixer (800 rpm) at 37°C for the indicated time: 10, 5, 3, 1, or 0 min. After incubation, the sample was centrifuged at 16,100 g for 10 min to generate a supernatant fraction that contains soluble Akita-Myc and a pellet fraction that harbors aggregated Akita-Myc. Aggregated Akita-Myc was resuspended in a SDS sample buffer. Samples containing soluble or aggregated Akita-Myc were subjected to reducing SDS-PAGE followed by immunoblotting.
35S-amino-acid Pulse Labeling
24 h post-transfection, HEK 293T cells were incubated in media lacking Cys and Met for 30 min, and then 35S pulse-labeled for 10 min at 37°C using Trans 35S label (Perkin Elmer). Cells were washed with ice-cold PBS (containing 10 mM NEM) and were then lysed in RIPA buffer (10 mM Tris pH 7.4, 150 mM NaCl, 0.1% SDS, 1% NP-40, 2 mM EDTA) containing 2 mM NEM. The lysates were precleared using Pansorbin for 30 min at 4°C, and then immunoprecipitated with guinea pig anti-Insulin antibody on protein A agarose beads overnight at 4°C. A small portion of the lysate (~10 μl) prior to preclearing was saved to quantitatively measure radioactive amino acid incorporation in each sample. Reducing SDS-PAGE of all the immunoprecipitated samples was carried out, normalized to TCA-precipitable cpm in the lysates. The gel was fixed, dried, exposed to X-ray film, and the proinsulin bands quantified by scanning densitometry.
XBP1 Splicing Assay
HEK 293T cells transfected with plasmids encoding Akita-Myc or an empty vector (pcDNA3.1) were co-transfected with either scrambled, CCPG1, Sec62, FAM134B, or RTN3 siRNA. XBP1 splicing assay was performed as described in (Uemura et al., 2009).
Immunofluorescence Staining and Imaging
HeLa cells were fixed with 4% PFA for 20 minutes, permeabilized with 0.2% Triton X-100/TBS/3% BSA for 20 minutes, and blocked with 0.2% Tween-20/TBS/3% BSA for one hour at room temperature. Primary antibodies were diluted in 0.2% Tween-20/TBS/3% BSA and incubated overnight at 4°C. Secon dary antibodies were also diluted in 0.2% Tween-20/TBS/3% BSA and incubated at room temperature for one hour. Cells were mounted with Prolong Gold anti-fade reagent with DAPI. Microscopy was performed using a Zeiss LSM 800 confocal laser scanning microscope with a Plan-Apochromat 40×/1.4 oil differential interference contrast (DIC) M27 objective. Images were airyscan processed in Zen 2.3 software.
Proinsulin Secretion Assay
Proinsulin secretion assay was previously described (Cunningham et al., 2017), with few modifications indicated below. Briefly, HEK 293T cells were transfected with a plasmid encoding WT proinsulin-sfGFP. 8 h post transfection, the medium was replaced with 500 μL of fresh medium and incubated for 16 h. Medium, the whole cell extract, and the insoluble samples were collected and subjected to SDS-PAGE and analyzed by immunoblotting.
Quantification and Statistical Analysis
Details on statistical analyses can be found in each figure legend. Each experiment shown was replicated at least n=3 times. Additionally, western blot quantification was performed using NIH ImageJ v1.52i with significance p<0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Myc | Immunology Consultants Laboratory | Cat#RMYC-45A |
| Mouse anti-Myc | Laboratory of Kristen Verhey | N/A |
| Rabbit anti-Orp150/Grp170 | Abcam | Cat# ab124884, RRID:AB_10973544 |
| Rabbit anti-BiP | Abcam | Cat# ab21685, RRID:AB_2119834 |
| Mouse anti-Hsp90alpha/beta (F-8) | Santa Cruz Biotechnology | Cat# sc-13119, RRID:AB_675659 |
| Mouse anti-PDI | Abcam | Cat# ab2792, RRID:AB_303304 |
| Mouse anti-GFP | Proteintech Group | Cat# 66002-1-Ig, RRID:AB_11182611 |
| Rabbit anti-Hrd1 | Proteintech Group | Cat# 13473-1-AP, RRID:AB_2287023 |
| Rabbit anti-Beclin1 | MBL International | Cat# PD017, RRID:AB_1278767 |
| Rabbit anti-CCPG1 | Proteintech Group | Cat# 13861-1-AP, RRID:AB_2074010 |
| Rabbit anti-Sec62 [EPR9212] | Abcam | Cat# ab137022 |
| Rabbit anti-RTN3 | Bethyl Laboratories | Cat# A302-860A |
| Rabbit anti-RTN3 | Boster Biological Technology | Cat# PA2256, RRID:AB_2665372 |
| Rabbit anti-FAM134B | Proteintech Group | Cat# 21537-1-AP |
| Rabbit anti-RTN4 | Santa Cruz Biotechnology | Cat# sc-271878, RRID:AB_10709573 |
| Rabbit anti-FLAG | Millipore Sigma | Cat# F7425, RRID:AB_439687 |
| Mouse anti-FLAG | Millipore Sigma | Cat# F3165, RRID:AB_259529 |
| Rabbit anti-LC3 | MBL International | Cat# PM036, RRID:AB_2274121 |
| Guinea Pig anti-Insulin | Covance Inc. (Custom Antibody Service) | N/A |
| Goat anti-Rabbit IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-11037, RRID:AB_2534095 |
| Goat anti-Mouse IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11029, RRID:AB_2534088 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Phenylmethylsulfonyl fluoride (PMSF) | Acros Organics | Cat# AC215740010; Cas: 329-98-6 |
| N-ethylmaleimide (NEM) | Millipore Sigma | Cat# E3876; Cas: 128-53-0 |
| Dithiobis-succinimidyl-propionate (DSP) Crosslinker | Thermo Fisher Scientific | Cat# 22585; Cas: 57757-57-0 |
| Cycloheximide | VWR | Cat# 94271; Cas 66-81-9 |
| MG132 | Cayman Chemicals | Cat# 10012628; Cas: 133407-82-6 |
| DL-Dithiothreitol | Millipore Sigma | Cat# D0632; Cas: 3483-12-3 |
| BSA | Millipore Sigma | Cat# A6003; Cas: 9048-46-8 |
| Grp170-FLAG | (Inoue and Tsai, 2015); This Paper | N/A |
| G41L Grp170-FLAG | (Inoue and Tsai, 2015); This Paper | N/A |
| BiP-FLAG | This Paper | N/A |
| Ponceau-S | Fisher Scientific | Cat# BP103-10; Cas: 6226-79-5 |
| Bafilomycin A1 | Millipore Sigma | Cat# 508409 CAS: 88899-55-2 |
| Polyethylenimine | Fisher Scientific | Cat# AC178571000; Cas: 9002-98-6; 7732-18-5 |
| Prolong Gold Antifade Mountant with DAPI | Thermo Fisher Scientific | Cat# P36931 |
| Experimental Models: Cell Lines | ||
| Human: HEK 293T | ATCC | Cat# CRL-3216, RRID:CVCL_0063 |
| Human: HeLa | ATCC | Cat# CCL-2 RRID:CVCL_0030 |
| Rat: INS1 832/13 | Laboratory of Christopher Newgard | RRID:CVCL_7226 |
| Oligonucleotides | ||
| siGrp170: GCUCAAUAAGGCCAAGUUUdTdT | Millipore Sigma | N/A |
| siHrd1: GGAGACUGCCACUACAGUUGUdTdT | Millipore Sigma | N/A |
| Human siBeclin1: GGUCUAAGACGUCCAACAAdTdT | Millipore Sigma | N/A |
| Rat siAtg5: UCAACGAAAUGCAGAGAAAdTdT | Millipore Sigma | N/A |
| siCCPG1: UUCCAUGUACCAGCUUAGUUAdTdT | Millipore Sigma | N/A |
| siSec62: GAAGGAUGAGAAAUCUGAAUUdTdT | Millipore Sigma | N/A |
| siRTN3: UCAGUGUCAUCAGUGUGGUUUCUUAdTdT | Millipore Sigma | N/A |
| siFAM134B: CAAAGATGACAGTGAATTAdTdT | Millipore Sigma | N/A |
| siRTN4: GUUCAGAAGUACAGUAAUUdTdT | Millipore Sigma | N/A |
| siULK2: GAGCAUAGGAACAGUGAUAUUdTdT | Millipore Sigma | N/A |
| Recombinant DNA | ||
| pTarget-hProCpepMyc | (Liu et al., 2010a) | N/A |
| pTarget-hProC(A7)Y-CpepMyc | (Liu et al., 2010a) | N/A |
| pTarget-hProCpepSfGFP | (Haataja et al., 2013) | N/A |
| pTarget-hProC(A7)Y-CpepSfGFP | (Haataja et al., 2013) | N/A |
| pcDNA3.1-FLAG-RTN3L | (Grumati et al., 2017); Laboratory of Ivan Dikic | N/A |
| pcDNA3.1-FLAG-RTN3LΔ6LIRs | (Grumati et al., 2017); Laboratory of Ivan Dikic | N/A |
| pcDNA3.1-GFP-Sec61β | Laboratory of Tom Rapoport | N/A |
| pAc-GFPC1-Rtn3c | Laboratory of Tom Rapoport | N/A |
| pAc-GFPC1-Rtn4a | Laboratory of Tom Rapoport | N/A |
| pAc-GFPC1-Rtn4HD | Laboratory of Tom Rapoport | N/A |
| pcDNA3.1-Grp170-FLAG | (Inoue and Tsai, 2015); this paper | N/A |
| pcDNA3.1-G41L Grp170-FLAG | (Inoue and Tsai, 2015); this paper | N/A |
| pcDNA3.1-FLAG-BiP | This paper | N/A |
| pcDNA3.1-C28F POMC-FLAG | (Kim et al., 2018) Laboratory of Ling Qi | N/A |
| pRc/RSV-G57S Pro-AVP-FLAG | (Shi et al., 2017) Laboratory of Ling Qi | N/A |
| pcDNA3.1-LAMP1-YFP | (Qian et al., 2009) Laboratory of Joel Swanson | N/A |
| pcDNA3-HA-AMPKα1 | (Inoki et al., 2003) Laboratory of Ken lonki | N/A |
| pRK7-Myc-AMPKβ1 | (Suzuki et al., 2013) Laboratory of Ken lonki | N/A |
| pRK7-Myc-AMPKγ1 | (Suzuki et al., 2013) Laboratory of Ken lonki | N/A |
ACKNOWLEDGEMENTS
We would like to thank Ryan Baldridge (University of Michigan) for use of the FPLC apparatus, Kaylee Steen (University of Michigan) for her assistance in confocal microscope, Ling Qi (University of Michigan) for the mutant pro-AVP and POMC constructs, and Diane Fingar (University of Michigan) for reagents. We would also like to thank members of the Tsai and Arvan laboratories for their helpful suggestions and feedback. B.T. and P.A. are funded by the National Institutes of Health (RO1 DK111174). C.N.C. is supported by NIH National Institute of Diabetes and Digestive and Kidney Diseases NRSA F31-DK-113653 and partially supported by University of Michigan Endowment for Basic Sciences EDGE Award and Rackham Graduate School Predoctoral Research Grant.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare there is no conflict of interest relevant to this manuscript.
REFERENCES
- Allen JR, Nguyen LX, Sargent KE, Lipson KL, Hackett A, and Urano F (2004). High ER stress in beta-cells stimulates intracellular degradation of misfolded insulin. Biochem Biophys Res Commun 324, 166–170. [DOI] [PubMed] [Google Scholar]
- Arunagiri A, Haataja L, Cunningham CN, Shrestha N, Tsai B, Qi L, Liu M, and Arvan P (2018). Misfolded proinsulin in the endoplasmic reticulum during development of beta cell failure in diabetes. Ann N Y Acad Sci 1418, 5–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates G (2003). Huntingtin aggregation and toxicity in Huntington’s disease. Lancet 361, 1642–1644. [DOI] [PubMed] [Google Scholar]
- Bays NW, Gardner RG, Seelig LP, Joazeiro CA, and Hampton RY (2001). Hrd1p/Der3p is a membrane-anchored ubiquitin ligase required for ER-associated degradation. Nat Cell Biol 3, 24–29. [DOI] [PubMed] [Google Scholar]
- Behnke J, Mann MJ, Scruggs FL, Feige MJ, and Hendershot LM (2016). Members of the Hsp70 Family Recognize Distinct Types of Sequences to Execute ER Quality Control. Mol Cell 63, 739–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S, McDonald KL, and Walter P (2006). Autophagy counterbalances endoplasmic reticulum expansion during the unfolded protein response. PLoS Biol 4, e423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernales S, Schuck S, and Walter P (2007). ER-phagy: selective autophagy of the endoplasmic reticulum. Autophagy 3, 285–287. [DOI] [PubMed] [Google Scholar]
- Bordallo J, Plemper RK, Finger A, and Wolf DH (1998). Der3p/Hrd1p is required for endoplasmic reticulum-associated degradation of misfolded lumenal and integral membrane proteins. Mol Biol Cell 9, 209–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, and Klionsky DJ (2007). Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res 17, 839–849. [DOI] [PubMed] [Google Scholar]
- Chan SJ, Keim P, and Steiner DF (1976). Cell-free synthesis of rat preproinsulins: characterization and partial amino acid sequence determination. Proc Natl Acad Sci U S A 73, 1964–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CN, He K, Arunagiri A, Paton AW, Paton JC, Arvan P, and Tsai B (2017). Chaperone-Driven Degradation of a Misfolded Proinsulin Mutant in Parallel With Restoration of Wild-Type Insulin Secretion. Diabetes 66, 741–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, et al. (2011). Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331, 456–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester A, De Leonibus C, Grumati P, Fasana E, Piemontese M, Staiano L, Fregno I, Raimondi A, Marazza A, Bruno G, et al. (2019). A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fregno I, Fasana E, Bergmann TJ, Raimondi A, Loi M, Solda T, Galli C, D’Antuono R, Morone D, Danieli A, et al. (2018). ER-to-lysosome-associated degradation of proteasome-resistant ATZ polymers occurs via receptor-mediated vesicular transport. EMBO J 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fumagalli F, Noack J, Bergmann TJ, Cebollero E, Pisoni GB, Fasana E, Fregno I, Galli C, Loi M, Solda T, et al. (2016). Translocon component Sec62 acts in endoplasmic reticulum turnover during stress recovery. Nat Cell Biol 18, 1173–1184. [DOI] [PubMed] [Google Scholar]
- Grumati P, Morozzi G, Holper S, Mari M, Harwardt MI, Yan R, Muller S, Reggiori F, Heilemann M, and Dikic I (2017). Full length RTN3 regulates turnover of tubular endoplasmic reticulum via selective autophagy. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haataja L, Snapp E, Wright J, Liu M, Hardy AB, Wheeler MB, Markwardt ML, Rizzo M, and Arvan P (2013). Proinsulin intermolecular interactions during secretory trafficking in pancreatic beta cells. J Biol Chem 288, 1896–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampton RY, Gardner RG, and Rine J (1996). Role of 26S proteasome and HRD genes in the degradation of 3-hydroxy-3-methylglutaryl-CoA reductase, an integral endoplasmic reticulum membrane protein. Mol Biol Cell 7, 2029–2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Cunningham CN, Manickam N, Liu M, Arvan P, and Tsai B (2015). PDI reductase acts on Akita mutant proinsulin to initiate retrotranslocation along the Hrd1/Sel1L-p97 axis. Mol Biol Cell 26, 3413–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodish I, Liu M, Rajpal G, Larkin D, Holz RW, Adams A, Liu L, and Arvan P (2010). Misfolded proinsulin affects bystander proinsulin in neonatal diabetes. J Biol Chem 285, 685–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoki K, Zhu T, and Guan KL (2003). TSC2 mediates cellular energy response to control cell growth and survival. Cell 115, 577–590. [DOI] [PubMed] [Google Scholar]
- Inoue T, and Tsai B (2015). A nucleotide exchange factor promotes endoplasmic reticulum-to-cytosol membrane penetration of the nonenveloped virus simian virus 40. J Virol 89, 4069–4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine GB, El-Agnaf OM, Shankar GM, and Walsh DM (2008). Protein aggregation in the brain: the molecular basis for Alzheimer’s and Parkinson’s diseases. Mol Med 14, 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang R, Zeh HJ, Lotze MT, and Tang D (2011). The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ 18, 571–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaminets A, Heinrich T, Mari M, Grumati P, Huebner AK, Akutsu M, Liebmann L, Stolz A, Nietzsche S, Koch N, et al. (2015). Regulation of endoplasmic reticulum turnover by selective autophagy. Nature 522, 354–358. [DOI] [PubMed] [Google Scholar]
- Kim GH, Shi G, Somlo DR, Haataja L, Song S, Long Q, Nillni EA, Low MJ, Arvan P, Myers MG Jr., et al. (2018). Hypothalamic ER-associated degradation regulates POMC maturation, feeding, and age-associated obesity. J Clin Invest 128, 1125–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kundu M, Viollet B, and Guan KL (2011). AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol 13, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Haataja L, Wright J, Wickramasinghe NP, Hua QX, Phillips NF, Barbetti F, Weiss MA, and Arvan P (2010a). Mutant INS-gene induced diabetes of youth: proinsulin cysteine residues impose dominant-negative inhibition on wild-type proinsulin transport. PLoS One 5, e13333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hodish I, Haataja L, Lara-Lemus R, Rajpal G, Wright J, and Arvan P (2010b). Proinsulin misfolding and diabetes: mutant INS gene-induced diabetes of youth. Trends Endocrinol Metab 21, 652–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Hodish I, Rhodes CJ, and Arvan P (2007). Proinsulin maturation, misfolding, and proteotoxicity. Proc Natl Acad Sci U S A 104, 15841–15846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee A, Morales-Scheihing D, Butler PC, and Soto C (2015). Type 2 diabetes as a protein misfolding disease. Trends Mol Med 21, 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki J, Kubota H, Yoshida H, Naitoh M, Goji J, Yoshinaga T, Mori K, Koizumi A, and Nagata K (2004). The endoplasmic reticulum stress response is stimulated through the continuous activation of transcription factors ATF6 and XBP1 in Ins2+/Akita pancreatic beta cells. Genes Cells 9, 261–270. [DOI] [PubMed] [Google Scholar]
- Omari S, Makareeva E, Roberts-Pilgrim A, Mirigian L, Jarnik M, Ott C, Lippincott-Schwartz J, and Leikin S (2018). Noncanonical autophagy at ER exit sites regulates procollagen turnover. Proc Natl Acad Sci U S A 115, E10099–E10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Easton DP, Chen X, MacDonald IJ, Wang XY, and Subjeck JR (2003). The chaperoning properties of mouse grp170, a member of the third family of hsp70 related proteins. Biochemistry 42, 14893–14902. [DOI] [PubMed] [Google Scholar]
- Qian M, Cai D, Verhey KJ, and Tsai B (2009). A lipid receptor sorts polyomavirus from the endolysosome to the endoplasmic reticulum to cause infection. PLoS Pathog 5, e1000465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruggiano A, Foresti O, and Carvalho P (2014). Quality control: ER-associated degradation: protein quality control and beyond. J Cell Biol 204, 869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz ML, Krus KL, Kaushik S, Dang D, Chopra R, Qi L, Shakkottai VG, Cuervo AM, and Lieberman AP (2018). Coordinate regulation of mutant NPC1 degradation by selective ER autophagy and MARCH6-dependent ERAD. Nat Commun 9, 3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi G, Somlo DRM, Kim GH, Prescianotto-Baschong C, Sun S, Beuret N, Long Q, Rutishauser J, Arvan P, Spiess M, et al. (2017). ER-associated degradation is required for vasopressin prohormone processing and systemic water homeostasis. J Clin Invest 127, 3897–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata Y, Voss C, Rist JM, Hu J, Rapoport TA, Prinz WA, and Voeltz GK (2008). The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J Biol Chem 283, 18892–18904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MD, Harley ME, Kemp AJ, Wills J, Lee M, Arends M, von Kriegsheim A, Behrends C, and Wilkinson S (2018). CCPG1 Is a Non-canonical Autophagy Cargo Receptor Essential for ER-Phagy and Pancreatic ER Proteostasis. Dev Cell 44, 217–232 e211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MH, Ploegh HL, and Weissman JS (2011). Road to ruin: targeting proteins for degradation in the endoplasmic reticulum. Science 334, 1086–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steiner DF, Cunningham D, Spigelman L, and Aten B (1967). Insulin biosynthesis: evidence for a precursor. Science 157, 697–700. [DOI] [PubMed] [Google Scholar]
- Stoy J, Edghill EL, Flanagan SE, Ye H, Paz VP, Pluzhnikov A, Below JE, Hayes MG, Cox NJ, Lipkind GM, et al. (2007). Insulin gene mutations as a cause of permanent neonatal diabetes. Proc Natl Acad Sci U S A 104, 15040–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Bridges D, Nakada D, Skiniotis G, Morrison SJ, Lin JD, Saltiel AR, and Inoki K (2013). Inhibition of AMPK catabolic action by GSK3. Mol Cell 50, 407–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai B, Ye Y, and Rapoport TA (2002). Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat Rev Mol Cell Biol 3, 246–255. [DOI] [PubMed] [Google Scholar]
- Uemura A, Oku M, Mori K, and Yoshida H (2009). Unconventional splicing of XBP1 mRNA occurs in the cytoplasm during the mammalian unfolded protein response. J Cell Sci 122, 2877–2886. [DOI] [PubMed] [Google Scholar]
- Wang J, Takeuchi T, Tanaka S, Kubo SK, Kayo T, Lu D, Takata K, Koizumi A, and Izumi T (1999). A mutation in the insulin 2 gene induces diabetes with severe pancreatic beta-cell dysfunction in the Mody mouse. J Clin Invest 103, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss MA (2009). Proinsulin and the genetics of diabetes mellitus. J Biol Chem 284, 19159–19163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzmann A, Volkmer J, and Zimmermann R (2006). The nucleotide exchange factor activity of Grp170 may explain the non-lethal phenotype of loss of Sil1 function in man and mouse. FEBS Lett 580, 5237–5240. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.