

Abstract
(–)-Podophyllotoxin is one of the most potent microtubule depolymerizing agents and has served as an important lead compound in antineoplastic drug discovery. Here, we report a short chemoenzymatic total synthesis of (–)-podophyllotoxin and related aryltetralin lignans. Vital to this approach is the use of an enzymatic oxidative C–C coupling reaction to construct the tetracyclic core of the natural product in a diastereoselective fashion. This strategy allows gram scale access to (–)-deoxypodophyllotoxin and is readily adaptable to the preparation of related aryltetralin lignans.
Keywords: biocatalysis, total synthesis, podophyllotoxin, aryltetralin lignans, non-heme dioxygenase
Graphical Abstract
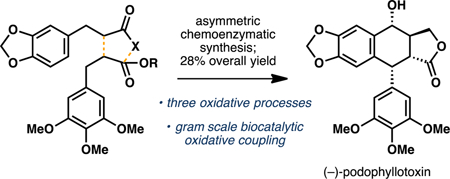
Biocatalytic C–C coupling: Podophyllotoxin, a potent microtubule depolymerizing agent, was synthesized in a concise manner through a strategic use of three oxidative processes. Highlights of the route include a gram-scale biocatalytic C–C coupling to construct the tetracyclic core of the natural product and a chemoselective late-stage installation of the secondary alcohol at C7.
The aryltetralin lignans are an important family of secondary metabolites possessing a wealth of antiviral, antibacterial, and antineoplastic activities.1 The most prominent member of the family, (–)-podophyllotoxin (1, Figure 1A), is a potent microtubule depolymerizer that binds to the colchicine site of the tubulin subunit.2 The use of 1 as a lead compound in anticancer drug development has led to the identification of several synthetic derivatives, including etoposide (2) and teniposide, which are currently used to treat several types of cancer.3 In contrast to 1, the antineoplastic properties of these synthetic derivatives arise from inhibition of topoisomerase II during the late S and early G2 stage of the cell cycle. Several other aryltetralin lignans with alternative substitution patterns on the aryl rings have also been identified in nature (e.g., 3–6).4 Interestingly, investigations into the bioactivities of these lignans have suggested that subtle changes in the substitution patterns of the aromatic rings could significantly affect the microtubule depolymerizing activity and therapeutic window of the scaffold.4b,d,e The medicinal importance of this natural product family has spurred numerous chemical approaches,5,6 including a recent Pd-catalyzed C–H arylation strategy to 1 by Maimone.6c Despite this progress, an efficient enantioselective approach to aryltetralin lignans has proven elusive thus far. Herein, we describe an asymmetric, chemoenzymatic approach to 1 that features enantioselective construction of the key dibenzylbutyrolactone precursor and harnesses the biocatalytic utility of a non-heme dioxygenase from podophyllotoxin biosynthesis to forge the key C ring of the natural product. We showcase the scalability and practicality of this approach by conducting the biocatalytic transformation on gram scale in excellent yield. Finally, we demonstrate the general utility of this synthetic strategy in the concise synthesis of related aryltetralin lignin natural products. During the preparation of this manuscript, Kroutil, Fuchs and coworkers reported a complementary chemoenzymatic synthesis of (–)-deoxypodophyllotoxin, (–)-epi-podophyllotoxin and (–)-podophyllotoxin via biocatalytic kinetic resolution of racemic dibenzylbutyrolactone precursors.7 In contrast, our approach establishes the absolute stereochemistry of the dibenzylbutyrolactone precursor at an early stage and does not require a kinetic resolution process to provide the enantioenriched tetracyclic product in the key enzymatic cyclization step.
Figure 1.
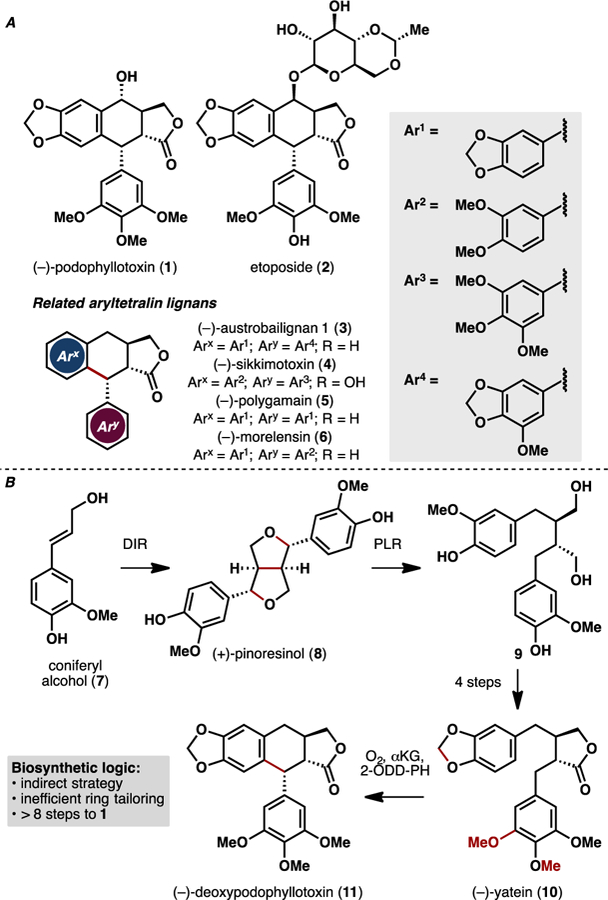
A. Chemical structures of (–)-podophyllotoxin (1), etoposide (2), and related aryltetralin lignans. B. Biosynthetic pathway for the production of (–)-deoxypodophyllotoxin (11) from coniferyl alcohol (7) in Podophyllum hexandrum.
Biosynthetically, podophyllotoxin arises from oxidative dimerization of coniferyl alcohol (7, Figure 1B).8 A proton-coupled electron transfer event in the presence of dirigent protein (DIR) converts 7 to enantioenriched (+)-pinoresinol (8), which is subsequently reduced by pinocresol lariciresinol reductase (PLR) to the corresponding diol (9). Oxidation to the butyrolactone and several ring tailoring events by two P450s and a SAM-dependent methyltransferase affords (–)-yatein (10). A non-heme dioxygenase, referred to as 2-ODD-PH, effects an oxidative C–C coupling to convert 10 to (–)-deoxypodophyllotoxin (11). While the enzymes that are responsible for the conversion of 11 to 1 have not been identified to date, the accumulation of (–)-epi-podophyllotoxin in tobacco leaves has been observed to be dependent on CYP82D61 expression, implicating the potential role of this enzyme in late-stage tailoring. More recently, in vitro studies by Chang, Chien and coworkers provided biochemical confirmation for the role of 2-ODD-PH in the oxidative benzylic coupling step and shed some light on the mechanistic nature of this transformation.9
From synthetic economy perspective,10 the biosynthetic logic involved in podophyllotoxin production suffers from several non-strategic bond-forming steps. Several steps are expended on the oxidation state adjustments of the butyrolactone moiety as well as on the alteration of the substitution pattern on the aryl rings. We hypothesized that a chemoenzymatic approach that involves rapid asymmetric construction of 10 and harnesses the synthetic utility of 2-ODD-PH in the key C ring formation would enable a more efficient approach towards 11 (Figure 2). A late-stage chemical oxidation on 11 would introduce the requisite secondary alcohol and complete the synthesis of 1. To this end, we conducted an asymmetric synthesis of 10 through the use of oxidative enolate coupling methodology developed by Baran and coworkers (Scheme 1A).11 Coupling of oxazolidinone 12 and ester 13 in the presence of LDA and CuII salt effected the formation of the dicarbonyl product as an inconsequential mixture of diastereomers. Subjection of this intermediate to chemoselective reduction with LiBH4 and thermal equilibration with DBU afforded 10, the native substrate of 2-ODD-PH, as a single diastereomer in moderate overall yield and set the stage for the key enzymatic coupling step.
Figure 2.
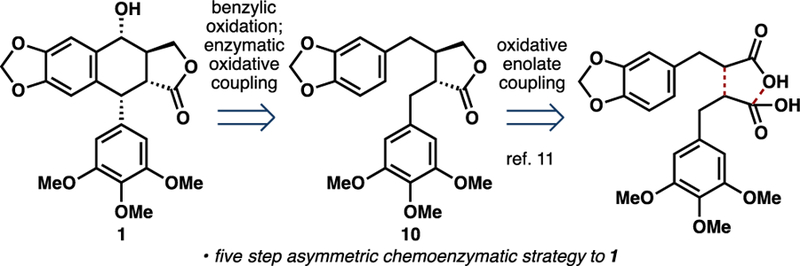
Retrosynthetic analysis of (–)-podophyllotoxin (1) employing chemoenzymatic disconnections.
Scheme 1.
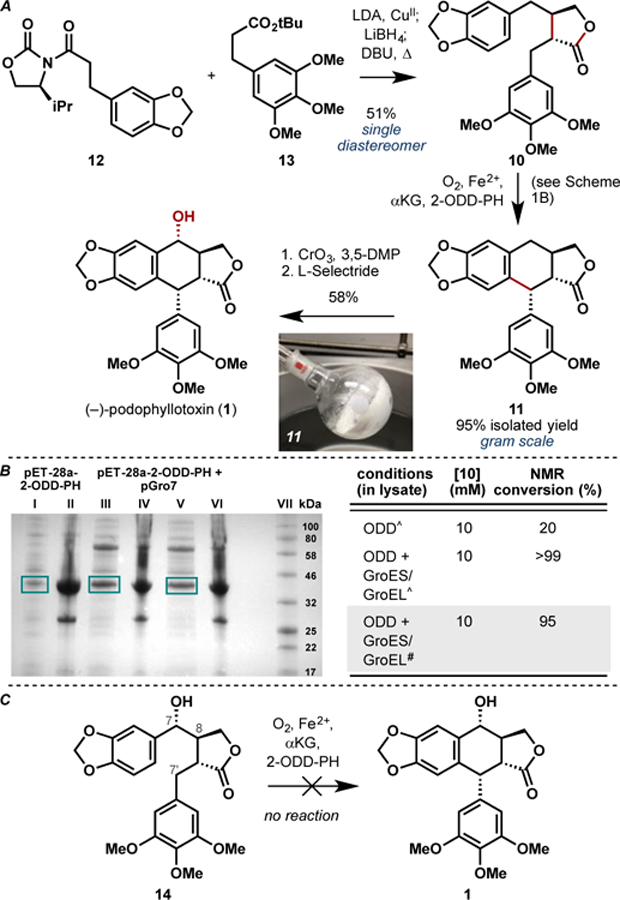
A. Chemoenzymatic total synthesis of (–)-podophyllotoxin (1) starting from 12 and 13. B. Improvement in soluble expression of 2-ODD-PH in the presence of chaperones GroES/GroEL and corresponding improvement in the conversion of 10 to 11 using clarified lysate. (I) Induction at OD600 = 0.8 with 25 µM IPTG; (II) Insoluble fraction; (III) Induction at OD600 = 0.8 with 25 µM IPTG; (IV) Insoluble fraction; (V) Induction at OD600 = 1.4 with 25 µM IPTG; (VI) Insoluble fraction; (VII) Protein ladder. ^Reaction conducted at 10 mg scale. #Reaction conducted at 1.5 g scale. C. Attempted conversion of advanced intermediate 14 to 1 using 2-ODD-PH as oxidation biocatalyst. See Supporting Information for full experimental details.
For practical reasons, we elected to conduct the enzymatic transformation using clarified lysate of E. coli expressing 2-ODD-PH. When expressed as N-His6-tagged protein, 2-ODD-PH was observed to exist predominantly in insoluble form (Scheme 1B). Despite the suboptimal enzyme expression, clarified lysate of E. coli expressing N-His6-tagged 2-ODD-PH could be used to catalyze the conversion of 10 to 11 in moderate conversion (ca. 20% conversion on 10 mg scale). To ensure adequate material throughput in our synthesis, we embarked on further enzyme expression optimization. Co-expression of chaperones GroEL and GroES12 was found to boost the soluble expression level of N-His6-tagged 2-ODD-PH and in turn enabled full conversion of 10 to 11 using clarified lysate. Gratifyingly, the biotransformation could be conducted on gram scale without any appreciable loss in conversion to afford 95% isolated yield of 11. Under these conditions, 400 mL of clarified lysate could be produced consistently from 500 mL of expression culture (pre-lysis OD600 = 30) and ca. 1.6 g of 10 could be oxidized in a single pass, translating to a titer yield of more than 3 g/L. With 11 in hand, an efficient strategy to introduce the secondary alcohol at C7 was sought. Prior synthesis of related aryltetralin lignans suggest that a two-step process with NBS and PCC could be employed to introduce ketone functionality at C7.6g,13 We found that the use of CrO314 as oxidant allowed for direct access to the C7 ketone product, which in turn could be reduced with L-Selectride6g to complete our chemoenzymatic synthesis of (–)-podophyllotoxin. Taken together, this route proceeded in five steps in asymmetric fashion with 28% overall yield. It is worth noting that an alternative route to 1 starting from alcohol 14 was also explored as it would hypothetically enable a three-step asymmetric synthesis of 1 from commercial materials (Scheme 1C).15 However, the use of 2-ODD-PH on 14 failed to elicit the desired C–C coupling to forge the C ring of 1 and led only to recovery of unreacted 14 in our hands.
To further explore the synthetic utility of this approach, we chose to target the chemoenzymatic preparation of several related aryltetralin lignans. For optimal divergency, α-alkylation16 of enantiopure butyrolactones 15–17 and S4 was employed to prepare the enzymatic cyclization precursors (Figure 3A). Among the eleven additional dibenzylbutyrolactones tested (18–22, S2, S3, S5–S8, see Table S3 in the Supporting Information for a full list of substrates tested), five (18–22) were found to produce the desired oxidative coupling products when submitted to the enzymatic reaction without any formation. For these five substrates, no undesired diastereomers or side products arising from benzylic hydroxylation were observed.
Figure 3.
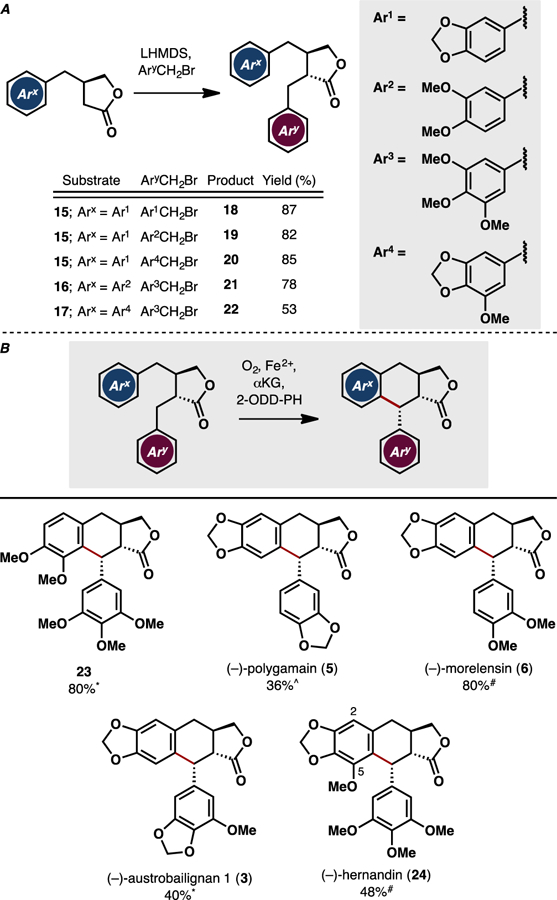
A. Diversity-oriented synthesis of dibenzylbutyrolactones 18–22 via α-alkylation. B. Chemoenzymatic synthesis of 23, (–)-polygamain (5), (–)-morelensin (6), (–)-austrobailignan I (3), and (–)-hernandin (24) via late-stage C–C coupling with 2-ODD-PH. *^Reaction conducted at 5 mM substrate concentration with pre-lysis OD600 = 30. ^Reaction conducted at 1.5 mM substrate concentration with pre-lysis OD600 = 30. #Reaction conducted at 4 mM substrate concentration with pre-lysis OD600 = 30.
Despite bearing high structural similarity to yatein, 21 was unexpectedly converted by 2-ODD-PH to a product (23) that is regioisomeric to deoxypodophyllotoxin and deoxysikkimotoxin. While 18 and 19 could be converted to (–)-polygamain (5) and (–)-morelensin (6) by 2-ODD-PH, the reactions had to be performed at lower substrate:enzyme ratio to provide synthetically useful yields of the desired products. A similar trend was also observed when lactones 20 and 22 were employed in the enzymatic reaction to produce (–)-austrobailignan 1 (3) and (–)-hernandin (24), respectively. To properly benchmark the oxidative coupling efficiency, we also measured total turnover numbers (TTNs) and specific activities of 2-ODD-PH on substrates 18–22 and these data were found to be in good agreement with the yields obtained in the preparative scale reactions (See Table S4 for TTN and specific activity measurements). Comparing the relative reactivity of 18 versus 19 and 10 versus 20, we noted that a simple switch of 1,2-dimethoxy motif to 1,2-methylenedioxy bridge in the southern fragment led to several fold reduction in reaction conversion. Furthermore, introduction of an additional methoxy group at C2 (substrates S5–S8) completely abolished enzymatic activity while an additional methoxy group at C5 (substrate 22) was tolerated in the reaction. These results indicate that the active site of 2-ODD-PH is highly sensitive to changes in the substitution patterns of the aryl rings. Additionally, Kroutil and Fuchs showed that the trimethoxybenzene moiety in the southern fragment of the scaffold is essential for successful C–C coupling when substrates containing C7-OH were used. In contrast, our results suggest that substrates containing less electron-rich aromatic rings could still be employed in the reaction when C7-OH is absent. Taken together, these observations point to subtle changes in substrate positioning in the active site when C7-OH is present, which in turn affects the outcome of the oxidative process. Future structural studies on 2-ODD-PH are expected to provide some insight on the key interactions that govern the substrate specificity of the enzyme and inform subsequent engineering work to expand its substrate scope.
In summary, we have developed a concise chemoenzymatic synthesis of (–)-podophyllotoxin and related aryltetralin lignans through the use of three oxidative transformations. A key oxidative enolate coupling between oxazolidinone 12 and ester 13 efficiently furnished (–)-yatein (10) while establishing the absolute stereoconfiguration at C8 of the butyrolactone motif. In the subsequent key step, biocatalytic C–C coupling catalyzed by 2-ODD-PH constructed the tetracyclic core of podophyllotoxin with complete diastereoselectivity and excellent isolated yield on gram scale. Finally, a late-stage chemoselective oxidation enabled rapid introduction of the final secondary alcohol at C7. Overall, our chemoenzymatic approach towards (–)-podophyllotoxin constitutes one of the shortest and highest yielding synthetic route towards this venerable target (five steps, 28% overall yield, see Table S5 for summary of recent approaches towards 1). The modularity and the scalability of the route, coupled with the complete avoidance of precious metal catalysts, should render it attractive for both scale-up production of 1 and future medicinal chemistry exploration. Similar to our previous work in the use of non-heme dioxygenases in the synthesis of complex alkaloids and morphed peptides,12b,17 this works provides further evidence for the biocatalytic utility of non-heme dioxygenases in streamlining the synthesis of medicinally relevant natural product scaffolds.
Supplementary Material
Acknowledgements
Financial support for this work was generously provided by The Scripps Research Institute and the National Institutes of Health (grant GM128895). We acknowledge the Shen lab and the Roush lab for generous access to their instrumentation.
Footnotes
Experimental Section
See Supporting Information for Experimental Details.
References
- [1].Botta B, Monache GD, Misiti D, Vitali A, Zappia G, Curr. Med. Chem 2001, 8, 1363–1381. [DOI] [PubMed] [Google Scholar]
- [2].(a) Gordaliza M, Castro MA, del Corral JMM, San Feliciano A, Curr. Pharm. Des 2000, 6, 1811–1839; [DOI] [PubMed] [Google Scholar]; (b) Wolff J, Knipling L, Cahnmann HJ, Palumbo G, Proc. Natl. Acad. Sci. USA 1991, 88, 2820–2824; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Loike JD, Horwitz SB, Biochemistry 1976, 15, 5435–5443; [DOI] [PubMed] [Google Scholar]; (d) Schilstra MJ, Martin SR, Bayley PM, J. Biol. Chem 1989, 264, 8827–8834. [PubMed] [Google Scholar]
- [3].(a) Clark PI, Slevin ML, Clin. Pharmacokinet 1987, 223–252; [DOI] [PubMed]; (b) Long BH, Musial ST, Brattain MG, Cancer Res 1985, 45, 3106–3112; [PubMed] [Google Scholar]; (c) Smith PJ, Soues S, Gottlueb T, Falk SJ, Watson JV, Osborne RJ, Bleehen MM, Br. J. Cancer 1994, 70, 914–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].(a) Jolad SD, Wiedhopf RM, Cole JR, J. Pharm. Sci 1977, 66, 892–893; [DOI] [PubMed] [Google Scholar]; (b) Wu CC, Huang KF, Yang TY, Li YL, Wen CL, Chu SL, Chen TH, PLoS One 2015, 10, e0132052; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tran TD, Pham NB, Booth R, Forster PI, Quinn RJ, J. Nat. Prod 2016, 79, 1514–1523; [DOI] [PubMed] [Google Scholar]; (d) Hartley RM, Peng J, Fest GA, Dakshanamurthy S, Frantz DE, Brown ML, Mooberry SL, Mol. Pharmacol 2012, 81, 431–439; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Pena-Moran OA, Villarreal MK, Alvarez-Berber L, Meneses-Acosta A, Rodriguez-Lopez V, Molecules 2016, 21, E1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].For recent reviews on this topic, see: (a) Sellars JD, Steel PG, Eur. J. Org. Chem 2007, 3815–3828;; (b) Sun J-S, Liu H, Guo X-H, Liao J-X, Org. Biomol. Chem 2016, 14, 1188–1200; [DOI] [PubMed] [Google Scholar]; (c) Yu X, Che Z, Xu H, Chem. Eur. J 2017, 23, 4467–4526. [DOI] [PubMed] [Google Scholar]
- [6].For recent total syntheses of 1, see: (a) Stadler D, Bach T, Angew. Chem. Int. Ed 2008, 47, 7557–7559; [DOI] [PubMed] [Google Scholar]; (b) Wu Y, Zhao J, Chen J, Pan C, Zhang H, Org. Lett 2009, 11, 597–600; [DOI] [PubMed] [Google Scholar]; (c) Ting CP, Maimone TJ, Angew. Chem. Int. Ed 2014, 53, 3115–3119; [DOI] [PubMed] [Google Scholar]; (d) Lisiecki K, Krawczyk KK, Roszkowski P, Maurin JK, Czarnocki Z, Org. Biomol. Chem 2016, 14, 460–469; [DOI] [PubMed] [Google Scholar]; (e) Hajra S, Garai S, Hazra S, Org. Lett 2017, 19, 6530–6533; [DOI] [PubMed] [Google Scholar]; (f) Xiao J, Cong X-W, Yang G-Z, Wang Y-W, Peng Y, Org. Lett 2018, 20, 1651–1654. [DOI] [PubMed] [Google Scholar]
- [7].Lazzarotto M, Hammerer L, Hetmann M, Borg A, Schmermund L, Steiner L, Hartmann P, Belaj F, Kroutil W, Gruber K, Fuchs M, Angew. Chem. Int. Ed 2019, accepted, doi: 10.1002/anie.201900926. [DOI] [PMC free article] [PubMed]
- [8].Lau W, Sattely ES, Science 2015, 349, 1224–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Chang W-C, Yang Z-J, Tu Y-H, Chien T-C, Org. Lett 2019, 21, 228–232. [DOI] [PubMed] [Google Scholar]
- [10].Newhouse TR, Baran PS, Hoffmann RW, Chem. Soc. Rev 2009, 38, 3010–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].DeMartino MP, Chen K, Baran PS J. Am. Chem. Soc 2008, 130, 11546–11560. [DOI] [PubMed] [Google Scholar]
- [12].(a) Thomas JG, Ayling A, Baneyx F, Appl. Biochem. Biotechnol 1997, 66, 197–238; [DOI] [PubMed] [Google Scholar]; (b) Zhang X, King-Smith E, Renata H, Angew. Chem. Int. Ed 2018, 57, 5037–5041. [DOI] [PubMed] [Google Scholar]
- [13].Takano S, Sato N, Otaki S, Ogasawara K, Heterocycles 1987, 25, 69–73. [Google Scholar]
- [14].Dauben WG, Lorber M, Fullerton DS, J. Org. Chem 1969, 34, 3587–3592. [Google Scholar]
- [15].Compound 14 could be prepared in 2 steps using protocols developed in the following report: Hajra S, Giri AK, Hazra S, J. Org. Chem 2009, 74, 7978–7981. [DOI] [PubMed] [Google Scholar]
- [16].Charlton JL, Chee G-L, Can. J Chem 1997, 75, 1076–1083. [Google Scholar]
- [17].(a) Zwick CR III, Renata H, J. Am. Chem. Soc 2018, 140, 1165–1169; [DOI] [PubMed] [Google Scholar]; (b) Zwick CR III, Renata H, J. Org. Chem 2018, 83, 7407–7415; [DOI] [PubMed] [Google Scholar]; (c) Ziwck CR III, Renata H, Tetrahedron 2018, 74, 6469–6473. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.