

SUMMARY
The expression of some proteins in the autophagy pathway decline with age, which may impact neurodegeneration in diseases, including Alzheimer’s Disease. We have identified a novel non-canonical function of several autophagy proteins in the conjugation of LC3 to Rab5+, clathrin+ endosomes containing β-amyloid in a process of LC3-associated endocytosis (LANDO). We found that LANDO in microglia is a critical regulator of immune-mediated aggregate removal and microglial activation in a murine model of AD. Mice lacking LANDO but not canonical autophagy in the myeloid compartment or specifically in microglia have a robust increase in pro-inflammatory cytokine production in the hippocampus and increased levels of neurotoxic β-amyloid. This inflammation and β-amyloid deposition was associated with reactive microgliosis and tau hyperphosphorylation. LANDO-deficient AD mice displayed accelerated neurodegeneration, impaired neuronal signaling, and memory deficits. Our data support a protective role for LANDO in microglia in neurodegenerative pathologies resulting from β-amyloid deposition.
Keywords: LC3-associated endocytosis, LANDO, autophagy, LC3-associated phagocytosis, β-amyloid, Alzheimer’s Disease, neurodegeneration, neuroinflammation, microglia, microgliosis, tau pathology, receptor-mediated endocytosis
Graphical Abstract
INTRODUCTION
Microglial cells are the primary immune cell of the central nervous system (CNS). Although distinct from peripheral macrophages (Lenz and Nelson, 2018), microglia have the ability to recognize pathogens and other inflammatory stimulants by virtue of a host of receptors including toll-like receptors (TLRs) (Gurley et al., 2008), Fc receptors (Fuller et al., 2014), Ig-superfamily receptors including TREM2 (Ulrich et al., 2014; Wang et al., 2016; Zhao et al., 2018), scavenger receptors (SR) (Wilkinson and El Khoury, 2012) and complement receptors (Doens and Fernandez, 2014). It is currently believed that cooperation between several of these receptor families is responsible for the recognition of and response to Aβ-peptide oligomers and amyloids (Aβ) by microglial cells (Doens and Fernandez, 2014; Liu et al., 2012). Upon binding of ligands such as Aβ, microglial cells internalize the target by receptor-mediated endocytosis (RME), leading to activation of signaling pathways and cytokine production in a ligand-dependent manner (Dheen et al., 2007). Microglia can undergo pro-inflammatory, classical (M1) or anti-inflammatory, alternative (M2) activation depending on which immune receptor is engaged, resulting in the activation of multiple downstream intracellular signaling pathways (Wang et al., 2014).
Microglia are the principal mediators of inflammation in response to Aβ accumulation (Machado et al., 2016; Perry and Holmes, 2014; Wang et al., 2015a), and their contribution to neuroinflammation is highly correlated to the progression of neurodegeneration and synaptic dysfunction, particularly with respect to Alzheimer’s disease (AD). Pro-inflammatory cytokines and chemokines secreted into the immediate neurological environment accelerate neuronal injury and eventually neuron death (Aktas et al., 2007; Heckmann et al., 2018; Morales et al.,2014).
The process of macro-autophagy (herein, autophagy) functions to sustain cell survival under conditions of nutrient deprivation and to remove damaged organelles, protein aggregates, and other cellular components to maintain “quality control”. The expression of several autophagy proteins, in particular Beclin1, ATG5, and ATG7, decline with age, and is exacerbated in AD (Lipinski et al., 2010; Lucin et al., 2013; Pickford et al., 2008; Rubinsztein et al., 2011). Roles for autophagy have been explored in the context of neuronal function, homeostasis, and alterations leading to disease pathology and neuronal dysfunction (Colacurcio et al., 2018; Ejlerskov et al., 2019; Harris and Rubinsztein, 2011; Menzies et al., 2017; Rubinsztein et al., 2015). While axonal autophagy has been implicated as a protective mechanism in the early stages of neurodegeneration (Maday, 2016), the functions of autophagy proteins are less well characterized in the context of microglia and immune activation.
We hypothesized that defects in autophagy or autophagy proteins might influence microglial behavior in the context of Aβ deposition. We employed an established murine model of AD and found that in the myeloid compartment (including microglia), the autophagy protein, ATG5, promoted protection from Aβ accumulation, neuroinflammation, and neurodegeneration. In contrast, another requisite autophagy protein, RB1-inducible coiled-coil protein 1, also called FIP200 (herein, FIP200), has no such function. Strikingly, a protein that constrains canonical autophagy, Rubicon, was similarly required for the protective effects. In the absence of Rubicon either globally or conditionally in microglia, or myeloid ATG5 (but not myeloid or microglial FIP200), we observed accelerated disease pathology and neurodegeneration, reactive microgliosis, tau pathology, and behavioral impairment.
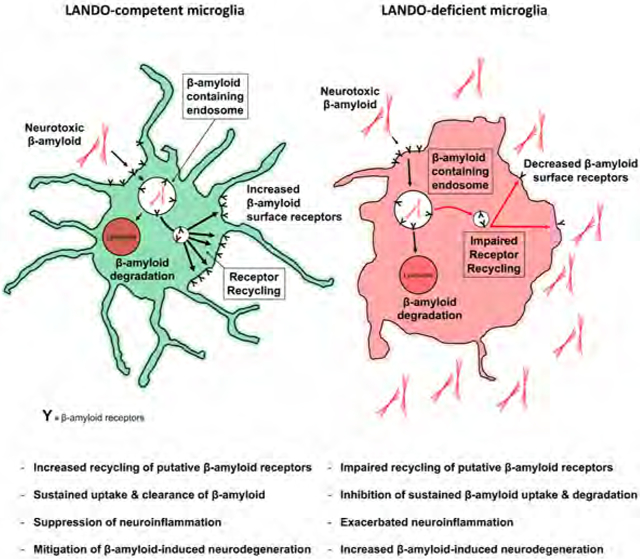
In dissecting these effects, we identified a novel form of RME and receptor recycling characterized by association of LC3/GABARAP-family proteins (herein, “LC3”) with endosomal membranes, a process we call LC3-Associated eNDOcytosis (LANDO). In response to Aβ, LANDO supports the clearance of Aβ deposits and prevents microglial activation, such that in its absence, inflammatory cytokine production and neurodegeneration proceed unchecked. This alternative function of some components of the autophagy pathway support the idea that changes in the expression of autophagy proteins can influence AD pathology in a manner that does not involve canonical autophagy.
RESULTS
ATG5 and Rubicon-deficiency exacerbate Aβ deposition and pathology
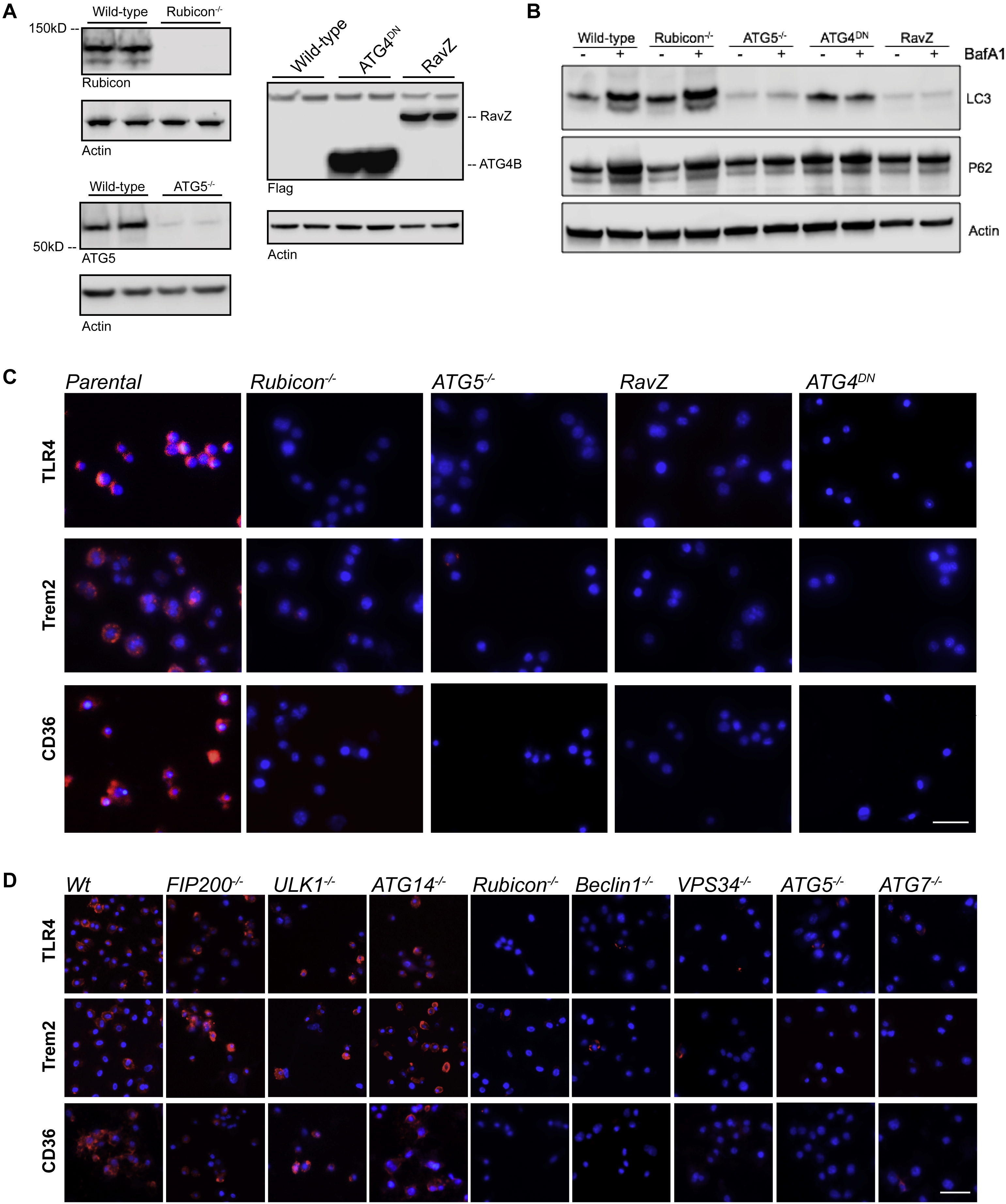
To ascertain the involvement of microglial autophagy proteins in the establishment and progression of Aβ deposition and neuroinflammation, we employed a murine model in which animals express transgenes of APP and Presenilin1 containing several mutations associated with human familial AD, the 5xFAD (B6.Cg-Tg (APPSwFILon, PSEN1 *M146L*L286V) 6799Vas) model (Oakley et al., 2006). The 5xFAD transgene was crossed into mice with conditional ablation of the key autophagy regulators FIP200 and ATG5 using lysozyme M (LysM/Lyz2)-Cre-lox recombination, which targets cells of the myeloid lineage and microglia, with an efficiency ranging from 40-90% in microglia (Abram et al., 2014; Ferro et al., 2018; Pulido-Salgado et al., 2017). Primary microglia isolated from LysM-Cre+ FIP200fl/fl and ATG5fl/fl mice (referred to as FIP200,cre+ and ATG5,cre+) showed a significant reduction in mRNA and protein expression when compared to LysM-Cre− littermates (Fig. S1A,B). It has also been suggested that LysM-cre can produce off-target effects in neurons (Orthgiess et al., 2016). Therefore, we analyzed FIP200 and ATG5 protein expression in primary neurons from both FIP200 and ATG5 genotypes using LysM-cre and observed no changes in endogenous expression levels (Fig. S1C). Moreover, primary microglia from these genotypes displayed a dramatic reduction in both autophagic activation and autophagic flux when compared to cells isolated from cre− littermates, as measured by LC3-II generation following treatment with rapamycin or LC3-II and P62 accumulation following treatment with the lysosomal inhibitor bafilomycin A1 (Fig. S1D,E). In addition to conditional ablation of FIP200 and ATG5 in myeloid cells, we also examined 5xFAD mice with germline-deficiency of Rubicon (Rubicon−/−). Rubicon has been shown to be an inhibitor of canonical autophagy (Matsunaga et al., 2009) and a key component of a non-canonical function of autophagy proteins that modulates inflammatory immune activation (Cunha et al., 2018; Martinez et al., 2016).
To establish mice that have conditional deletion of FIP200 or Rubicon in microglia, we generated 5xFAD, FIP200fl/fl or 5xFAD, Rubiconfl/fl mice with a CX3CR1-creERT2 transgene (Goldmann et al., 2013) (See STAR Methods). Primary microglia isolated from 5xFAD, Rubicon and FIP200, CX3CR1-creERT2 mice treated with tamoxifen at one month of age and allowed to reconstitute for 3 months (herein referred to as MG-cre+) showed ablation of Rubicon (Fig. S1F,G) or FIP200 (Fig. S1G), as indicated. In contrast, expression of each gene was restored in peripheral, splenic macrophages (Fig. S1H), as expected.
Mice that are autophagy-deficient (5xFAD, FIP200, LysM- or MG-cre+) displayed no differences in Aβ deposition when compared to LysM- or MG-cre− littermates (Fig. 1A-D). In contrast, deletion of ATG5 in the myeloid compartment (5xFAD, ATG5, LysM-cre+) led to a dramatic increase in Aβ plaque number and plaque size within the hippocampus of young 5xFAD mice, at a time point (4 mos) when the 5xFAD model showed little Aβ accumulation (Fig. 1A-D). This difference between myeloid deficiency of these two autophagy genes suggested an alternative pathway responsible for regulating Aβ deposition, separate from canonical autophagy. Consistent with this notion and similar to mice deficient in myeloid ATG5, 5xFAD Rubicon−/− mice and 5xFAD Rubicon MG-cre+ mice displayed an increase in Aβ plaque number and plaque size compared to either Rubicon+/− or MG-cre− littermates (Fig. 1A-D and S1I). Likewise, both 5xFAD ATG5 LysM-cre+, 5xFAD Rubicon−/−, and 5xFAD Rubicon MG-cre+ mice showed early accumulation of Aβ within the cortex, whereas mice deficient for myeloid or microglial FIP200 were unaffected compared to LysM- or MG-cre− littermates (Fig. 1E,F and Fig. S1J,K). Whole brain immunoblot analysis for Aβ confirmed these increases in deposition (Fig. 1D). We also noted lower Aβ accumulation in the FIP200 MG-cre+or− and the Rubicon MG-cre− (control) animals than we observed in our LysM-cre− or Rubicon+/− animals (Fig. 1B,C,D). It is possible that this was due to the described ability of tamoxifen, given to all of the MG-cre animals, to reduce oligomerization of Aβ (Park et al., 2016). Despite this, the effects on Aβ accumulation upon microglial-specific deletion of Rubicon were similar to those observed in Rubicon−/− mice (Fig. 1B,C,D).
Figure 1. ATG5 and Rubicon-deficiency exacerbates Aβ deposition.

A. Representative images for Aβ (red) in the hippocampus of 4m-old 5xFAD mice with indicated genetic alterations. Scale bars, 100μm
B. and C. Quantification of Aβ plaque # (C) and plaque area (D) in the hippocampus of 4m-old 5xFAD mice. Each point represents average quantification from one mouse.
D. Whole brain Aβ analysis by immunoblot in 4m-old mice from the indicated 5xFAD genotypes.
E. Representative images for Aβ (red) deposition in the 5th cortical layer in 4m-old 5xFAD mice with microglia-specific deletion (MG-cre+) of FIP200 or Rubicon. Scale bars, 50μm.
F. Quantification of Aβ plaque # in the cortex of 4m-old 5xFAD, microglia FIP200 or Rubicon-deficient mice. Each point represents average quantification from one mouse.
Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. **p<0.01, ****p<0.0001.
The exacerbation of Aβ accumulation within the hippocampus and cortex of the myeloid ATG5 and Rubicon-deficient mice but not in mice deficient in myeloid FIP200 is suggestive of an alternative function of canonical autophagy proteins in the regulation of Aβ deposition.
LC3 is recruited to endosomes containing Aβ.
In an attempt to delineate the differences in Aβ accumulation in the animal models presented above and the role of the autophagy proteins in this process, we engineered BV2 murine microglial cells expressing GFP-LC3 that are deficient in FIP200, ATG5, or Rubicon using CRISPR/Cas9 (Fig. S2A). Cells were cultured in the presence of oligomeric Aβ1-42 labeled with TAMRA. Parental cells and those lacking FIP200 displayed recruitment of LC3 to Aβ-containing vesicles (Fig. 2A). In stark contrast, ATG5 and Rubicon-deficient cells cultured with Aβ had a marked reduction in such recruitment (Fig. 2A), as well as membrane-associated LC3 (Fig. 2B) as measured by flow cytometry and described previously (Eng et al., 2010; He et al., 2009; Martinez et al., 2011; Martinez et al., 2015). Inhibition of actin polymerization and phagocytosis using latrunculin A (de Oliveira and Mantovani, 1988; Oliveira et al., 1996), prevented the internalization of the phagocytic substrate zymosan but had no effect on either the endocytic substrate dextran or Aβ (Fig. 2C), suggesting that uptake of Aβ in our model occurs primarily through endocytosis (RME). Consistent with this hypothesis, LC3+ Aβ-containing vesicles showed co-localization of clathrin and the endosomal marker Rab5 with LC3 (Fig. S2B). Inhibition of phagocytosis with latrunculin A also did not alter the association of LC3 to endosomal membranes in response to Aβ (Fig. S2C). Taken together, these data suggest that both ATG5 and Rubicon are necessary for recruitment of LC3 to Aβ-containing endosomes, whereas FIP200 is dispensable.
Figure 2. ATG5 and Rubicon-deficiency impairs LANDO and recycling of Aβ receptors.

A. Representative images showing that GFP-LC3-recruitment to Aβ (red) containing vesicles in BV2 microglia is dependent on ATG5 and Rubicon, but not FIP200. White arrows indicate LC3+ endosomes. Cells were treated with 1μM oligomeric TAMRA-Aβ1-42 for 6h. Scale bars, 5μm. Values are (# of cells containing LC3+, Aβ+ vesicles/100 Aβ-containing cells).
B. Quantification of membrane-associated GFP-LC3 in BV2 microglia following 6h stimulation with 1μM oligomeric TAMRA-Aβ1-42. GFP-LC3 was assayed using flow cytometry. Each point represents one independent experiment performed in triplicate.
C. Quantification of zymosan (4:1, particle:cell), dextran (500ng/ml), or Aβ (1μM) uptake in BV2 microglia treated with either a vehicle or 50mM latrunculin A (LA). n=3 per condition performed in duplicate.
D. Aβ clearance assay performed in BV2 microglia treated with oligomeric TAMRA-Aβ1-42 for 1h. n=4 per genotype performed in duplicate.
E. Quantification of fluorescent pH-rodo signal from BV2 microglia of the indicated genotypes stimulated with pH-rodo Zymosan (5:1, particle:cell) or pH-rodo Aβ (1μM) for 3h. n=3 per genotype performed in duplicate.
F. Quantification of the co-localization between zymosan or Aβ and LAMP1 labeled lysosomes in BV2 microglia (see Fig. S2F). Cells were treated with zymosan (4:1, particle:cell), or Aβ (1μM) for 3h. n=3 per genotype performed in duplicate.
G. Primary and secondary uptake of Aβ measured in BV2 microglia. Each point represents one independent experiment performed in duplicate.
H. Representative images of receptor recycling for TLR4, TREM2, and CD36 in BV2 microglia. Scale bars, 50mm.
I. Quantification of recycled receptors in BV2 microglia (See STAR Methods). Each point is one independent experiment performed in duplicate.
J. Representative images of TREM2 recycling in primary microglia from Rubicon+/− or Rubicon−/− mice. Scale bars, 10μm.
K. Quantification of TREM2 recycling in primary microglia from indicated genotypes. Each point is one independent experiment performed in duplicate.
Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. *p<0.05, **p<0.01, ***p<0.001.
Our findings with ATG5, Rubicon, and FIP200 are consistent with a well-established non-canonical function of autophagy proteins in the LC3-associated phagocytosis (LAP) pathway (Heckmann et al., 2017). Deficiencies in LAP have been shown to reduce the ability of a cell to degrade phagosome cargo, including dying cells, fungi, and bacteria by impairing phagosome maturation and lysosomal interaction (Heckmann et al., 2017; Heckmann and Green, 2019; Martinez et al., 2011; Martinez et al., 2016; Martinez et al., 2015). We hypothesized that this may in part explain the increased Aβ accumulation observed in our deficient murine models. To test this idea, we performed a pulse-chase assay using oligomeric TAMRA-labeled Aβ1-42. Contrary to our assumptions, however, ablation of either ATG5 or Rubicon had no effect on either the endocytosis or degradation of Aβ (Fig. 2D). Indeed, earlier studies had shown no role for LAP in the maturation of dextran-containing endosomes or in the degradation of internalized EGFR following ligation (Cunha et al., 2018). Consistent with these reports, in the absence of Rubicon and ATG5, maturation of phagosomes was diminished as determined using pH-rodo-labeled zymosan, while maturation of endosomes containing pH-rodo-labeled Aβ was unaffected (Fig. 2E, S2E). Likewise, in cells lacking ATG5 or Rubicon, but not FIP200, phagosomes containing zymosan showed a reduced association with the lysosome marker LAMP1, while LAMP1 association with Aβ-containing endosomes was unaffected (Fig. 2F, S2F). These results argue against a role for LAP in regulating Aβ endocytosis and degradation. Based on the association of LC3 to Aβ-containing endosomes, we propose the term LC3-Associated eNDOcytosis (LANDO) to describe this effect and suggest that LANDO is distinct from LAP and may have a unique role in Aβ clearance in vivo.
Recycling of the putative Aβ receptors TREM2 and CD36 to the plasma membrane, following RME, require two autophagy proteins, Beclin1 and VPS34 (Lucin et al., 2013). Therefore, we examined the role of FIP200, ATG5, and Rubicon in recycling of TREM2, CD36, and TLR4 (suggested to be a putative Aβ receptor (Reed-Geaghan et al., 2009; Song et al., 2011). We first asked if subsequent rounds of Aβ endocytosis would be impacted in our model. We treated BV2 cells with AF488-Aβ1-42 and measured primary Aβ uptake. Consistent with our clearance assay, the loss of FIP200, ATG5, or Rubicon had no effect on primary uptake of Aβ (Fig. 2G). We next added TAMRA-labeled Aβ1-42 and allowed for a second round of RME to occur. Again, we quantified the amount of internalized TAMR-1-42 and observed a reduction in secondary uptake in ATG5 and Rubicon-deficient cells but not those deficient in FIP200 (Fig. 2G). These results support a failure to return receptors to the plasma membrane following initial internalization, since secondary rounds of uptake were decreased but primary uptake was unaffected.
To better understand the abrogation of secondary uptake, we evaluated the impact of loss of FIP200, ATG5, and Rubicon on receptor recycling, using an established method (Lucin et al., 2013) (Fig. S3). In BV2 microglia, depletion of FIP200 had no effect on either the surface expression, internalization (Fig. S2D), or recycling of CD36, TLR4, or TREM2 (Fig. 2H,I). Strikingly, ablation of either ATG5 and Rubicon led to abrogation of recycling for all three receptors (Fig. 2H,I) while initial surface expression and internalization were unaffected (Fig. S2D). Furthermore, since TREM2 is the most well-characterized Aβ receptor that activates Aβ endocytosis (Doens and Fernandez, 2014; Ries and Sastre, 2016; Ulland et al., 2017; Wang et al., 2015b; Zhao et al., 2018), we evaluated TREM2 recycling in primary microglia cells isolated from Rubicon−/− mice. Rubicon-deficiency dramatically reduced recycling of TREM2 (Fig. 2J,K). These findings indicate a role for ATG5 and Rubicon in the regulation of receptor recycling upon internalization of Aβ, as assessed by this method (Lucin, et al., 2013). Moreover, when taken together, the abrogation of receptor recycling but not internalization can explain why we did not see altered primary uptake but observed an impact on secondary rounds of endocytosis that are ATG5 and Rubicon dependent, but FIP200 independent. Therefore, LANDO facilitates the recycling of internalized Aβ receptors to the plasma membrane, supporting continued rounds of endocytosis.
To further explore this phenomenon, we employed the RAW264.7 myeloid cell line, and found that TLR4, TREM2, and CD36 effectively recycled to the plasma membrane following antibody-induced internalization. This recycling was dramatically impaired upon CRISPR/Cas9-mediated ablation of Rubicon or ATG5, upon expression of a dominant negative ATG4, or upon expression of the LC3-specific protease, RavZ (Fig. 3A, S4A-C). Expression of ATG4DN or RavZ, or ablation of ATG5 abrogated LC3-lipidation and autophagic flux (seen as accumulation of p62 upon treatment with bafilomycin A), as expected (Fig. S4B). The requirement for ATG4, which converts LC3 to LC3-I (Kabeya et al., 2004) and the effect of RavZ, which irreversibly cleaves lipidated LC3 (Choy et al., 2012; Kwon et al., 2017), strongly suggest that lipidation of LC3-family proteins is required for recycling of these receptors. We then extended our analysis using primary, bone marrow-derived macrophages (BMDMs) from animals with myeloid ablation of a number of genes required for autophagy and/or LAP (Fig. 3B, Fig. S4D). Ablation of Beclin1, VPS34, ATG5, or ATG7, required for both autophagy and LAP, prevented recycling of TLR4, TREM2, and CD36, as did Rubicon, required for LAP but not autophagy (Martinez et al., 2015). In contrast, no effects were seen upon ablation of ULK1, FIP200, or ATG14, all required for autophagy, but dispensable for LAP (Martinez et al., 2015). Therefore, the requirements for recycling of these receptors to the plasma membrane appear to be identical to those for LAP and distinct from those of autophagy. However, since we observed no effects on degradation of Aβ (Fig. 2D), and since antibody-induced receptor internalization is via endocytosis and not phagocytosis, we conclude that it is the association of LC3 with endosomes (LANDO) that displays the genetic requirements for recycling of receptors for Aβ. While we cannot formally exclude functions for LAP in the clearance and response to Aβ in vivo, our results strongly suggest that LANDO plays a more central role in this process.
Figure 3. Abrogation of LANDO promotes Aβ-induced inflammation.

A. Quantification of receptor recycling in RAW264.7 cells deficient in the indicated genes as shown or overexpressing RavZ or dominant-negative ATG4 as shown. Each data point represents a unique experiment performed in duplicate.
B. Quantification of receptor recycling in BMDMs isolated from the indicated genotypes and for the indicated receptors. Each data point represents a unique experiment performed in duplicate.
C. and D. Pro-inflammatory cytokine expression in (C) BMDMs or (D) BV2 microglia in response to 1μM oligomeric Aβ1-42 measured by qPCR. Cells were treated for 12h. For C., n=3 per genotypes performed in duplicate. For D., n=4 per genotype performed in triplicate.
E. qPCR analysis of pro-inflammatory gene expression in primary microglia following 1μM oligomeric Aβ1-42 exposure for 12h. n=3 per genotype performed in triplicate.
F. Cytokine production by primary microglia in response to 1μM oligomeric Aβ1-42 measured 12h post-incubation by ELISA. n=3 per genotype performed in duplicate.
Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. *p<0.05, **p<0.01, ***p<0.001.
LANDO protects against Aβ-induced microglial activation
Our data suggest that the exacerbation of Aβ accumulation in our LANDO-deficient mice is a result of impaired recycling of putative Aβ receptors leading to extracellular deposition. Aβ, especially the Aβ1-42 oligomer and fibril, is an established instigator of neuroinflammation (Cai et al., 2014). Since we have previously shown a vital role for LAP in regulating inflammatory immune responses (Martinez et al., 2016), we hypothesized that defective LANDO may have a similar effect. We therefore analyzed inflammatory cytokines in the BMDMs exposed to Aβ1-42 oligomers in the above experiment. Strikingly, those genotypes that displayed defective receptor recycling showed a dramatically elevated TNFα, IL-1β, and IL-6 response to Aβ1-42 (Fig. 3C).
We similarly examined the effects of Aβ1-42 on microglia. As expected, BV2 cells treated with Aβ1-42 had elevated pro-inflammatory gene expression, including IL-1β, IL-6, CCL5, and TNFα as reported (Pan et al., 2011) and consistent with human disease. FIP200-deficiency failed to have any impact on cytokine expression in response to Aβ1-42 (Fig. 3D). However, loss of LANDO in both ATG5 and Rubicon-deficient cells resulted in a robust increase in all four pro-inflammatory genes evaluated (Fig. 3D).
These findings were further substantiated in primary microglia from Rubicon-deficient mice. A potent hyperactivation of TNFα, IL1-β and IL-6 was observed at both the mRNA (Fig. 3E) and protein levels (Fig. 3F) upon Aβ1-42 exposure in Rubicon−/− microglia. Together, these results suggest a role for LANDO in the mitigation of inflammatory activation in response to Aβ.
We next asked if reactive microglial activation was present in our murine models. Using Iba1 as a marker for microglial activation (Hoogland et al., 2015), we assessed hippocampal and cortical sections to evaluate microglial activation. Consistent with our in vitro findings, myeloid or microglia-specific FIP200-deficiency had no effect on the extent of Iba1 expression in either the hippocampus or cortex of 5xFAD mice (Fig. 4A-C). In contrast, microglial hyperactivation was present in 5xFAD mice deficient in either Rubicon or myeloid ATG5 (Fig. 4A,B). This activation was not constrained to the hippocampus and was also present in the cerebral cortex (Fig. 4A,C).
Figure 4. LANDO decreases Aβ-induced reactive microgliosis.

A. Representative images showing microglial activation/expansion (green-Iba1 positive) in the hippocampus and the 5th cortical layer (cortex) of the indicated 4m-old 5xFAD genotypes. Scale bars, 100μm.
B. and C. Quantification of activated microglia in the hippocampus (B) and cortex (C) respectively. Data shown is the MFI of Iba1 staining for each genotype relative to Wt, 5xFAD littermate controls. Each point represents an individual mouse.
D. Representative images indicating microglial morphology in 4m-old mice. Scale bars, 10μm.
E. Quantification of ramified vs. ameboid microglia in the indicated 5xFAD genotypes at 4m of age. Each point represents an individual mouse.
F. Representative images and quantification of microglia/plaque-association in 4m-old 5xFAD, Rubicon+/− or Rubicon−/− mice. (See STAR Methods). Each point represents an individual mouse. Scale bars, 5μm.
G. qPCR analysis of inflammatory gene expression in hippocampal slices from 4m-old 5xFAD mice of the indicated genotypes. n=7 mice per genotype, qPCR performed in triplicate.
Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. *p<0.05, **p<0.01, ***p<0.001.
Hyperactivation of microglia leads to the morphological transition of cells from the ramified to the inflammatory ameboid state (Kim and Joh, 2006). Morphological analysis of microglia in the hippocampus of 5xFAD LANDO-deficient mice revealed a ramified-to-ameboid transition, with Rubicon-deficient (both global and microglia-specific) and myeloid ATG5-deficient mice having a reduction in ramified microglia in the hippocampus when normalized to total microglial numbers and compared to control littermates (Fig. 4D,E). FIP200-deficiency had no effect on microglial state transition when compared to control littermates. Furthermore, in 5xFAD Rubicon-deficient mice, analysis of microglia-plaque association revealed an increase in plaque-associated microglia (Fig. 4F), suggestive of progressive gliosis in response to Aβ.
Due to the high level of microglia activation in ATG5 and Rubicon-deficient mice, and the transition to the more reactive ameboid morphology, we decided to profile the most clinically relevant pro-inflammatory cytokines implicated in neuroinflammation in AD (Shaftel et al., 2008). Consistent with what we observed with cultured cells (Fig. 3), Rubicon- or myeloid ATG5-deficiency led to significant upregulation of pro-inflammatory cytokines at the transcriptional level in the brain, whereas myeloid FIP200-deficiency had no effect. Using Iba1 expression as a positive control, we observed significant increases in TNFα, IL-1β, IL-6, and a marginal increase in CCL5 within the hippocampus of 5xFAD Rubicon−/− and myeloid ATG5-deficient mice compared to control littermates (Fig. 4G). Neither microglial activation nor pro-inflammatory cytokine expression were altered in mice deficient in the 5xFAD transgene (Fig. S5A,B). We observed that over 90% of CD11b+ cells in brains from the 5xFAD Rubicon+/− and−/− mice were TMEM119+ (a microglia-specific marker (Bennett et al., 2016)) (Fig. S5C,D). Furthermore, the TMEM119+ cells isolated from 5xFAD Rubicon−/− mice had higher Iba1 expression compared to Rubicon+/− animals (Fig. S5E). Therefore, we conclude that the vast majority of inflammatory cells we observed were microglia rather than brain-infiltrating peripheral monocytes, and that genetic manipulation of either Rubicon or ATG5 does not affect basal cytokine expression in the absence of 5xFAD.
These results suggest a critical role for Rubicon and myeloid ATG5 in mitigating reactive/inflammatory microglial activation in the 5xFAD model. Taken as a whole, these data suggest LANDO may contribute to not only the regulation of aberrant Aβ deposition but also the immune activation of microglial cells in response to Aβ exposure.
LANDO-deficient 5xFAD mice have robust tau pathology
In agreement with human AD, LANDO-deficient 5xFAD animals displayed severe Aβ accumulation that promoted reactive microgliosis. A marker of progressive AD in both humans and mice is the hyperphosphorylation of the microtubule-stabilizing protein tau. The incidence of tau phosphorylation increases as disease progresses and leads to microtubule de-stabilization and failure of the microtubule architecture, especially within neuronal axons (Frost et al., 2015; Gong and Iqbal, 2008; Noble et al., 2013). We evaluated the phosphorylation state of tau and identified the presence of phospho-tau which paralleled both our Aβ and microglial phenotypes. Myeloid or microglia-specific FIP200 depletion had no impact on tau phosphorylation, however loss of either Rubicon (global or microglia-specific) or myeloid ATG5 promoted tau phosphorylation throughout the hippocampus (Fig. 5A,C, S6A) and the cerebral cortex (Fig. 5B,D, S6B). These data suggest loss of LANDO promotes rapid alterations to tau that are indicative of highly progressive disease.
Figure 5. LANDO mitigates tau hyperphosphorylation.

A. and B. Representative images showing phosphorylation of Tau at S202/T205 in the hippocampus (A) and cortex (B) of 4m-old LANDO-deficient 5xFAD mice. Scale bars, 100μm for A, 30μm for B. Upper images are combined DAPI and anti-pTau, lower are anti-pTau only.
C. and D. Quantification of phospho-tau in the hippocampus (C) and cortex (D) of the indicated 5xFAD genotypes at 4m of age. The MFI of phospho-tau staining for each genotype is shown as relative to Wt, 5xFAD littermates as described for Iba1 staining (See Fig. 4 legend). Each point represents an individual mouse.
Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. *p<0.05, **p<0.01, ***p<0.001.
LANDO-deficient 5XFAD mice display accelerated neuronal death and impaired neuronal function
Because tau phosphorylation is likely to promote axonal degeneration and eventual neuronal death, we analyzed cell death within the brains of our 5xFAD models. 5xFAD Rubicon−/− microglia-specific Rubicon-deficient, and myeloid ATG5-deficient mice all displayed a decrease in NeuN+ hippocampal neurons (Fig. 6A,B). FIP200-deficiency had no impact on neuronal number.
Figure 6. LANDO-deficiency promotes Aβ-induced neuronal death.

A. Representative images showing neurons (NeuN-green) in the hippocampus of the indicated 4m-old 5xFAD genotypes. Scale bars, 100μm.
B. Quantification of neuronal content (#) within the hippocampus at 4m of age. Each point represents an individual mouse.
C. Representative images identifying neuronal apoptosis within the CA3-region of the hippocampus of 4m-old 5xFAD Rubicon-deficient mice. Scale bars, 30μm.
D. Quantification of apoptotic neurons within the hippocampus of 4m-old 5xFAD Rubicon-deficient mice. Each point represents an individual mouse.
E. and F. Analysis of hippocampal synaptic transmission (E) and long-term potentiation (F) in 5xFAD Rubicon-deficient mice. n=9 mice per genotype with a minimum of 5 slices per mouse.
Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. *p<0.05, **p<0.01, ***p<0.001.
Since we observed a reduction in the number of neurons in LANDO-deficient, but not autophagy-deficient models, we evaluated cell death in brains isolated from 5xFAD Rubicon−/− mice. To measure neuronal apoptosis, we stained for cleaved-caspase 3. There was a robust increase in cleaved-caspase 3-positive neurons in 5xFAD Rubicon-deficient mice compared to littermate controls in the CA3-field of the hippocampus (Fig. 6C,D). This region has been implicated as one of the first sites of neuronal dysfunction and neuronal death in AD (Belvindrah et al., 2014; Padurariu et al., 2012; Zhang et al., 2013). In combination, these results support the idea that control of Aβ deposition and microglial activation by LANDO is critical for preventing hyperphosphorylation of tau, neuronal apoptosis, and degeneration.
We next performed electrophysiological assessment of neuronal function in an effort to substantiate and refine our observations of neuron loss in the CA3-field. We found that 5xFAD Rubicon−/− mice showed a reduction in hippocampal synaptic transmission and, as a consequence, impaired long-term potentiation (LTP) when compared to littermate controls (Fig. 6E,F). The neuronal death and major impairment in neuronal physiology was surprising, as 5xFAD mice do not usually show signs of cell death and functional impairment until at least 5-6 months of age, and to a lesser extent than we observed in 4-month old 5xFAD Rubicon−/− mice (Eimer and Vassar, 2013). Therefore, when microglial LANDO is defective, neuronal cell death induced by Aβ is accelerated, particularly within the pre-synaptic neurons of the hippocampus. Reduction of this neuronal population was confirmed by inhibition of pre-synaptic transmission and LTP.
LANDO-deficiency accelerates behavioral and memory impairment in 5xFAD mice
Our data support a physiological role for LANDO in microglia in the mitigation and protection against microglial activation and immune-mediated aggregate Aβ) removal. We therefore subjected mice to a variety of well-characterized behavioral tests known to be affected at late stages in the 5xFAD model. In advanced AD, patients often complain of anhedonia, or the inability to sense or experience pleasure (Naudin et al., 2015; Reichman and Coyne, 1995), which can be analyzed in mice using a sucrose preference test (SPT) (Briones et al., 2012; Liu et al., 2018). 5xFAD mice deficient in myeloid FIP200 showed no variation in their preference for sucrose water when compared to wild-type 5xFAD animals, suggesting they have intact reward behavior (Fig. 7A). In contrast, both Rubicon and myeloid ATG5-deficient mice presented with anhedonia. Both genotypes were at approximately 50% sucrose preference, or simple chance (Fig. 7A) by 4 months of age. Interestingly, behavioral and memory deficits do not typically begin to show significant differences in 5xFAD animals until at least 5-7 months of age (Girard et al., 2014; Ohno, 2009). 5xFAD, Rubicon-deficient mice presented with anhedonia as young as 2.5 months old. No variations in total fluid intake between genotypes was observed (Fig. 7B).
Figure 7. Loss of CA3 neurons in LANDO-deficient mice leads to behavior and memory impairment.

A. and B. Sucrose preference test (A) and fluid intake measurement (B) for the indicated 5xFAD genotypes at both 2.5 and 4m of age. Each data point represents an individual mouse.
C. and D. Y-maze test for short-term memory measuring spontaneous arm alternation (C) and total arm entries (D) in the indicated 5xFAD genotypes at 4m of age. Each data point represents an individual mouse.
E. – G. Analysis of novel object recognition measuring total exploration time (E), preference for the novel object (F), and the ability to discriminate (G) in 4m-old 5xFAD Rubicon+/− or Rubicon−/− mice. Each data point represents an individual mouse.
Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. *p<0.05, ***p<0.001, ****p<0.0001.
Results from the SPT suggested a pervasive memory impairment. Therefore, we employed two routine tests for short-term and working short-term memory, the novel object recognition test (NOR) and the Y-maze test, respectively. Consistent with their performance in the SPT, 5xFAD Rubicon−/− and myeloid ATG5-deficient mice had a reduction in spontaneous alternation (Fig. 7C) without having a decrease in total arm entries (Fig. 7D) in the Y-maze test. Moreover, short-to-medium term memory was drastically reduced in the 5XFAD Rubicon−/− mice, as measured by NOR. Rubicon-deficiency resulted in a decrease in novel object preference, and an almost complete reduction in their discrimination index (Fig 7F,G). These analyses illustrate the importance for the molecular regulation of microglial function by LANDO in maintaining CNS integrity and immune function upon Aβ deposition, allowing for homeostasis in memory and behavior.
DISCUSSION
Our studies investigating the role of the autophagy proteins in the regulation of immune function in the CNS are reliant upon genetic manipulation of key autophagy regulators FIP200, ATG5, and the negative regulator Rubicon. We employed a well-characterized model of Aβ aggregate deposition, the 5xFAD AD mouse model with genetic manipulation of our target autophagy-related genes. We observed that both global and microglia-specific deletion of Rubicon and myeloid-specific deletion of ATG5 resulted in exacerbated Aβ deposition and plaque formation, reactive microgliosis, tau hyperphosphorylation, and neuronal cell death and dysfunction, leading to significant memory impairment. Myeloid or microglial deficiency in FIP200 failed to have an effect on the above parameters when compared to littermate controls. Moreover, all animals were generated on the C57BL/6J background which was confirmed by microsatellite analysis, therefore the differences observed do not reflect a background effect between strains. Therefore, our results strongly suggest a role for microglial Rubicon and ATG5 in the clearance of Aβ and mitigation of microglial activation that is distinct from their functions in canonical autophagy.
The observed roles for ATG5 and Rubicon, but not FIP200, is reminiscent of LAP. However, while LAP functions to promote phagosome maturation and cargo destruction (Heckmann et al., 2017), we observed no effects of our deletions on the rate of Aβ degradation, endosome maturation, or lysosome association (Fig. 2). Because we observed the association of LC3 and the endosomal markers Rab5 and clathrin at the membranes of Aβ-containing endosomes that was not altered by inhibition of phagocytosis, we called this LANDO. According to the receptor-recycling method we employed and established by others (Lucin et al., 2013; Yin et al., 2016), we found that LANDO is required for the recycling of putative Aβ receptors (CD36, TREM2, and TLR4) from internalized endosomes to the plasma membrane. A previous study had shown that this recycling of CD36 and TREM2 is dependent on Beclin1 and VPS34 (Lucin et al., 2013), which we confirmed in primary BMDM (Fig. 3). We further found that Rubicon, ATG5, ATG7, and ATG4 were required, while ULK1, FIP200, and ATG14 were dispensable for this effect. The Legionella-derived protease, RavZ, which irreversibly cleaves lipidated LC3 (Choy et al., 2012; Kwon et al., 2017) also prevented this receptor recycling, again, as defined by this method (Lucin et al., 2013; Yin et al., 2016). The role for ATG4, which processes LC3 proteins to LC3-I for lipidation, and the effect of RavZ, as well as the roles for the ligation machinery (ATG7, ATG5) strongly suggest that lipidation of LC3 at the endosome functions in the recycling of these receptors. We conclude that LANDO is distinct from canonical autophagy and plays a requisite role in the recycling of putative Aβ receptors. The assay used herein does not evaluate the role of LANDO in receptor degradation, or other potential perturbations in endosome trafficking, and it remains possible that such alterations alter receptor signaling to promote cytokine expression in response to Aβ. In turn, increased cytokines, such as interleukin-1, can influence Aβ uptake (Mrak and Griffin, 2000; Shaftel et al., 2007). Nevertheless, our findings support the notion that LANDO in the myeloid compartment of the CNS and more specifically in microglia, functions to protect neurons from the neuroinflammatory and neurodegenerative effects of Aβ deposition.
The reactive microgliosis we observed in the 5xFAD Rubicon and myeloid ATG5-deficient mice resulted in robust increases in neuroinflammatory activation. Based on our findings using myeloid ATG5-deficiency and conditional ablation of Rubicon in microglia, we suggest that LANDO functions in microglia not only to promote Aβ clearance but also to promote an anti-inflammatory immune response. One caveat to the 5xFAD models used herein is the inability to fully exclude a role for infiltrating peripheral monocytes in Aβ clearance and neuroinflammation. We found, however, no increase in peripheral macrophage infiltration at 4-months of age in our 5xFAD Rubicon-deficient mice (Figure S5). A previous study utilized parabiosis to evaluate the contribution of circulating monocytes to the microglial pool in 5xFAD mice and demonstrated a negligible contribution of peripheral cells (Wang et al., 2016). Moreover, the use of CX3CR1- creERT2 to transiently ablate Rubicon resulted in Rubicon-deficient microglia but Rubicon-sufficient splenic macrophages at 4-months of age, but does not completely exclude the possibility that Rubicon-deficient peripheral monocytes may have entered the brain prior to reconstitution of this population from the bone marrow. Therefore, although our results suggest LANDO is required in microglia for imparting the protection observed in our 5xFAD models, we cannot completely exclude any involvement of peripheral immune cells in the effects we observed.
Our data strongly suggest that microglial Rubicon and myeloid ATG5 are protective against Aβ-induced pro-inflammatory cytokine production and its consequences (Fig. 3 and 4). The reasons for this are not known. We had previously suggested that persistence of cargo in LAP-deficient cells enables continued signaling at the phagosome membrane, however LANDO-deficient cells did not display increased persistence of endosomal Aβ. However, loss of LANDO affected secondary uptake of Aβ, leading to increased extracellular deposition, and it is plausible that these deposits signal at the cell surface via other receptors to promote inflammatory signaling. Similar effects were observed in response to dying cells in macrophages lacking Rac1 or the efferocytosis receptors merTK and TIM4 (Cai et al., 2018; Camenisch et al., 1999; Martinez et al., 2011; Scott et al., 2001; Thorp et al., 2008), consistent with this idea.
We have found that a subset of the autophagy proteins including Beclin1, ATG5, and ATG7 are obligatory for LANDO function. The expression of these three proteins decreases with age (Lipinski et al., 2010), which may lead to an insufficiency in LANDO, establishing a putative risk factor for pathology. Whole body acute deletion of ATG7 causes lethal neuronal degeneration (Karsli-Uzunbas et al., 2014). While autophagy proteins have been shown to impact the clearance of protein aggregates in neurons, most likely by the canonical autophagic process of aggrephagy (Frake et al., 2015; Menzies et al., 2017; Qaisiya et al., 2017; Sarkar et al., 2007; Sarkar et al., 2009; Webb et al., 2003; Yamamoto and Simonsen, 2011), effects in microglia of the CNS are more likely due to LANDO. Finally, the dramatically accelerated disease observed in our 5xFAD animals may facilitate the testing of neuroprotective agents in vivo.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-TLR4 | Abcam | Cat# ab22048 |
| Anti-CD36 | Abcam | Cat# ab23680 |
| Anti-NeuN | Abcam | Cat# ab104224 |
| Anti-LAMP1 | Abcam | Cat# ab25630 |
| Anti-CD11b | Abcam | Cat# ab8878 |
| Anti-TMEM119 | Abcam | Cat# ab209064 |
| Anti-cleaved-caspase 3 | Cell Signaling | Cat# 9664 |
| Anti-FIP200 | Cell Signaling | Cat# 12436 |
| Anti-ATG5 | Cell Signaling | Cat# 12994 |
| Anti-Rubicon (D9F7) | Cell Signaling | Cat# 8465 |
| Anti-Aβ 82E1 | IBL | Cat# 10326 |
| Anti-Iba1 | Novus | Cat# NB100-1028 |
| Anti-TREM2 | R&D Systems | Cat# MAB17291 |
| Anti-FLAG M2 | Sigma-Aldrich | Cat# F3165 |
| Anti-β-actin | Thermo | Cat# MA1-91399 |
| Anti-phospho-tau (S202/T205) | Thermo | Cat# 44-768G |
| Bacterial and Virus Strains | ||
| pLenti-V2 lentivirus (for CRISPR) | This Paper | N/A |
| lentiBrite-GFP-LC3 Sensor | Millipore | Cat# 17-10193 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| β-amyloid (1-42) peptide - unlabeled | Anaspec | Cat# AS-20276 |
| β-amyloid (1-42) peptide - TAMRA | Anaspec | Cat# AS-60476 |
| β-amyloid (1-42) peptide – Hilyte 488 | Anaspec | Cat# AS-60479-01 |
| β-amyloid (1-42) peptide - TAMRA | SJCRH Core | Custom Synthesis |
| β-amyloid (1-42) scrambled peptide – TAMRA | SJCRH Core | Custom Synthesis |
| Dextran – Texas Red | Invitrogen | Cat# D1863 |
| Zymosan A – AF594 | Invitrogen | Cat# Z23374 |
| ProLong Diamond DAPI mounting media | Invitrogen | Cat# P36971 |
| pHRodo Zymosan Bioparticles | Invitrogen | Cat# P35364 |
| Rapamycin | Sigma-Aldrich | Cat# R8781 |
| Latrunculin A | Sigma-Aldrich | Cat# L5163 |
| Critical Commercial Assays | ||
| Neural Tissue Dissociation Kit | Miltenyi | Cat# 130-092-628 |
| Microglia Isolation Kit | Miltenyi | Cat# 130-093-634 |
| Universal SYBR Green | Bio-Rad | Cat# 1725271 |
| M-MLV Kit | Invitrogen | Cat# 28025013 |
| pHRodo iFL Labeling Kit | Invitrogen | Cat# P36014 |
| Mouse Multianalyte Inflammatory Cytokine ELISA Kit | Qiagen | Cat# MEM-004A |
| RNeasy Mini Kit | Qiagen | Cat# 74104 |
| Experimental Models: Cell Lines | ||
| BV2 murine microglia | Washington University | Herbert Virgin |
| RAW264.7 murine macrophage | ATCC | Cat# SC-6003 |
| Primary murine BMDM | This Paper | N/A |
| Primary murine microglia | This Paper | N/A |
| Experimental Models: Organisms/Strains | ||
| 5xFAD transgenic mice | Jackson Laboratory | Cat# 34840-JAX |
| CX3CR1-creERT2 | Jackson Laboratory | Cat# 021160 |
| FIP200fl/fl LysM-Cre+, 5xFAD | This Paper | N/A |
| ATG5fl/fl LysM-Cre+, 5xFAD | This Paper | N/A |
| Rubicon−/−, 5xFAD | This Paper | N/A |
| Rubiconfl/fl CX3CR1-creERT2, 5xFAD | This Paper | N/A |
| FIP200fl/fl CX3CR1-creERT2, 5xFAD | This Paper | N/A |
| Beclin1fl/fl LysM-Cre+ | University of Kentucky | Edmund Rucker |
| ATG7fl/fl LysM-Cre+ | TMIMS | Masaaki Komatsu |
| ATG14fl/fl LysM-Cre+ | Washington University | Herbert Virgin |
| VPS34fl/fl LysM-Cre+ | Yale University | Richard Flavell |
| ULK1−/− LysM-Cre+ | SJCRH | Mondira Kundu |
| Oligonucleotides | ||
| Rubicon-sgRNA1 5’ – CACCGAGGAGACTCGTCCATACACG – 3’ 3’ – AAACCGTGTATGGACGAGTCTCCTC – 5’ |
This paper | N/A |
| Rubicon-sgRNA2 5’ – CACCGTGATGAGGAACGGGCGAAGA – 3’ 3’ – AAACTCTTCGCCCGTTCCTCATCAC – 5’ |
This paper | N/A |
| ATG5-sgRNA1 5’ – GTGAGCCTCAACCGCATCCT – 3’ 3’ – CACTCGGAGTTGGCGTAGGA – 5’ |
This paper | N/A |
| ATG5-sgRNA2 5’ – CGGAACAGCTTCTGGATGAA – 3’ 3’ – GCCTTGTCGAAGACCTACTT – 5’ |
This paper | N/A |
| FIP200-sgRNA1 5’ – AGAGTGTGTACTTACAGCGC – 3’ 3’ – TCTCACACATGAATGTCGCG – 5’ |
This paper | N/A |
| FIP200-sgRNA2 5’ – GAGGATCATGCTCCTAGAAC – 3’ 3’ – CTCCTAGTACGAGGATCTTG – 5’ |
This paper | N/A |
| Actin qPCR F: ATGGAGGGGAATACAGCCC R: TTCTTTGCAGCTCCTTCGTT |
This paper | N/A |
| TNFα qPCR F: CCTGTAGCCCACGTCGTAGC R: AGCAATGACTCCAAAGTAGACC |
This paper | N/A |
| IL1β qPCR F: CACAGCAGCACATCAACAAG R: GTGCTCATGTCCTCATCCTG |
This paper | N/A |
| IL6 qPCR F: GAGGATACCACTCCCAACAGACC R: AAGTGCATCATCGTTGTTCATACA |
This paper | N/A |
| CCL5 qPCR F: CCAATCTTGCAGTCGTGTTTGT R: CATCTCCAAATAGTTGATGTATTCTTGAAC |
This paper | N/A |
| Iba1 qPCR F: CAGACTGCCAGCCTAAGACA R: AGGAATTGCTTGTTGATCCC |
This paper | N/A |
| Recombinant DNA | ||
| pLenti-V2 plasmid | Addgene | Cat# 52961 |
| psPAX plasmid | Addgene | Cat# 12260 |
| pVSVg plasmid | Addgene | Cat# 8454 |
| pMXs-FLAG plasmid | Cell Biolabs | Cat# RTV-016 |
| pMXs-FLAG-mATG4B-C74A | This Paper | N/A |
| pMXs-FLAG-RavZ | This Paper | N/A |
| Software and Algorithms | ||
| Slidebook 6 | 3i | https://www.intelligent-imaging.com/slidebook |
| Nikon NIS-elements Advanced Research | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| FlowJo v10.4 | Tree Star | https://www.flowjo.com/solutions/flowjo |
| LiCOR Image Studio | LiCOR | https://www.licor.com/bio/image-studio/ |
| GraphPad Prism 7.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| G*Power | University of Dusseldorf | http://www.gpower.hhu.de/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Douglas R. Green (douglas.green@stjude.org).
MATERIALS & REAGENTS
See Key Resources Table.
EXPERIMENTAL MODEL & SUBJECT DETAILS
Mice
The 5xFAD transgenic mice carrying the following five mutations: Swedish (K670N and M671L), Florida (I716V) and London (V717I) in human APP695 and human PS1 cDNA (M146L and L286V) under the transcriptional control of the neuron-specific Thy-1 promoter and were purchased from The Jackson Laboratory. 5xFAD mice were crossed to FIP200fl/fl LysM-Cre+ (kindly provided by Jun-Lin Guan, University of Michigan), ATG5fl/fl LysM-Cre+ (kindly provided by Thomas A. Ferguson, Washington University), Rubicon−/− mice which were generated by our laboratory as described previously (Martinez et al., 2015), and Rubiconfl/fl mice (kindly provided by Jennifer Martinez, NIEHS). CX3CR1-creERT2 mice were purchased from the Jackson Laboratory and conditional deletion of FIP200 or Rubicon was achieved using tamoxifen induction and peripheral reconstitution as described previously (Goldmann et al., 2013). CX3CR1-creERT2 positive mice that have been injected with tamoxifen and have reconstituted the monocytic pool are referred to herein as MG-cre+cre mice. MG-cre− animals used in the present study have been exposed to tamoxifen and allowed the same reconstitution period as the MG-cre+ mice to ensure no off-target effects from tamoxifen administration. Mice used for bone marrow isolation and BMDM culture were kindly provided as follows; Beclin1fl/fl LysM-Cre+ (Edmund Rucker, University of Kentucky), ATG7fl/fl LysM-Cre+ (Masaaki Komatsu at The Tokyo Metropolitan Institute of Medical Science), ATG14fl/fl LysM-Cre+ (Herbert Virgin, Washington University), VPS34fl/fl LysM-Cre+ (Richard Flavell, Yale University), and ULK1−/− LysM-Cre+ (Mondira Kundu, St. Jude Children’s Research Hospital).
Unless otherwise noted, all experiments were performed on mixed sex cohorts at 4-months of age. Depending on genotype, either LysM-cre−, MG-cre−or Rubicon+/− littermates were used as controls. The St. Jude Institutional Animal Care and Use Committee approved all procedures in accordance with the Guide for the Care and Use of Animals. All mice were housed in pathogen-free facilities, in a 12-hour light/dark cycle in ventilated cages, with chow and water supply ad libitum.
The genetic backgrounds of all mice used were assessed at the DartMouse™ Speed Congenic Core Facility at the Geisel School of Medicine at Dartmouth. DartMouse uses the Illumina, Inc. (San Diego, CA) Infinium Genotyping Assay to interrogate a custom panel of 5307 SNPs spread throughout the genome. The raw SNP data were analyzed using DartMouse’s SNaP-Map™ and Map-Synth™ software, allowing the determination for each mouse of the genetic background at each SNP location. All strains used had an overall background identity of greater than 97%.
Cells
BV2 murine microglia and RAW264.7 cells were obtained from ATCC. Cells were maintained in complete DMEM media (10% fetal bovine serum (FBS), 200 mM L-glutamine and 100 units/ml penicillin-streptomycin). All the cell lines used were confirmed as mycoplasma negative using MycoAlert Mycoplasma Detection kit (Lonza #LT07).
For preparation of bone marrow-derived macrophages (BMDM), male or female mice at 6 to 12 weeks of age were euthanized and bone marrow cells were harvested from the femurs and differentiated in DMEM containing 20% FBS, 200 mM L-glutamine, 100 units/ml penicillin-streptomycin, 20 ng/ml recombinant human M-CSF for 10 days. BMDMs were harvested and seeded on tissue culture plates 1 day before stimulation and maintained in complete DMEM media. All cells used in this study were cultivated at 37°C with 5% CO2.
METHOD DETAILS
Generation and maintenance of cell lines
BV2 microglia deficient in FIP200, ATG5, and Rubicon were generated using CRISPR/Cas9 technology by lentiviral transduction and puromycin selection. Two guide RNAs (gRNA) were designed for each gene (See Reagents List for sequences) and cloned into the pLenti-V2 plasmid (Addgene). Lentivirus was produced using HEK293T cells co-expressing pPAX and pVSVg plasmids (Addgene) and our CRISPR pLenti-V2 plasmids using Lipofectamine 2000 (Invitrogen). BV2 cells were subsequently transduced and transduction efficiency was confirmed by immunoblot analysis following two weeks of puromycin selection. An empty pLenti-V2 vector was transduced to establish a parental cell line. Once confirmed, cells were then exposed to LentiBrite GFP-tagged LC3 lentivirus (Millipore) to establish GFP-LC3 positive lines. BV2 microglia deficient in TREM2 were kindly provided by Marco Colonna (Washington University).
RAW264.7 lines deficient in Rubicon or ATG5 were established using CRISPR/Cas9 viral transduction as described above for BV2 cells. RAW264.7 cells that overexpress either RavZ or a dominant-negative ATG4 were generated by transduction using a retrovirus carrying pMXs-Flag-mATG4B-C74A (Blasticidin) or pMXs-Flag-RavZ (Blasticidin) or the empty vector. The retroviral vectors were creased as follows. Mouse ATG4B was cloned from a mouse cDNA library into pMXs retroviral vector. The active site Cysteine (C74) was mutated to Alanine using site-directed mutagenesis. The original vector expressing RavZ was a gift from Craig Roy (Yale University), the ORF was subcloned into pMXs.
All lentiviral and retroviral work was performed in accordance with the guidelines set forth by the SJCRH Institutional Biosafety Committee and within the scope of our approved Biosafety protocol.
β-amyloid preparation and treatment
Both labeled and unlabeled Aβ1-42 was purchased in lyophilized form and resuspended according to the manufacturer’s recommendation at a concentration of 100μM (Anaspec). In brief, Aβ1-42 was resuspended to 5mM in DMSO and then adjusted to 100μM using DMEM/F12 culture media. Oligomerization was allowed to occur for 24h at 4°C prior to addition to cells at 1μM unless otherwise indicated. Incubation times for each individual experiment is detailed in the respective figure legends.
Primary microglia and neuron isolation
Mice were anesthetized with isoflurane and perfused with 1% BSA in PBS. Brains were subsequently harvested and immediately processed using the papain-based Neural Dissociation Kit (Miltenyi). Myelin was removed using myelin removal beads and microglia were purified using CD11b microglia beads (Miltenyi). Primary neurons were purified from dissociated whole brain described above using the mouse neuron isolation kit (Miltenyi). Purified cells were then used as indicated. All steps were performed per the manufacturer’s instructions.
Microscopy and image analysis
For all non-live cell-based imaging, cells were cultured in 4-well chamber slides (Ibidi) and were fixed and stained as indicated. In brief, cells were fixed with 4% PFA for 10 min. followed by permeabilization using 0.1% Triton-X 100 for 5 min. Cells were blocked in 0.5% BSA in PBS for 30min prior to staining with primary antibodies overnight at 4°C. Cells were then washed 3x in PBS and then stained with the indicated fluorescent secondary antibodies for 30 min. Cells were subsequently washed 3x with PBS and post-fixed in 1% PFA for 10 min. prior to imaging. For all live cell-based imaging, cells were immediately transferred to an environment controlled, live-cell imaging chamber (Ibidi).
For preparation of brain tissue see “Preparation of brain samples” below. Slides were subjected to antigen retrieval using 1% sodium citrate boiling for 20 min. followed by 3x PBS washing. Slides were blocked in 0.5% BSA in PBS. Antibody staining was carried out as described above. Following final washing, slides were mounted using ProLong Diamond Anti-Fade mounting media with DAPI.
All imaging was performed on either an Eclipse Ti-E TIRF/N-Storm/epifluorescence microscope (Nikon) or a Marianis spinning disk confocal microscope (Intelligent Imaging Innovations (3i)) equipped with an EMCCD camera. Image analysis including all quantification was performed using either Nikon NIS-elements Advanced Research Imaging software or Slidebook 6 (3i).
Image analysis for relative Iba1 and phospho-tau staining was achieved by quantifying the mean fluorescent intensity (MFI) of either Iba1 or phospho-tau signal using NIS-elements. MFI data for each genotype was subsequently compared to MFI signal obtained from Wt, 5xFAD littermate controls to achieve a relative comparison.
Analysis of microglial/plaque-association was defined as the % of microglia per field that are co-localizing with Aβ plaques, normalized to total plaque # per field.
Analysis and quantification of microglial morphology was achieved using Slidebook 6 software. Morphological state was determined by measuring cell diameter following 3D reconstruction and confirmed by manual counting/analysis of microglia shape per defined field across multiple areas of each slide.
Flow cytometry
For all uptake assays cells were analyzed without fixation. For membrane-associated GFP-LC3 analysis, cells were processed as described below. For brain infiltrating monocytes, cells were isolated as described using the Neural Tissue Dissociation Kit (Miltenyi). Primary cells were fixed, permeabilized, and staining using the Cyto Fix/Perm Staining Kit (BD Bioscience) and the indicated, conjugated primary antibodies. For all experiments, cells were analyzed using a Sony SP6800 Spectral Analyzer (Sony). All analyses were performed using FlowJo v10.4 (Tree Star). Fluorescent compensation was performed using BD compensation beads (BD Bioscience).
Preparation of brain samples
Mice were anesthetized with isoflurane and perfused with ice-cold PBS containing 1 U/ml of heparin. Right brain hemispheres were fixed in 4% PFA overnight at 4°C, rinsed in PBS, and incubated overnight at 4°C in 30% sucrose before fr eezing in a 2:1 mixture of 30% sucrose and optimal cutting temperature compound (OCT). Serial 20 μm coronal sections were cut on a cryo-sliding microtome. Cortices and hippocampi of the left-brain hemispheres were carefully dissected out and flash frozen for biochemical analysis or processed for RNA isolation.
Membrane-associated LC3 analysis
To quantify membrane association of GFP-LC3, cells were harvested and permeabilized using 200μg/ml digitonin for 15 min. on ice. Cytosolic GFP-LC3 was removed by washing cells 5x in cold PBS. Cells were then resuspended in 0.5% BSA in PBS for analysis by flow cytometry as described above. This assay has been well-established and verified previously (Eng et al., 2010; He et al., 2009; Martinez et al., 2015).
Cell & Tissue Lysis and Immunoblot
Cells were lysed in RIPA buffer for 30 min on ice [50 mM Tris (pH 7.5), 150 mM NaCl, 1% Triton X100, 0.5% deoxycholate (DOC), 0.1% SDS, protease inhibitor tablet (Roche), 1 mM NaF, 1 mM Na3VO4, and 1 mM PMSF]. Brain samples were mechanically homogenized in RIPA buffer. After centrifugation, supernatants were analyzed by SDS/PAGE. All blots were imaged using HRP-conjugated secondary antibodies and ECL using a LiCOR Odyssey Fx imaging system (LiCOR). All immunoblot analysis was performed using LiCOR Image Studio software.
Real-Time RT-PCR
Total RNA was isolated from cells or tissue using the RNeasy Kit (Qiagen) according to the manufacturer’s instructions. First-strand synthesis was performed using M-MLV reverse transcriptase (Invitrogen). Realtime PCR was performed using SYBR GREEN PCR master mix (Applied Biosystems) in an Applied Biosystems 7900HT thermocycler using SyBr Green detection protocol as outlined by the manufacturer using the following PCR conditions: 50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 1 5s and 60°C for 1 min. mRNA was normalized to actin allowing for comparison of mRNA levels. Please see key reagents table for qPCR primer sequences.
Vesicle maturation using pH-rodo substrates
Quantification of vesicle maturation was achieved using pH-rodo labeled zymosan and pH-rodo labeled β-amyloid. pH-rodo zymosan was purchased from Invitrogen. pH-rodo β-amyloid was produced by conjugating pH-rodo to unlabeled Aβ1-42 using the pH-rodo iFL labeling kit (Invitrogen) per the manufacturer’s instructions. Labeled β-amyloid was then oligomerized as described above. BV2 microglia were subsequently exposed to either pH-rodo labeled zymosan or β-amyloid pH-rodo zymosan (5:1, particle:cell) or pH-rodo β-amyloid (1μ) was added to cells for 3h and cells were imaged as described above. Quantification was performed as described for MFI.
Receptor Recycling
For receptor recycling, cells were plated on 4-well Ibidi tissue culture-coated chamber slides and allowed to reach 50% confluence. Cells were then blocked for 15 min. in the presence of 10% normal donkey-serum at 37°C. Primary antibodies tar geting the indicated receptor (see reagent list) were then added at a dilution of 1:100 in 1% donkey-serum in DMEM and cells were incubated at 37°C for 1h. Antibody-containing media was aspirated and cells were acid washed with cold-DMEM, pH 2.0. Cells were returned to 10% donkey-serum in DMEM for 1h. Alexa Fluor 568-labeled secondary antibodies were diluted 1:1000 in 1% donkey-serum in DMEM and added to cells for 1h at 37°C to label recycled receptors. Cells were subsequently acid washed as described above and then fixed in 4% PFA in PBS for 15 min. Cell permeable Hoechst dye was added to label nuclei.
Following the above protocol, fluorescent signal from recycled receptors is intracellular and readily determined by fluorescent microscopy as described above. Quantification of recycling was achieved by calculating the sum of AF568-fluorescent area divided by the total number of cells. Nikon NIS-Elements AR software was used for all image analyses and quantification. This method is well-established and used as previously reported (Lucin et al., 2013).
Furthermore, validated commercially available antibodies for CD36, TLR4, and TREM2 were used for all recycling experiments. Specificity of the antibody targeting the primary β-amyloid receptor TREM2 was confirmed by staining wild-type and TREM2 −/− BV2 microglial cells (data not shown).
Amyloid Uptake
Primary and secondary β-amyloid uptake was assayed as follows. BV2 clones were treated with 1μM Alexa Fluor 488-labeled Aβ1-42. Mean fluorescent intensity (MFI) for AF-488 was determined by flow cytometry after 12h and considered the primary uptake. 1μM TAMRA-labeled Aβ1-42 was subsequently added to the medium following the primary uptake phase. MFI for TAMRA was assessed by flow cytometry 12h following the primary uptake timepoint. This timepoint constitutes the secondary uptake.
Phagocytosis and endocytosis analysis
To delineate between phagocytosis and endocytosis, cells were treated as indicated with the phagocytic inhibitor latrunculin A. The following control substrates were used, zymosan (phagocytosis) and dextran (endocytosis), Cells were pre-treated for 1h with LA and then treated for 1h with zymosan or 3h with dextran or Aβ. All substrates were fluorescently labeled as follows, zymosan (AF594), dextran (Texas Red), and Aβ (TAMRA). Co-incubation with specific substrates was carried out at 37°C for 1h or 3h as indicated. Cells were either fixed for imaging or analyzed by flow cytometry as described above respectively.
Electrophysiology
Acute transverse hippocampal slices (400 μm) were prepared as previously described (Gingras et al., 2015). Briefly, mouse brains were quickly removed and placed in cold (4°C) dissecting ACSF containing 125 mm choline-Cl, 2.5 mm KCl, 0.4 mm CaCl2, 6 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 20 mm glucose (285–295 mOsm) under 95% O2 and 5% CO2. After dissection, slices were incubated for 1 h in ACSF containing 125 mm NaCl, 2.5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 10 mm glucose (285–295 mOsm) under 95% O2 and 5% CO2 at room temperature and then transferred into the submerged recording chamber and superfused (2–3 ml/min) with warm (30°C-32<C) ACSF. The field recordings were performed by using a setup with 8 submerged recording chambers (Campden Instruments). The fEPSPs were recorded from the CA1 stratum radiatum by using an extracellular glass pipette (3–5 MΩ) filled with ACSF. Schaffer collateral/commissural fibers in the stratum radiatum were stimulated with a bipolar tungsten electrode placed 200–300 μm away from the recording pipette.
Behavior & memory analysis
For sucrose preference tests, mice were individually house and allowed to acclimate to the testing room for 48h prior to starting the experiment. A dual bottle setup was introduced where both bottles contained only standard water. Again, mice were allowed to acclimate to the dual bottle setup for 3 days. After acclimation, one bottle was replaced with a 2% sucrose solution. Water consumption was monitored daily for 4 days. Bottles were rotated daily to minimize side bias and normalized for leakage. All results are shown as the averaged consumption and preference over the 4-day test period.
For Y-maze spontaneous alternation analysis, mice were housed in the testing room and allowed to acclimate for 48h. The Y-maze test consisted of a single 5 min. trial per mouse. Spontaneous Alternation [%] was defined as consecutive entries in 3 different arms (ABC), divided by the number of possible alternations (total arm entries minus 2). Mice with less than 5 arm entries during the 5 min. trial were excluded from the analysis.
Novel object recognition (NOR) was performed in an open-field box (40cm × 40cm). Mice were allowed to acclimate to the testing room for 48h. For habituation, mice were allowed to explore the open-field for 15 min. per day for two days. Mice were then exposed to two identical objects for 10 min. on the day of testing. 2h later a novel object was introduced, and mice were allowed to explore for 5 min. during the test phase. The time spent exploring each object was quantified manually. Novel object preference (%) and the discrimination index ((time with novel)/(novel + familiar) * 100) were calculated for each mouse.
Quantification & Statistical Analysis
Please refer to the legend of the figures for description of sample sizes and statistical test performed. Data were plotted and analyzed with GraphPad Prism 7.0 software. All experiments were designed and are powered to a minimum of 0.8 as calculated using G*Power. Differences were considered statistically significant when the p-value was less than 0.05.
Supplementary Material
Figure S1. LysM-cre and CX3CR1-creERT2 mediated abrogation of FIP200 and ATG5. Related to Figure 1. A. and B. LysM-cre mediated reduction of FIP200 and ATG5 in primary microglia isolated from the indicated genotypes as measured by immunoblot (A) and qPCR (B). For B., n=4 mice for both cre−, n=5 mice for FIP200fl/flcre+ and ATG5fl/flcre+. qPCR performed in triplicate. C. Representative immunoblot analysis showing minimal off-target effects of LysM-cre in purified neurons isolated from the indicated 5xFAD genotypes. D and E. Analysis of autophagic activation and autophagic flux in primary microglia isolated from FIP200fl/fl and ATG5fl/fl LysM-cre+ or − mice as indicated. Cells were treated with either rapamycin (100nm) or bafilomycin A1 (25nm) for 12 hours. Autophagic activation was determined by LC3-lipidation (D and E) and autophagic flux by P62 accumulation (E) by immunoblot. F and G. CX3CR1-creERT2 mice of the indicated 5xFAD genotypes were injected with tamoxifen at 21 and 23 days of age and peripheral expression in monocytes was allowed to reconstitute for 3m, leading to microglia-specific deletion of the indicated genes. Microglia were purified from each genotype and expression for either FIP200 or Rubicon was analyzed by immunoblot, mice were 4m of age. H. To confirm microglia-specific deletion, FIP200 and Rubicon reconstitution was analyzed in splenic macrophages from 4m-old mice. I. Representative images showing Aβ accumulation in the cortex of myeloid ATG5-deficient 5xFAD mice. Scale bars, 100μm. J. Representative images of cortical Aβ deposition in 4m-old myeloid-specific FIP200 and ATG5-deficient 5xFAD mice and Rubicon-deficient mice. Each point represents an individual mouse. Scale bars, 50μm. K. Quantification of cortical Aβ content as measured by plaque # from the 5th cortical layer of the indicated genotypes. All mice were 4m of age and each point represents an individual mouse. Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. *p<0.05, ***p<0.001.
{kind=link}
Figure S2. Characterization of BV2 microglia lacking FIP200, ATG5, and Rubicon. Related to Figure 2. A. Immunoblot analysis showing successful depletion of FIP200, ATG5, or Rubicon as indicated in BV2 microglia by CRISPR/Cas9. Two samples of each are shown. B. Representative imaging showing co-localization between LC3-positive, Aβ-containing vesicles and the endosomal markers clathrin and Rab5 in BV2 murine microglia. Cells were treated for 6h with 1μM TAMRA-Aβ1-42. Aβ is false-colored blue to facilitate imaging. Scale bars, 5μm. Values represent Mander’s co-localization coefficient for LC3/Clathrin or LC3/Rab5 respectively. C. Quantification of membrane-associated LC3 by flow cytometry in vehicle or latrunculin A (LA) treated parental BV2 microglia in response to either zymosan or Aβ. n=3 for each condition performed in duplicate. D. Flow cytometry-based quantification (MFI) of receptor surface expression (left) and receptor internalization (right) for receptor recycling assays (Fig. 2) in BV2 microglia. Each point represents a unique experiment performed in duplicate. E. Representative images showing uptake and maturation of vesicles containing pH-rodo labeled zymosan or Aβ as indicated. (See Fig. 2E for quantification and experimental description). Scale bars, 10μm. F. Representative images showing zymosan or Aβ co-localization with LAMP1 labeled lysosomes in BV2 microglia of the indicated genotypes (See Fig. 2F for quantification and experimental details). Scale bars, 2μm. Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. **p<0.01.
{kind=link}
Figure S3. Receptor recycling assay. Related to Figure 2. Schematic representation showing the method for quantifying receptor recycling (Lucin et al., 2013; Yin et al., 2016). (1) Following blocking as described in the STAR methods, cells are incubated with primary antibodies against the receptors of interest. (2) Any remaining cell-surface primary antibodies are then removed by acid washing. (3) Internalized receptors bound to their cognate primary antibody are allowed to cycle to the plasma membrane for 1h. (4) Cells are incubated with AF568-labeled anti-immunoglobulin (Ig) antibody to label recycled primary antibody and allowed to re-internalize carrying the AF568-tag. (5) Any remaining cell-surface AF568-secondary antibodies are removed by acid washing. Cells are then fixed and nuclei staining for cell # quantification and imaging for fully recycled receptors (intracellular signal) is performed as described in the STAR methods.
{kind=link}
Figure S4. Recycling of CD36, TREM2, and TLR4 in RAW264.7 and BMDMs is LANDO-dependent. Related to Figure 3. A. Immunoblot analysis showing either CRISPR/Cas9-mediated depletion or retroviral mediated overexpression of the indicated genes in RAW264.7 cells. B. Analysis of autophagic flux in RAW264.7 cells of the indicated genotypes. Cells were treated with 25nM bafilomycin A1 (BafA1) for 12h. Autophagic activation was determined by LC3-lipidation and P62 accumulation by immunoblot. C. and D. Representative imaging of receptor recycling in (C) RAW264.7 cells and (D) primary BMDMs. Scale bars, 50μm.
{kind=link}
Figure S5. Analysis of infiltrating monocytes versus resident microglia in 5xFAD Rubicon-deficient mice. Related to Figure 4. A. Quantification of whole brain Iba1 expression in 6m-old 5xFADnull Rubicon or ATG5 mice as indicated by flow cytometry. n=5 mice per genotype. B. Quantification of whole brain pro-inflammatory cytokine expression in 6m-old 5xFADnu Rubicon or ATG5 mice as indicated by flow cytometry. n=5 mice per genotype, performed in triplicate. C. Representative flow cytometric analysis of resident microglia versus peripheral monocytes. Expression of the microglia-specific receptor TMEM119 was analyzed on the total CD11b monocytic pool (containing all monocytes present in the brain) to delineate peripheral cells (TMEM119−) versus resident microglia (TMEM119+). D. Quantification of the percentage of infiltrating monocytes as measured by flow cytometry. n=4 mice per genotype. E. Representative histogram and quantification of microglia activation by Iba1 expression on TMEM119+ cells from 5xFAD Rubicon+/− and Rubicon−/− mice. n=4 mice per genotype. Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. ***p<0.001.
{kind=link}
Figure S6. Rubicon-deficiency promotes tau hyperphosphorylation in 5xFAD mice. Related to Figure 5. A and B. Representative images showing hyperphosphorylation of tau at S202/T205 in 4m-old Rubicon-deficient mice in both (A) the hippocampus and (B) the cortex. (See Fig. 5C,D for quantification). Scale bars, 100μm for A, 30μm for B.
{kind=link}
Highlights.
LC3-associated endocytosis (LANDO) requires Rubicon and ATG5, but not FIP200
LANDO is required for recycling of Aβ receptors including TREM2 in microglia.
LANDO confers protection against Aβ deposition and murine Alzheimer’s disease (AD)
Microglial LANDO protects against neuronal loss and memory impairment in murine AD
An LC3-associated endocytosis pathway involved in the recycling of non-canonical members of the autophagy machinery is essential for the clearance of amyloid aggregates by microglia in a model of AD
ACKNOWLEDGEMENTS
The authors thank Patrick Fitzgerald, Junmin Peng, and Donnie Eddins (SJCRH) for thoughtful discussions, suggestions, and technical assistance, and Marco Colonna for providing TREM2−/− BV2 cells. This work was supported by grants from the US National Institutes of Health; AI40646 and CA231620 to D.R.G., AI138492 and CA231423 to B.L.H, a Distinguished Innovator award from the Lupus Research Alliance to D.R.G, ALSAC, and the John H. Sununu Endowed Fellowship to B.L.H. B.T. is supported by the Paul Barrett Endowed Fellowship.
Footnotes
COMPETING INTERESTS
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Abram CL, Roberge GL, Hu Y, and Lowell CA (2014). Comparative analysis of the efficiency and specificity of myeloid-Cre deleting strains using ROSA-EYFP reporter mice. J Immunol Methods 408, 89–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aktas O, Ullrich O, Infante-Duarte C, Nitsch R, and Zipp F (2007). Neuronal damage in brain inflammation. Arch Neurol 64, 185–189. [DOI] [PubMed] [Google Scholar]
- Belvindrah R, Nosten-Bertrand M, and Francis F (2014). Neuronal migration and its disorders affecting the CA3 region. Front Cell Neurosci 8, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, Mulinyawe SB, Bohlen CJ, Adil A, Tucker A, et al. (2016). New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A 113, E1738–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briones A, Gagno S, Martisova E, Dobarro M, Aisa B, Solas M, Tordera R, and Ramirez M (2012). Stress-induced anhedonia is associated with an increase in Alzheimer's disease-related markers. Br J Pharmacol 165, 897–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai B, Kasikara C, Doran AC, Ramakrishnan R, Birge RB, and Tabas I (2018). MerTK signaling in macrophages promotes the synthesis of inflammation resolution mediators by suppressing CaMKII activity. Sci Signal 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Z, Hussain MD, and Yan LJ (2014). Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer's disease. Int J Neurosci 124, 307–321. [DOI] [PubMed] [Google Scholar]
- Camenisch TD, Koller BH, Earp HS, and Matsushima GK (1999). A novel receptor tyrosine kinase, Mer, inhibits TNF-alpha production and lipopolysaccharide-induced endotoxic shock. J Immunol 162, 3498–3503. [PubMed] [Google Scholar]
- Choy A, Dancourt J, Mugo B, O'Connor TJ, Isberg RR, Melia TJ, and Roy CR (2012). The Legionella effector RavZ inhibits host autophagy through irreversible Atg8 deconjugation. Science 338, 1072–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colacurcio DJ, Pensalfini A, Jiang Y, and Nixon RA (2018). Dysfunction of autophagy and endosomal-lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer's Disease. Free Radic Biol Med 114, 40–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, Quarato G, Boada-Romero E, Kalkavan H, Johnson MDL, et al. (2018). LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 175, 429–441 e416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira CA, and Mantovani B (1988). Latrunculin A is a potent inhibitor of phagocytosis by macrophages. Life Sci 43, 1825–1830. [DOI] [PubMed] [Google Scholar]
- Dheen ST, Kaur C, and Ling EA (2007). Microglial activation and its implications in the brain diseases. Curr Med Chem 14, 1189–1197. [DOI] [PubMed] [Google Scholar]
- Doens D, and Fernandez PL (2014). Microglia receptors and their implications in the response to amyloid beta for Alzheimer's disease pathogenesis. J Neuroinflammation 11, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eimer WA, and Vassar R (2013). Neuron loss in the 5XFAD mouse model of Alzheimer's disease correlates with intraneuronal Abeta42 accumulation and Caspase-3 activation. Mol Neurodegener 8, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ejlerskov P, Ashkenazi A, and Rubinsztein DC (2019). Genetic enhancement of macroautophagy in vertebrate models of neurodegenerative diseases. Neurobiol Dis 122, 3–8. [DOI] [PubMed] [Google Scholar]
- Eng KE, Panas MD, Karlsson Hedestam GB, and McInerney GM (2010). A novel quantitative flow cytometry-based assay for autophagy. Autophagy 6, 634–641. [DOI] [PubMed] [Google Scholar]
- Ferro A, Qu W, Lukowicz A, Svedberg D, Johnson A, and Cvetanovic M (2018). Inhibition of NF-kappaB signaling in IKKbetaF/F;LysM Cre mice causes motor deficits but does not alter pathogenesis of Spinocerebellar ataxia type 1. PLoS One 13, e0200013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frake RA, Ricketts T, Menzies FM, and Rubinsztein DC (2015). Autophagy and neurodegeneration. J Clin Invest 125, 65–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Gotz J, and Feany MB (2015). Connecting the dots between tau dysfunction and neurodegeneration. Trends Cell Biol 25, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller JP, Stavenhagen JB, and Teeling JL (2014). New roles for Fc receptors in neurodegeneration-the impact on Immunotherapy for Alzheimer's Disease. Front Neurosci 8, 235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras S, Earls LR, Howell S, Smeyne RJ, Zakharenko SS, and Pelletier S (2015). SCYL2 Protects CA3 Pyramidal Neurons from Excitotoxicity during Functional Maturation of the Mouse Hippocampus. J Neurosci 35, 10510–10522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girard SD, Jacquet M, Baranger K, Migliorati M, Escoffier G, Bernard A, Khrestchatisky M, Feron F, Rivera S, Roman FS, et al. (2014). Onset of hippocampus-dependent memory impairments in 5XFAD transgenic mouse model of Alzheimer's disease. Hippocampus 24, 762–772. [DOI] [PubMed] [Google Scholar]
- Goldmann T, Wieghofer P, Muller PF, Wolf Y, Varol D, Yona S, Brendecke SM, Kierdorf K, Staszewski O, Datta M, et al. (2013). A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat Neurosci 16, 1618–1626. [DOI] [PubMed] [Google Scholar]
- Gong CX, and Iqbal K (2008). Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem 15, 2321–2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurley C, Nichols J, Liu S, Phulwani NK, Esen N, and Kielian T (2008). Microglia and Astrocyte Activation by Toll-Like Receptor Ligands: Modulation by PPAR-gamma Agonists. PPAR Res 2008, 453120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris H, and Rubinsztein DC (2011). Control of autophagy as a therapy for neurodegenerative disease. Nat Rev Neurol 8, 108–117. [DOI] [PubMed] [Google Scholar]
- He C, Bartholomew CR, Zhou W, and Klionsky DJ (2009). Assaying autophagic activity in transgenic GFP-Lc3 and GFP-Gabarap zebrafish embryos. Autophagy 5, 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann BL, Boada-Romero E, Cunha LD, Magne J, and Green DR (2017). LC3-Associated Phagocytosis and Inflammation. J Mol Biol 429, 3561–3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann BL, and Green DR (2019). LC3-associated phagocytosis at a glance. J Cell Sci 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heckmann BL, Tummers B, and Green DR (2018). Crashing the computer: apoptosis vs. necroptosis in neuroinflammation. Cell Death Differ. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoogland IC, Houbolt C, van Westerloo DJ, van Gool WA, and van de Beek D (2015). Systemic inflammation and microglial activation: systematic review of animal experiments. J Neuroinflammation 12, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, and Yoshimori T (2004). LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci 117, 2805–2812. [DOI] [PubMed] [Google Scholar]
- Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, Kalaany NY, Jacks T, Chan CS, Rabinowitz JD, et al. (2014). Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 4, 914–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YS, and Joh TH (2006). Microglia, major player in the brain inflammation: their roles in the pathogenesis of Parkinson's disease. Exp Mol Med 38, 333–347. [DOI] [PubMed] [Google Scholar]
- Kwon DH, Kim S, Jung YO, Roh KH, Kim L, Kim BW, Hong SB, Lee IY, Song JH, Lee WC, et al. (2017). The 1:2 complex between RavZ and LC3 reveals a mechanism for deconjugation of LC3 on the phagophore membrane. Autophagy 13, 70–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenz KM, and Nelson LH (2018). Microglia and Beyond: Innate Immune Cells As Regulators of Brain Development and Behavioral Function. Front Immunol 9, 698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, et al. (2010). Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer's disease. Proc Natl Acad Sci U S A 107, 14164–14169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu MY, Yin CY, Zhu LJ, Zhu XH, Xu C, Luo CX, Chen H, Zhu DY, and Zhou QG (2018). Sucrose preference test for measurement of stress-induced anhedonia in mice. Nat Protoc 13, 1686–1698. [DOI] [PubMed] [Google Scholar]
- Liu S, Liu Y, Hao W, Wolf L, Kiliaan AJ, Penke B, Rube CE, Walter J, Heneka MT, Hartmann T, et al. (2012). TLR2 is a primary receptor for Alzheimer's amyloid beta peptide to trigger neuroinflammatory activation. J Immunol 188, 1098–1107. [DOI] [PubMed] [Google Scholar]
- Lucin KM, O'Brien CE, Bieri G, Czirr E, Mosher KI, Abbey RJ, Mastroeni DF, Rogers J, Spencer B, Masliah E, et al. (2013). Microglial beclin 1 regulates retromer trafficking and phagocytosis and is impaired in Alzheimer's disease. Neuron 79, 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado V, Zoller T, Attaai A, and Spittau B (2016). Microglia-Mediated Neuroinflammation and Neurotrophic Factor-Induced Protection in the MPTP Mouse Model of Parkinson's Disease-Lessons from Transgenic Mice. Int J Mol Sci 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maday S (2016). Mechanisms of neuronal homeostasis: Autophagy in the axon. Brain Res 1649, 143–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez J, Almendinger J, Oberst A, Ness R, Dillon CP, Fitzgerald P, Hengartner MO, and Green DR (2011). Microtubule-associated protein 1 light chain 3 alpha (LC3)-associated phagocytosis is required for the efficient clearance of dead cells. Proc Natl Acad Sci U S A 108, 17396–17401. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Martinez J, Cunha LD, Park S, Yang M, Lu Q, Orchard R, Li QZ, Yan M, Janke L, Guy C, et al. (2016). Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature 533, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Martinez J, Malireddi RK, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan JL, Tan H, Peng J, et al. (2015). Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat Cell Biol 17, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Matsunaga K, Saitoh T, Tabata K, Omori H, Satoh T, Kurotori N, Maejima I, Shirahama-Noda K, Ichimura T, Isobe T, et al. (2009). Two Beclin 1-binding proteins, Atg14L and Rubicon, reciprocally regulate autophagy at different stages. Nat Cell Biol 11, 385–396. [DOI] [PubMed] [Google Scholar]
- Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Fullgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, et al. (2017). Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 93, 1015–1034. [DOI] [PubMed] [Google Scholar]
- Morales I, Guzman-Martinez L, Cerda-Troncoso C, Farias GA, and Maccioni RB (2014). Neuroinflammation in the pathogenesis of Alzheimer's disease. A rational framework for the search of novel therapeutic approaches. Front Cell Neurosci 8, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mrak RE, and Griffin WS (2000). Interleukin-1 and the immunogenetics of Alzheimer disease. J Neuropathol Exp Neurol 59, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naudin M, Mondon K, El-Hage W, Perriot E, Boudjarane M, Desmidt T, Lorette A, Belzung C, Hommet C, and Atanasova B (2015). Taste identification used as a potential discriminative test among depression and Alzheimers disease in elderly: A pilot study. Psychiatry Res 228, 228–232. [DOI] [PubMed] [Google Scholar]
- Noble W, Hanger DP, Miller CC, and Lovestone S (2013). The importance of tau phosphorylation for neurodegenerative diseases. Front Neurol 4, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, et al. (2006). Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci 26, 10129–10140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno M (2009). Failures to reconsolidate memory in a mouse model of Alzheimer's disease. Neurobiol Learn Mem 92, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira CA, Kashman Y, and Mantovani B (1996). Effects of latrunculin A on immunological phagocytosis and macrophage spreading-associated changes in the F-actin/G-actin content of the cells. Chem Biol Interact 100, 141–153. [DOI] [PubMed] [Google Scholar]
- Orthgiess J, Gericke M, Immig K, Schulz A, Hirrlinger J, Bechmann I, and Eilers J (2016). Neurons exhibit Lyz2 promoter activity in vivo: Implications for using LysM-Cre mice in myeloid cell research. Eur J Immunol 46, 1529–1532. [DOI] [PubMed] [Google Scholar]
- Padurariu M, Ciobica A, Mavroudis I, Fotiou D, and Baloyannis S (2012). Hippocampal neuronal loss in the CA1 and CA3 areas of Alzheimer's disease patients. Psychiatr Danub 24, 152–158. [PubMed] [Google Scholar]
- Pan XD, Zhu YG, Lin N, Zhang J, Ye QY, Huang HP, and Chen XC (2011). Microglial phagocytosis induced by fibrillar beta-amyloid is attenuated by oligomeric beta-amyloid: implications for Alzheimer's disease. Mol Neurodegener 6, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SK, Ratia K, Ba M, Valencik M, and Liebman SW (2016). Inhibition of Abeta42 oligomerization in yeast by a PICALM ortholog and certain FDA approved drugs. Microb Cell 3, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry VH, and Holmes C (2014). Microglial priming in neurodegenerative disease. Nat Rev Neurol 10, 217–224. [DOI] [PubMed] [Google Scholar]
- Pickford F, Masliah E, Britschgi M, Lucin K, Narasimhan R, Jaeger PA, Small S, Spencer B, Rockenstein E, Levine B, et al. (2008). The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid beta accumulation in mice. J Clin Invest 118, 2190–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido-Salgado M, Vidal-Taboada JM, Garcia Diaz-Barriga G, Serratosa J, Valente T, Castillo P, Matalonga J, Straccia M, Canals JM, Valledor A, et al. (2017). Myeloid C/EBPbeta deficiency reshapes microglial gene expression and is protective in experimental autoimmune encephalomyelitis. J Neuroinflammation 14, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qaisiya M, Mardesic P, Pastore B, Tiribelli C, and Bellarosa C (2017). The activation of autophagy protects neurons and astrocytes against bilirubin-induced cytotoxicity. Neurosci Lett 661, 96–103. [DOI] [PubMed] [Google Scholar]
- Reed-Geaghan EG, Savage JC, Hise AG, and Landreth GE (2009). CD14 and toll-like receptors 2 and 4 are required for fibrillar A{beta}-stimulated microglial activation. J Neurosci 29, 11982–11992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichman WE, and Coyne AC (1995). Depressive symptoms in Alzheimer's disease and multi-infarct dementia. J Geriatr Psychiatry Neurol 8, 96–99. [DOI] [PubMed] [Google Scholar]
- Ries M, and Sastre M (2016). Mechanisms of Abeta Clearance and Degradation by Glial Cells. Front Aging Neurosci 8, 160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Bento CF, and Deretic V (2015). Therapeutic targeting of autophagy in neurodegenerative and infectious diseases. J Exp Med 212, 979–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, Marino G, and Kroemer G (2011). Autophagy and aging. Cell 146, 682–695. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Davies JE, Huang Z, Tunnacliffe A, and Rubinsztein DC (2007). Trehalose, a novel mTOR-independent autophagy enhancer, accelerates the clearance of mutant huntingtin and alpha-synuclein. J Biol Chem 282, 5641–5652. [DOI] [PubMed] [Google Scholar]
- Sarkar S, Ravikumar B, and Rubinsztein DC (2009). Autophagic clearance of aggregate-prone proteins associated with neurodegeneration. Methods Enzymol 453, 83–110. [DOI] [PubMed] [Google Scholar]
- Scott RS, McMahon EJ, Pop SM, Reap EA, Caricchio R, Cohen PL, Earp HS, and Matsushima GK (2001). Phagocytosis and clearance of apoptotic cells is mediated by MER. Nature 411, 207–211. [DOI] [PubMed] [Google Scholar]
- Shaftel SS, Griffin WS, and O'Banion MK (2008). The role of interleukin-1 in neuroinflammation and Alzheimer disease: an evolving perspective. J Neuroinflammation 5, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaftel SS, Kyrkanides S, Olschowka JA, Miller JN, Johnson RE, and O'Banion MK (2007). Sustained hippocampal IL-1 beta overexpression mediates chronic neuroinflammation and ameliorates Alzheimer plaque pathology. J Clin Invest 117, 1595–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M, Jin J, Lim JE, Kou J, Pattanayak A, Rehman JA, Kim HD, Tahara K, Lalonde R, and Fukuchi K (2011). TLR4 mutation reduces microglial activation, increases Abeta deposits and exacerbates cognitive deficits in a mouse model of Alzheimer's disease. J Neuroinflammation 8, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorp E, Cui D, Schrijvers DM, Kuriakose G, and Tabas I (2008). Mertk receptor mutation reduces efferocytosis efficiency and promotes apoptotic cell accumulation and plaque necrosis in atherosclerotic lesions of apoe−/− mice. Arterioscler Thromb Vasc Biol 28, 1421–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulland TK, Song WM, Huang SC, Ulrich JD, Sergushichev A, Beatty WL, Loboda AA, Zhou Y, Cairns NJ, Kambal A, et al. (2017). TREM2 Maintains Microglial Metabolic Fitness in Alzheimer's Disease. Cell 170, 649–663 e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H, Stewart FR, Piccio L, Colonna M, and Holtzman DM (2014). Altered microglial response to Abeta plaques in APPPS1-21 mice heterozygous for TREM2. Mol Neurodegener 9, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N, Liang H, and Zen K (2014). Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front Immunol 5, 614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WY, Tan MS, Yu JT, and Tan L (2015a). Role of pro-inflammatory cytokines released from microglia in Alzheimer's disease. Ann Transl Med 3, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Cella M, Mallinson K, Ulrich JD, Young KL, Robinette ML, Gilfillan S, Krishnan GM, Sudhakar S, Zinselmeyer BH, et al. (2015b). TREM2 lipid sensing sustains the microglial response in an Alzheimer's disease model. Cell 160, 1061–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Ulland TK, Ulrich JD, Song W, Tzaferis JA, Hole JT, Yuan P, Mahan TE, Shi Y, Gilfillan S, et al. (2016). TREM2-mediated early microglial response limits diffusion and toxicity of amyloid plaques. J Exp Med 213, 667–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb JL, Ravikumar B, Atkins J, Skepper JN, and Rubinsztein DC (2003). Alpha-Synuclein is degraded by both autophagy and the proteasome. J Biol Chem 278, 25009–25013. [DOI] [PubMed] [Google Scholar]
- Wilkinson K, and El Khoury J (2012). Microglial scavenger receptors and their roles in the pathogenesis of Alzheimer's disease. Int J Alzheimers Dis 2012, 489456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto A, and Simonsen A (2011). The elimination of accumulated and aggregated proteins: a role for aggrephagy in neurodegeneration. Neurobiol Dis 43, 17–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin J, Liu X, He Q, Zhou L, Yuan Z, and Zhao S (2016). Vps35-dependent recycling of Trem2 regulates microglial function. Traffic 17, 1286–1296. [DOI] [PubMed] [Google Scholar]
- Zhang QG, Wang RM, Scott E, Han D, Dong Y, Tu JY, Yang F, Reddy Sareddy G, Vadlamudi RK, and Brann DW (2013). Hypersensitivity of the hippocampal CA3 region to stress-induced neurodegeneration and amyloidogenesis in a rat model of surgical menopause. Brain 136, 1432–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Wu X, Li X, Jiang LL, Gui X, Liu Y, Sun Y, Zhu B, Pina-Crespo JC, Zhang M, et al. (2018). TREM2 Is a Receptor for beta-Amyloid that Mediates Microglial Function. Neuron 97, 1023–1031 e1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. LysM-cre and CX3CR1-creERT2 mediated abrogation of FIP200 and ATG5. Related to Figure 1. A. and B. LysM-cre mediated reduction of FIP200 and ATG5 in primary microglia isolated from the indicated genotypes as measured by immunoblot (A) and qPCR (B). For B., n=4 mice for both cre−, n=5 mice for FIP200fl/flcre+ and ATG5fl/flcre+. qPCR performed in triplicate. C. Representative immunoblot analysis showing minimal off-target effects of LysM-cre in purified neurons isolated from the indicated 5xFAD genotypes. D and E. Analysis of autophagic activation and autophagic flux in primary microglia isolated from FIP200fl/fl and ATG5fl/fl LysM-cre+ or − mice as indicated. Cells were treated with either rapamycin (100nm) or bafilomycin A1 (25nm) for 12 hours. Autophagic activation was determined by LC3-lipidation (D and E) and autophagic flux by P62 accumulation (E) by immunoblot. F and G. CX3CR1-creERT2 mice of the indicated 5xFAD genotypes were injected with tamoxifen at 21 and 23 days of age and peripheral expression in monocytes was allowed to reconstitute for 3m, leading to microglia-specific deletion of the indicated genes. Microglia were purified from each genotype and expression for either FIP200 or Rubicon was analyzed by immunoblot, mice were 4m of age. H. To confirm microglia-specific deletion, FIP200 and Rubicon reconstitution was analyzed in splenic macrophages from 4m-old mice. I. Representative images showing Aβ accumulation in the cortex of myeloid ATG5-deficient 5xFAD mice. Scale bars, 100μm. J. Representative images of cortical Aβ deposition in 4m-old myeloid-specific FIP200 and ATG5-deficient 5xFAD mice and Rubicon-deficient mice. Each point represents an individual mouse. Scale bars, 50μm. K. Quantification of cortical Aβ content as measured by plaque # from the 5th cortical layer of the indicated genotypes. All mice were 4m of age and each point represents an individual mouse. Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. *p<0.05, ***p<0.001.
Figure S2. Characterization of BV2 microglia lacking FIP200, ATG5, and Rubicon. Related to Figure 2. A. Immunoblot analysis showing successful depletion of FIP200, ATG5, or Rubicon as indicated in BV2 microglia by CRISPR/Cas9. Two samples of each are shown. B. Representative imaging showing co-localization between LC3-positive, Aβ-containing vesicles and the endosomal markers clathrin and Rab5 in BV2 murine microglia. Cells were treated for 6h with 1μM TAMRA-Aβ1-42. Aβ is false-colored blue to facilitate imaging. Scale bars, 5μm. Values represent Mander’s co-localization coefficient for LC3/Clathrin or LC3/Rab5 respectively. C. Quantification of membrane-associated LC3 by flow cytometry in vehicle or latrunculin A (LA) treated parental BV2 microglia in response to either zymosan or Aβ. n=3 for each condition performed in duplicate. D. Flow cytometry-based quantification (MFI) of receptor surface expression (left) and receptor internalization (right) for receptor recycling assays (Fig. 2) in BV2 microglia. Each point represents a unique experiment performed in duplicate. E. Representative images showing uptake and maturation of vesicles containing pH-rodo labeled zymosan or Aβ as indicated. (See Fig. 2E for quantification and experimental description). Scale bars, 10μm. F. Representative images showing zymosan or Aβ co-localization with LAMP1 labeled lysosomes in BV2 microglia of the indicated genotypes (See Fig. 2F for quantification and experimental details). Scale bars, 2μm. Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. **p<0.01.
Figure S3. Receptor recycling assay. Related to Figure 2. Schematic representation showing the method for quantifying receptor recycling (Lucin et al., 2013; Yin et al., 2016). (1) Following blocking as described in the STAR methods, cells are incubated with primary antibodies against the receptors of interest. (2) Any remaining cell-surface primary antibodies are then removed by acid washing. (3) Internalized receptors bound to their cognate primary antibody are allowed to cycle to the plasma membrane for 1h. (4) Cells are incubated with AF568-labeled anti-immunoglobulin (Ig) antibody to label recycled primary antibody and allowed to re-internalize carrying the AF568-tag. (5) Any remaining cell-surface AF568-secondary antibodies are removed by acid washing. Cells are then fixed and nuclei staining for cell # quantification and imaging for fully recycled receptors (intracellular signal) is performed as described in the STAR methods.
Figure S4. Recycling of CD36, TREM2, and TLR4 in RAW264.7 and BMDMs is LANDO-dependent. Related to Figure 3. A. Immunoblot analysis showing either CRISPR/Cas9-mediated depletion or retroviral mediated overexpression of the indicated genes in RAW264.7 cells. B. Analysis of autophagic flux in RAW264.7 cells of the indicated genotypes. Cells were treated with 25nM bafilomycin A1 (BafA1) for 12h. Autophagic activation was determined by LC3-lipidation and P62 accumulation by immunoblot. C. and D. Representative imaging of receptor recycling in (C) RAW264.7 cells and (D) primary BMDMs. Scale bars, 50μm.
Figure S5. Analysis of infiltrating monocytes versus resident microglia in 5xFAD Rubicon-deficient mice. Related to Figure 4. A. Quantification of whole brain Iba1 expression in 6m-old 5xFADnull Rubicon or ATG5 mice as indicated by flow cytometry. n=5 mice per genotype. B. Quantification of whole brain pro-inflammatory cytokine expression in 6m-old 5xFADnu Rubicon or ATG5 mice as indicated by flow cytometry. n=5 mice per genotype, performed in triplicate. C. Representative flow cytometric analysis of resident microglia versus peripheral monocytes. Expression of the microglia-specific receptor TMEM119 was analyzed on the total CD11b monocytic pool (containing all monocytes present in the brain) to delineate peripheral cells (TMEM119−) versus resident microglia (TMEM119+). D. Quantification of the percentage of infiltrating monocytes as measured by flow cytometry. n=4 mice per genotype. E. Representative histogram and quantification of microglia activation by Iba1 expression on TMEM119+ cells from 5xFAD Rubicon+/− and Rubicon−/− mice. n=4 mice per genotype. Data are represented as mean ± SEM. Significance was calculated using Student’s t-test. ***p<0.001.
Figure S6. Rubicon-deficiency promotes tau hyperphosphorylation in 5xFAD mice. Related to Figure 5. A and B. Representative images showing hyperphosphorylation of tau at S202/T205 in 4m-old Rubicon-deficient mice in both (A) the hippocampus and (B) the cortex. (See Fig. 5C,D for quantification). Scale bars, 100μm for A, 30μm for B.