

Abstract
Aims:
Electric lighting is beneficial to modern society; however, it is becoming apparent that light at night (LAN) is not without biological consequences. Several studies have reported negative effects of LAN on health and behavior in humans and nonhuman animals. Exposure of non-diabetic mice to dim LAN impairs glucose tolerance, whereas a return to dark nights (LD) reverses this impairment. We predicted that exposure to LAN would exacerbate the metabolic abnormalities in TALLYHO/JngJ (TH) mice, a polygenic model of type 2 diabetes mellitus (T2DM).
Materials and Methods:
We exposed 7-week old male TH mice to either LD or LAN for 8–10 weeks in two separate experiments. After 8 weeks of light treatment, we conducted intraperitoneal glucose tolerance testing (ipGTT) followed by intraperitoneal insulin tolerance testing (ipITT). In Experiment 1, all mice were returned to LD for 4 weeks, and ipITT was repeated.
Key Findings:
The major results of this study are i) LAN exposure for 8 weeks exacerbates glucose intolerance and insulin resistance ii) the effects of LAN on insulin resistance are reversed upon return to LD, iii) LAN exposure results in a greater increase in body weight compared to LD exposure, iv) LAN increases the incidence of mice developing overt T2DM, and v) LAN exposure decreases survival of mice with T2DM.
Significance:
In conclusion, LAN exacerbated metabolic abnormalities in a polygenic mouse model of T2DM, and these effects were reversed upon return to dark nights. The applicability of these findings to humans with T2DM needs to be determined.
Keywords: Diabetes, light at night, glucose tolerance, insulin resistance, metabolic dysfunction
Introduction
Exposure to light at night (LAN) is pervasive in urban regions, as well as in many rural settings. A recent atlas of light pollution indicates that approximately 50% of the United States is exposed to at least dim levels of LAN [1]. The invention of electric lighting allowed for extended daytime, and thus more time for human productivity. However, it is becoming apparent that exposure to LAN is not without consequences [2–4].
Physiology and behavior is intimately tied with the 24-hour solar day via the circadian system [5,6]. The major vertebrate circadian clock is in the suprachiasmatic nucleus (SCN) of the hypothalamus, but most peripheral tissues also maintain circadian rhythmicity, synchronized by the SCN [7,8]. A molecular network in the SCN responds to light relayed by intrinsically photosensitive retinal ganglion cells (ipRGCs), ensuring coordination with the light cycle [9,10]. Aberrant light can disrupt entrainment of the molecular clock and dysregulate circadian rhythms [11].
Consequences of exposure to LAN are becoming better understood through laboratory studies. LAN is implicated in the exacerbation of many cancers, mood disorders, obesity, and metabolic abnormalities [3,4,12]. Mice exposed to 5 lux of LAN for 4 weeks, a level similar to a nightlight approximately 3 meters away, increased body weight compared with mice housed in dark nights [11]. Mice also developed impaired glucose tolerance when exposed to LAN (5 lux) [13]. When these mice were returned to dark nights, glucose tolerance returned to baseline. Thus, metabolic activity can be altered by even dim levels of LAN.
Impaired glucose tolerance, along with insulin resistance and hyperglycemia, is a characteristic of type 2 diabetes mellitus (T2DM) [14,15]. Since 1980 the prevalence of T2DM has nearly quadrupled [16,17]. Nearly 9.3% of the U.S. population has T2DM [16], and recent estimates suggest that 1/3 of Americans born after 2000 will develop T2DM in their lifetime [18]. T2DM is associated with an array of secondary complications including vision loss, pregnancy complications, mental health issues, kidney disease, increased risk for stroke and hypertension, and degeneration of peripheral nerves [19], and it more than doubles the risk of cardiovascular disease [20]. T2DM also increases the risk for early mortality and morbidity [21]. The estimated total cost of T2DM and related complications was $327 billion in total medical costs, lost work and wages in 2017 [22], thus T2DM is a major public health concern that promises to worsen.
The precise cause for T2DM remains unknown, but it likely involves an interaction between genetic predisposition and environmental factors such as high calorie diets, excess weight, and inactivity [23–25]. Because exposure to LAN alters key characteristics of T2DM, we predicted that exposure to LAN would exacerbate metabolic abnormalities of the disease. To assess this, we exposed a population of TALLYHO/JngJ (TH) mice, a polygenic model of T2DM, to either dark nights or LAN (40 lux), and measured body weight, glucose tolerance, and insulin resistance. Forty lux is a common night-time exposure level in humans and comparable to a television or tablet screen at night [26,27].
Materials and Methods
Six-week old male TALLYHO/JngJ mice (TH) were purchased from The Jackson Laboratory (Bar Harbor, ME). TH mice are an inbred, polygenic model of T2DM and develop many characteristics that mimic human T2DM including hyperglycemia, hyperinsulinemia, hyperlipidemia, moderate obesity, and enlargement of the islets of Langerhans [28–32]. The TH mouse strain originated from two outbred Theiler Original mice that spontaneously developed polyuria and glucosuria. The research colony was established by selective breeding for hyperglycemia (29). The phenotype is less than 100% penetrant, and only male TH mice manifest diabetes. According to the documentation provided by Jackson Laboratory, the T2DM phenotype develops between weeks 10 and 14 of age. Thus, mice were exposed to LAN during the time these mice develop this phenotype. Upon arrival, mice were housed in light-controlled chambers at The Ohio State University. Following 1 week of habituation, at seven weeks of age, mice were weighed, fasting (4 h) blood glucose was tested, and they were assigned to two groups with equivalent body weight and blood glucose concentrations (n=11/group in experiment 1, n=18/group in experiment 2). Mice were tagged via ear-punch, group-housed (3–4/cage) and assigned to one of two lighting regimens: LD = 14h light (150 lux):10h dark (0 lux) or LAN = 14h light (150 lux):10h dim (40 lux). All other housing conditions remained constant. Forty lux of light was used because it is a common exposure level in humans and similar to the amount of light given off by electronic devices at night [26, 27]. Mice had access to food (Harlan Teklad 8640; Madison, WI, USA) and filtered tap water ad libitum, except where noted below. Body weight was measured once per week at the same day and time throughout the experiments. All procedures had prior approval of The Ohio State University Institutional Animals Care and Use Committee, and mice were maintained in accordance with the recommendations of the National Institutes of Health and The Guide for the Care and Use of Laboratory Animals. This study was conducted as two treatment-balanced experiments. The first experiment was to establish a physiological phenotype and to reverse the phenotype by replacing the LAN-exposed mice back into dark nights. The second experiment was to increase sample size (based on the penetrance of the disease in experiment 1) and to collect tissues following light exposure.
Experiment 1
After 4 weeks in light treatments, an intraperitoneal glucose tolerance test (ipGTT) was conducted following a 6 h fast. Initial fasting blood glucose concentrations were measured via the tail vein of each mouse using a Contour glucose meter (Bayer). Dextrose (1g/kg; Hospira) was then administered to each mouse by intraperitoneal injection, and glucose concentrations were measured 15, 30, 60, 90, and 120 min following injection. Mice were then returned to their respective lighting conditions. After 8 weeks in light treatments, mice were fasted overnight (16 h), and the ipGTT was repeated. Mice were again returned to their respective lighting conditions, and following 2 days of recovery, mice were fasted for 4 h, and an intraperitoneal insulin tolerance test (ipITT) was conducted. Initial fasting blood glucose concentrations were measured for each mouse, followed by an intraperitoneal insulin (2 U/kg; Humalin R, Eli Lilly) injection. Blood glucose concentrations were measured 30, 60, 90, and 120 min following injection. After 2 additional weeks in LAN, mice were moved into the LD chamber. All mice were housed in LD conditions for 4 weeks, at which time an ipITT, the most direct method of testing for insulin resistance, the dominant pathogenic mechanism of T2DM, was repeated, and the mice were euthanized (Fig. 1). All ipGTTs and ipITTs were conducted between 9:00 and 13:00, the last 4 hours of the light phase.
Figure 1.
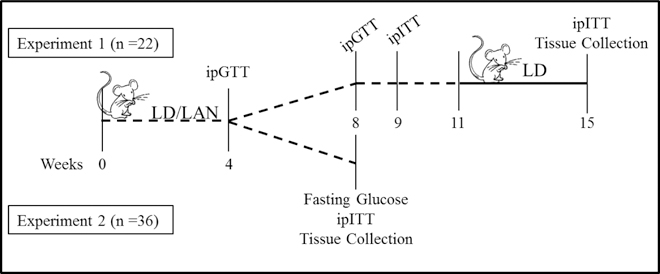
Timeline of metabolic tests conducted during experiments 1 and 2. LD = 14h light (150 lux):10h dark (0 lux); LAN = 14h light (150 lux):10h dim (40 lux); ipGTT = intraperitoneal glucose tolerance test; ipITT = intraperitoneal insulin tolerance test.
Experiment 2
After 4 weeks in light treatments, an ipGTT was conducted following a 6 h fast. Mice were then returned to their respective lighting conditions. Eight weeks after beginning light treatments, the mice were fasted overnight, and 16-h fasting blood glucose concentrations were measured. After allowing the mice to recover for 2 days, an ipITT was conducted following a 4 h fast (Fig. 1). Mice were removed from the study if they lost more than 20% of their initial body mass or displayed sickness behaviors (i.e., hunched posture, decreased grooming, etc.).
Statistical Analyses
Mice with a fasting (16 h) blood glucose concentration over 200 mg/dL were considered to have overt T2DM (experiment 1: n =10; experiment 2: n =27). This definition was based on previous literature [28,31–36]. Body weight, ipGTT, and ipITT results are reported for mice with overt T2DM. The prevalence of T2DM in each experiment was calculated. Mice that expired prior to the 16-h fast were included in the T2DM group because they were all in the top range of blood glucose concentrations measured at 4 weeks.
Change in body weight was calculated each week as a percentage of each mouse’s initial body weight, and the mean change was compared between LD and LAN. Body weight was compared using a 2-way ANOVA followed by a Dunn-Sidak test for multiple comparisons. All ipGTT and ipITT results were analyzed by calculating the area under the curve (AUC) for each individual mouse, and the mean AUC was calculated for LD and LAN groups. If the AUC results met the assumptions of parametric testing, then a student’s 2-tailed t-test was used. If the AUC results did not meet the assumptions of parametric testing, the nonparametric Mann-Whitney test was used to compare groups. Fasting glucose concentrations were analyzed using a 2-way ANOVA with experiment number and light treatment as main effects, followed by multiple student’s t-tests. Survival data were analyzed using the log-rank test, and the disease prevalence was analyzed with Fisher’s exact test. Results were considered significantly different if p<0.05, and all results are reported as a mean and standard deviation (SD). All statistical analyses were conducted using JMP Pro 13 (SAS) and GraphPad Prism 7.
Results
Experiment 1
The change in body weight over the first 11 weeks of light treatment was compared between mice housed in LD or LAN. There was a significant difference between treatment groups (p = 0.048), over time (p<0.001), and there was also a significant interaction between time and light treatment (p<0.001, n=22). At week 5, mice housed in LAN (113.7, SD 5.8) gained significantly more weight than mice housed in LD (102.0, SD 5.0, p = 0.002). Also, after 10 weeks, mice housed in LAN (108.4, SD 4.7) had gained significantly more weight than mice housed in LD (99.6, SD 2.6, p = 0.04) as a percent change from baseline (Fig. 2a). The change in body weight from weeks 11 – 15, the period of time mice originally housed in LAN were moved to dark nights, was significantly changed over time (p = 0.006), however there was no difference between treatment groups (p = 0.27), and there was no interaction effect (p = 0.89, Fig. 2a).
Figure 2.
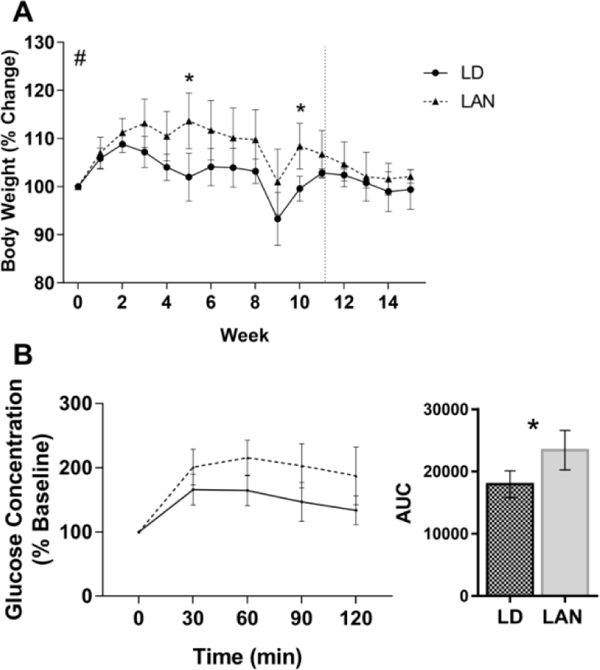
Metabolic phenotype of mice in experiment 1. A) Percent change in body mass of Tallyho/JngJ mice housed in either dark nights (LD) or light at night (LAN) for 11 weeks (2-way ANOVA by treatment, p = 0.048; time, p < 0.001, and treatment x time interaction, p < 0.001). At weeks 5 (p = 0.002) and 10 (p = 0.04), mice housed in LAN gained significantly more weight than mice housed in LD (Dunn-Sidak Post Hoc Test). After 11 weeks, the LAN group was moved to LD through week 15. There was a significant effect of time on body weight (p = 0.006), but no significant differences in the percent change in body mass between light treatments (p = 0.27) or a significant interaction (p = 0.89; 2-way ANOVA) (n=22). B) Glucose concentration as a % of baseline concentration during an intraperitoneal glucose tolerance test. The area under the curve was significantly higher for mice housed in LAN for 8 weeks compared with mice housed in LD (student’s t-test; p = 0.03). Error bars represent the standard deviation of the mean, n = 4 (LD) and 6 (LAN).
After 4 weeks in light treatments, mice housed in LAN (n=6) tended to have a larger AUC during ipGTT compared with mice housed in LD (n=4), but the differences were not statistically significant (p = 0.11). However, after 8 weeks in light treatments, mice housed in LAN had a larger AUC during ipGTT compared with mice housed in LD (p = 0.03, n = 4 (LD) and 6 (LAN), Fig. 2b). Additionally, mice housed in LAN had a larger AUC during ipITT compared with mice housed in LD (p = 0.03, Fig. 3a). Following 4 weeks of light reversal to dark nights, mice originally housed in LAN no longer differed from mice that were housed in LD for the entire duration of the experiment (p = 0.64, n = 4 (LD) and 6 (LAN), Fig. 3b).
Figure 3.
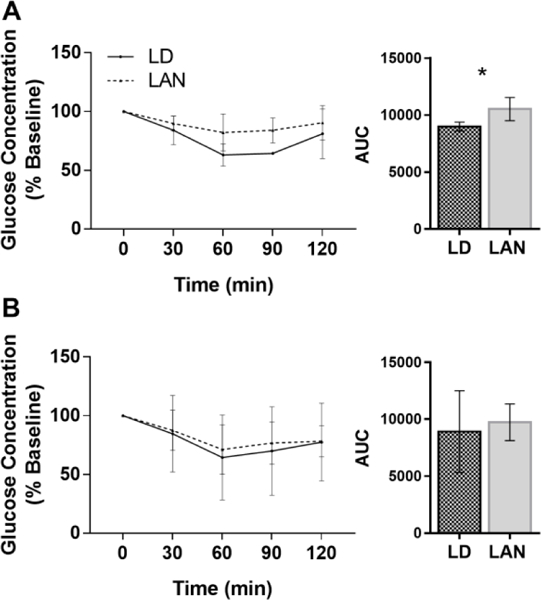
Glucose concentration as a % of baseline concentration during an intraperitoneal insulin tolerance test in experiment 1. A) The area under the curve was significantly higher for mice housed in LAN for 9 weeks compared with mice housed in LD (student’s t-test; p = 0.03). B) The area under the curve was not significantly different between mice housed in LD throughout the duration of the experiments and mice housed in LAN, followed by 4 weeks of light reversal (student’s t-test; p=0.73). Error bars represent the standard deviation of the mean, n = 4 (LD) and 6 (LAN).
Experiment 2
There was a significant difference in the change in body weight over time between light treatments (p < 0.001) and over time (p < 0.001), but no significant interaction between time and light treatment (p = 0.19, Fig. 4a). The percentage of weight gain was higher in mice housed in LAN at weeks 1, 2, 3, 5, and 8 compared with mice housed in LD. At week 1, mice housed in LAN (112.1, SD 4.5) gained significantly more weight than mice housed in LD (103.9, SD 3.28, p = 0.01). At week 2, mice housed in LAN (110.7, SD 6.2) gained significantly more weight than mice housed in LD (103.2, SD 4.1, p = 0.03). At week 3, mice housed in LAN (113.3, SD 6.7) gained significantly more weight than mice housed in LD (105.04, SD 4.7, p = 0.01). At week 5, mice began to lose weight, but mice housed in LAN (109.5, SD 7.0) continued to weigh significantly more than mice housed in LD (101.6, SD 4.6, p = 0.03). At week 8, mice housed in LAN (105.5, SD 10.3) also weighed significantly more than mice housed in LD (95.2, SD 10.5, p = 0.02, Fig. 4a).
Figure 4.
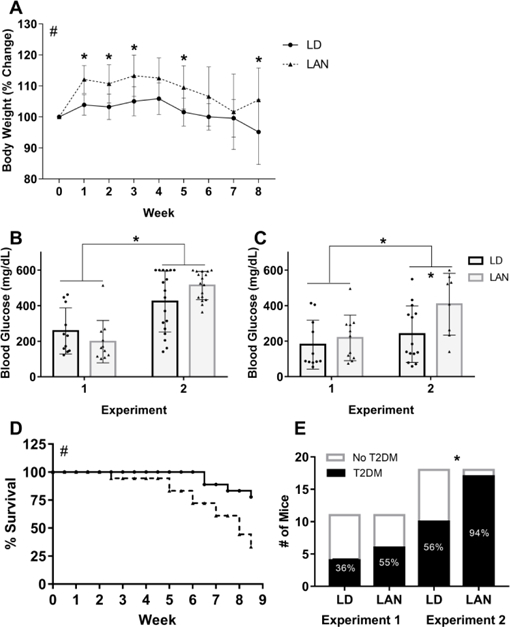
A) Percent change in body mass of Tallyho/JngJ mice housed in either dark nights (LD) or light at night (LAN) for 8 weeks (2-way ANOVA by treatment, p < 0.0018; time, p < 0.001, and treatment x time interaction, p=0.19). Mice housed in LAN gained significantly more weight than mice housed in LD at weeks 1 (n = 10 (LD) and 17 (LAN), p = 0.01), 2 (p = 0.03), 3 (p = 0.01), 5 (p = 0.03), and 8 (n = 7 (LD) and 7 (LAN), p = 0.02) (Dunn-Sidak Post Hoc Test) in experiment 2. B) After 4 weeks in light treatments, mice used in experiment 2 had higher fasting blood glucose than the mice used in experiment 1 (student’s t-test, p < 0.001). C) After 8 weeks in light treatments, mice used in experiment 2 had higher fasting blood glucose than mice used in experiment 1 (student’s t-test, p = 0.04), and in experiment 2, mice housed in LAN had higher fasting blood glucose than mice housed in LD (student’s t-test, p = 0.01). D) In experiment 2, survival was impaired for mice housed in LAN compared with mice housed in LD (log-rank test, p = 0.007). E) In experiment 2 and in both experiments combined, more mice housed in LAN developed type 2 diabetes mellitus compared with mice housed in LD (Fisher’s exact test, p = 0.0008). Error bars represent the standard deviation of the mean, n = 18/group at the beginning of the experiment.
After 4 weeks in light treatment, there were no differences between mice housed in LD (n = 10) and LAN (n = 17) in the ipGTT (p = 0.56). After both 4 weeks (p < 0.001) and 8 weeks (p = 0.01), the mice used in experiment 2 had higher fasting glucose concentrations compared with mice used in experiment 1 (Figs. 4b and 4c). In experiment 2, after 8 weeks, mice housed in LAN (n = 10) had higher fasting glucose concentrations than mice housed in LD (n = 7) (p = 0.01, Fig. 4c). More mice met endpoint criteria and were euthanized early or had early mortality when housed in LAN compared with LD (p = 0.007). After 8.5 weeks, 33.33% of mice housed in LAN survived, whereas 77.8% of mice housed in LD survived (Fig. 4d). The mice that had early mortality had significantly higher fasting (6 hr) blood glucose concentrations (578.5, SD 29.5) after 4 weeks in light treatment than mice that survived the duration of the experiment (401.2, SD 138.9, p = 0.0003). Additionally, the number of mice that developed T2DM was greater when housed in LAN. In experiment 1, the number of mice to develop T2DM was not significantly different from mice housed in LD (Fig. 4e). However, in experiment 2, the number of mice to develop T2DM when housed in LAN was significantly higher than the number of mice to develop T2DM when housed in LD (p = 0.008, Fig. 4e).
Discussion
The major findings of the current study in a rodent model of T2DM are: i) LAN exposure for 8 weeks exacerbates glucose intolerance and insulin resistance, ii) the effects of LAN on insulin resistance are reversed upon return to LD, iii) LAN exposure results in a greater increase in body mass compared to LD exposure, iv) LAN increases the incidence of mice developing overt T2DM, and v) LAN exposure decreases survival of mice with T2DM. This study represents an essential next step in understanding the metabolic effects of LAN as non-diabetic mice were previously shown to develop impaired glucose tolerance when exposed to LAN [13]. To our knowledge, this is the first study that examines the metabolic consequences of LAN exposure in a rodent model of T2DM.
Characteristics of T2DM include hyperglycemia, impaired glucose tolerance, and insulin resistance [14,15]. TH mice housed in LAN developed more severe hyperglycemia than mice housed in dark nights. Although this effect was not significant after 8 weeks in experiment 1, with increased sample size (experiment 2), the effect was statistically significant. Blood glucose concentrations in male C57BL/6J mice fasted overnight are typically under 100 mg/dL [28], but definitions of hyperglycemia and T2DM in mice vary among sources. Hyperglycemia has been defined as a non-fasting blood glucose concentration >250 mg/dL [29] and with an average overnight fasting blood glucose concentration of 163 mg/dL in another study [33]. T2DM was defined as a non-fasting glucose concentration between 300 and 400 mg/dL [28], overnight fasting levels >300 mg/dL [34], a 6-h fasting blood glucose >250 mg/dL [35], and >200 mg/dL [36]. Here, T2DM was defined as a fasting (16 h) blood glucose concentration over 200 mg/dL based on these previous studies and glucose concentrations in this specific population of mice. Male TH mice typically develop hyperglycemia around 10 weeks of age, and the severity increases with age [28]. However, because the phenotype is not 100% penetrant, the presence and incidence of hyperglycemia varies among the population. Among the mice used here, some remained normoglycemic, with a fasting blood glucose concentration under 100 mg/dL. A second group had elevated fasting blood glucose concentrations, between 100 and 200 mg/dL, and a third group developed overt T2DM, with fasting blood glucose concentrations over 200 mg/dL. A greater proportion of mice developed overt T2DM when housed in LAN, suggesting exposure to LAN raises blood glucose concentrations and increases the risk of developing T2DM in a susceptible population.
Hyperglycemia develops when insulin secretion can no longer compensate for insulin resistance. Thus, insulin resistance is the dominant pathogenesis mechanism of T2DM [37,38], and is the earliest defect [39]. After 8 weeks in LAN, mice with T2DM had increased resistance to insulin. In experiment 1, mice originally housed in LAN were placed into LD conditions for 4 weeks. Returning the mice to dark nights was sufficient to restore insulin tolerance to a similar level as mice housed in LD, strongly supporting LAN as the causative factor and suggesting the phenotype might be reversible. In experiment 2, severely hyperglycemic mice had early mortality, and thus accurate insulin resistance testing in this group of mice could not be conducted. However, these mice were more hyperglycemic when housed in LAN, thus, considering insulin resistance is the mechanism underlying hyperglycemia, it is likely these mice were insulin resistant as well. The severity of hyperglycemia was greater and the onset was earlier in the experiment 2 mice compared with experiment 1. Although hyperglycemia and glucose intolerance in male TH mice is consistent, the onset and extent varies considerably among litters [32]. A cohort of TH male mice housed at Jackson Laboratories had earlier onset of hyperglycemia than the 10–12 weeks reported in literature [31]. Additionally, glucose intolerance was not evident until after puberty, around 8 weeks of age, in one study [28], but another study reported mild glucose intolerance at only 4 weeks of age [44]. It is commonly understood that most forms of T2DM follow a polygenic inheritance, and the pathologic pathways that underlie the disease are different among affected individuals [23,24]. Thus, despite variability among studies, pathogenesis of T2DM in TH mice closely simulates that of human T2DM [31].
Glucose tolerance reflects the ability to regulate blood glucose levels. Humans with impaired glucose tolerance are at the highest risk of developing T2DM [40,41]. Impaired glucose tolerance is caused by a decrease in glucose transport and metabolism in muscle and adipocytes [42,43]. In experiment 1, TH mice with T2DM had greater impairment of glucose tolerance when housed in LAN. The ipGTT results in experiment 2 were restricted because the upper limit of the glucose meter was 600 mg/dL. After 4 weeks in lighting conditions, the fasting glucose concentrations were elevated and thus plateaued during the ipGTT after glucose injection. However, it is probable that without this limitation, the mice in experiment 2 would have had significantly impaired glucose tolerance after just 4 weeks of LAN treatment, similar to the glucose intolerance in experiment 1 after 8 weeks.
An unintended result of experiment 2, likely due to severe hyperglycemia in this group of mice, was that survival was impaired. Although loss of mice was present in both LD and LAN groups, impaired survival was significantly greater when mice were housed in LAN. Additionally, the mice that died early were more severely hyperglycemic after 4 weeks of light treatment compared with mice who survived. Although, to our knowledge, decreased survival in TH mice have not been previously reported in the literature, early mortality is common in other diabetic mouse models such as the commonly studied leptin receptor-deficient C57BLKS/J-Leprdb/Leprdb mouse. These mice display early mortality as hyperglycemia progresses [31]. The TH mice in this study displayed polyuria, which likely led to dehydration or hyperosmolar coma [34]. The effect of LAN on survival in mice with T2DM needs to be confirmed in future studies.
T2DM is considered a disease of chronic, systemic inflammation [45]. Increased circulating inflammatory markers such as IL-6 and C-reactive protein (CRP) are associated with the development of T2DM [46], and circulating TNFα and IL-6 concentrations positively correlate with insulin resistance [47,48]. In rats, in vivo administration of human recombinant IL-6 induced gluconeogenesis, hyperglycemia, and hyperinsulinemia [49]. Although we could not test for inflammatory markers in our experiments (due to light reversal in experiment 1 and the loss of the severely affected half of the mice in experiment 2, which limited our statistical power and skewed any results to a less severe phenotype), there are compelling arguments supporting inflammation as a possible mechanism of increased T2DM severity due to LAN exposure. Increased body fat in humans increases inflammation and is likely involved in T2DM pathogenesis [15,45,48]. Obesity is linked to an imbalance of homeostatic and proinflammatory immune responses. Several inflammatory effector proteins derive from adipose tissue including TNFα, IL-6, IL-1β, leptin, adiponectin, resistin, and acylation-stimulating protein [50]. Furthermore, IL-6 and TNFα production increase with increased adiposity [51]. Activation of inflammatory pathways in adipose tissue and the brain can alter metabolic homeostasis over time and dysregulate physiological responses that maintain insulin and leptin sensitivity [52]. Although the precise mechanism of obesity-related insulin sensitivity remains unclear, it appears that within adipose tissue, TNFα phosphorylates and deactivates insulin receptor (IR) and insulin receptor substrate 1 (IRS-1) [50,53]. Low insulin receptor substrate 1 (IRS1) expression and protein levels have been associated with the development of insulin resistance in humans [4] and mice [30]. Stress and inflammatory response pathways, which can be activated by free fatty acids, are key signaling mechanisms through which IRS1 can be phosphorylated at Ser307 [55,56]. LAN has been previously implicated in increasing body weight in mice [13,57], and indeed, TH mice housed in LAN gained significantly more body weight over time compared with mice housed in LD. Furthermore, reversing the light treatment eliminated the difference in body weight concomitant with restoring insulin resistance to a similar level as mice housed in LD. Taken together, LAN could lead to insulin resistance and exacerbated metabolic dysregulation via obesity-induced inflammatory pathways This mechanism warrants further investigation.
Although circadian rhythms were not measured here, LAN has been previously implicated in circadian dysregulation. Mice housed in constant light become arrhythmic and have increased body mass [13,58,59] Although dim LAN (5 lux) does not necessarily alter circadian locomotor activity [60], it is sufficient to alter circadian temperature rhythms [61] and suppress rhythmic expression of clock genes in the SCN [11,62]. Likewise, 5 lux of light impairs glucose tolerance, increases carbohydrate over fat oxidation, decreases whole body energy expenditure, and increases body mass without altering food intake [61]. Circadian dysregulation induces peripheral inflammation [63,64]. Although there is limited information regarding peripheral inflammation and LAN-induced circadian dysregulation, exposure to just 5 lux of LAN for 4 weeks was sufficient to increase expression of Mac1 and TNFα in white adipose tissue of Swiss-Webster mice in one study [57]. Whether LAN increases inflammation and directly exacerbates metabolic dysfunction, or LAN increases body mass, which in turn increases inflammatory processes and exacerbates T2DM metabolic characteristics remains to be determined.
Conclusion
In conclusion, metabolic dysfunction was exacerbated in TH mice when housed in 40 lux of LAN. Although the precise mechanism remains unknown, it is likely that increased inflammation from circadian dysregulation, increased body weight, or a combination of both increases insulin resistance, characteristic of T2DM. In humans, worsening of T2DM metabolic dysfunction would also increase the likelihood of developing secondary complications of the disease, thus negatively affecting quality of life. Exposure to LAN also increased the prevalence of T2DM in a susceptible population (i.e. polygenic TH mice). Thus, exposure to LAN may also be an important consideration for humans with pre-diabetes. Reducing LAN exposure while sleeping could be a cost-effective method to reduce metabolic dysfunction in patients with pre-diabetes or T2DM. Further studies are required to clarify the translational potential of this research to human populations.
Acknowledgements
The authors thank Dana Shaw for her technical assistance. This work was supported by the Schulte Research Foundation and the National Institutes of Health [grant numbers R21CA202745 and R01NS092388].
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
The authors have no personal, professional, or financial relationships that could potentially be construed as a conflict of interest.
Datasets are available upon request:
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
References
- [1].Falchi F, Cinzano P, Duriscoe D, Kyba CCM, Elvidge CD, Baugh K, Portnov BA, Rybnikova NA, Furgoni R, The new world atlas of artificial night sky brightness, Sci. Adv 2 (2016) e1600377–e1600377. doi: 10.1126/sciadv.1600377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fonken LK, Nelson RJ, Effects of light exposure at night during development, Curr. Opin. Behav. Sci 7 (2016) 33–39. doi: 10.1016/j.cobeha.2015.10.008. [DOI] [Google Scholar]
- [3].Dominoni DM, Borniger JC, Nelson RJ, Light at night, clocks and health: from humans to wild organisms, Biol. Lett 12 (2016) 20160015. doi: 10.1098/rsbl.2016.0015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Falchi F, Cinzano P, Elvidge CD, Keith DM, Haim A, Limiting the impact of light pollution on human health, environment and stellar visibility, J. Environ. Manage (2011). doi: 10.1016/j.jenvman.2011.06.029. [DOI] [PubMed] [Google Scholar]
- [5].Sen A, Sellix MT, The Circadian Timing System and Environmental Circadian Disruption: From Follicles to Fertility, Endocrinology 157 (2016) 3366–3373. doi: 10.1210/en.2016-1450. [DOI] [PubMed] [Google Scholar]
- [6].Czeisler CA, Duffy JF, Shanahan TL, Brown EN, Mitchell JF, Rimmer DW, Ronda JM, Silva EJ, Allan JS, Emens JS, Dijk D-J, Kronauer RE, Stability, Precision, and Near-24-Hour Period of the Human Circadian Pacemaker, Science (80-. ) 284 (1999). http://science.sciencemag.org/content/284/5423/2177 (accessed August 14, 2017). [DOI] [PubMed] [Google Scholar]
- [7].Van Den Pol AN, Dudek FE, Cellular communication in the circadian clock, the suprachiasmatic nucleus, Neuroscience 56 (1993) 793–811. doi: 10.1016/0306-4522(93)90128-3. [DOI] [PubMed] [Google Scholar]
- [8].Guo H, Brewer JM, Champhekar A, Harris RBS, Bittman EL, Differential control of peripheral circadian rhythms by suprachiasmatic-dependent neural signals., Proc. Natl. Acad. Sci. U. S. A 102 (2005) 3111–3116. doi: 10.1073/pnas.0409734102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Benarroch EE, Suprachiasmatic nucleus and melatonin: Reciprocal interactions and clinical correlations, Neurology 71 (2008) 594–598. doi: 10.1212/01.wnl.0000324283.57261.37. [DOI] [PubMed] [Google Scholar]
- [10].Partch CL, Green CB, Takahashi JS, Molecular architecture of the mammalian circadian clock, Trends Cell Biol 24 (2014) 90–99. doi: 10.1016/J.TCB.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Fonken LK, Aubrecht TG, Meléndez-Fernández OH, Weil ZM, Nelson RJ, Dim light at night disrupts molecular circadian rhythms and increases body weight., J. Biol. Rhythms 28 (2013) 262–71. doi: 10.1177/0748730413493862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Russart KLG, Nelson RJ, Light at night as an environmental endocrine disruptor, Physiol. Behav 190 (2018) 82–89. doi: 10.1016/j.physbeh.2017.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fonken LK, Workman JL, Walton JC, Weil ZM, Morris JS, Haim A, Nelson RJ, Light at night increases body mass by shifting the time of food intake., Proc. Natl. Acad. Sci. U. S. A 107 (2010) 18664–9. doi: 10.1073/pnas.1008734107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Group NDD, Classification and Diagnosis of Diabetes Mellitus and Other Categories of Glucose Intolerance, Diabetes 28 (1995) 1039–1057. doi: 10.2337/diab.28.12.1039. [DOI] [PubMed] [Google Scholar]
- [15].Abdul-Ghani MA, DeFronzo RA, Pathophysiology of prediabetes., Curr. Diab. Rep 9 (2009) 193–9. http://www.ncbi.nlm.nih.gov/pubmed/19490820 (accessed July 6, 2018). [DOI] [PubMed] [Google Scholar]
- [16].Centers for Disease Control and Prevention, National Diabetes Statistics Report: Estimates of Diabetes and its Burden in the United States, US Dep. Heal. Hum. Serv (2014) 2014. doi: 10.1177/1527154408322560. [DOI] [Google Scholar]
- [17].Zimmet PZ, Magliano DJ, Herman WH, Shaw JE, Diabetes: A 21st century challenge, Lancet Diabetes Endocrinol 2 (2014) 56–64. doi: 10.1016/S2213-8587(13)70112-8. [DOI] [PubMed] [Google Scholar]
- [18].Facts F, Diabetes ON, National Diabetes Fact Sheet, Centers Dis. Control Prev. U.S. Dep. Heal. Hum. Serv CS217080A (2011) 1–12. doi: [Google Scholar]; Centers for Disease Control and Prevention. National diabetes fact sheet: national estimates and general information on diabetes and prediabetes in the United States, 2011. Atlanta, GA: U.S. Department of Health and Human Services, Centers for Disease Con. [Google Scholar]
- [19].Israili ZH, Advances in the treatment of type 2 diabetes mellitus, Am. J. Ther 18 (2011) 117–152. doi: 10.1097/MJT.0b013e3181afbf51. [DOI] [PubMed] [Google Scholar]
- [20].Moss SE, Klein R, Klein BE, Cause-specific mortality in a population-based study of diabetes., Am. J. Public Health 81 (1991) 1158–1162. doi: 10.2105/AJPH.81.9.1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gregg EW, Gu Q, Cheng YJ, Narayan KMV, Cowie CC, Mortality trends in men and women with diabetes, 1971 to 2000, Ann. Intern. Med 147 (2007) 149–155. doi: 10.7326/0003-4819-147-3-200708070-00167. [DOI] [PubMed] [Google Scholar]
- [22].Yang W, Dall TM, Beronjia K, Lin J, Semilla AP, Chakrabarti R, Hogan PF, Petersen MP, Economic costs of diabetes in the U.S. in 2017, Diabetes Care 41 (2018) 917–928. doi: 10.2337/dci18-0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Leahy JL, Pathogenesis of type 2 diabetes mellitus, Arch. Med. Res 36 (2005) 197–209. doi: 10.1016/j.arcmed.2005.01.003. [DOI] [PubMed] [Google Scholar]
- [24].O’Rahilly S, Barroso I, Wareham NJ, Genetic factors in type 2 diabetes: The end of the beginning?, Science (80-. ) 307 (2005) 370–373. doi: 10.1126/science.1104346. [DOI] [PubMed] [Google Scholar]
- [25].Tuomi T, Santoro N, Caprio S, Cai M, Weng J, Groop L, The many faces of diabetes: A disease with increasing heterogeneity, Lancet 383 (2014) 1084–1094. doi: 10.1016/S0140-6736(13)62219-9. [DOI] [PubMed] [Google Scholar]
- [26].Komada Y, Aoki K, Gohshi S, Ichioka H, Shibata S, Effects of television luminance and wavelength at habitual bedtime on melatonin and cortisol secretion in humans, Sleep Biol. Rhythms 13 (2015) 316–322. doi: 10.1111/sbr.12121. [DOI] [Google Scholar]
- [27].Wood B, Rea MS, Plitnick B, Figueiro MG, Light level and duration of exposure determine the impact of self-luminous tablets on melatonin suppression, Appl. Ergon 44 (2013) 237–240. doi: 10.1016/j.apergo.2012.07.008. [DOI] [PubMed] [Google Scholar]
- [28].Kim JH, Stewart TP, Soltani-Bejnood M, Wang L, Fortuna JM, Mostafa OA, Moustaid-Moussa N, Shoieb AM, McEntee MF, Wang Y, Bechtel L, Naggert JK, Phenotypic characterization of polygenic type 2 diabetes in TALLYHO/JngJ mice., J. Endocrinol 191 (2006) 437–46. doi: 10.1677/joe.1.06647. [DOI] [PubMed] [Google Scholar]
- [29].Kim JH, Sen Ś, Avery CS, Simpson E, Chandler P, Nishina PM, Churchill GA, Naggert JK, Genetic Analysis of a New Mouse Model for Non-Insulin-Dependent Diabetes, Genomics 74 (2001) 273–286. doi: 10.1006/GENO.2001.6569. [DOI] [PubMed] [Google Scholar]
- [30].Wang Y, Nishina PM, Naggert JK, Degradation of IRS1 leads to impaired glucose uptake in adipose tissue of the type 2 diabetes mouse model TALLYHO/Jng, J. Endocrinol 203 (2009) 65–74. doi: 10.1677/JOE-09-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Leiter EH, Strobel M, O’Neill A, Schultz D, Schile A, Reifsnyder PC, Comparison of two new mouse models of polygenic type 2 diabetes at the Jackson Laboratory, NONcNZO10Lt/J and TALLYHO/JngJ, J. Diabetes Res 2013 (2013) 165327. doi: 10.1155/2013/165327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kim JH, Saxton AM, The TALLYHO mouse as a model of human type 2 diabetes., Methods Mol. Biol 933 (2012) 75–87. doi: 10.1007/978-1-62703-068-7_6. [DOI] [PubMed] [Google Scholar]
- [33].Steppan CM, Bailey ST, Bhat S, Brown EJ, Banerjee RR, Wright CM, Patel HR, Ahima RS, Lazar MA, The hormone resistin links obesity to diabetes, Nature 409 (2001) 307–312. doi: 10.1038/35053000. [DOI] [PubMed] [Google Scholar]
- [34].Withers DJ, Gutierrez JS, Towery H, Burks DJ, Ren J-M, Previs S, Zhang Y, Bernal D, Pons v S., Shulman GI, Bonner-Weir S, White MF, Disruption of IRS-2 causes type 2 diabetes in mice, Nature 391 (1998) 900–904. doi: 10.1038/36116. [DOI] [PubMed] [Google Scholar]
- [35].Han BG, Hao C-M, Tchekneva EE, Wang Y-Y, Lee CA, Ebrahim B, Harris RC, Kern TS, Wasserman DH, Breyer MD, Qi Z, Markers of glycemic control in the mouse: comparisons of 6-h- and overnight-fasted blood glucoses to Hb A1c., Am. J. Physiol. Endocrinol. Metab 295 (2008) E981–6. doi: 10.1152/ajpendo.90283.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Polotsky VY, Wilson JA, Haines AS, Scharf MT, Soutiere SE, Tankersley CG, Smith PL, Schwartz AR, O’Donnell CP, The Impact of Insulin-Dependent Diabetes on Ventilatory Control in the Mouse, Am. J. Respir. Crit. Care Med 163 (2001) 624–632. doi: 10.1164/ajrccm.163.3.2007120. [DOI] [PubMed] [Google Scholar]
- [37].Taylor SI, Deconstructing type 2 diabetes, Cell 97 (1999) 9–12. doi: 10.1016/S0092-8674(00)80709-6. [DOI] [PubMed] [Google Scholar]
- [38].Conde SV, Ribeiro MJ, Melo BF, Guarino MP, Sacramento JF, Insulin resistance: a new consequence of altered carotid body chemoreflex?, J. Physiol 595 (2017) 31–41. doi: 10.1113/JP271684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Cline GW, Petersen KF, Krssak M, Shen J, Hundal RS, Trajanoski Z, Inzucchi S, Dresner A, Rothman DL, Shulman GI, Impaired Glucose Transport as a Cause of Decreased Insulin-Stimulated Muscle Glycogen Synthesis in Type 2 Diabetes, N. Engl. J. Med 341 (1999) 240–246. doi: 10.1056/NEJM199907223410404. [DOI] [PubMed] [Google Scholar]
- [40].Tabák AG, Herder C, Rathmann W, Brunner EJ, Kivimäki M, Prediabetes: A high-risk state for diabetes development, Lancet 379 (2012) 2279–2290. doi: 10.1016/S0140-6736(12)60283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Kong APS, Luk AOY, Chan JCN, Detecting people at high risk of type 2 diabetes- How do we find them and who should be treated?, Best Pract. Res. Clin. Endocrinol. Metab 30 (2016) 345–355. doi: 10.1016/j.beem.2016.06.003. [DOI] [PubMed] [Google Scholar]
- [42].DeFronzo RA, Insulin resistance: A multifaceted syndrome responsible for NIDDM, obesity, hypertension, dyslipidaemia and atherosclerosis, Neth. J. Med 50 (1997) 191–197. doi: 10.1016/S0300-2977(97)00012-0. [DOI] [PubMed] [Google Scholar]
- [43].Shepherd PR, Kahn BB, Glucose Transporters and Insulin Action — Implications for Insulin Resistance and Diabetes Mellitus, N. Engl. J. Med 341 (1999) 248–257. doi: 10.1056/NEJM199907223410406. [DOI] [PubMed] [Google Scholar]
- [44].Sung YY, Lee YS, Jung WH, Kim HY, Cheon HG, Yang SD, Rhee SD, Glucose intolerance in young TallyHo mice is induced by leptin-mediated inhibition of insulin secretion, Biochem. Biophys. Res. Commun 338 (2005) 1779–1787. doi: 10.1016/j.bbrc.2005.10.160. [DOI] [PubMed] [Google Scholar]
- [45].Festa A, D’Agostino R, Howard G, Mykkänen L, Tracy RP, Haffner SM, Chronic subclinical inflammation as part of the insulin resistance syndrome: The insulin resistance atherosclerosis study (IRAS), Circulation 102 (2000) 42–47. doi: 10.1161/01.CIR.102.1.42. [DOI] [PubMed] [Google Scholar]
- [46].Pradhan AD, Manson JE, Rifai N, Buring JE, Ridker PM, C-Reactive Protein, Interleukin 6, and Risk of Developing Type 2 Diabetes Mellitus, JAMA 286 (2001) 327. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- [47].Tsigos C, Kyrou I, Chala E, Tsapogas P, Stavridis JC, Raptis SA, Katsilambros N, Circulating tumor necrosis factor alpha concentrations are higher in abdominal versus peripheral obesity, Metabolism 48 (1999) 1332–1335. doi: 10.1016/S0026-0495(99)90277-9. [DOI] [PubMed] [Google Scholar]
- [48].Bastard JP, Jardel C, Bruckert E, Blondy P, Capeau J, Laville M, Vidal H, Hainque B, Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss., J. Clin. Endocrinol. Metab 85 (2000) 3338–3342. doi: 10.1210/jcem.85.9.6839. [DOI] [PubMed] [Google Scholar]
- [49].Stith RD, Luo J, Endocrine and carbohydrate responses to interleukin-6 in vivo, Circ Shock 44 (1994) 210–215. http://www.ncbi.nlm.nih.gov/pubmed/7628063 (accessed July 6, 2018). [PubMed] [Google Scholar]
- [50].Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM, Increased adipose tissue expression of tumor necrosis factor-alpha in human obesity and insulin resistance., J. Clin. Invest 95 (1995) 2409–2415. doi: 10.1172/JCI117936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Fried SK, Bunkin DA, Greenberg AS, Omental and subcutaneous adipose tissues of obese subjects release interleukin-6: Depot difference and regulation by glucocorticoid, J. Clin. Endocrinol. Metab 83 (1998) 847–850. doi: 10.1210/jc.83.3.847. [DOI] [PubMed] [Google Scholar]
- [52].Lumeng CN, Saltiel AR, Inflammatory links between obesity and metabolic disease, J. Clin. Invest 121 (2011) 2111–2117. doi: 10.1172/JCI57132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Boucher J, Kleinridders A, Ronald Kahn C, Insulin receptor signaling in normal and insulin-resistant states, Cold Spring Harb. Perspect. Biol 6 (2014). doi: 10.1101/cshperspect.a009191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Carvalho E, Jansson PA, Axelsen M, Eriksson JW, Huang X, Groop L, Rondinone C, Sjöström L, Smith U, Low cellular IRS 1 gene and protein expression predict insulin resistance and NIDDM., FASEB J 13 (1999) 2173–2178. http://www.ncbi.nlm.nih.gov/pubmed/10593864 (accessed July 9, 2018). [DOI] [PubMed] [Google Scholar]
- [55].Nguyen MTA, Satoh H, Favelyukis S, Babendure JL, Imamura T, Sbodio JI, Zalevsky J, Dahiyat BI, Chi N-W, Olefsky JM, JNK and Tumor Necrosis Factor-α Mediate Free Fatty Acid-induced Insulin Resistance in 3T3-L1 Adipocytes, J. Biol. Chem 280 (2005) 35361–35371. doi: 10.1074/jbc.M504611200. [DOI] [PubMed] [Google Scholar]
- [56].Kim JK, Kim YJ, Fillmore JJ, Chen Y, Moore I, Lee J, Yuan M, Li ZW, Karin M, Perret P, Shoelson SE, Shulman GI, Prevention of fat-induced insulin resistance by salicylate, J. Clin. Invest 108 (2001) 437–446. doi: 10.1172/JCI11559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Fonken LK, Lieberman RA, Weil ZM, Nelson RJ, Dim Light at Night Exaggerates Weight Gain and Inflammation Associated With a High-Fat Diet in Male Mice, Endocrinology 154 (2013) 3817–3825. doi: 10.1210/en.2013-1121. [DOI] [PubMed] [Google Scholar]
- [58].Moriya T, Yoshinobu Y, Kouzu Y, Katoh A, Gomi H, Ikeda M, Yoshioka T, Itohara S, Shibata S, Involvement of glial fibrillary acidic protein (GFAP) expressed in astroglial cells in circadian rhythm under constant lighting conditions in mice, J. Neurosci. Res 60 (2000) 212–218. doi: 10.1002/(SICI)1097-4547(20000415)60:2<212::AID-JNR10>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- [59].Ohta H, Yamazaki S, McMahon DG, Constant light desynchronizes mammalian clock neurons, Nat. Neurosci 8 (2005) 267–269. doi: 10.1038/nn1395. [DOI] [PubMed] [Google Scholar]
- [60].Fonken LK, Hecmarie Meléndez-Fernández O, Weil ZM, Nelson RJ, Exercise attenuates the metabolic effects of dim light at night, Physiol. Behav 124 (2014) 33–36. doi: 10.1016/j.physbeh.2013.10.022. [DOI] [PubMed] [Google Scholar]
- [61].Borniger JC, Maurya SK, Periasamy M, Nelson RJ, Acute dim light at night increases body mass, alters metabolism, and shifts core body temperature circadian rhythms, Chronobiol. Int 00 (2014) 1–9. doi: 10.3109/07420528.2014.926911. [DOI] [PubMed]
- [62].Bedrosian TA, Galan A, Vaughn CA, Weil ZM, Nelson RJ, Light at Night Alters Daily Patterns of Cortisol and Clock Proteins in Female Siberian Hamsters, J. Neuroendocrinol 25 (2013) 590–596. doi: 10.1111/JNE.12036. [DOI] [PubMed] [Google Scholar]
- [63].Castanon-Cervantes O, Wu M, Ehlen JC, Paul K, Gamble KL, Johnson RL, Besing RC, Menaker M, Gewirtz AT, Davidson AJ, Dysregulation of inflammatory responses by chronic circadian disruption., J. Immunol 185 (2010) 5796–805. doi: 10.4049/jimmunol.1001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Cermakian N, Westfall S, Kiessling S, Circadian Clocks and Inflammation: Reciprocal Regulation and Shared Mediators, Arch. Immunol. Ther. Exp. (Warsz) 62 (2014) 303–318. doi: 10.1007/s00005-014-0286-x. [DOI] [PubMed] [Google Scholar]