

Abstract
Introduction
Adverse event (AE) under-reporting has been a recurrent issue raised during health authorities Good Clinical Practices (GCP) inspections and audits. Moreover, safety under-reporting poses a risk to patient safety and data integrity. The current clinical Quality Assurance (QA) practices used to detect AE under-reporting rely heavily on investigator site and study audits. Yet several sponsors and institutions have had repeated findings related to safety reporting, and this has led to delays in regulatory submissions. Recent developments in data management and IT systems allow data scientists to apply techniques such as machine learning to detect AE under-reporting in an automated fashion.
Objective
In this project, we developed a predictive model that enables Roche/Genentech Quality Program Leads oversight of AE reporting at the program, study, site, and patient level. This project was part of a broader effort at Roche/Genentech Product Development Quality to apply advanced analytics to augment and complement traditional clinical QA approaches.
Method
We used a curated data set from 104 completed Roche/Genentech sponsored clinical studies to train a machine learning model to predict the expected number of AEs. Our final model used 54 features built on patient (e.g., demographics, vitals) and study attributes (e.g., molecule class, disease area).
Results
In order to evaluate model performance, we tested how well it would detect simulated test cases based on data not used for model training. For relevant simulation scenarios of 25%, 50%, and 75% under-reporting on the site level, our model scored an area under the curve (AUC) of the receiver operating characteristic (ROC) curve of 0.62, 0.79, and 0.92, respectively.
Conclusion
The model has been deployed to evaluate safety reporting performance in a set of ongoing studies in the form of a QA/dashboard cockpit available to Roche Quality Program Leads. Applicability and production performance will be assessed over the next 12–24 months in which we will develop a validation strategy to fully integrate our model into Roche QA processes.
Electronic supplementary material
The online version of this article (10.1007/s40264-019-00831-4) contains supplementary material, which is available to authorized users.
Key Points
| Safety under-reporting is a recurrent issue in clinical trials. |
| We built a machine learning model that detects under-reporting of adverse events. |
| This model is used to trigger quality assurance activities to protect patient safety and to avoid delayed filing. |
Introduction
Compliance with the fundamental principles of Good Clinical Practice (GCP) ensures the rights, safety, and wellbeing of research subjects and ensures the integrity of clinical research data. Trial sponsors are required by the International Conference on Harmonization (ICH) guidelines to implement and maintain quality assurance (QA) and quality control systems to achieve these objectives [1].
One of the main issues reported in GCP health authority inspections and sponsor audits is the lack of adverse event (AE) reporting from the investigator sites to the sponsor [2, 3]. Under-reporting poses a significant risk to data integrity and to patient safety [1, 4–6]. Furthermore, several sponsors have failed to mitigate AE under-reporting and this has led to delays in regulatory submission or to non-approval of new drugs [6, 7].
Finally, there are also some concerns that safety data collected in randomized controlled trials (RCTs) have limitations [8] that could be aggravated by AE under-reporting. First of all, the sample size of RCTs is tailored to detect drug efficacy but not to statistically detect incidents that occur with a lower rate than a positive drug response. Furthermore, RCT AE reporting and analysis standards (lack of time-to-event reporting, using standardized incidence ratios, and normalizing by patient-years) favor the detection of AEs that can occur with a uniform risk rate over the entire observation window over detection of those AEs that have high risk rates at a specific time frame inside the observation window [8]. In the light of these limitations, which make statistical AE detection in the setting of RCTs very challenging, it becomes evident that AE under-reporting poses a great risk to the detection of AEs and to patient safety.
Current clinical QA practices heavily rely on audits to detect sites or studies with quality issues, including AE under-reporting [9]. The increasing number of clinical trials and sites and the growing complexity of study designs make it challenging to detect AE under-reporting. Current site monitoring strategies, which rely on on-site source data verification (SDV) and on risk-based approaches, are attempting to address the issue [10, 11], yet AE under-reporting remains as a common audit and inspection finding [2].
A holistic QA approach that addresses the above raised issues concerning AE reporting is not currently available. However, the industry has recently been trying to leverage modern developments in data management and IT systems that facilitate the cross-analysis of clinical studies. Statistical analysis can be performed on this data based on certain attributes to help identify issues in safety reporting and to be able to estimate or predict the number of AEs reported per patient. We used our combined, historical clinical study data to develop a predictive model for the expected number of AEs per patient based on study and patient attributes including but not limited to therapeutic area, study design, mechanism of drug action, mode of administration, vitals, commonly assessed laboratory measurements, medical history and concomitant medications. We propose a model that will provide insight to clinical QA professionals to detect and mitigate safety reporting risks more holistically and efficiently.
The development of a predictive model that can help detect under-reporting requires a deep understanding of data science, clinical safety, and QA. The project has been conducted by the Roche/Genentech quality data analytics team, a team of data scientists, in collaboration with Roche/Genentech clinical and QA subject matter experts (SMEs).
The mission of the Roche/Genentech quality data analytics team is to build data-driven solutions for clinical QA at Roche/Genentech to complement and augment traditional QA approaches to improve the quality and oversight of GCP—and Good Pharmacovigilance Practices (GVP)—regulated activities.
Method
Outline and Assumptions
The objective of this proof-of-concept (PoC) effort was to develop and assess the performance of a predictive model that can help detect AE under-reporting and to develop a visual interface for QA professionals. The scope of this PoC was to predict AE under-reporting, not predicting adverse drug reactions that should occur in clinical trials. GCPs require all AEs, whether or not there might be a causal relationship between the intake of the drug and the events, to be reported timely to the sponsor [1].
The identification of study investigator sites suspected of under-reporting amounts to an unsupervised anomaly detection problem [12]. In this class of problems, one tries to identify which elements of a data set are anomalous; for example, which objects in a production line show a defect, or which study sites are not compliant with GCP. The main difference from a classification task is that the data points are unlabeled. Under the assumption that a majority of them behave normally, a possible approach to solve these problems is to fit a probability distribution to the data and flag as anomalous those data points that have a likelihood below a certain threshold. The performance of the anomaly detector can then be assessed with a small sample of anomalous points, either manually detected or simulated, and regular ones in the same way as one would assess a classifier, namely with metrics such as the area under the receiver operating characteristic (ROC) curve, precision, recall, or accuracy.
Working on the assumption that the curated data set of finished and completed studies used for model training contained a majority of compliant study sites (see also Sect. 2.2.1.), we could build a probabilistic model for the random variable describing the number of AEs reported by a given study site. We collected data from each site, which were modeled as a random variable , a feature vector that we believed had a direct influence on .
When we considered a new study site and made observations of the feature vector and of the number of reported AEs, from the conditional probability density of our model we computed the probability of observing this number of AEs or less, that we defined as the significance level. We then picked a threshold and we decided to act for significance levels below it.
Clinical trial data can be interpreted as a set of multivariate time series of measurements for each patient in the study (some of them being constant, for instance the demographic data). Furthermore, this data is typically collected during the patient visits, which is when AEs are reported to the investigator [13]. Therefore, we decomposed the number of AEs reported by a site into the sum of the numbers of AEs reported by the corresponding patients,
and similarly, the number of AEs reported by a patient into the sum of the numbers of AEs reported at each visit,
We could make predictions either at the site level, patient level, or visit level. Given the granularity of clinical data, we decided to focus on the visit level. A sudden change in vital parameters such as the weight could be indicative of health deterioration and thus the occurrence of AEs [14]. Moreover, once we used this model on ongoing studies, we wanted to be able to update our predictions as new data from the sites came in, which was easier to do if we started at the visit level.
We were thus interested in the probability density conditioned on the feature vector that summarizes information on the patient known at the time of the visit. To estimate the relation between and , given the amount of historical data at our disposal, we decided to apply machine learning algorithms. The usual least squared error regression was ill-advised in this situation as it would imply that predicting zero AE instead of five costs the same as predicting 95 instead of 100, which was not the case. We could have considered logarithmic least squares, but since we were dealing with a count variable, it was best to minimize the Poisson deviance. In this class of models, the random variable was interpreted as a Poisson process,
where we had to express the Poisson parameter as a function of . Due to the complexity of the underlying biology of AEs, the empirical approach seemed more promising than theoretical modeling and we decided to use machine learning for this task. The advantage of this approach was that Poisson processes are additive in their parameters, so we immediately obtained:
Furthermore, assuming our estimate of was accurate, we could calculate the significance level of an observation of adverse events,
Even if these assumptions did not hold perfectly and was thus not a well-calibrated probability, we could still use it as a scoring function to detect under-reporting and evaluate its discriminating power with a ROC curve.
Data
Raw Data
The raw data set we used came from Roche/Genentech-sponsored clinical trials. We used common data attributes from 104 completed studies that covered various molecule types and disease areas. The data set included 3231 individual investigator sites, with 18,682 study subjects that underwent 288,254 study visits. Of note, any study subject data was used in a de-identified format. To mitigate the risk of having studies with under-reporting in our data set, we used only data from completed and terminated clinical trials, where AE reconciliation and SDV had been performed as part of the study closure activities. The six common patient data attributes across the studies that we selected in our curated data set were demographics, medical history, concomitant medications, vitals, visits, and adverse events, following the Study Data Tabulation Model (SDTM) standard [15]. As mentioned above, we focused on the visits, which we labeled by study code, patient number, and visit date. We also considered study attributes available in the Roche Clinical Trial Management System (CTMS) and included them in our data set: study type, route of administration, concomitant agents, disease area, blinding, randomization, and study phase. We used a different classification for the molecule classes and the disease areas from the one used in the Roche CTMS to ensure their clinical relevance in terms of AE reporting. Molecules were classified using the Anatomical Therapeutic Chemical (ATC) classification system [16]. For the disease areas, we used a simple classification that reflects the populations enrolled in our clinical trials (healthy participants, malignancies, autoimmune diseases, neurodegenerative diseases, respiratory diseases, skin disorders, lung diseases, infectious diseases, others). As we needed to have a model that can generalize to the diversity and volume of clinical studies we run at Roche/Genentech, we purposely chose study and patient attributes that are systematically captured in our clinical programs. See Table 1 below for an overview of our curated data set.
Table 1.
Attributes available in our curated data-set
| Level | Source | Extracted data |
|---|---|---|
| Patient | SDTM demographics | Age, sex, ethnicity |
| Visit | SDTM medical history | Number of co-occurring conditions |
| Visit | SDTM concomitant medications | Number of concomitant medications |
| Visit | SDTM vitals | Height, weight, blood pressure |
| Visit | SDTM visits | Number of previous visits |
| Visit | SDTM adverse events | Number of reported AEs |
| Study | Clinical Trial Management System | Intervention type, route of administration, use of concomitant agents, phase, randomization, blinding, molecule class, disease type |
AEs adverse events, SDTM study data tabulation model
Features and Targets
Each AE was assigned to the first visit following the onset date and all AEs assigned to a specific visit were aggregated into the observation , that we tried to predict.
To construct features, we needed to project all data attributes to the visit level. For demographic characteristics that were constant, such as sex and ethnicity, or had a direct dependence on the date, such as age, this was straightforward. For medical history, we counted the events that occurred before every visit. Since new entries from screening in the medical history section of the electronic case report form (eCRF) normally correspond to AEs that should get reported, they provide a strong signal. Similarly, we counted concomitant medications, because the more drugs a patient receives, the more AEs he will likely experience [14, 17]. From the vitals reported at each visit, we included blood pressure and its relative variation since the previous visit. We also used patient weight, its relative variation since the previous visit, and the trend over the last 3 weeks as attributes, as a change in weight could be linked to a worsening of health and hence the occurrence of AEs. The disease area, the molecule class and mechanism of action, and the route of administration were also included as categorical features, as these characteristics have a strong influence on the type and number of AEs [14]. We picked the drug class instead of the molecule itself as a feature to ensure generalization to previously unseen drugs, consenting to increase the bias in order to reduce the variance. For a selection of the created features and how they correlate with the number of reported AEs, see Electronic Supplementary Material 1.
Before regrouping the features in the vector , we used one-hot encoding on the categorical variables, we raised the age variable to the power 1.4 in order to have a roughly normal distribution, and we standardized the continuous variables.
Once the set of features was selected, we relied on machine learning algorithms to pick the best ones through optimization of a loss function.
In our model, we used 54 features, with the highest contribution coming from the following ones:
Number of previous visits made by the patient
Cumulative count of concomitant medications up to the current visit
Disease is a malignancy (Boolean)
Disease is pulmonary but non-malignant (Boolean)
Administration is oral (Boolean)
See Electronic Supplementary Material 2 for the full list of features used in the final model.
Training, Validation, and Test Sets
As in most machine learning projects, we split our data into a training, a validation, and a test set. The training set was used to minimize the loss function with respect to the parameters of the model, the validation set to control for overfitting and to pick the hyper-parameters of the model via grid search, and the test set finally to assess the generalization performance to new data [18]. In our case, the test set was also used for the simulation of under-reporting introduced in the outline.
It should be noted that we could not randomly assign each pair to one of the three sets as we were ultimately interested in , the count of adverse events reported by a single site. We needed to work on subsets and assign each of them to one of the training, validation, and test sets. At the level of the prediction for , this prevented data leakage due to a patient finding himself in two different sets.
We assumed that the molecule class had a significant influence on the number of AEs [17]; therefore, we decided to stratify the sites by this factor when splitting them into the training, validation, and test sets, to ensure a representation of every class in each set.
While respecting these constraints, we tried to assign roughly 60% of the sites to the training set and 20% each to the validation and test sets.
Under-Reporting Simulation
In order to evaluate how the significance level discriminates under-reporting anomalies from normal behavior, we had to simulate under-reporting sites due to the lack of real-world examples where all necessary data attributes had been captured. To do so, we picked a sample of the test set where we artificially lowered the AE count to simulate under-reporting. Explicitly, for each pair from this sample of the test set, we built an under-reporting pair , with How much smaller than depended on how we wanted to define under-reporting, which required input from subject matter experts. We defined three types of scenarios (described below), one following a statistical approach, one reducing all AEs by a fixed ratio, and one simulating absence of reporting.
The negative cases of under-reporting, where denotes the label for the classification problem, from the test set could then be merged with the positive cases of under-reporting from the simulated under-reporting set to form the classification test set, from which we could build a ROC curve for the significance levels and . We selected a sample instead of the whole test set to exclude sites where the difference between and would be too low to be worrisome from a quality perspective and would therefore add unnecessary noise in the evaluation of the models. In defining the under-reporting scenarios, we thus had to specify as a function of and which sites to keep in
Statistical Scenario
The ‘statistical scenario’ relied on the assumption that the total number of AEs reported by a single site followed a Poisson distribution, . Our best estimate for was given by the observed number of AEs, and a low number of reported AEs could be defined as the first percentile of this distribution ,
where denotes the quantile function of probability distribution . Table 2 summarizes a few values of this function. We kept in the under-reporting sample only the sites with .
Table 2.
Examples of simulated values of under-reporting in the statistical scenario
| 1 | 5 | 10 | 50 | 100 | 500 | 1000 | ||
| 0 | 1 | 3 | 34 | 77 | 449 | 927 | ||
Ratio Scenarios
In the ‘ratio scenarios’, we arbitrarily kept a fixed fraction of AEs. We tried several values, namely (25% under-reporting), (50% under-reporting), (67% under-reporting), (75% under-reporting) and (90% under-reporting), and again we kept in the under-reporting sample only the sites with .
Zero Scenario
The ‘zero scenario’ simulated the absence of reporting from the smaller sites, so we set and retained only those with 10 patients or fewer but at least six reported AEs in total for the positive cases. In our test set, those represented 329 sites out of 643.
Machine Learning Algorithm
The problem of modeling the number of adverse events reported on a given visit as a Poisson process, , could be tackled with machine learning. Given observations and of the features and numbers of reported AEs, the goal was to find an approximation of that minimizes a loss function,
where the sum runs over all visits in the training set and the individual loss penalizes inaccuracy in the individual prediction of . Its exact form depends on the type of modeling. For Poisson processes, it is the Poisson deviance
Several algorithms are suitable to optimize this loss function, the most commonly used are generalized linear models [19], gradient boosting machines [20], and neural networks. We dismissed neural networks as we felt the limited signal to noise ratio did not justify the investment in computational power and architecture design. We tried the other two algorithms and obtained the best performance with gradient boosting machines, so we settled for this one. A thorough introduction can be found in The elements of statistical learning: data mining, inference and prediction [21], but we provide a brief overview of the algorithm here.
A regression tree would try to solve this optimization problem by successively splitting regions of the feature space in halves and assigning a value for to each region of the final partition. While the accuracy of a single tree is fairly low, ensemble methods such as gradient boosting machines or random forests aggregate the predictions of many trees in a weighted average and achieve a much better performance. A gradient boosting machine constructs this average iteratively: it starts with a simple estimate and successively updates its current prediction with a new tree that tries to replicate the current gradient of the loss function. This approach was inspired by the gradient descent methods widely used in optimization, which gave the name of the algorithm.
Implementation
We stored our data in a Hadoop [22] cluster to ensure scalability to an arbitrary number of studies, with the data preprocessing and feature engineering coded in PySpark. Several software packages offer more or less sophisticated implementations of gradient boosting machines. They mainly differ by the way single trees are fit to the current gradient of the loss function and by different performance optimizations. We used the Sparkling Water [23] implementation of H2O, which would allow our entire pipeline to be easily exported as a Spark application if we decided, for instance, to move to a cloud-based solution.
Results
Based on the simulated under-reporting scenarios described in Sect. 2.3 and the predictions of our trained gradient boosting machines on the test set, we obtained the following ROC curves for the task of detecting under-reporting with a score function given by the significance levels of the observations and the simulated reduced values. For the statistical scenario (see Sect. 2.3.2), our model scored an area under the ROC curve of 0.67 (see Fig. 1).
Fig. 1.
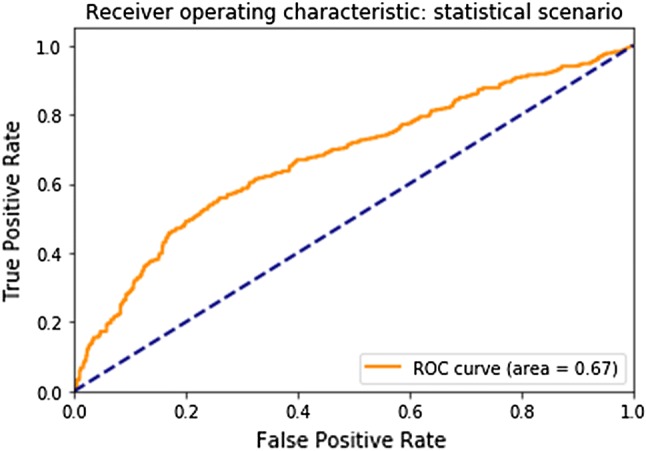
Receiver operating characteristic (ROC) curve for the statistical scenario
For the zero scenario (small investigator site), our model scored an area under the ROC curve of 0.97 (see Fig. 2). For the scenarios of 25%, 50%, 67%, and 75% under-reporting on the site level, our model scored an area under the ROC curve of 0.62, 0.79, 0.89, and 0.92, respectively (see Fig. 3).
Fig. 2.
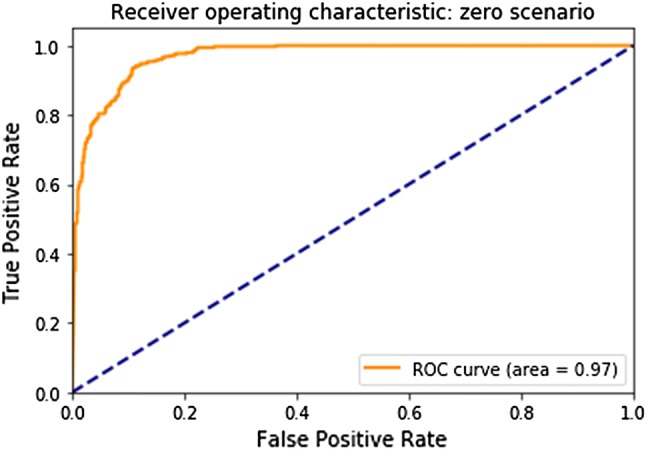
Receiver operating characteristic (ROC) curve for the zero scenario (for small investigator sites)
Fig. 3.
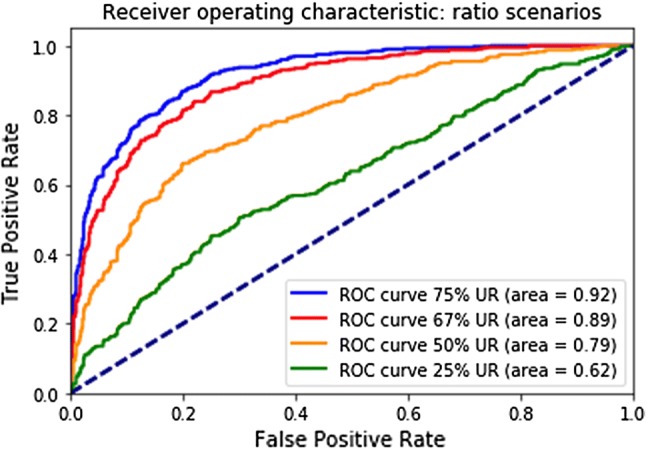
Receiver operating characteristic (ROC) curves for the percentage scenarios. UR under-reporting
We used a trade-off between true positive rates and false positive rates to define alert levels in order to prioritize the study sites to be further investigated. See Sect. 4 for more details.
Discussion
We built a visual and interactive dashboard using Tableau®. Data from ongoing clinical studies were collected on a monthly basis and fed to our model in order to get updated values and predictions for the volume of reported AEs. Of note, it is planned to feed our model with data from ongoing clinical studies on a daily basis to generate updated predictions in real time.
In order to detect sites that were at risk of under-reporting, we used the findings from the simulation scenarios to derive an alert level (AL) flagging system. The significance level score for each site allowed us to rank the sites by risk of under-reporting; however, a reasonable cut-off had to be found to determine which of the high-risk sites needed to be flagged for the end user. In order to calculate the best trade-off between maximizing true positive rate (tpr) and minimizing false positive rate (fpr), we used Youden’s J statistics [24] on each simulation scenario. We identified three consecutive threshold values that allowed us to group all sites into four groups (AL3, AL2, AL1, AL0), with AL3 indicating the highest risk and AL0 the lowest risk for under-reporting. The tpr for all simulation scenarios and the corresponding fpr are listed in Table 3.
Table 3.
Performance metrics for sites grouped by different alert levels
| Alert level 3 | Alert level 2–3 | Alert level 1–3 | Alert level 0 | |
|---|---|---|---|---|
| fpr | 0.14 | 0.22 | 0.25 | 0.75 |
| Zero scenario tpr | 0.95 | 0.99 | 0.99 | 0.01 |
| 75% under-reporting tpr | 0.80 | 0.90 | 0.91 | 0.09 |
| 67% under-reporting tpr | 0.72 | 0.84 | 0.86 | 0.14 |
| 50% under-reporting tpr | 0.50 | 0.64 | 0.66 | 0.36 |
| 25% under-reporting tpr | 0.31 | 0.37 | 0.39 | 0.61 |
fpr false positive rate, tpr true positive rate
The fpr of each alert level is indicative of the minimum percentage of sites that will be flagged in a set of data from ongoing studies, all of which will need to be screened in order to detect true under-reporting sites with the indicated performance metrics. If the percentage of under-reporting sites in our sample were exceptionally high (> 1%), the percentage of sites being flagged would increase accordingly but without affecting the tpr metric. Based on those assumptions, we can interpret the performance for AL3 as follows: by reviewing the top ~ 14% of the sites with the highest under-reporting risk predicted by our model, we will identify 95% of small sites not reporting any AEs, 80% of all sites with 75% under-reporting, 72% of all sites with 67% under-reporting, 50% of all sites with 50% under-reporting and 31% of all sites with 25% AE under-reporting. We can reasonably increase these detection rates by including sites flagged with AL2 and AL1 into our reviewing process.
The alert levels are displayed on the dashboard along with other important site parameters. Access to it has been granted to quality program leads at Roche/Genentech. It allows a holistic and nearly real-time quality oversight for safety reporting. Studies and sites that are suspected of under-reporting will be considered at risk and will trigger additional quality activities (e.g., audits). The tool will also be used by auditors to select sites and/or patients for review during study or investigator site audits.
As explained in the introduction, current clinical QA practices heavily rely on investigator and study audits [9]. For quality oversight activities, our predictive model has a significant advantage, as it enables holistic and real-time monitoring of safety reporting at various levels, which had not been possible when solely relying upon audits. With the current performance, a site that reported significantly fewer AEs than predicted (e.g., 67% under-reporting and more, see Sect. 3) would very likely be detected early enough that quality program leads can trigger mitigation activities. For audit selection and planning, risk factors such as high recruiting sites/studies and other quality indicators were used to prioritize audits. The output of our predictive model could be integrated to refine the current risk assessment process. During audits, the current practice for site or patient selection was mainly based on random sampling and adherence to defined quality indicators; hence our model enables data-driven selection of patients (during site audits) and of study sites (during study audits).
Health Authorities inspectors and GCP requirements do not provide any defined threshold on what is considered AE under-reporting. However, the regulatory guidelines emphasize that a risk-based approach should be applied and quality assurance teams must focus on the highest risk areas [1–3]. Hence, we proposed various thresholds of under-reporting (Sect. 2.3.1) to assess if the model performance will enable detection of the most problematic investigator sites. In this context, our model allows us to pursue a risk-based approach when screening sites for safety reporting issues. By focusing on 14% of the high-risk study sites as ranked by our model, we will be able to detect 95% of small sites with no under-reporting, 80% of sites with 75% under-reporting, 72% of sites with 67% under-reporting, 50% of sites with 50% under-reporting, and 31% of sites with 25% under-reporting. The level of performance of our predictive model was perfectly acceptable in the context of being used by quality program leads (program, study, and site oversight), quality strategists (audit selection and planning) and auditors (site and patient review selection), as it provided a more robust quantification of the risk of under-reporting than the current standard. Our predictive model and its associated visualization have been designed to be fit for purpose for clinical QA. However, we will share our approach, our models, and the associated tool with other teams responsible for study oversights, namely study teams and clinical monitors. Such a tool and approach could also be used for site monitoring, especially in the context of centralized and/or risk-based monitoring.
Limitations
The main obstacle we had to overcome in this work was the absence of labeled positive cases of under-reporting to evaluate our models. As a work-around, we simulated under-reporting at the site level because our end-goal was the identification of suspicious sites, and summation across several patients made our somehow simplistic statistical scenarios more likely than if we had applied them at patient level. We picked our approach of combining machine learning with a probabilistic interpretation of the results for computational reasons and the immediate availability of off-the-shelf products. The price to pay was that the significance levels we computed were not well-calibrated probabilities, because they failed to capture the uncertainty in the prediction of and by extension of . As a result, we could not be certain that a good performance at detecting under-reporting at the site level would translate well to the patient level. A fully probabilistic, well-calibrated model would be more reliable. Potential approaches include probabilistic graphical models and Bayesian neural networks.
Our models have been trained solely on Roche/Genentech-sponsored clinical trial data. Access to clinical trial data from other sponsors would be a prerequisite to assess the performance of our models on non-Roche/Genentech clinical studies. We are considering approaching other sponsors and regulators to further assess the performance of our models and possibly teaming up to build the next version of the model to detect AE under-reporting. Further analysis using real-world data will also be performed with an upcoming collaboration effort with Flatiron Health that provide curated real-world data. Once we have extended our data corpus, we will seek to develop a new modeling strategy that allows us to differentiate between study types during the evaluation of model performance.
At the time of the experiment, we did not have access to a curated data set that would allow us to map clinical investigator sites to specific countries/regions. As AE reporting culture might differ from one country/region to another [25], we are considering the integration of geographical locations of studies and sites as a feature in the next version of our model.
Conclusions
In this paper, we presented the development of a predictive model that enabled detection of suspected AE under-reporting. Our model scored an AUC of the ROC curve of 0.62, 0.79, and 0.92 when tested at different scenarios: 25%, 50%, and 75% of AE under-reporting, respectively. The model is now being used by Quality Program Leads at Roche/Genentech on a limited number of ongoing studies. It will be deployed in production in the course of 2019/2020 and will be applied to all ongoing clinical studies. This is part of a broader effort at Roche/Genentech Product Quality to leverage advanced analytics to augment and complement traditional clinical QA approaches. With regards to the model itself, there are plans to enhance it in the coming months. The next version will assess alternative machine learning models (as explained in Sect. 4.). It will also integrate additional clinical study data sets and other data sources, such as—but not limited to—site/study geographical location.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
Content review was provided by Antoinette Chan and Elaine (Min) Zou, who were employed by Roche/Genentech at the time this research was completed.
Compliance with Ethical Standards
Funding
Funding for development and testing of the anomaly detection system was supplied by Roche/Genentech.
Conflict of Interest
Timothé Ménard, Yves Barmaz, Björn Koneswarakantha, Rich Bowling, and Leszek Popko were employed by Roche/Genentech at the time this research was completed.
Ethics Statement
All human subject data used in this analysis were used in a de-identified format.
References
- 1.International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. E26(R2) Guideline for Good Clinical Practices. 2016. https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E6/E6_R2__Step_4_2016_1109.pdf. Accessed 10 Dec 2018.
- 2.Medicine and Healthcare products Regulatory Agency. GCP inspection metrics report. 2018. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/706356/GCP_INSPECTIONS_METRICS_2016-2017__final_11-05-18_.pdf. Accessed 10 Dec 2018.
- 3.Food and Drug Administration. 2018 inspection data set. https://www.fda.gov/downloads/ICECI/Inspections/UCM628362.xlsx. Accessed 10 Dec 2018.
- 4.Food and Drug Administration. Guidance for Industry: Investigator Responsibilities — Protecting the Rights, Safety, and Welfare of Study Subjects. 2009. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM187772.pdf. Accessed 10 Dec 2018.
- 5.Pitrou I, Boutron I, Ahmad N, Ravaud P. Reporting of safety results in published reports of randomized controlled trials. Arch Intern Med. 2009;169(19):1756–1761. doi: 10.1001/archinternmed.2009.306. [DOI] [PubMed] [Google Scholar]
- 6.Food and Drug Administration. Warning letter to AB Science 6/16/15 https://www.fda.gov/ICECI/EnforcementActions/WarningLetters/ucm453979.htm. Accessed 10 Dec 2018.
- 7.Sacks L, Shamsuddin H, Yasinskaya Y, Bouri K, Lanthier M, Sherman R. Scientific and regulatory reasons for delay and denial of fda approval of initial applications for new drugs, 2000–2012. JAMA. 2014;311(4):378–384. doi: 10.1001/jama.2013.282542. [DOI] [PubMed] [Google Scholar]
- 8.Yazici Y. Some concerns about adverse event reporting in randomized clinical trials. Bull NYU Hosp Jt Dis. 2008;66(2):143–145. [PubMed] [Google Scholar]
- 9.Li H, Hawlk S, Hanna K, Klein G, Petteway S. Developing and implementing a comprehensive clinical QA audit program. Qual Assur J. 2007;11:128–137. doi: 10.1002/qaj.420. [DOI] [Google Scholar]
- 10.Food and Drug Administration. Guidance for industry: oversight of clinical investigations—a risk-based approach to monitoring. 2013. https://www.fda.gov/downloads/Drugs/Guidances/UCM269919.pdf. Accessed 10 Dec 2018.
- 11.Hurley C, Sinnott C, Clarke M, Kearney P, Racine E, Eustace J, et al. Perceived barriers and facilitators to risk based monitoring in academic-led clinical trials: a mixed methods study. Trials. 2017;18:423. doi: 10.1186/s13063-017-2148-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chandola V, Banerjee A, Kumar V. Anomaly detection: a survey. ACM Comput Surveys. 2009;41(3):1–58. doi: 10.1145/1541880.1541882. [DOI] [Google Scholar]
- 13.Ceh, E. Adverse event reporting: during the study. 2009. https://firstclinical.com/journal/2009/0908_Adverse_Reporting.pdf. Accessed 10 Dec 2018.
- 14.Chaboyer W, Thalib L, Foster M, Ball C, Richards B. Predictors of adverse events in patients after discharge from the intensive care unit. Am Assoc Crit Care Nurs. 2008;17(3):255–263. [PubMed] [Google Scholar]
- 15.Study Data Tabulation Model (SDTM), v 1.6. 2017. https://www.cdisc.org/standards/foundational/sdtm. Accessed 10 Dec 2018.
- 16.World Health Organisation. Anatomical therapeutic chemical (ATC) classification system. https://www.whocc.no/atc_ddd_index/. Accessed 10 Dec 2018.
- 17.Singh S, Dhasmana DC, Bisht M, Singh PK. Pattern of adverse drug reactions to anticancer drugs: a quantitative and qualitative analysis. Indian J Med Paediatr Oncol. 2017;38(2):140–145. doi: 10.4103/ijmpo.ijmpo_18_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Handelma GS, Kok HK, Chandra RV, Razavi AH, Huang S, Brooks M, et al. Peering into the black box of artificial intelligence: evaluation metrics of machine learning methods. AJR Am J Roentgenol. 2018;17:1–6. doi: 10.2214/AJR.18.20224. [DOI] [PubMed] [Google Scholar]
- 19.Nelder J, Wedderburn R. Generalized linear models. J R Stat Soc. 1972;135(3):370–384. [Google Scholar]
- 20.Friedman J. Greedy function approximation: a gradient boosting machine. Ann Stat. 2001;29(5):1189–1232. doi: 10.1214/aos/1013203451. [DOI] [Google Scholar]
- 21.Hastie T, Tibshirani R, Friedman J. The elements of statistical learning: data mining, inference and prediction. New York: Springer; 2009. [Google Scholar]
- 22.Apache Hadoop. https://hadoop.apache.org. Accessed on 10 Dec 2018.
- 23.H2O Sparkling Water. https://www.h2o.ai/products/h2o-sparkling-water. Accessed on 10 Dec 2018.
- 24.Youden WJ. Index for rating diagnostic tests. Cancer. 1950;3(1):32–35. doi: 10.1002/1097-0142(1950)3:1<32::AID-CNCR2820030106>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 25.Fujita S, Seto K, Ito S, Wu Y, Huang CC, Hasegawa T. The characteristics of patient safety culture in Japan, Taiwan and the United States. BMC Health Serv Res. 2013;14(13):20. doi: 10.1186/1472-6963-13-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.