

The work presented here describes a positive genetic selection strategy for isolating mutations that destabilize the outer membrane permeability barrier of E. coli. Given the importance of the outer membrane in restricting the entry of antibiotics, characterization of the genes and their products that affect outer membrane integrity will enhance the understanding of bacterial membranes and the development of strategies to bypass the outer membrane barrier for improved drug efficacy.
KEYWORDS: iron transport, lipid transport, suppressor mutations
ABSTRACT
The intake of certain nutrients, including ferric ion, is facilitated by the outer membrane-localized transporters. Due to ferric insolubility at physiological pH, Escherichia coli secretes a chelator, enterobactin, outside the cell and then transports back the enterobactin-ferric complex via an outer membrane receptor protein, FepA, whose activity is dependent on the proton motive force energy transduced by the TonB-ExbBD complex of the inner membrane. Consequently, ΔtonB mutant cells grow poorly on a medium low in iron. Prolonged incubation of ΔtonB cells on low-iron medium yields faster-growing colonies that acquired suppressor mutations in the yejM (pbgA) gene, which codes for a putative inner-to-outer membrane cardiolipin transporter. Further characterization of suppressors revealed that they display hypersusceptibility to vancomycin, a large hydrophilic antibiotic normally precluded from entering E. coli cells, and leak periplasmic proteins into the culture supernatant, indicating a compromised outer membrane permeability barrier. All phenotypes were reversed by supplying the wild-type copy of yejM on a plasmid, suggesting that yejM mutations are solely responsible for the observed phenotypes. The deletion of all known cardiolipin synthase genes (clsABC) did not produce the phenotypes similar to mutations in the yejM gene, suggesting that the absence of cardiolipin from the outer membrane per se is not responsible for increased outer membrane permeability. Elevated lysophosphatidylethanolamine levels and the synthetic growth phenotype without pldA indicated that defective lipid homeostasis in the yejM mutant compromises outer membrane lipid asymmetry and permeability barrier to allow enterobactin intake, and that YejM has additional roles other than transporting cardiolipin.
IMPORTANCE The work presented here describes a positive genetic selection strategy for isolating mutations that destabilize the outer membrane permeability barrier of E. coli. Given the importance of the outer membrane in restricting the entry of antibiotics, characterization of the genes and their products that affect outer membrane integrity will enhance the understanding of bacterial membranes and the development of strategies to bypass the outer membrane barrier for improved drug efficacy.
INTRODUCTION
The outer membrane (OM) of Gram-negative bacteria serves as a selective permeability barrier by letting small (500 Da or less) hydrophilic compounds cross the membrane but impeding the entry of large hydrophobic compounds (1, 2). Two constitutively expressed channel-forming proteins, OmpC and OmpF (also known as porins), establish a general diffusion pathway for small hydrophilic solutes to cross the OM. Unlike the OmpC and OmpF porins, LamB and Tsx form OM channels specific to maltodextrins and nucleosides, respectively (1). A different class of OM proteins (OMPs) controls the flow of solutes in an energy-dependent manner. These include BtuB and FepA, which form channels specifically for vitamin B12 (cobalamin) and enterobactin (enterochelin), respectively (3). Transport through these proteins requires an inner membrane protein complex composed of TonB, ExbB, and ExbD, which transduces energy from the proton motive force to the OM-localized transporters (3). Consequently, without the TonB-ExbBD energy complex, the OM-localized transporters are functionally inactive, i.e., the substrate can still bind to the transporter from the cell exterior but is unable to translocate through the channel without it undergoing an energy-dependent conformational transition.
Aside from the channel-forming OMPs, lipopolysaccharide (LPS), present exclusively in the outer leaflet of the OM, contributes to the OM permeability barrier in most Gram-negative bacteria (4). The cell surface-exposed regions of LPS, the O antigen and the outer core, are rich in sugars and their phosphorylated forms that prevent hydrophobic compounds from crossing the OM (4). Moreover, various components of LPS can undergo chemical modifications that alter cell surface properties and susceptibility toward cationic and hydrophobic antibiotics in response to environmental conditions (4). Phospholipids (PLs) are another major lipid component of the OM that, unlike LPS, are present primarily in the inner leaflet of the OM (5, 6). Such uneven distribution of LPS and PL makes the OM of Gram-negative bacteria an asymmetrical lipid bilayer. The three major PL species, phosphatidylethanolamine (PE), phosphatidylglycerol (PG), and cardiolipin (CL), are present at a ratio of roughly 75:20:5, respectively (7). Certain OMPs (lipoproteins) are covalently modified by PLs. Lipoproteins contribute either to the mechanical strength of the OM or serve as a component of the OM biogenesis machinery (8). Substantial progress in the last 2 decades has led to a clearer understanding of the process of OM biogenesis. The most noteworthy has been the discovery of machineries that assemble the channel-forming β-barrel OMPs (BAM) (9–11) and that transport LPS (Lpt) (12, 13) to the OM.
Mutations that affect the synthesis or assembly of the OM components can compromise the integrity of the OM permeability barrier. Given the importance of the OM in bacterial physiology and survival against antimicrobial compounds, past efforts have been aimed at devising genetic strategies to obtain mutants with increased OM permeability. One such strategy entailed positive selection for mutants that can grow on maltodextrins (oligomers of maltose of four to seven glucose units) as the sole carbon source in the absence of LamB, an OMP specific for maltodextrins (14). Accordingly, without LamB, E. coli cells are unable to grow on a minimal medium supplemented with maltodextrins as the sole carbon source and hence display a Dex− phenotype. However, Dex+ colonies appear at low frequencies, carrying suppressor mutations in either of the porin genes (ompC and ompF [15–17]), a locus that causes the expression of a novel porin (OmpG [18, 19]), or the OM-localized LPS transporter gene (lptD [20]). A common feature of these mutations is they either enlarge the porin channel size, express a novel OmpG porin with large channels, or unplug the LptD channel to accommodate maltodextrins. Inspired in part by the Dex+ porin mutants, the in vitro-constructed FepA mutants with deletions of loops that gate the channel were shown to transform FepA into a porin-like OMP (21). Interestingly, these FepA loop deletion mutants allowed enterobactin transport independent of TonB (21).
The work presented here is based on the poor growth phenotype of a tonB deletion mutant (ΔtonB) on medium without extra iron supplementation. We exploited this phenotype to isolate faster-growing revertants in an attempt to determine TonB-independent routes by which enterobactin can cross the OM. Our data show that ΔtonB revertants, harboring mutations in the yejM gene, can bypass the need for FepA by increasing OM permeability. Since limiting iron intake can be one of the strategies to curtail bacterial growth in the host, understanding the means by which bacteria can bypass the TonB-dependent iron intake pathway will be critical in achieving the goal of controlling bacterial pathogenicity.
RESULTS AND DISCUSSION
Growth defect of ΔtonB mutants on rich medium and isolation of suppressor mutations.
During an attempt to determine the significance of OmpF and OmpC porins in iron intake in the absence of the high-affinity TonB-dependent iron uptake system, we noted that a MC4100-derived ΔtonB mutant strain grew poorly on a rich medium (lysogeny broth agar [LBA]) not supplemented with additional ferric chloride (Fig. 1A, sector 2). Supplementation of the medium with 40 μM ferric chloride, however, fully reversed the growth defect (Fig. 1B, sector 2), presumably by allowing low-affinity transporters to bring the iron inside the ΔtonB cell. To test whether this phenomenon is also observed in other E. coli K-12 strain backgrounds, we transduced ΔtonB::Kmr into BW25113 and W3110 and found that the resulting ΔtonB derivatives also grew poorly, albeit not as poorly as the MC4100-derived strain, on unsupplemented LBA plates (Fig. 1A, sectors 4 and 6) but not on ferric chloride-supplemented plates (Fig. 1B). To ensure that the observed growth defect is due solely to the loss of TonB function, we transformed strains with a tonB+ plasmid and found that the plasmid fully reversed the iron-dependent growth defect in all three strain backgrounds (data not shown).
FIG 1.
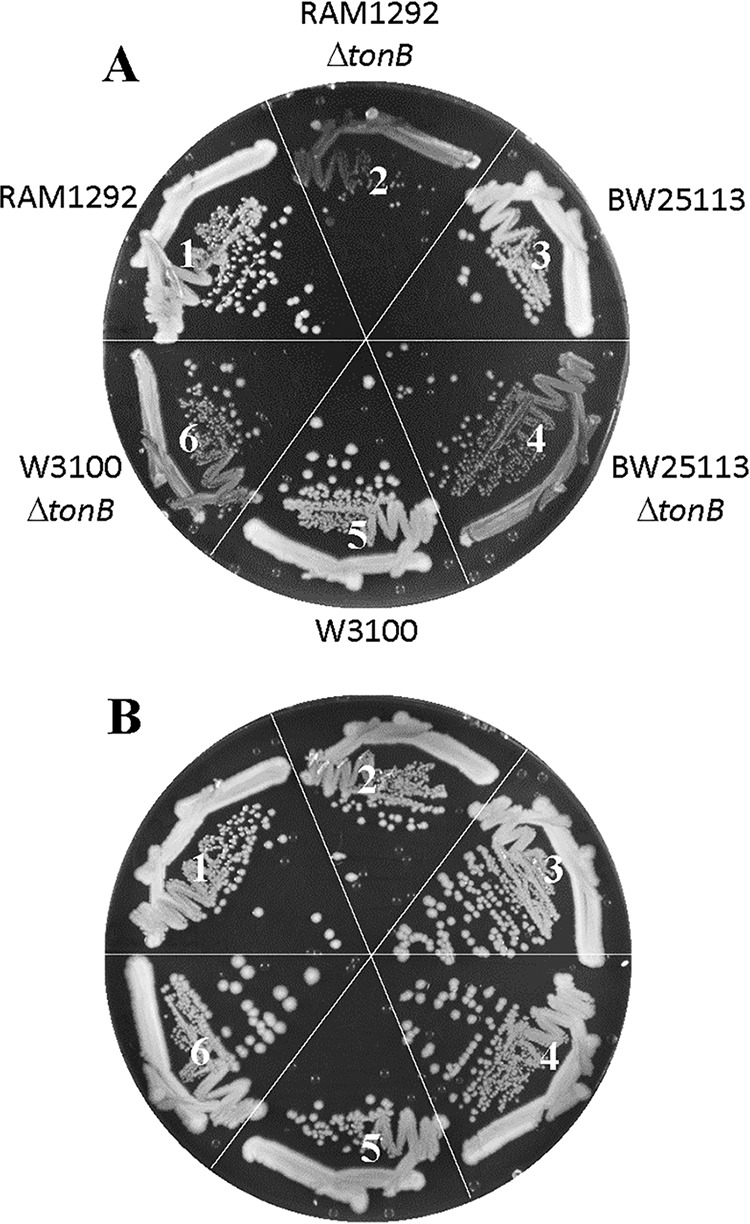
Effect of the ΔtonB mutation on bacterial growth. Bacterial growth on LBA (A) or LBA supplemented with 40 μM FeCl3 (B) was recorded after incubation of petri plates at 37°C for 24 h. The bacterial strains used are as follows: 1, RAM1292 (wild type); 2, RAM2572 (RAM1292 ΔtonB::scar); 3, BW25113 (wild type); 4, RAM2596 (BW25113 ΔtonB::scar); 5, W3100 (wild type); and 6, RAM2595 (W3100 ΔtonB::scar).
When the MC4100-based ΔtonB mutant strain was incubated for 36 to 48 h on LBA at 37°C, faster-growing colonies (revertants) emerged over the slow background growth (Fig. 2A, sector 2). These revertants were purified (Fig. 2A, sectors 3 and 4), and their growth was compared to those of tonB+ and ΔtonB strains. Revertants grew significantly better than did the ΔtonB parental strain on LBA but slightly poorer than the tonB+ strain, and they no longer produced faster-growing colonies (Fig. 2A). All strains displayed robust and comparable growth on LBA supplemented with ferric chloride (Fig. 2B).
FIG 2.
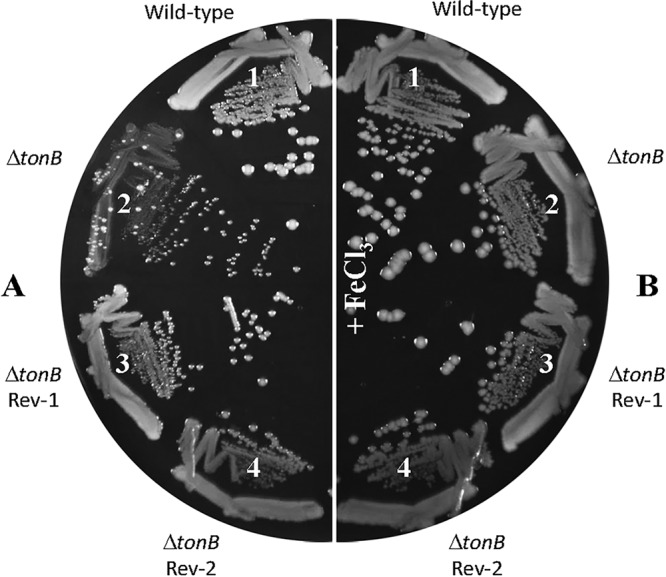
Improved growth of ΔtonB revertants. Bacterial growth on LBA (A) or LBA supplemented with 40 μM FeCl3 (B) was recorded after incubation of petri plates at 37°C for 48 h. The bacterial strains used are as follows: 1, RAM1292 (wild type); 2, RAM2505 (RAM1292 ΔtonB::Kmr); 3, RAM2485 (RAM2505 revertant 1 [Rev-1]); and 4, RAM2486 (RAM2505 revertant 2 [Rev-2]).
Characterization of the faster-growing revertants.
We tested whether the suppressor mutations, which allow the revertants to grow faster on LBA without TonB, can also bypass the need for other components of the high-affinity iron transport pathway. For this, we deleted fepA, fepB, or entF from the ΔtonB and ΔtonB plus suppressor backgrounds. FepA is the OM receptor for the enterobactin-ferric complex, FepB is the periplasmic enterobactin-ferric binding protein, and EntF is involved in enterobactin synthesis in the cytoplasm (see reference 22 for a review on bacterial iron homeostasis). The kanamycin resistance (Kmr) marker, which replaced the deletion alleles of the three aforementioned genes, was transduced into the parent (ΔtonB::scar) and mutant (ΔtonB::scar plus suppressor) backgrounds, and the growth of the resulting strains was recorded after incubation at 37°C for 24 h on LBA and LBA supplemented with ferric chloride (Fig. 3). The absence of FepA from the parent and revertant backgrounds had no effect on growth on LBA (Fig. 3A, compare sectors 1 and 2 to 3 and 4), indicating that both the lingering growth of the ΔtonB mutant strain and the faster growth of the revertant are independent of the OM receptor protein. Thus, the suppressor mutation appears to bypass TonB and FepA in improving growth on the unsupplemented LBA medium. It is worth noting that TonB-independent FepA mutants have been previously shown to allow nonspecific diffusion of enterobactin-ferric and other unrelated chemicals, including SDS (21). However, our genetic data eliminate fepA as the site of the suppressor mutation.
FIG 3.
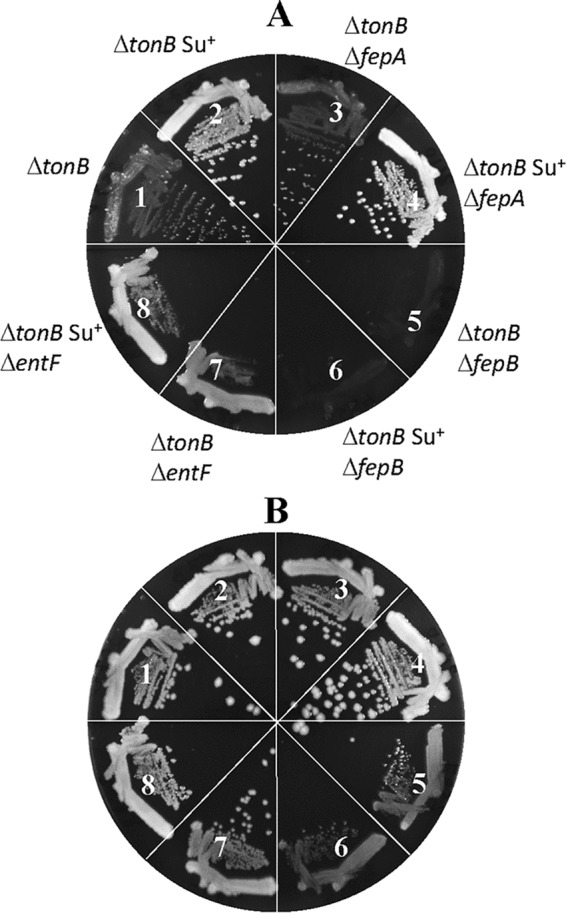
Effects of mutations disabling enterobactin synthesis or transport on ΔtonB suppressor mutations. Bacterial growth on LBA (A) or LBA supplemented with 40 μM FeCl3 (B) was recorded after incubation of petri plates at 37°C for 36 h. The bacterial strains used are as follows: 1, RAM2572 (ΔtonB::scar); 2, RAM2573 (RAM2572 revertant 1, suppressor+); 3, RAM2608 (RAM2572 ΔfepA::Kmr); 4, RAM2610 (RAM2573 ΔfepA::Kmr); 5, RAM2587 (RAM2572 ΔfepB::Kmr); 6, RAM2589 (RAM2573 ΔfepB::Kmr); 7, RAM2586 (RAM2572 ΔentF::Kmr); and 8, RAM2588 (RAM2573 ΔentF::Kmr).
Without FepB, growth of the ΔtonB and ΔtonB plus suppressor mutant strains was severely compromised on LBA (Fig. 3A, sectors 5 and 6). Their growth improved somewhat on medium supplemented with ferric chloride (Fig. 3B, sectors 5 and 6), albeit to a much lower degree than when only TonB was absent (Fig. 3A and B, sector 1). A severe growth defect of the ΔtonB ΔfepB mutant strain, regardless of the suppressor mutation, reflected the dependence of the suppressor mutation on FepB for its faster-growing phenotype. Moreover, the resumption of modest growth upon the addition of ferric chloride indicated the existence of a TonB/FepB-independent iron transport pathway, which is presumably overwhelmed by the excessive accumulation of secreted iron-chelating enterobactin outside the cell in the absence of TonB and FepB. The deletion of entF, which halted enterobactin production, caused an interesting growth pattern, that of robust and confluent growth but with no single colonies on LBA (Fig. 3A, sectors 7 and 8). The robust confluence is presumably due to the absence of iron sequestration by enterobactin, thus allowing enterobactin-independent pathways to transport iron. The lack of single-colony formation is presumably associated with the recently reported anti-reactive oxygen species (anti-ROS) activity of enterobactin (23). Indeed, single-colony formation of the ΔtonB ΔentF mutant was rescued when LBA plates were incubated microaerobically or when supplemented with ascorbic acid or catalase (Fig. 4). Due to the production of copious amounts of enterobactin in the ΔtonB ΔfepB mutant strain, incubation under microaerobic conditions did not appreciably improve the growth of this strain (Fig. 4D). The identical growth behaviors of the ΔtonB ΔentF and ΔtonB ΔentF plus suppressor mutant strains in Fig. 3 indicated that the suppressor phenotype is not independent of enterobactin. Together, these data show that the suppressor mutation can bypass the OM-localized enterobactin receptor for its enhanced growth phenotype on LBA.
FIG 4.

Effects of low oxygen, ascorbic acid, or catalase on bacterial growth. (A to C) Bacterial growth on LBA (A) and LBA supplemented with 1 mM ascorbic acid (B) or catalase (C) was recorded after incubation of petri plates under aerobic conditions at 37°C. (D) Growth on LBA was also recorded after incubation of the plate under the microaerobic condition at 37°C. One hundred microliters of a 150-U/mg catalase solution was spread on an LBA plate. Plates were incubated for either 24 h (A and B) or 36 h (C and D). Two colonies of each strain were tested. The bacterial strains used are RAM2572 (ΔtonB::scar), RAM1292 (wild type), RAM2587 (RAM2572 ΔfepB::Kmr), and RAM2586 (RAM2572 ΔentF::Kmr).
Mapping and identification of the suppressor mutations.
The genetic location of the suppressor mutations was found serendipitously while testing whether they map within the porin genes. P1-mediated transduction of ΔompC::Cmr (Cmr, chloramphenicol resistance) reversed the suppressor phenotype nearly 50% of the time, indicating that the suppressor mutations do not map in ompC but are rather genetically linked to it. Subsequent P1 transductional crosses involving additional linked markers, including napA::Kmr, confirmed the location of suppressor mutations to be at the 49-minute region of the chromosome. The precise location of the suppressor mutations was ascertained by whole-genome sequence analysis. The final confirmation of the mutation responsible for the phenotype came from Sanger sequencing of the PCR-amplified DNA fragment and reestablishment of the faster-growth phenotype on LBA when the mutation was moved by P1 transduction into a fresh ΔtonB background.
All three suppressor mutations were located within the yejM gene, which codes for a 586-residue-long protein. In Salmonella enterica subsp. enterica serovar Typhimurium (24) and Shigella flexneri (25), YejM (called PbgA in these bacteria) is reported to transport CL from the inner membrane (IM) to the OM. YejM is predicted to fold into the following two domains: the IM-localized transmembrane (TM) domain, composed of the first 190 residues, and a periplasmically exposed globular domain derived from the last 326 residues. A 70-residue linker region connects the two domains. Recently, high-resolution structures of the PbgA/YejM globular domain were solved (26). The TM domain of PbgA/YejM is shown to be essential, while a complete deletion of the globular domain produces pleiotropic phenotypes, including high-temperature-sensitive growth defects, increased sensitivity toward hydrophobic antibiotics, leakage of the periplasmic enzymes, and an apparent reduction in lipid A level (26–28).
All three yejM mutations isolated in this study caused a frameshift in the region of YejM corresponding to the periplasmic globular domain (Fig. 5) after nucleotide (nt) 1163, 1214, or 1624 of the gene, respectively, and hence were given allele names of yejM1163, yejM1214, and yejM1624. The yejM1163 allele, which frameshifted the open reading frame after codon 388, introduced a premature stop codon after the nonnative codon 390. In the subsequent analysis, we primarily employed yejM1163 due to its robust phenotypes. A complementation test using the wild-type copy of yejM cloned into pBAD24 showed full reversal of the faster-growth phenotype of yejM1163 (Fig. 6), indicating that yejM1163 is a loss-of-function recessive mutation solely responsible for the improved growth phenotype of the ΔtonB mutant strain.
FIG 5.

A cartoon showing wild-type and mutant YejM proteins. The majority of the wild-type YejM protein is composed of a transmembrane domain (residues 20 to 190) and a globular periplasmic domain (residues 260 to 586). The presence of the frameshift mutation in the three yejM suppressor alleles (yejM1163, yejM1214, and yejM1624) causes frameshift after nt 1163, 1214, and 1624, corresponding to residues Q388, I404, and K542, respectively, of the YejM protein, respectively. The stop codon appears after the nonnative residues R390, W444, and Y546 (shown in parentheses).
FIG 6.
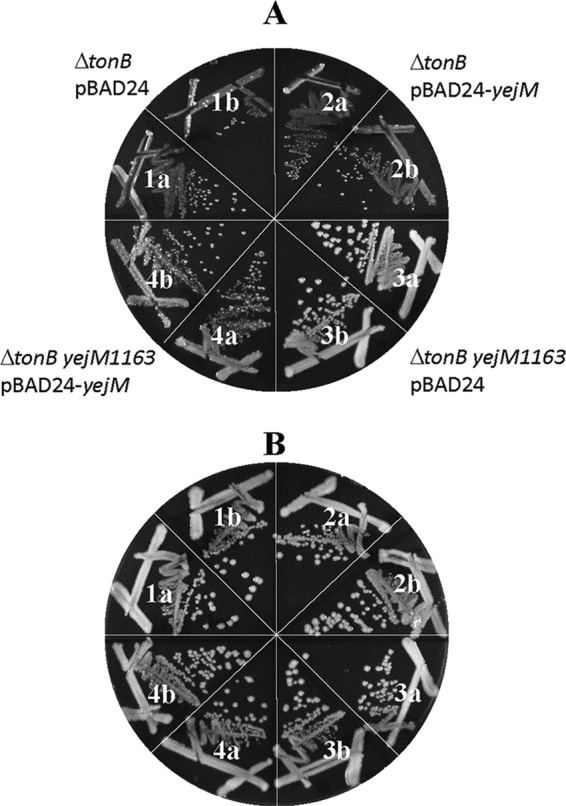
Genetic complementation of yejM1163. The ΔtonB and ΔtonB yejM1163 mutant strains containing an empty vector plasmid (pBAD24) or a yejM clone (pBAD24-yejM) were grown on LBA plus ampicillin (A) or LBA plus ampicillin and ferric chloride (B). Growth was recorded after incubation at 37°C for 24 h. The bacterial strains used are as follows: 1a and b, RAM3144 [ΔtonB::scar(pBAD24)]; 2a and b, RAM3145 [ΔtonB::scar(pBAD24-yejM)]; 3a and b, RAM3146 [ΔtonB::scar yejM1163(pBAD24)]; and 4a and b, RAM3147 [(ΔtonB::scar yejM1163(pBAD24-yejM)].
How does yejM1163 overcome the growth defect caused by the ΔtonB mutation?
Based on our observation that yejM1163 bypasses the OM receptor requirement (Fig. 3) and the prior knowledge that a truncation of YejM causes defects associated with the OM (26–28), we surmise that yejM1163 destabilizes the OM permeability barrier to allow enterobactin-Fe3+ to cross the OM. This hypothesis was tested by two independent methods. First, we determined the MIC of a large (1,450 Da) hydrophilic antibiotic, vancomycin, which cannot readily cross the OM of Gram-negative bacteria. However, a compromised OM permeability barrier may allow vancomycin to enter and kill the cell by preventing peptidoglycan cross-linking. MICs were determined by the 2-fold serial dilution method, and the MIC was recorded as the lowest concentration at which the antibiotic prevented bacterial growth (optical density at 600 nm [OD600] <0.1). The presence of yejM1163 in both the tonB+ and ΔtonB backgrounds lowered the vancomycin MIC by 4-fold, indicating a significant breach in the OM permeability barrier (Table 1). The presence of a plasmid expressing wild-type yejM reversed vancomycin susceptibility (Table 1), showing that, like the elevated growth phenotype, the vancomycin susceptibility phenotype stems from the loss-of-function nature of the yejM1163 allele.
TABLE 1.
MICs of vancomycin in various genetic backgrounds
| Relevant genotype | Vancomycin MIC (μg/ml) |
|---|---|
| Wild type (tonB+) | 160 |
| yejM1163 | 40 |
| ΔtonB | 80 |
| ΔtonB yejM1163 | 20 |
| yejM1163(pBAD24) | 40 |
| yejM1163(pBAD24-yejM) | 120 |
| ΔtonB yejM1163(pBAD24) | 20 |
| ΔtonB yejM1163(pBAD24-yejM) | 80 |
| ΔtolA | 10–20 |
| ΔtolA ΔtonB | 5–10 |
| ΔclsABC | 120 |
| ΔclsABC yejM1163 | 30 |
| ΔtonB ΔclsABC | 60 |
| ΔtonB ΔclsABC yejM1163 | <20 |
We then asked whether another mutation, unrelated to yejM, known to breach the OM permeability barrier would also reverse the poor-growth phenotype of the ΔtonB mutant on LBA. To test this, we employed a null allele of tolA. TolA forms a multiprotein transenvelope Tol-Pal complex (29). The absence of any one member of the Tol-Pal complex compromises OM permeability, resulting in hypersusceptibility to various antibiotics and leakage of periplasmic proteins (30, 31). Indeed, the absence of TolA lowered the vancomycin MIC by 8- to 16-fold (Table 1). Moreover, the ΔtolA mutation improved the growth of the ΔtonB mutant strain on LBA, and no faster-growing revertants appeared (Fig. 7, compare sectors 3 and 6). It should be noted that since the absence of TolA causes pleiotropic membrane defects, growth of the ΔtolA cells is somewhat compromised (Fig. 7, compare sectors 1 and 5).
FIG 7.
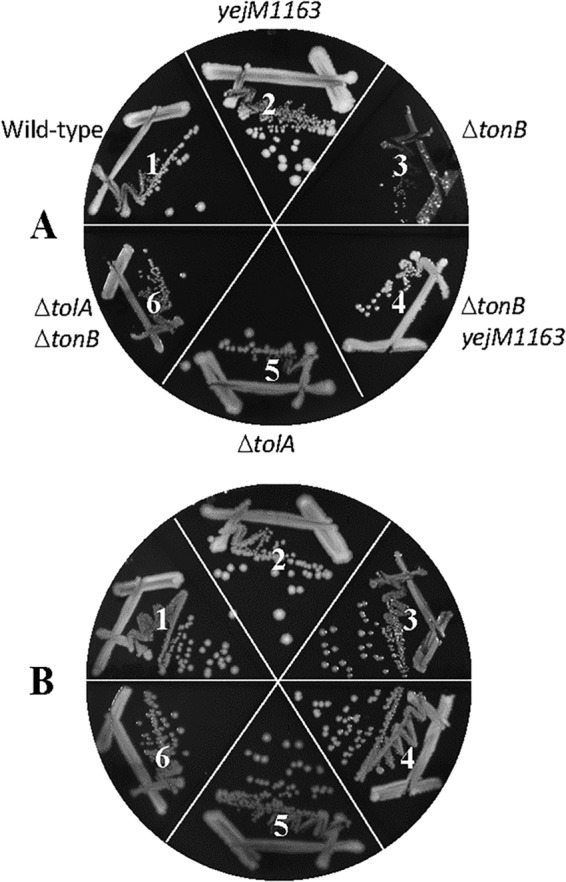
Effect of the ΔtolA mutation on ΔtonB mutant growth. Bacterial growth on LBA (A) or LBA supplemented with 40 μM FeCl3 (B) was recorded after incubation of petri plates at 37°C for 36 h. The bacterial strains used are as follows: 1, RAM3148 (wild type, ΔnapA::Kmr); 2, RAM3149 (ΔnapA::Kmr yejM1163); 3, RAM3150 (ΔnapA::Kmr ΔtonB::scar); 4, RAM3151 (ΔnapA::Kmr ΔtonB::scar yejM1163); 5, RAM2815 (ΔtolA::Cmr); and 6, RAM3152 (ΔtonB::scar ΔtolA::Cmr).
The second method we employed to assess the yejM1163-mediated OM permeability defect involved direct examination of leaked cellular proteins in the culture supernatant. The cell-free culture supernatant was concentrated 10-fold and analyzed by SDS-PAGE, and proteins were visualized after staining with Coomassie brilliant blue (Fig. 8). As expected, no protein bands were detected from the wild-type sample. However, the presence of yejM1163 in both wild-type and ΔtonB backgrounds caused a significant leakage of proteins in the culture supernatant. Similarly, the ΔtolA mutant displayed a dramatic leaky phenotype (Fig. 8). To rule out the possibility that cell lysis caused the release of cellular proteins in the culture supernatant, we assayed for the presence β-galactosidase, a cytoplasmic enzyme, from the cell-free culture supernatant of the wild-type and yejM1163 mutant strains carrying rpoHP3::lacZ. About 1%, or 3 Miller units, of the total whole-cell β-galactosidase activity (360 Miller units) was present in the supernatant of both strains, showing the absence of any significant and yejM1163-specific cell lysis. Together, these data support the hypothesis that yejM1163-mediated perturbation in the OM permeability barrier causes the entry of the enterobactin-ferric complex into the periplasm independent of TonB and the OM receptor FepA.
FIG 8.
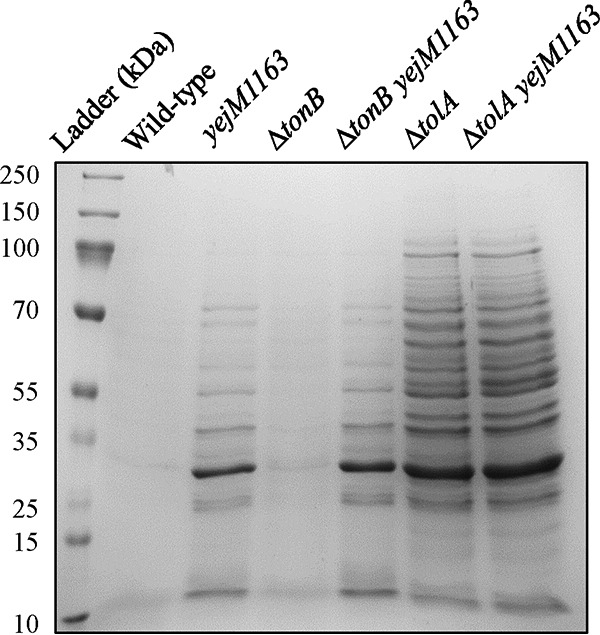
SDS-PAGE analysis of the cell-free culture supernatants. Overnight-grown cultures were spun down, and the supernatants, after filtration and concentration, were analyzed by SDS-PAGE. The proteins were visualized after staining with Coomassie brilliant blue. The bacterial strains used are the same ones described in the Fig. 7 legend.
yejM1163 causes an OM permeability defect independent of CL.
Recent studies have implicated PbgA, a homolog of YejM in Salmonella and Shigella spp., in CL binding and transport from the IM to OM (24–26). Therefore, it is conceivable that the OM permeability defect of yejM1163 stems from a defect in CL transport to the OM. If so, a defect in CL synthesis may also result in increased OM permeability and promote growth of the ΔtonB mutant strain on LBA. Moreover, if the phenotypes of yejM1163 stem solely from its defect in CL transport to the OM, these phenotypes should be concealed in a background deficient in CL synthesis due to epistasis. To test these possibilities, we deleted all three CL synthase genes, clsABC (32), from the wild-type, yejM1163, ΔtonB, and yejM1163 ΔtonB backgrounds and determined vancomycin MIC and growth on unsupplemented LBA. The deletion of clsABC in wild-type and ΔtonB backgrounds caused a slight reduction (25%) in vancomycin MIC (Table 1), indicating that CL is not a significant contributor to the OM permeability barrier. Consistent with these data, the absence of CL from the ΔtonB background did not improve growth on LBA (Fig. 9, sectors 4 to 7). More importantly, yejM1163 continued to display its phenotypes, both lower vancomycin MICs (Table 1) and improved growth of the ΔtonB mutant strain on LBA (Fig. 9, sector 8), even in the absence of CL. Therefore, it is unlikely that a potential CL transport defect of yejM1163 is the sole cause of the OM permeability defect and the improved growth phenotype of the ΔtonB mutant strain. This suggests that YejM may have a role independent of CL transport, which is responsible for the permeability phenotype.
FIG 9.
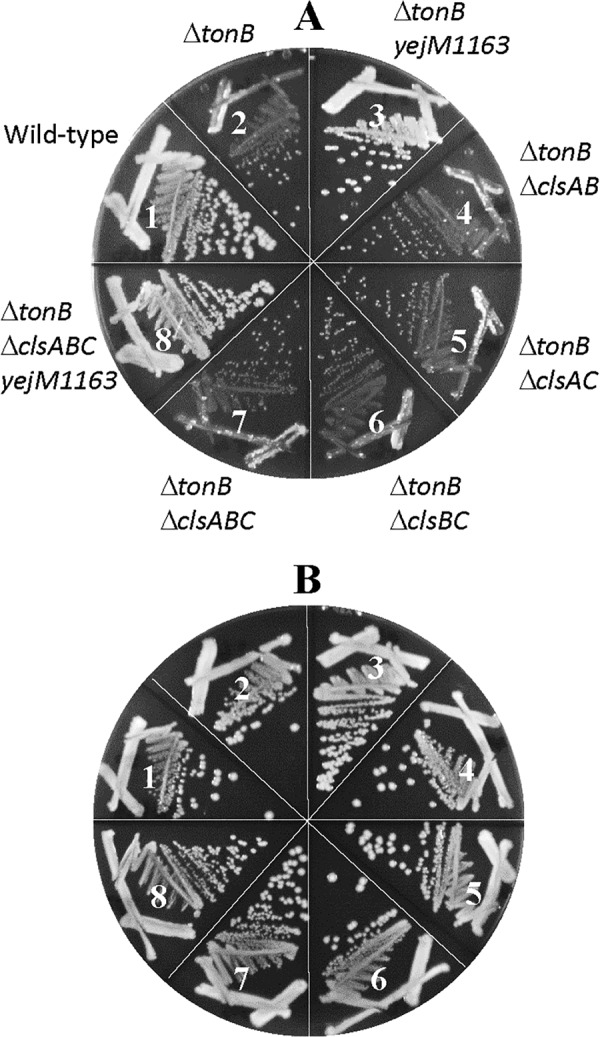
Effect of cardiolipin on a ΔtonB mutant growth. Bacterial growth on LBA (A) or LBA supplemented with 40 μM FeCl3 (B) was recorded after incubation of petri plates at 37°C for 36 h. The bacterial strains used are as follows: 1, RAM3148 (wild type, ΔnapA::Kmr); 2, RAM3150 (ΔnapA::Kmr ΔtonB::scar); 3, RAM3151 (ΔnapA::Kmr ΔtonB::scar yejM1163); 4, RAM3153 (ΔtonB::scar ΔclsAB); 5, RAM3154 (ΔtonB::scar ΔclsAC); 6, RAM3155 (ΔtonB::scar ΔclsBC); 7, RAM3156 (ΔtonB::scar ΔclsABC); and 8, RAM3157 (ΔtonB::scar ΔclsABC napA::Kmr yejM1163).
What causes OM permeability increase in the yejM1163 mutant?
The data presented above show that the lack of CL per se does not significantly increase OM permeability. Moreover, the persistence of the yejM1163 phenotypes in the ΔclsABC background eliminates the possibility that the accumulation of CL in the IM of the yejM1163 mutant is somehow responsible for the OM permeability defect. Finally, given the recessive nature of yejM1163, it is unlikely that the truncated YejM1163 polypeptide disrupts membrane structure to influence the permeability barrier. It is worth mentioning that the increased OM permeability is not unique to the yejM1163 mutant, as all three yejM mutant alleles isolated in this study lower vancomycin MIC (data not shown). Similarly, previously characterized yejM truncations and the original missense mutation, resulting in a G205A substitution, also displayed elevated OM permeability phenotypes (27, 28). Therefore, the increased OM permeability phenotypes are commonly associated with the loss of function of the nonessential periplasmic domain of YejM.
To gain further insights into the effects of yejM1163 in the lipid composition, we extracted lipids from whole cells and analyzed them by thin-layer chromatography. No significant differences were apparent in the three major PL species, PE, PG, and CL, between the wild-type and yejM1163 strains (Fig. 10). In the ΔclsABC and yejM1163 ΔclsABC backgrounds, where CL was absent, the levels of its direct precursor, PG, went up significantly (Fig. 10). To reveal finer differences in PL composition at the subspecies level, we subsequently analyzed the lipids by liquid chromatography-tandem mass spectrometry (LC-MS/MS). The data provided a few interesting clues (Table 2). In the yejM1163 mutant, unlike phosphatidic acid (PA), the precursor of most lipids, the overall levels of PE, PG, and CL were slightly down relative to that of the wild type. In contrast, the level of the lyso-form of PE (LPE), lacking one of the acyl chains, went up 3- to 4-fold. The two most common LPE species were 16:1 and 18:1, whose increase correlated with a significant decrease in the PE 16:1_18:1 species (Table 2). In the ΔclsABC mutant, CL was expectedly absent, and this accompanied an almost-2-fold increase in the PG level and a modest 25% increase in the PE level (Table 2). These increases are consistent with the fact that both PG and PE are CL precursors (32). In the yejM1163 ΔclsABC mutant, there was a dramatic increase in the LPE level, and this increase was much greater than that observed in the mutant with the yejM1163 mutation alone (Table 2). The fact that the absence of CL synthases by themselves only slightly increases the LPE level indicates that yejM1163 is the main driving force behind such an overproduction of LPE. Nevertheless, the synergistic nature of the yejM1163 and ΔclsABC mutants concerning increased LPE levels indicates that an imbalance in lipid synthesis and homeostasis further contribute to LPE accumulation in the yejM1163 mutant.
FIG 10.
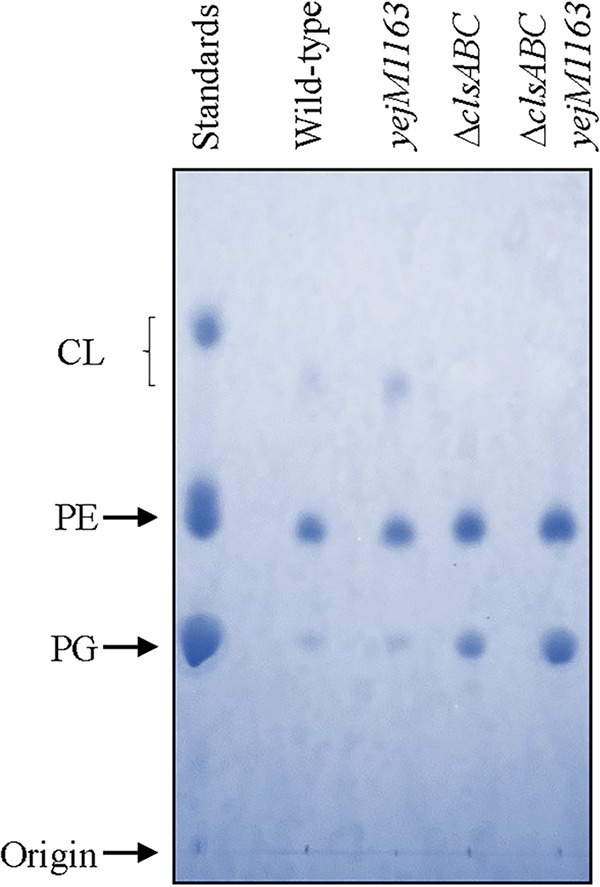
Analysis of phospholipids by thin-layer chromatography. After chromatography, phospholipid spots were visualized by spraying the dried plate with molybdenum blue reagent. The standards contain a mixture of PE, PG, and CL. The strains used are RAM3148 (wild type, ΔnapA::Kmr), RAM3149 (ΔnapA::Kmr yejM1163), RAM3158 (ΔclsABC ΔnapA::Kmr), and RAM3159 (ΔclsABC ΔnapA::Kmr yejM1163).
TABLE 2.
Total phospholipid analysis by liquid chromatography-tandem mass spectroscopya
| PL type (ionization mode) | Mol wt | PL content (ng) ± SD |
Change relative to WTb |
|||||
|---|---|---|---|---|---|---|---|---|
| Wild type (1) | yejM1163 mutant (2) | ΔclsABC mutant (3) | yejM1163 ΔclsABC mutant (4) | 2/1 | 3/1 | 4/1 | ||
| PA 43:2 | 798.61 | 52.15 ± 5.97 | 50.61 ± 4.15 | 49.27 ± 2.77 | 48.13 ± 2.96 | 0.97 | 0.94 | 0.92 |
| PE 18:1/18:1 (+) | 743.55 | 21.64 ± 11.73 | 12.30 ± 4.24 | 26.14 ± 6.84 | 8.19 ± 2.72 | 0.57 | 1.21 | 0.38 |
| PE 16:0_18:1 and PE 16:1_18:0 (+) | 717.53 | 29.15 ± 15.87 | 22.68 ± 7.84 | 30.73 ± 4.84 | 17.98 ± 7.03 | 0.78 | 1.05 | 0.62 |
| PE 16:1_18:1 (+) | 715.51 | 54.14 ± 31.44 | 35.27 ± 14.51 | 64.23 ± 7.46 | 41.42 ± 13.99 | 0.65 | 1.19 | 0.77 |
| PE 16:0_17:1 (+) | 703.51 | 6.37 ± 2.90 | 4.92 ± 1.89 | 11.75 ± 3.17 | 4.87 ± 1.74 | 0.77 | 1.84 | 0.76 |
| PE 16:0_16:1 (+) | 689.50 | 74.70 ± 37.24 | 69.77 ± 28.09 | 90.08 ± 19.75 | 74.12 ± 32.08 | 0.93 | 1.21 | 0.99 |
| PE 16:1/16:1 (+) | 687.48 | 8.60 ± 5.26 | 6.29 ± 3.58 | 19.24 ± 7.35 | 9.23 ± 3.52 | 0.73 | 2.24 | 1.07 |
| PE (+) total | 194.59 | 151.23 | 242.20 | 155.81 | 0.78 | 1.24 | 0.80 | |
| LPE 16:0 (+) | 453.28 | 0.10 ± 0.06 | 0.77 ± 0.46 | 0.11 ± 0.02 | 2.71 ± 0.57 | 7.80 | 1.08 | 27.57 |
| LPE 16:1 (+) | 451.27 | 0.44 ± 0.10 | 0.55 ± 0.36 | 0.28 ± 0.09 | 2.59 ± 0.56 | 1.27 | 0.65 | 5.91 |
| LPE 16:1 (+) | 451.27 | 0.09 ± 0.05 | 0.90 ± 0.50 | 0.15 ± 0.04 | 2.02 ± 0.30 | 9.97 | 1.68 | 22.45 |
| LPE (+) total | 0.63 | 2.22 | 0.54 | 7.30 | 3.54 | 0.86 | 11.68 | |
| PE 18:1/18:1 (−) | 743.55 | 32.53 ± 12.24 | 24.99 ± 2.74 | 39.02 ± 9.06 | 19.07 ± 6.15 | 0.77 | 1.20 | 0.59 |
| PE 16:1_18:0 and PE 16:0_18:1 (−) | 717.53 | 14.84 ± 8.32 | 18.82 ± 0.15 | 16.82 ± 5.90 | 15.39 ± 2.98 | 1.27 | 1.13 | 1.04 |
| PE 16:0_18:1 (−) | 717.53 | 48.14 ± 17.37 | 50.09 ± 6.49 | 51.15 ± 7.21 | 43.12 ± 10.04 | 1.04 | 1.06 | 0.90 |
| PE 16:1_18:1 (−) | 715.52 | 51.53 ± 18.92 | 41.38 ± 2.58 | 67.35 ± 13.71 | 45.53 ± 8.96 | 0.81 | 1.31 | 0.88 |
| PE 15:0_18:1 (−) | 703.52 | 9.81 ± 4.14 | 11.85 ± 1.23 | 10.06 ± 3.06 | 6.83 ± 2.47 | 1.21 | 1.03 | 0.70 |
| PE 16:0_17:1 (−) | 703.52 | 14.05 ± 5.28 | 14.85 ± 0.96 | 20.53 ± 5.19 | 13.80 ± 4.59 | 1.06 | 1.46 | 0.98 |
| PE 16:1/16:1 (−) | 687.48 | 17.04 ± 8.10 | 15.57 ± 3.38 | 39.23 ± 17.36 | 25.11 ± 8.35 | 0.91 | 2.30 | 1.47 |
| PE 14:0_16:0 (−) | 663.48 | 7.37 ± 3.32 | 8.61 ± 1.93 | 6.69 ± 1.59 | 5.95 ± 2.09 | 1.11 | 0.86 | 0.77 |
| PE 14:0_16:1 (−) | 661.47 | 11.60 ± 6.56 | 11.27 ± 0.57 | 14.73 ± 5.32 | 17.40 ± 6.77 | 0.97 | 1.27 | 1.50 |
| PE (−) total | 207.23 | 197.87 | 265.58 | 192.19 | 0.95 | 1.28 | 0.93 | |
| LPE 18:1 (−) | 479.30 | 0.43 ± 0.18 | 1.54 ± 0.08 | 0.49 ± 0.19 | 6.62 ± 4.27 | 3.55 | 1.12 | 15.28 |
| LPE 16:0 (−) | 453.29 | 0.30 ± 0.04 | 1.38 ± 0.16 | 0.32 ± 0.09 | 6.67 ± 1.71 | 4.55 | 1.05 | 21.99 |
| LPE 16:1 (−) | 451.27 | 0.46 ± 0.12 | 2.00 ± 0.59 | 0.63 ± 0.20 | 6.80 ± 2.77 | 4.35 | 1.36 | 14.78 |
| LPE 16:1 (−) | 451.27 | 0.39 ± 0.12 | 1.49 ± 0.44 | 0.59 ± 0.20 | 6.45 ± 2.81 | 3.78 | 1.50 | 16.39 |
| LPE (−) total | 1.59 | 6.41 | 2.02 | 26.53 | 4.03 | 1.27 | 16.69 | |
| PG 18:1/18:1 (−) | 774.54 | 16.10 ± 5.93 | 10.73 ± 1.44 | 28.00 ± 5.09 | 16.14 ± 5.56 | 0.67 | 1.74 | 1.00 |
| PG 16:0_18:1 (−) | 748.53 | 18.24 ± 7.41 | 17.30 ± 1.60 | 30.33 ± 3.09 | 28.45 ± 6.56 | 0.95 | 1.66 | 1.56 |
| PG 16:1_18:1 (−) | 746.51 | 13.07 ± 7.55 | 8.39 ± 2.39 | 28.71 ± 9.55 | 27.51 ± 12.28 | 0.64 | 2.20 | 2.11 |
| PG 16:0_16:1 (−) | 720.50 | 14.46 ± 5.56 | 16.95 ± 0.68 | 27.63 ± 5.81 | 33.43 ± 8.04 | 1.17 | 1.91 | 2.31 |
| PG 16:1/16:1 (−) | 718.48 | 2.42 ± 1.32 | 2.21 ± 0.39 | 8.33 ± 1.26 | 8.70 ± 2.14 | 0.91 | 3.44 | 3.59 |
| PG 14:0_16:1 (−) | 692.46 | 1.20 ± 0.48 | 1.42 ± 0.41 | 3.76 ± 1.12 | 5.15 ± 1.25 | 1.19 | 3.14 | 4.30 |
| PG (−) total | 61.86 | 53.37 | 114.67 | 105.54 | 0.87 | 1.94 | 1.82 | |
| CL 70:3 (−) | 1,431.02 | 0.14 ± 0.12 | 0.08 ± 0.03 | 0.00 | 0.00 | 0.53 | 0.00 | 0.00 |
| CL 70:4 (−) | 1,429.00 | 0.22 ± 0.18 | 0.10 ± 0.03 | 0.00 | 0.00 | 0.44 | 0.00 | 0.00 |
| CL 68:4 (−) | 1,400.97 | 0.24 ± 0.18 | 0.12 ± 0.04 | 0.00 | 0.00 | 0.52 | 0.00 | 0.00 |
| CL 64:3 (−) | 1,346.92 | 0.16 ± 0.12 | 0.13 ± 0.04 | 0.00 | 0.00 | 0.82 | 0.00 | 0.00 |
| CL 66:2 (−) | 1,376.97 | 0.27 ± 0.25 | 0.23 ± 0.06 | 0.00 | 0.00 | 0.85 | 0.00 | 0.00 |
| CL 64:2 (−) | 1,348.94 | 0.31 ± 0.26 | 0.29 ± 0.08 | 0.00 | 0.00 | 0.94 | 0.00 | 0.00 |
| CL 68:3 (−) | 1,402.99 | 0.54 ± 0.45 | 0.36 ± 0.13 | 0.00 | 0.00 | 0.66 | 0.00 | 0.00 |
| CL 66:3 (−) | 1,374.96 | 0.55 ± 0.46 | 0.42 ± 0.14 | 0.00 | 0.00 | 0.76 | 0.00 | 0.00 |
| CL (−) total | 2.42 | 1.71 | 0.00 | 0.00 | 0.71 | 0.00 | 0.00 | |
PL quantities were determined by dividing PL peak areas with the peak area of the closest standard and then multiplying by the mass of the injected standard in nanograms. SDs were determined from three independent biological samples. PL species shown represent 94% (PE and PG) to 100% (LPE and CL) of all subtypes detected.
Column heading numbers (e.g., “2/1”) refer to column numbers under “PL content.”
LPE, which is present only in trace amounts in E. coli, is a metabolic intermediate of enzymatic actions resulting from the acylation of lipid A or lipoproteins or from the degradative action of activated phospholipases (33). Owing to its detergent-like structure, LPE is deemed not suitable in the lipid bilayer structure and hence is rapidly extracted and converted to PE through lipid homeostasis processes that are not fully understood (33). While not a stable part of the E. coli membranes, LPE is intrinsic to several bacterial membranes where it plays an important role in physiology and pathogenesis (34). Therefore, increased LPE levels in the yejM1163 and yejM1163 ΔclsABC mutants of E. coli are certainly intriguing and reflect aberrant lipid homeostasis.
Effects of PldA and PagP on the yejM1163-mediated suppression.
Elevated LPE levels in the yejM1163 background could be caused by the action of two OM enzymes, PldA (35) and PagP (36), which generate LPE either by PE hydrolysis (PldA) or during the transfer of a palmitate chain from PE to hexa-acylated LPS (PagP). These enzymes are normally dormant but are activated in response to the perturbation of the OM structure (e.g., lipid asymmetry) due to the mislocalization of PL to the outer leaflet of the OM (37–39). It is conceivable that aberrant lipid homeostasis in the yejM mutant perturbs OM lipid asymmetry, leading to the activation of PldA/PagP. If so, null mutations in the pldA or pagP gene could further interfere with lipid homeostasis by not removing mislocalized PLs from the OM outer leaflet. The absence of pldA in the ΔtonB background produced no growth phenotype (Fig. 11). However, deletion of pldA from the yejM1163 ΔtonB background produced a significant growth defect, regardless of the presence of supplemented iron in the medium (Fig. 11, compare panels A and B), showing that the synthetic phenotype of the ΔpldA mutant is linked to yejM1163 mutation and not ΔtonB mutant-mediated iron deficiency. In contrast to the pldA mutation, the absence of pagP had no negative effect on growth in either genetic background (Fig. 11). These data support the hypothesis that a yejM1163-mediated defect in lipid homeostasis disturbs OM lipid asymmetry, increasing OM permeability and activating PldA which attempts to normalize OM lipid asymmetry.
FIG 11.
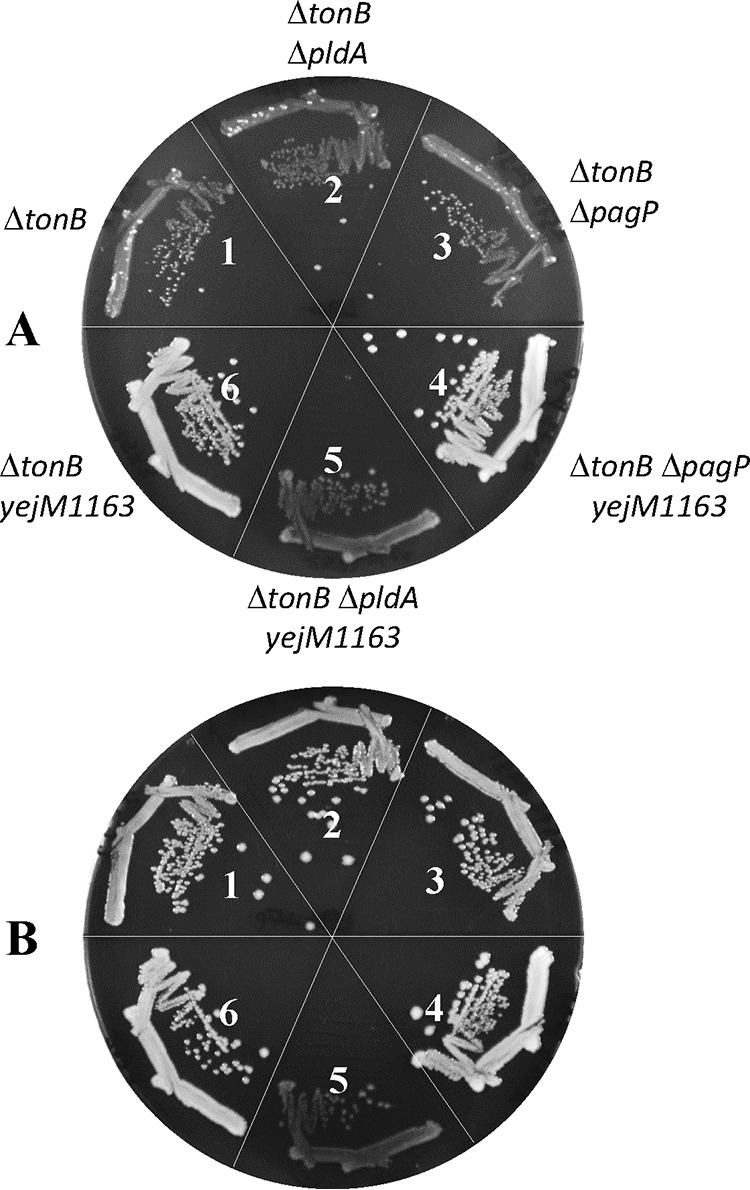
Growth effects of the ΔpldA and ΔpagP mutations in ΔtonB and ΔtonB yejM1163 backgrounds. Bacterial growth on LBA (A) or LBA supplemented with 40 μM FeCl3 (B) was recorded after incubation of petri plates at 37°C for 24 h. The bacterial strains used are as follows: 1, RAM2572 (ΔtonB::scar); 2, RAM3160 (ΔtonB::scar pldA::Kmr); 3, RAM3161 (ΔtonB::scar ΔpagP::Kmr); 4, RAM2562 (ΔtonB::scar yejM1163); 5, RAM3162 (ΔtonB::scar yejM1163 ΔpldA::Kmr); and 6, RAM3163 (ΔtonB::scar yejM1163 ΔpagP::Kmr).
A previous analysis with a mutant yejM allele, with much stronger phenotypes than those displayed by the three yejM alleles described here, reported reduced lipid A (LPS) levels based on lower LPS-to-PL ratios in the mutant strain (27). However, this or the work presented here cannot discern which of the two lipid molecules are directly impacted by the yejM mutation. Moreover, YejM/PbgA is reported to transport CL from the IM to the OM (24, 25). The fact that CL in E. coli is not essential, but YejM is, at least its N-terminal TM domain, suggests that YejM, in addition to CL transport, plays a role in lipid homeostasis that makes it essential for cell viability. Mutations that disrupt the periplasmic domain of YejM to some degree also interfere with lipid homeostasis, thus disrupting OM lipid asymmetry and the OM permeability barrier to allow molecules like enterobactin and vancomycin to leak through the OM either to promote (enterobactin) or suppress (vancomycin) bacterial growth. Clearly, more work is needed to determine the precise reason for YejM/PbgA essentiality and its role in lipid homeostasis.
MATERIALS AND METHODS
Bacterial strains, culture conditions, and antibiotic susceptibility assays.
The Escherichia coli K-12 strains used in this study are listed in Table 3. All bacterial strains are derived from RAM1292 (MC4100 Δara; referred to as wild type [40]) or RAM2572 (MC4100 Δara714 ΔtonB::scar). Lysogenic broth (LB) was prepared from Difco LB EZMix powder. LB agar (LBA) was prepared using LB plus 1.5% agar (Becton, Dickinson). When needed, kanamycin (25 μg/ml), ampicillin (50 μg/ml), and FeCl3 (40 μM) were added to LB or LBA. Unless specified, all cultures were incubated at 37°C from 24 to 36 h. Faster-growing revertants of ΔtonB cells were isolated on LBA after 48 h of incubation. All chemicals were of analytical grade. Antibiotic susceptibility was assessed by determining the MICs of vancomycin by the 2-fold serial dilution method using 96-well microtiter plates. Approximately 105 cells were seeded in each well and grown for 18 h with gentle aeration in 200 μl LB supplemented with various concentrations of vancomycin. The OD600 was read by a VersaMax microplate reader. The MIC was determined to be the lowest antibiotic concentration that corresponded to an OD600 of <0.1. All MIC experiments were carried out with three or more biological replicates.
TABLE 3.
Escherichia coli K-12 strains used in the study
| Strain name | Relevant genotype | Reference or source |
|---|---|---|
| RAM1292 | MC4100 Δara714 | 40 |
| RAM2505 | RAM1292 ΔtonB::Kmr | This study |
| RAM2572 | RAM1292 ΔtonB::scar | This study |
| RAM2485 | RAM2505 yejM1214 (revertant 1) | This study |
| RAM2486 | RAM2505 yejM1624 (revertant 2) | This study |
| RAM2562 | RAM2505 yejM1163 (revertant 3) | This study |
| RAM2573 | RAM2485 ΔtonB::scar | This study |
| RAM2586 | RAM2572 ΔentF::Kmr | This study |
| RAM2587 | RAM2572 ΔfepB::Kmr | This study |
| RAM2588 | RAM2573 ΔentF::Kmr | This study |
| RAM2589 | RAM2573 ΔfepB::Kmr | This study |
| W3100 | Wild type | Coli Genetic Stock Center |
| RAM2595 | W3100 ΔtonB::scar | This study |
| BW25113 | (ΔaraD-araB)567 ΔlacZ4787 | Coli Genetic Stock Center |
| RAM2596 | BW25115 ΔtonB::scar | This study |
| RAM2608 | RAM2572 ΔfepA::Kmr | This study |
| RAM2610 | RAM2573 ΔfepA::Kmr | This study |
| RAM2815 | RAM1292 ΔtolA::Cmr | Lab stock |
| RAM3144 | RAM3150 [ΔtonB::scar ΔnapA::Kmr)(pBAD24)] | This study |
| RAM3145 | RAM3150 [ΔtonB::scar ΔnapA::Kmr)(pBAD24-yejM)] | This study |
| RAM3146 | RAM3151 [ΔtonB::scar ΔnapA::Kmr yejM1163)(pBAD24)] | This study |
| RAM3147 | RAM3151 [ΔtonB::scar ΔnapA::Kmr yejM1163)(pBAD24-yejM)] | This study |
| RAM3148 | RAM1292 ΔnapA::Kmr | This study |
| RAM3149 | RAM1292 ΔnapA::Kmr yejM1163 | This study |
| RAM3150 | RAM2572 (ΔtonB::scar) ΔnapA::Kmr | This study |
| RAM3151 | RAM2572 (ΔtonB::scar) ΔnapA::Kmr yejM1163 | This study |
| RAM3152 | RAM2572 (ΔtonB::scar) ΔtolA::Cmr | This study |
| RAM3153 | RAM2572 (ΔtonB::scar) ΔclsA::scar ΔclsB::Kmr | This study |
| RAM3154 | RAM2572 (ΔtonB::scar) ΔclsA::scar ΔclsC::Kmr | This study |
| RAM3155 | RAM2572 (ΔtonB::scar) ΔclsB::scar ΔclsC::Kmr | This study |
| RAM3156 | RAM2572 (ΔtonB::scar) ΔclsA::scar ΔclsB::scar ΔclsC::scar ΔnapA::Kmr | This study |
| RAM3157 | RAM2572 (ΔtonB::scar) ΔclsA::scar ΔclsB::scar ΔnapA::Kmr yejM1163 | This study |
| RAM3158 | ΔclsABC ΔnapA::Kmr | This study |
| RAM3159 | ΔclsABC ΔnapA::Kmr yejM1163 | This study |
| RAM3160 | RAM2572 ΔpldA::Kmr | This study |
| RAM3161 | RAM2572 ΔpagP::Kmr | This study |
| RAM3162 | RAM2562 ΔpldA::Kmr | This study |
| RAM3163 | RAM2562 ΔpagP::Kmr | This study |
Genetic and DNA methods.
Bacteriophage P1-mediated transductions, using antibiotic resistance markers, were carried out as described previously (41). When desired, the antibiotic resistance marker from the gene deletion site was removed using pCP20, which expresses FLP recombinase (42). The promoterless yejM gene was cloned into pBAD24 (43) by ligating a 1.8-kb PCR-amplified yejM open reading frame (ORF). Cloning was facilitated by the introduction of NcoI and HindIII restriction sites during PCR amplification of the yejM ORF and ligating into pBAD24 that was also restricted with NcoI and HindIII. The ligated mixture was transformed into transformation-competent E. coli JM109 cells, and transformed colonies were selected for LBA containing ampicillin. Plasmids containing the correct size yejM gene clone were extracted using QIAprep spin miniprep kit (Qiagen) and sequenced. pBAD24-yejM was then transformed into appropriate strains for complementation analysis. The primer sequences used for yejM cloning are available on request. Sanger sequencing and whole-genome sequencing, as described previously (31), were carried out at the Arizona State University DNA Core Facility.
Protein methods.
β-Galactosidase assays were performed to determine cytosolic leakage of proteins due to cell lysis. The β-galactosidase activities of rpoHP3::lacZ (44) were measured from three independent cultures in duplicates using the method described by Miller (45). No membrane-permeabilizing agent, such as chloroform or SDS, was used in determining β-galactosidase activity from the cell-free culture supernatant. To isolate cell-free supernatant, cells from 1-ml overnight cultures were pelleted by centrifugation at 16,000 × g for 5 min. The supernatant was collected and passed through a 0.22-μm filter. To analyze periplasmic protein leakage from the cell, 800 μl of cell-free supernatant described above was dried in a SpeedVac and reconstituted in 50 μl Tris-HCl (pH 7.8). Concentrated supernatants (9 μl) were analyzed by SDS-PAGE, and proteins were visualized by staining the gel with Coomassie brilliant blue (Sigma).
Lipid extraction for thin-layer chromatography.
Lipid extraction was accomplished through the original Bligh-Dyer method (46). Briefly, cells were pelleted from 50 ml of overnight cultures and resuspended in 6 ml of 10 mM Tris-HCl–1 mM EDTA solution (pH 7.8). For 1 ml of resuspension, the sample was centrifuge pelleted and reconstituted in 250 μl of distilled water (dH2O). Two hundred fifty microliters of methanol and 500 μl of chloroform were added to the solution, which was then incubated at room temperature with rotation for 30 min. After centrifugation at 16,000 × g for 10 min, the organic layer was collected and dried under a stream of nitrogen gas. The dried lipid extract was resuspended in 20 μl of chloroform-methanol (2:1 [vol/vol]) and dissolved by gentle vortexing. A silica gel 60 (Millipore) thin-layer chromatography (TLC) plate was prerun with chloroform-methanol solution (1:1 [vol/vol]) and dried overnight. The plate was then impregnated with 1.8% boric acid in 95% ethanol and activated at 100°C for 10 min. Between 1 and 5 μl of sample or 5 μl of PE and PG and 10 μl of CL standards (Sigma) were spotted on the dried plate, and TLC was performed using a solvent system of chloroform-methanol-water-1 M ammonium hydroxide (60:37.5:3:2 [vol/vol/vol/vol]). After drying the plate, the lipids were visualized by molybdenum blue spray reagent (Sigma).
Lipid extraction for LC-MS/MS.
Three independently grown overnight cultures per strain were diluted hundredfold in 5 ml LB and grown with aeration until an OD600 of approximately 0.5. The cells were pelleted by centrifugation, washed twice in phosphate buffer (PB) (pH 7.0), and resuspended in 1 ml of PB (pH 7.0). The exact volume of resuspension was adjusted to the OD600 of each sample to represent approximately 5 × 108 cells. Total lipids were extracted from each sample by adding 1 ml of ice-cold isopropanol (47). The contents were vortexed and then snap-frozen using liquid nitrogen followed by a 3-min sonication in an ice bath. Finally, the samples were vortexed and precipitated by centrifugation for 5 min at 21,100 × g. The supernatant was dried using a SpeedVac. For positive-mode MS, a 300-μl aliquot of the supernatant was dried and reconstituted with 60 μl of the reconstitution solvent, which was prepared by incorporating isotopically labeled standards (lysophosphatidylcholine [LPC], lysophosphatidylethanolamine [LPE], phosphatidylcholine [PC], PE, phosphatidylserine [PS], PG, phosphatidylinositol [PI], cholesteryl ester [ChE], diacylglycerol [DG], triacylglycerol [TG], sphingomyelin [SM], and cholesterol [Chol] in CHCl3) diluted in isopropanol. Ten microliters was combined from each sample to create a pooled sample for quality control and identification. For negative-mode MS, a 200-μl aliquot was dried, and the sample was reconstituted with 20 μl of the aforementioned reconstitution solvent. One hundred microliters of the supernatant was combined from each sample to create a pooled sample for quality control (QC) and identification. All samples were vortexed for 5 min, and the supernatant was transferred to autosampler vials following centrifugation and stored at 4°C until analysis.
Ten microliters of each sample was injected into the Thermo Fisher Scientific Vanquish ultrahigh-performance liquid chromatography (UHPLC) system coupled to a Thermo Scientific Q Exactive HF-X quadrupole-Orbitrap mass spectrometer. The reverse-phase chromatography column (Accucore C30) dimensions were 150 mm by 2.1 mm, with a 2.6-μm particle size. The composition of the mobile phase A was 40% H2O–60% acetonitrile, with 10 mM ammonium formate and 0.1% formic acid. The composition of mobile phase B was 10% acetonitrile–90% isopropanol with 10 mM ammonium formate and 0.1% formic acid. Full scans of MS1 were acquired at a resolution of 240,000 in the range from m/z 150 to 2,000. Full scans of MS2 were acquired at a resolution of 120,000. Data-dependent MS2 was acquired at normalized collision energy steps of 10, 30, and 50 with a resolution of 30,000.
ACKNOWLEDGMENTS
We are indebted to Candice Hanna, Trevor Holloway, and Ashley Jama for their assistance in various aspects of the project. We thank Marcin Grabowicz for the pldA and pagP null strains. We are grateful to David Gaul of the Georgia Institute of Technology for his assistance in lipid analysis by LC-MS/MS.
We thank the Research and Training Initiatives office of the School of Life Sciences and the NIH for grant R21 AI117150 for partly funding this work.
REFERENCES
- 1.Nikaido H. 2003. Molecular basis of bacterial outer membrane revisited. Microbiol Mol Biol Rev 67:593–656. doi: 10.1128/mmbr.67.4.593-656.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Silhavy TJ, Kahne D, Walker S. 2010. The bacterial cell envelope. Cold Spring Harb Perspect Biol 2:a000414. doi: 10.1101/cshperspect.a000414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noinaj N, Guillier M, Barnard TJ, Buchanan SK. 2010. TonB-dependent transporters: regulation, structure, and function. Annu Rev Microbiol 64:43–60. doi: 10.1146/annurev.micro.112408.134247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Raetz CRH, Whitfield C. 2002. Lipopolysaccharide endotoxins. Annu Rev Biochem 71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang Y-M, Rock CO. 2008. Membrane lipid homeostasis in bacteria. Nat Rev Microbiol 6:222–233. doi: 10.1038/nrmicro1839. [DOI] [PubMed] [Google Scholar]
- 6.Dowhan W. 2013. A retrospective: use of Escherichia coli as a vehicle to study phospholipid synthesis and function. Biochim Biophys Acta 1831:471–494. doi: 10.1016/j.bbalip.2012.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rowlett VW, Mallampalli V, Karlstaedt A, Dowhan W, Taegtmeyer H, Margolin W, Vitrac H. 2017. Impact of membrane phospholipid alterations in Escherichia coli on cellular function and bacterial stress adaptation. J Bacteriol 199:e00849-16. doi: 10.1128/JB.00849-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Okuda S, Tokuda H. 2011. Lipoprotein sorting in bacteria. Annu Rev Microbiol 65:239–259. doi: 10.1146/annurev-micro-090110-102859. [DOI] [PubMed] [Google Scholar]
- 9.Hagan CL, Silhavy TJ, Kahne D. 2011. β-Barrel membrane protein assembly by the Bam complex. Annu Rev Biochem 80:189–210. doi: 10.1146/annurev-biochem-061408-144611. [DOI] [PubMed] [Google Scholar]
- 10.Misra R. 2012. Assembly of the β-barrel outer membrane proteins in Gram-negative bacteria, mitochondria, and chloroplasts. ISRN Mol Biol 2012:708203. doi: 10.5402/2012/708203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noinaj N, Gumbart JC, Buchanan SK. 2017. The β-barrel assembly machine in motion. Nat Rev Microbiol 15:197–204. doi: 10.1038/nrmicro.2016.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu T, Malinverni J, Ruiz N, Kim S, Silhavy TJ, Kahne D. 2005. Identification of a multicomponent complex required for outer membrane biogenesis in Escherichia coli. Cell 121:235–245. doi: 10.1016/j.cell.2005.02.015. [DOI] [PubMed] [Google Scholar]
- 13.Okuda S, Sherman DJ, Silhavy TJ, Ruiz N, Kahne D. 2016. Lipopolysaccharide transport and assembly at the outer membrane: the PEZ model. Nat Rev Microbiol 14:337–345. doi: 10.1038/nrmicro.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wandersman C, Schwartz M, Ferenci T. 1979. Escherichia coli mutants impaired in maltodextrin transport. J Bacteriol 140:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Misra R, Benson SA. 1988. Isolation and characterization of OmpC porin mutants with altered pore properties. J Bacteriol 170:528–533. doi: 10.1128/jb.170.2.528-533.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Misra R, Benson SA. 1988. Genetic identification of the pore domain of the OmpC porin of Escherichia coli K-12. J Bacteriol 170:3611–3617. doi: 10.1128/jb.170.8.3611-3617.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Benson SA, Occi JL, Sampson BA. 1988. Mutations that alter the pore function of the OmpF porin of Escherichia coli K12. J Mol Biol 203:961–970. doi: 10.1016/0022-2836(88)90121-0. [DOI] [PubMed] [Google Scholar]
- 18.Misra R, Benson SA. 1989. A novel mutation, cog, which results in production of a new porin protein (OmpG) of Escherichia coli K-12. J Bacteriol 171:4105–4111. doi: 10.1128/jb.171.8.4105-4111.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fajardo DA, Cheung J, Ito C, Sugawara E, Nikaido H, Misra R. 1998. Biochemistry and regulation of a novel Escherichia coli K-12 porin protein, OmpG, which produces unusually large channels. J Bacteriol 180:4452–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sampson BA, Misra R, Benson SA. 1989. Identification and characterization of a new gene of Escherichia coli K-12 involved in outer membrane permeability. Genetics 122:491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rutz JM, Liu J, Lyons JA, Goranson J, Armstrong SK, Mcintosh MA, Feix JB, Klebba PE. 1992. Formation of a gated channel by a ligand-specific transport protein in the bacterial outer membrane. Science 258:471–475. doi: 10.1126/science.1411544. [DOI] [PubMed] [Google Scholar]
- 22.Andrews SC, Robinson AK, Rodríguez-Quiñones F. 2003. Bacterial iron homeostasis. FEMS Microbiol Rev 27:215–237. doi: 10.1016/S0168-6445(03)00055-X. [DOI] [PubMed] [Google Scholar]
- 23.Peralta DR, Adler C, Corbalán NS, García ECP, Pomares MF, Vincent PA. 2016. Enterobactin as part of the oxidative stress response repertoire. PLoS One 11:e0157799. doi: 10.1371/journal.pone.0157799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dalebroux ZD, Edrozo MB, Pfuetzner RA, Ress S, Kulasekara BR, Blanc M-P, Miller SI. 2015. Delivery of cardiolipins to the Salmonella outer membrane is necessary for survival within host tissues and virulence. Cell Host Microbe 17:441–451. doi: 10.1016/j.chom.2015.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rossi RM, Yum L, Agaisse H, Payne SM. 2017. Cardiolipin synthesis and outer membrane localization are required for Shigella flexneri virulence. mBio 8:e01199-17. doi: 10.1128/mBio.01199-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dong H, Zhang Z, Tang X, Huang S, Li H, Peng B, Dong C. 2016. Structural insights into cardiolipin transfer from the inner membrane to the outer membrane by PbgA in Gram-negative bacteria. Sci Rep 6:30815. doi: 10.1038/srep30815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nurminen M, Hirvas L, Vaara M. 1997. The outer membrane of lipid A-deficient Escherichia coli mutant LH530 has reduced levels of OmpF and leaks periplasmic enzymes. Microbiology 143:1533–1537. doi: 10.1099/00221287-143-5-1533. [DOI] [PubMed] [Google Scholar]
- 28.De Lay NR, Cronan JE. 2008. Genetic interaction between the Escherichia coli AcpT phosphopantetheinyl transferase and the YejM inner membrane protein. Genetics 178:1327–1337. doi: 10.1534/genetics.107.081836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lazzaroni JC, Germon P, Ray MC, Vianney A. 1999. The Tol proteins of Escherichia coli and their involvement in the uptake of biomolecules and outer membrane stability. FEMS Microbiol Lett 177:191–197. doi: 10.1111/j.1574-6968.1999.tb13731.x. [DOI] [PubMed] [Google Scholar]
- 30.Lopes J, Gottfried S, Rothfield L. 1972. Leakage of periplasmic enzymes by mutants of Escherichia coli and Salmonella Typhimurium: isolation of “periplasmic leaky” mutants. J Bacteriol 109:520–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kern B, Leiser OP, Misra R. 2019. Suppressor mutations in degS overcome the acute temperature-sensitive phenotype of ΔdegP and ΔdegP Δtol-pal mutants of Escherichia coli. J Bacteriol 201:e00742-18. doi: 10.1128/JB.00742-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tan BK, Bogdanov M, Zhao J, Dowhan W, Raetz CRH, Guan Z. 2012. Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as substrates. Proc Natl Acad Sci U S A 109:16504–16509. doi: 10.1073/pnas.1212797109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng L, Lin Y, Lu S, Zhang J, Bogdanov M. 2017. Biogenesis, transport and remodeling of lysophospholipids in Gram-negative bacteria. Biochim Biophys Acta 1862:1404–1413. doi: 10.1016/j.bbalip.2016.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sohlenkamp C, Geiger O. 2016. Bacterial membrane lipids: diversity in structures and pathways. FEMS Microbiol Rev 40:133–159. doi: 10.1093/femsre/fuv008. [DOI] [PubMed] [Google Scholar]
- 35.Snijder HJ, Ubarretxena-Belandia I, Blaauw M, Kalk KH, Verheij HM, Egmond MR, Dekker N, Dijkstra BW. 1999. Structural evidence for dimerization-regulated activation of an integral membrane phospholipase. Nature 401:717–721. doi: 10.1038/44890. [DOI] [PubMed] [Google Scholar]
- 36.Bishop RE, Gibbons HS, Guina T, Trent MS, Miller SI, Raetz C. 2000. Transfer of palmitate from phospholipids to lipid A in outer membranes of Gram-negative bacteria. EMBO J 19:5071–5080. doi: 10.1093/emboj/19.19.5071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bishop RE. 2005. The lipid A palmitoyltransferase PagP: molecular mechanisms and role in bacterial pathogenesis. Mol Microbiol 57:900–912. doi: 10.1111/j.1365-2958.2005.04711.x. [DOI] [PubMed] [Google Scholar]
- 38.Bishop RE. 2008. Structural biology of membrane-intrinsic beta-barrel enzymes: sentinels of the bacterial outer membrane. Biochim Biophys Acta 1778:1881–1896. doi: 10.1016/j.bbamem.2007.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.May KL, Silhavy TJ. 2018. The Escherichia coli phospholipase PldA regulates outer membrane homeostasis via lipid signaling. mBio 9:e00379-18. doi: 10.1128/mBio.00379-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Werner J, Misra R. 2005. YaeT affects the assembly of lipid-dependent and lipid-independent outer membrane proteins of Escherichia coli. Mol Microbiol 57:1450–1459. doi: 10.1111/j.1365-2958.2005.04775.x. [DOI] [PubMed] [Google Scholar]
- 41.Silhavy TJ, Berman ML, Enquist LW. 1984. Experiments with gene fusions. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 42.Datsenko KA, Wanner BL. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci U S A 97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guzman LM, Belin D, Carson MJ, Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol 177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mecsas J, Rouviere PE, Erickson JW, Donohue TI, Gross CA. 1993. The activity of σE, an Escherichia coli heat-inducible σ-factor, is modulated by expression of outer membrane proteins. Genes Dev 7:2618–2628. doi: 10.1101/gad.7.12b.2618. [DOI] [PubMed] [Google Scholar]
- 45.Miller JH. 1992. A short course in bacterial genetics: a laboratory manual and handbook for Escherichia coli and related bacteria, p 71–74. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- 46.Bligh EG, Dyer WJ. 1959. A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37:911–917. doi: 10.1139/y59-099. [DOI] [PubMed] [Google Scholar]
- 47.Hogan SR, Phan JH, Alvarado-Velez M, Wang MD, Bellamkonda RV, Fernández FM, LaPlaca MC. 2018. Discovery of lipidome alterations following traumatic brain injury via high-resolution metabolomics. J Proteome Res 17:2131–2143. doi: 10.1021/acs.jproteome.8b00068. [DOI] [PMC free article] [PubMed] [Google Scholar]