

Abstract
Pharmacologic inhibitors of cyclin-dependent kinases 4 and 6 (CDK4/6) have recently entered the therapeutic armamentarium of clinical oncologists, and show promising activity in patients with breast and other cancers. Although their chief mechanism of action is inhibition of Retinoblastoma (RB) protein phosphorylation and thus induction of cell cycle arrest, CDK4/6 inhibitors alter cancer cell biology in other ways that can also be leveraged for therapeutic benefit. These include modulation of mitogenic kinase signaling, induction of a senescence-like phenotype, and enhancement of cancer cell immunogenicity. Here, we describe the lesser appreciated effects of CDK4/6 inhibitors on cancer cells, and suggest ways by which they might be exploited to enhance the benefits of these agents for cancer patients.
Keywords: Cell cycle, CDK4/6, targeted therapy, immunotherapy
Development of pharmacologic CDK inhibitors
In order for a cell to divide, it must complete the cell cycle – a tightly regulated series of events governed in large part by the activity of cyclin proteins and their partner kinases, the cyclin-dependent kinases (CDKs). The CDKs that drive cell division are often hyperactive in cancers, and the sustained proliferative signaling that results is a well-recognized hallmark of malignancy [1]. Given the notion that inhibition of CDKs should halt otherwise uncontrolled cellular proliferation, scientists have long attempted to develop pharmacologic inhibitors of these CDKs. Nevertheless, many of the initial compounds lacked potency or selectivity. As a result, therapeutic targeting of the cell cycle machinery had remained an elusive goal.
The challenge stemming from a lack of specific CDK inhibitors has recently been met by the development of selective and potent inhibitors of cyclin-dependent kinases 4 and 6 (CDK4/6) [2–4]. CDKs 4 and 6 specifically regulate cellular transition from the G1 phase of the cell cycle to the S phase, and CDK4/6 inhibitors effectively block the proliferation of sensitive cancer cells by inducing G1 cell cycle arrest [5]. Consequently, these agents have moved rapidly from research laboratories into clinical trials, and three have now received FDA approval for the treatment of metastatic breast cancer [6]. These early clinical successes have been celebrated, but it is clear that only a fraction of the clinical potential of CDK4/6 inhibitors has been realized. In this article, we briefly introduce the CDK4/6 pathway, and then focus on novel therapeutic approaches that have the potential to maximize the utility of CDK4/6 inhibitors in the clinic.
Control of the G1/S transition
In order to enter the cell cycle and commence DNA replication, a cell must pass from the G1 phase into the S phase through a tightly regulated restriction point [7]. According to the “classical” cell cycle model, the G1/S transition begins in early G1 when the balance between mitogenic stimulation (via growth factor receptor activation) and inhibition tips in favor of the former, triggering an increase in the levels of D-type cyclins (Dl, D2, and D3) (Figure 1). D-type cyclins bind to CDK4 or CDK6, and the cyclin-CDK complexes then enter the nucleus where they are phosphorylated by the CDK-activating kinase (CAK) complex. In turn, activated CDK4/6 complexes phosphorylate the retinoblastoma (RB) tumor suppressor protein, as well as the related pocket proteins p107 and p130 [8–10]. RB phosphorylation by CDK4/6 partially derepresses activity of the E2F family of transcription factors [11], thus facilitating the expression of E2F target genes including those for the E-type cyclins (cyclins E1 and E2). Cyclin E then binds to and activates CDK2, which hyper-phosphorylates RB, further increasing the expression of E2F target genes that are critical for initiation of DNA synthesis and entry into S-phase [7, 11–13]. Notably, RB not only inhibits the expression of S phase genes by directly inhibiting E2F transactivation, but also by recruiting chromatin modifiers to DNA that can indirectly silence E2F-target gene expression [14, 15].
Figure 1. The classical model for regulation of the G1/S transition by cyclins and CDKs.
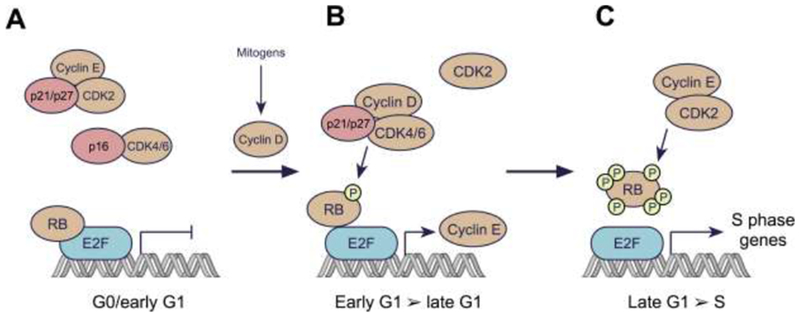
A: In resting cells, CDK4/6 and CDK2 are inactive. D-type cyclin levels are low due to the lack of mitogenic stimulus, limiting CDK4/6 activity. Moreover, CDKs 4 and 6 are bound by INK4 family members (e.g. p16), establishing binary complexes that lack kinase activity. CDK2 complexes are inhibited by the CIP/KIP proteins p21 and p27. Collectively, the suppression of CDK4/6 and CDK2 leads to RB hypo-phosphorylation, and hence repressed expression of E2F target genes. This repression is mediated by direct blockade of the E2F transactivation domain by RB, and by recruitment of histone modifiers to RB that further silence E2F target gene expression.
B: Levels of D-type cyclins increase in response to mitogenic stimuli, due to both enhancement of cyclin D gene expression and an increase in cyclin D protein stability. D-type cyclins bind to CDK4/6, forming complexes that are stabilized by p21 or p27. Cyclin D:CDK4/6 complexes then enter the nucleus and phosphorylate RB. This partially de-represses expression of E2F target genes, including those for the E-type cyclins. The partial phosphorylation of RB facilitates progression through G1.
C: As the levels of E-type cyclins rise in late G1, CDK2 is activated resulting in RB hyperphosphorylation and inactivation. Hyperphosphorylated RB is released from E2F, enabling increased transcription of E2F target genes necessary for the cell to proceed into S phase.
In reality, progression from G1 to S is not always regulated in such an orderly, step-wise manner. Indeed, RB contains several phosphorylation sites – amenable to phosphorylation by CDK4/6, CDK2, or both – and the consequences of phosphorylation at differing sites are distinct [16]. The capacity of these enzymes to facilitate the G1-S transition is probably a reflection of their net effects on RB phosphorylation at any given point in time. Indeed, the “alternate” cell cycle model proposes that the G1-S transition can be facilitated by CDK4/6 or CDK2 acting alone or in concert. As such, CDK4/6-mediated phosphorylation of RB is likely not an absolute pre-requisite for CDK2 activation [6].
Regulation of CDK4/6 and CDK2 activity is achieved in part by two families of endogenous inhibitory proteins. The first is the INK4 family, comprising p16INK4A, p15INK4B, p18INK4C, and p19INK4D. These proteins bind to CDKs 4 and 6, forming binary complexes that lack kinase activity. The second is the CIP/KIP family, which includes p27KIP1, p21CIP1, and p57KIP2. These proteins bind CDKs more promiscuously and have more diverse functions, potently inhibiting several CDKs (including CDK4/6, CDK2, and CDK1), but also in certain circumstances binding to and stabilizing the cyclin D-CDK4/6 holoenzyme. These divergent functions may be regulated by both the amount and the phosphorylation status of the CIP/KIP proteins [17–20].
The description above does not reflect the full biologic complexity of CDK4/6 activity. For example, although pre-clinical data suggests that different tissues harbor dependencies on particular D-type cyclins or on CDK4 or 6, these proteins can compensate for each other due to functional redundancy [21, 22]. More strikingly, cells within some organ compartments can proliferate in the absence of all D-type cyclins [23], or without both CDK4 and CDK6 [22]. In these instances the G1 transition may be led by complementary cyclin:CDK complexes, including those containing CDK2 or CDK1 [22, 24]. Although the notion that various cancer cells are particularly CDK4/6 dependent still renders the development of CDK4/6 inhibitors as an attractive strategy, these observations immediately suggest pathways by which cancer cells could escape the effects of CDK4/6 inhibition.
Dysregulation of CDK4/6 activity in cancer
Many human cancers harbor genomic or transcriptional aberrations that activate CDK4/6 [25], through a wide variety of mechanisms described below. In theory, these cancers are expected to show greater CDK4/6 dependency, and have traditionally been considered ideal candidates for treatment with CDK4/6 inhibitors. However, as is discussed later, not all of these alterations have been shown to confer heightened sensitivity to CDK4/6 inhibitors [26].
Alterations in cell cycle machinery genes
Amplification of genes encoding D-type cyclins is commonly observed in human cancer, and is correlated with increased levels of cyclin D protein. Based on data from large next-generation sequencing studies, cyclin D1 (CCND1) amplification is most commonly observed in cancers of the upper gastrointestinal tract, head and neck, breast, lung, and bladder [27, 28], and in breast cancer, it has been correlated with worse clinical outcomes [29]. CCND2 and CCND3 are also amplified in a variety of cancers, albeit less frequently [27]. In addition, numerous cancers demonstrate amplifications of CDK4 (most notably liposarcoma and glioblastoma) and CDK6 (upper gastrointestinal cancers and neuroendocrine carcinoma of the prostate) [27]. Certain genomic translocations and specific gene mutations can also markedly increase cyclin D levels in tumor cells. The resultant increase in CDK4/6 activity might be expected to confer sensitivity to CDK4/6 inhibitors, and this has indeed been shown to be the case [26]. For example, in mantle cell lymphoma the unique t11;14 translocation juxtaposes the CCND1 and IGH immunoglobulin heavy chain genes, leading to constitutive overexpression of cyclin D1 in a mitogen-independent fashion [30]. In addition, certain tumors carry mutations in the 3’-UTRs of CCND1, CCND2, and CCND3. These mutations can increase cyclin D levels by preventing degradation of cyclin D mRNA [26].
Loss of p16INK4A function
Loss of p16INK4A function is a relatively common event in cancer, usually driven by homozygous deletion of the CDKN2A locus which encodes for p16. Loss of p16 has been predicted to confer heightened CDK4/6 dependence in cancer cells [5]. Somatic CDKN2A deletion is most common in malignant peripheral nerve sheath tumors, glioblastoma, and carcinomas of the pancreas, head and neck, bladder, esophagus, and lung [27, 28]. Importantly, however, a study examining a large panel of human cancer cell lines has recently called into question whether loss of p16INK4A function is actually associated with heightened sensitivity to CDK4/6 inhibition [26]. The reasons for this discrepancy are unclear, but may relate to the fact that CDKN2A mutant tumors show a propensity to upregulate CDK2 activity rapidly after exposure to CDK4/6 inhibitors, thus restoring RB phosphorylation and G1 progression [26].
Upstream genomic events that increase cyclin D levels
In cancer cells that lack the genomic alterations described above, cyclin D levels and CDK4/6 activity are primarily regulated by mitogenic signaling. Notably, the mitogenic Ras-Raf-MEK-ERK pathway is a potent driver of CCND1 transcription [31]. A large number of cancers demonstrate elevated cyclin D1 levels due to a variety of aberrations that activate the Ras-Raf-MEK-ERK pathway – including activating mutations in signaling effector proteins and upstream growth factor receptors [32]. Increased mitogenic signaling through a hyperactive phosphoinositide-3-kinase (PI3K)-AKT pathway also increases cyclin D1 levels by preventing its nuclear export and increasing its translation [33–35]. In certain instances, the importance of mitogen-induced cyclin D1 upregulation for tumor growth has been unequivocally demonstrated – for example, CCND1 knockout mice are unable to form mammary tumors driven by ERBB2 or RAS [36]. However, it has not been definitively demonstrated that higher levels of cyclin D1 protein in cancer cells are associated with CDK4/6 inhibitor sensitivity more generally.
Clinical successes with CDK4/6 inhibitors to date
Three CDK4/6 inhibitors are currently approved for clinical use in the United States – palbociclib, ribociclib, and abemaciclib. All three agents are used as treatment for estrogen receptor (ER)-positive breast cancer based on the results of clinical trials demonstrating improvements in progression-free survival when CDK4/6 inhibitors are added to anti-estrogen therapy, the previous gold standard for treatment of ER-positive disease [37–39]. Abemaciclib is also approved for use as monotherapy in a similar patient population [40]. The rationale for focusing on breast cancer during early development of CDK4/6 inhibitors was strong. Studies in transgenic animals had shown that cyclin D1 deficiency strongly and specifically impairs mammary epithelial proliferation, suggesting a key role for the CDK4/6 pathway in this tissue [21]. Moreover, cyclin D1 and CDK4 are essential for the formation and growth of several murine mammary tumors [36, 41, 42]. The first preclinical study of CDK4/6 inhibitors in breast cancer showed that a large proportion of human breast cancer cell lines are sensitive to palbociclib, as evidenced by reduced RB phosphorylation and cell cycle arrest [5]. Interestingly, cells demonstrating a luminal pattern of gene expression (as defined in [43]) were particularly sensitive to palbociclib, and luminal breast cancers are much more likely to express ER, providing reason to develop CDK4/6 inhibitors in ER-positive cancers. Moreover, CCND1 is a known ER target gene, and combined inhibition of CDK4/6 and ER (with endocrine therapy) reduces tumor cell proliferation synergistically [5].
The clinical data beyond ER-positive breast cancer is sparser. Single-arm studies have been performed in a variety of cancer types bearing genomic abnormalities predicted to increase CDK4/6 inhibitor sensitivity including HER2-positive breast cancer, mantle cell lymphoma, liposarcoma, melanoma, non-small cell lung cancer, glioblastoma, neuroblastoma and malignant rhabdoid tumors [44–47]. Each study has shown evidence of clinical activity (partial response or prolonged stabilization of disease) in a subset of patients, but in the absence of randomized data, it is difficult to know how CDK4/6 inhibitor activity compares to existing standards of care.
Improving the efficacy of CDK4/6 inhibitors as cancer therapy
A wealth of preclinical data generated over decades describes the regulation of CDK4/6 activity in proliferating cells, and one key to broadening the clinical impact of CDK4/6 inhibitors lies in reviewing these old data for clues. In addition, the promise of clinical efficacy has rekindled interest in the CDK4/6 pathway and provided new research tools to understand it better. Based on both old and new data, a number of lines of investigation have begun to emerge in an effort to develop CDK4/6 inhibitors rationally and swiftly (Figure 2).
Figure 2. Key Figure. Strategies for deriving maximal clinical benefit from CDK4/6 inhibition.
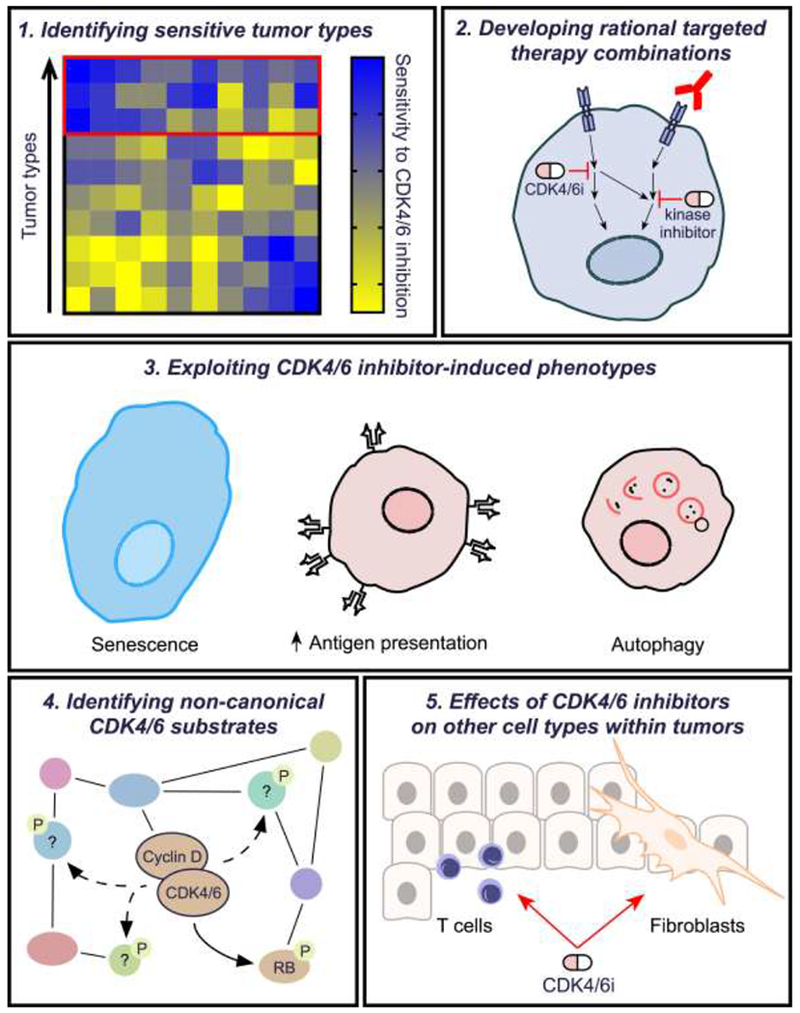
Although CDK4/6 inhibitors have shown impressive activity in the treatment of estrogen-receptor positive breast cancer, their full clinical potential has not been realized. This figure summarizes key areas that should be explored to expand the benefits of these agents in clinical practice.
1: Identification of sensitive tumor types using high throughput multi-omic analyses. In vitro studies to determine molecular predictors of cancer cell sensitivity and resistance to CDK4/6 inhibitors are critical for guiding patient selection in future clinical trials. Such studies should be performed using high-throughput techniques to maximize efficiency. Examples include CRISPR or RNAi screens (ideally performed both with and without concomitant CDK4/6 inhibitor treatment), pharmacologic compound screens that identify synergistic CDK4/6 inhibitor-containing combinations, and bioinformatic analyses that determine associations between cancer cells’ CDK4/6 inhibitor sensitivity and their genomic and transcriptomic profiles.
2: Understanding cross-talk and cooperativity between parallel pro-survival signaling pathways. The CDK4/6 signaling pathway intersects with other key mitogenic pathways (e.g. PI3K-AKT; Ras-Raf-MEK-ERK; steroid hormone signaling) in tumor cells. Synergistic anti-tumor effects have been observed through co-inhibition of CDK4/6 and these other pathways, using combination regimens incorporating either small molecule kinase inhibitors (depicted as pills) or inhibitory monoclonal antibodies (shown in red). Relatively few studies have systematically explored the impact of CDK4/6 inhibition on the phospho-proteome and signal transduction networks in cancer cells, and more are urgently required.
3. Exploiting biologic phenotypes induced by CDK4/6 inhibitors in cancer cells. Short-term treatment of cancer cells with CDK4/6 inhibitors induces RB-dependent G1 arrest. However, more prolonged exposure can induce profoundly different biological phenotypes in tumor cells, in a manner also reflective of sustained RB activation. Capitalizing upon these phenotypes might improve the efficacy of CDK4/6 inhibitors. For example, CDK4/6 inhibition invokes many hallmarks of cellular senescence (cellular enlargement, increased beta-galactosidase activity), raising the question of whether they should be combined with “senolytic” compounds (e.g. bcl-2/bcl-xl inhibitors). CDK4/6 inhibitors also increase tumor cell neoantigen presentation via cell surface MHC Class I, providing rationale for immunotherapy combinations. In addition, CDK4/6 inhibitors can induce tumor cell autophagy, and co-treatment with autophagy inhibitors (e.g. bafilomycin, hydroxychloroquine) can increase their efficacy.
4: Identification of non-canonical CDK4/6 substrates. Aside from their canonical substrate RB, CDKs 4 and 6 also bind to and phosphorylate a range of other protein substrates involved in diverse biologic processes. Key examples are the transcription factor FOXM1, certain glycolytic enzymes, and Nuclear Factor of Activated T cell (NFAT) family members. Inhibiting the phosphorylation of these substrates with CDK4/6 inhibitors can have wide-ranging effects on tumor cell biology, even in cells that lack RB function.
5. Understanding effects in cells of the tumor stroma. CDK4/6 inhibition can impact stromal cell biology within solid tumors. For example, fibroblasts (shown in yellow) can senesce in response to CDK4/6 inhibitor treatment, releasing cytokines that impair anti-tumor immunity and thus limit drug efficacy. Conversely, CDK4/6 inhibition can directly enhance effector functions in T lymphocytes (blue) to strengthen anti-tumor immune responses. Better understanding the effects of CDK4/6 inhibitors on these and other non-tumor cells could result in novel strategies that enhance drug activity and/or mitigate therapeutic resistance.
1. Identifying sensitive and resistant tumor types
Clinically, there is significant heterogeneity in the response of individual tumors to CDK4/6 inhibitors [48, 49] and it has therefore become imperative that we better understand biomarkers that predict sensitivity to these agents. Equally important is the problem of resistance to CDK4/6 inhibitors – for example, in breast cancer nearly all tumors acquire resistance to these agents after prolonged therapy, and the mechanisms underlying this are also poorly understood [50]. As described above, CDK4/6 activity is upregulated in cancers through a variety of genomic alterations including cyclin D amplification and loss of CDKN2A, and an assumption has been made that cancers harboring these alterations should be the most susceptible to pharmacologic CDK4/6 inhibition. However, recent analyses of clinical specimens have called this assumption into question – for example, in randomized clinical trials of palbociclib for breast cancer, CCND1 amplification and loss of p16 expression were not associated with enhanced CDK4/6 inhibitor response [51–53]. It is clear that more work is needed to identify predictors of sensitivity and resistance to these agents.
One approach is to select tumors originating from tissues wherein normal cells show a particular CDK4/6 dependence. A good example is the selection of breast cancers based on a mammary epithelial specific phenotype identified in CCND1 knockout mice. Similarly, preclinical studies have shown responses to CDK4/6 inhibitors amongst hematopoietic cancers [54, 55], in keeping with observations that CDK6 is expressed at high levels in lymphoid organs, and that CDK6 knockout mice show specific defects in hematopoiesis and thymic development [22]. Other approaches are also being used to determine correlations between tumor sensitivities to CDK4/6 inhibition and their molecular profiles. In a recently published study, the sensitivity of 560 human cancer cell lines to the CDK4/6 inhibitor abemaciclib was assessed [26]. First, the authors confirmed the previously described and somewhat intuitive notion that a lack of RB expression is associated with relative resistance to CDK4/6 inhibitors, and that response to these agents is almost always RB-dependent [26, 56]. This finding has also been strengthened through clinical reports describing emergence of RB1 mutations as tumors acquire resistance to CDK4/6 inhibitors [57]. Second, and in keeping with breast cancer clinical data, they did not find CCND1 amplification to be associated with greater drug sensitivity. Rather, they identified a specific group of other genomic features predicted to increase cyclin D levels that are strongly associated with CDK4/6 inhibitor sensitivity. These features, collectively termed “D Cyclin Activating Features” (DCAF) include CCND1 translocations (as seen in mantle cell lymphoma), amplification of CCND2 or CCND3, mutation in the 3’-UTR of CCND1–3, genomic loss of FBX031 (encoding for a D-type cyclin ubiquitin ligase), and expression of K-cyclin, a cyclin D1 variant from the Kaposi’s sarcoma virus [26]. Although particular DCAFs are more common in certain tumors types (e.g. CCND1 3’-UTR loss is more common in uterine and stomach cancers), many are distributed across a range of cancers. As a result of these findings, a clinical “basket trial” of abemaciclib therapy is currently underway, recruiting patients with any cancer type whose tumors demonstrate a DCAF upon genomic profiling ().
A number of intriguing questions arise from this work that are relevant to understanding the molecular predictors of CDK4/6 inhibitor response [26]. First, it is not clear why amplifications of CCND2 and CCND3, but not CCND1, were associated with sensitivity to CDK4/6 inhibitors. Second, and counter to expectations, cell lines with CDKN2A alterations were found to be relatively resistant to abemaciclib, due to their capacity to rapidly upregulate CDK2 activity after treatment. Third, TP53 mutations were strongly associated with CDK4/6 inhibitor resistance, and the mechanisms for this are unclear. Further mechanistic studies are required to strengthen the potential clinical implications of these findings.
One key challenge has been the development of accurate methods to identify tumors that have lost RB function, and which would therefore be expected to be CDK4/6 inhibitor-resistant. It is clear that tumors harboring genomic loss of RB1 alleles and/or loss of function RB1 mutations fall into this category, but these might only reflect a fraction of tumors lacking RB functionality [57, 58]. One proposed surrogate marker of RB dysfunction is a high level of pl6 as detected by immunohistochemistry. Indeed, RB and p16 levels are often inversely correlated in tumor cells, due to a previously described feedback loop by which p16 is expressed in response to dysregulated E2F target gene expression [58–60], and CDKN2A and RB1 lesions are mutually exclusive in most cancers. Despite this, clinical studies to date have failed to demonstrate that either the mRNA or protein levels of CDKN2A1p16 or RB1/RB are predictive of benefit from CDK4/6 inhibitors [51, 52]. More broadly, analysis of breast cancer specimens from randomized clinical trials have failed to consistently identify any biomarker of CDK4/6 inhibitor resistance at either the mRNA of protein level [50–52, 61, 62].
2. Developing rational targeted therapy combinations
The CDK4/6 pathway intersects with several key mitogenic signaling pathways in cancer cells, providing a strong rationale for combining CDK4/6 inhibitors with other targeted therapies. A prototypic case is the combination of CDK4/6 inhibitors and endocrine therapies (e.g. tamoxifen, aromatase inhibitors, fulvestrant) in ER-positive breast cancers [5, 63, 64]. Synergy between these agents can in part be explained by a “two-hit” mechanism, whereby endocrine therapy limits CCND1 transcription and CDK4/6 inhibitors block kinase activity directly (Figure 3) [65]. However, additional interactions between these pathways exist – for example in breast cancers that have acquired endocrine therapy resistance, ER can directly activate E2F target gene transcription and this can be reversed by CDK4/6 inhibitors [66]. Notably, androgen receptor (AR) signaling in prostate cancer shows several parallels to ER signaling in breast cancer. For example, both ER and AR drive CCND1 transcription, and CDK4/6 inhibitors show in vivo activity in both hormone-sensitive and castration-resistant prostate cancer [26, 67]. Clinical development of CDK4/6 inhibitors in prostate cancer is now emerging as an important priority, and phase II studies are currently underway (; ).
Figure 3. Cross-talk between the CDK4/6 and mitogenic signaling pathways in cancer.
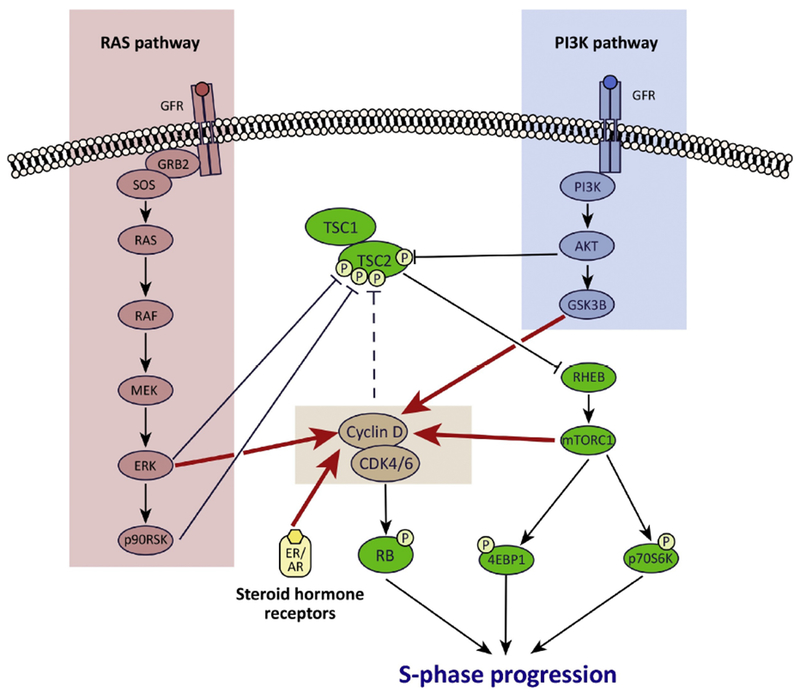
Extensive cross-talk exists between mitogenic signaling pathways and the CDK4/6 pathway in cancer cells. First, mitogenic signaling increases cyclin D1 levels to increase CDK4/6 activity via several mechanisms (red arrows): (a) PI3K pathway signaling reduces cyclin D1 turnover via GSK3β; (b) Ras pathway signaling promotes an ERK-dependent upregulation of transcription factors that drive cyclin D gene expression; (c) mTORC1 increases cyclin D protein translation; (d) CCND1 transcription is increased directly by activated ER (breast cancer) and AR (prostate cancer). In addition, these pathways also interact via their convergence on TSC2 and thus mTORCl: (a) ART, ERK, and p90RSK each directly phosphorylate TSC2 to activate mTORCl; (b) cyclin D-CDK4/6 also bind to and probably phosphorylate TSC2 to increase mTORCl activity (dashed line).
Rationale for synergistic combination therapy regimens: CDK4/6 inhibitors limit cell proliferation by reducing RB phosphorylation, but can also partially suppress TSC2 phosphorylation. Co-inhibiting the PI3K and/or Ras pathway not only reduces cyclin D1 levels (further enforcing RB activation), but also increases the suppression of TSC2 phosphorylation, maximizing mTORCl inhibition. Collectively, combined activation of RB and inhibition of mTORCl inhibition potently blocks progression of cells into S phase.
The CDK4/6 and PI3K-AKT-mTOR pathways also interact extensively (Figure 3). On the one hand, PI3K pathway activation (triggered by signaling through upstream receptor tyrosine kinases as well as mutations in PIK3CA or AKT [33, 34]) increases cyclin D1 levels. On the other hand, cyclin D-CDK4/6 can reciprocally stimulate mTORC1 signaling, which lies downstream of PI3K [68]. Combination regimens containing inhibitors of CDK4/6 and receptor tyrosine kinase-PI3K-AKT signaling have proven effective in a variety of preclinical tumor models, often exhibiting synergism. Examples include combinations of CDK4/6 inhibitors with HER2 inhibitors in breast cancer [5, 69, 70]; PI3K inhibitors in breast cancer, mesothelioma, and mantle cell lymphoma [71–73]; mTOR inhibitors in cancers of the breast or head and neck [74, 75]; IGF-1R inhibitors in pancreatic cancer [76]; and ALK inhibitors in neuroblastoma [77]. This synergy has complex mechanistic underpinnings: in brief, it is likely that the CDK4/6 and PI3K pathways both converge on the tumor suppressor, TSC2, a negative regulator of mTORC1 [68, 69, 78]. When TSC2 is phosphorylated, it degrades, which in turn increases mTORC1 activity. Activation of either the CDK4/6 or PI3K pathway can achieve this [68, 79, 80]. We have previously demonstrated an example wherein this interaction is responsible for therapeutic synergy: in HER2-positive breast cancer cells that are resistant to anti-HER2 agents, neither HER2 inhibitors nor CDK4/6 inhibitors can fully suppress TSC2 phosphorylation. When administered in combination, however, TSC2 phosphorylation is maximally suppressed, resulting in shutdown of mTORC1 activity, thus synergistically enhancing the cell cycle arrest induced by CDK4/6 inhibitors [69, 81]. Based on this mechanism, two randomized clinical trials are currently underway to determine whether CDK4/6 inhibitors can overcome therapeutic resistance in HER2-positive breast cancers (, ).
The RAS-RAF-MEK-ERK signaling pathway also enhances CDK4/6 activity by driving CCND1 transcription (Figure 3). This pathway is commonly activated by mutations in the KRAS, NRAS, and BRAF oncogenes [82]. Importantly, synergy between inhibitors of RAS pathway members and CDK4/6 is seen chiefly in cancer cells harboring activating mutations in KRAS, NRAS, or BRAF [83]. One important mechanism for this synergy is a double-hit on the CDK4/6 pathway, with co-ordinate lowering of cyclin D1 levels and inhibition of CDK4/6 resulting in maximal suppression of RB phosphorylation and hence G1 arrest [83, 84]. Preclinically, inhibiting both the CDK4/6 and Ras pathways has demonstrated success using a pan-RAF inhibitor (inhibiting ARAF, BRAF, and CRAF protein kinases) in multiple tumor types, as well as inhibitors of BRAF (in BRAF mutant melanoma) and MEK (in KRAS mutant lung or colorectal cancer and BRAF mutant melanoma) [83–91]. Interestingly, the RAS pathway, like the PI3K pathway, can also activate mTOR through downstream phosphorylation of TSC2 [92, 93] and combined CDK4/6 and MEK inhibition also leads to a synergistic suppression of mTORC1 signaling [94]. These results suggest that in addition to activating their own canonical substrates, the CDK4/6, PI3K, and RAS pathways all converge upon TSC2 such that the benefits of combination regimens extend to include mTORC1 inhibition. This notion has been strengthened by recent studies showing that acquired resistance to combined CDK4/6 and MEK inhibition in melanoma can be mediated by activating mutations in PIK3CA and heightened mTORC1 activity [95, 96].
3. Exploiting CDK4/6 inhibitor-induced cellular phenotypes
The initial effect of CDK4/6 inhibitors on responsive cancer cells is G1 cell cycle arrest. With more prolonged treatment, however, a range of other biological phenotypes have been observed. Each of these has its roots in the biology of the CDK4/6 pathway, and offers specific potential for the development of new CDK4/6 inhibitor-containing therapeutic combinations (Figure 2).
First, several investigators have reported that prolonged (several days or more) exposure of cancer cells to CDK4/6 inhibitors can induce a phenotype resembling cellular senescence. This is not altogether surprising, given the important role RB activation plays in mediating senescence [97, 98]. CDK4/6 inhibitor treatment can reproduce many of the morphological hallmarks of senescence including cellular enlargement, increased beta-galactosidase activity, and development of senescence-associated heterochromatin foci (SAHF) both in vitro and in vivo [54, 99, 100]. That said, it is not clear that CDK4/6 inhibitor-induced senescence is an exact phenocopy of classical senescence induced by DNA damage. For example, CDK4/6 inhibitor-induced senescence is reversible – as evidenced by resumption of tumor cell proliferation after prolonged drug treatment is withdrawn [99]. In addition, reports on whether CDK4/6 inhibition upregulates a transcriptional program for cytokines and chemokines known as the senescence-associated secretory phenotype (SASP) have been mixed, and the implications of this in vivo have not been explored [99, 100]. Classical cellular senescence is a unique state associated with dramatic changes in several fundamental aspects of cell biology [101]. It is important we now determine to what extent these features are recapitulated with CDK4/6 inhibition in order to understand how they might be exploited for therapeutic gain. For example, recent reports have described a series of candidate “senolytic” compounds, which show a specific capacity to induce apoptosis in cells that have undergone replicative senescence [101, 102]. The efficacy of these compounds in CDK4/6 inhibitor pre-treated cancer cells has not been assessed.
In certain models, CDK4/6 inhibitor treatment can also induce cancer cell autophagy, evidenced by the presence of autophagolysosomes and upregulation of various autophagic markers [103–105]. This is consistent with the fact that cyclin D-CDK4/6 activity restrains autophagy in normal epithelial compartments such as that of the mammary gland [106]. The mechanisms underlying CDK4/6 inhibitor-mediated autophagy induction are unclear, but autophagy may reflect a stress response triggered by a CDK4/6 inhibitor-mediated increase in reactive oxygen species (ROS) levels [103]. Importantly, inhibitors of autophagy (e.g. hydroxychloroquine, bafilomycin, and others) show synergy with CDK4/6 inhibitors in cancer cell lines, with the combination enhancing G1 arrest and the senescent phenotype [103, 105]. Evidence of CDK4/6 inhibitor-induced autophagy using in vivo systems is currently lacking, but such studies would help determine whether combination CDK4/6 inhibition and autophagy inhibition might have utility in clinical trials.
Another intriguing phenomenon is that CDK4/6 inhibitors can increase cancer cells’ presentation of tumor neoantigens on Major Histocompatibility Complex (MHC) Class I molecules, thus enhancing anti-tumor immune responses [72, 99, 107]. In cells expressing functional RB, CDK4/6 inhibitors reduce expression of the E2F target, DNMT1 (encoding a DNA methyltransferase). This results in DNA hypomethylation at certain transposable genomic elements, including those containing endogenous retroviral (ERV) sequences. The resultant ERV transcription increases tumor cell double-stranded RNA (dsRNA) content, provoking a dsRNA response that triggers tumor cell production of type III interferons and ultimately expression of interferon-stimulated genes including those encoding for the antigen presentation machinery [99]. Interestingly, upregulation of antigen presentation-related genes has also been observed in cells rendered senescent through other means, such as replicative senescence [108], and it is possible that CDK4/6 inhibitor-induced enhancement of tumor cell antigen presentation is but one component of the senescent phenotype. Moreover, previous studies have suggested that functional RB might also be required for tumor cells’ expression of MHC Class II molecules, although the mechanisms behind this are not known [109, 110]. In vivo, the increased tumor immunogenicity observed after CDK4/6 inhibition potentiates the activity of immune checkpoint blockade, a finding that has paved the way for clinical trials of combined CDK4/6 inhibition and immunotherapy in solid tumors [99, 107, 111].
Notably, although several groups have demonstrated enhanced anti-tumor immune responses after CDK4/6 inhibition in solid tumors [72, 99, 107, 112], one report has described the opposite. In that study, CDK4/6 inhibition reduced phosphorylation of the ubiquitin ligase adaptor SPOP, reducing ubiquitination of the programmed cell death protein ligand-1 (PD-L1). The resultant increase in tumor cell PD-L1 expression was associated with a reduction in effector T cell activity within murine mammary carcinomas [113]. Intriguingly, this suppression of anti-tumor immunity was associated with enhanced benefit of PD-1 blockade, thus also highlighting the promise of CDK4/6 inhibitor-immunotherapy combinations.
4. Identifying non-canonical CDK4/6 substrates
Given that RB is the canonical CDK4/6 substrate, the overwhelming majority of CDK4/6 inhibitor research in cancer has focused on RB-proficient cells. Moreover, tumor cells with low RB1 expression are generally insensitive to these agents in vitro, and loss of RB1 function is an emerging mechanism of CDK4/6 inhibitor resistance [26, 56, 57]. However, it is clear that CDKs 4 and 6 directly phosphorylate dozens if not hundreds of other proteins [114, 115], and that CDK4/6 inhibition can thus impact tumor cell biology in an RB-independent manner. Discussing the range of CDK4/6 substrates and their implications for cancer treatment is beyond the scope of this article, but some notable examples include the Forkhead Box 1 (FOXM1) transcription factor [115], key metabolic enzymes including 6-phosphofructokinase and pyruvate kinase M2 [116], the signal transduction protein IRS2 [117], the deubiquitinase DUB3 [118], and the ubiquitin ligase adaptor SPOP [113]. Given their protean cellular functions, the consequences of inhibiting the phosphorylation of these substrates are far-reaching, and have been shown to include modulation of tumor cell senescence, invasion and metastasis, metabolism, and immunogenicity [113, 115, 116, 118].
5. Effects of CDK4/6 inhibitors on other cell types
Finally, we must recognize that CDK4/6 inhibitor treatment in living organisms not only impacts tumor cells, but also other normal cell types within the tumor microenvironment and host, potentially impairing or enhancing tumor responses to therapy. For example, the importance of the CDK4/6 pathway in regulating both proliferation and senescence in fibroblasts has been understood for many years [98, 119], and a recent study demonstrated that CDK4/6 inhibitor-treated fibroblasts develop a senescent phenotype characterized by secretion of a large number of pro-inflammatory cytokines [120]. Hence, cancer-associated senescent fibroblasts may have the potential to enhance tumor growth by both directly stimulating tumor cells and also suppressing anti-tumor responses. Conversely, CDK4/6 inhibitors may have direct effects on specific lymphocyte populations that can potentiate anti-tumor immunity [99, 121]. First, CDK4/6 inhibitor treatment of human CD4+ T lymphocytes enhances their activity in vitro, as evidenced by upregulation of interleukin-2 secretion [112]. This occurs because CDK6 inhibition in particular enhances the nuclear translocation and activity of Nuclear Factor of Activated T cell (NFAT) family members [107, 112]. Second, CDK4/6 inhibitors suppress the proliferation of immunosuppressive regulatory T cells more than other T cell subsets, potentially shifting the immune balance in favor of an anti-tumor immune response [99]. Also noteworthy is the fact that endothelial cell proliferation and angiogenesis are CDK4/6 dependent phenomena [122], and the effects of CDK4/6 inhibition on tumor angiogenesis are not known. Finally, CDK4/6 inhibitors have been shown to inhibit the proliferation of bone marrow hematopoietic stem and progenitor cells, mitigating the damage these cells can incur from exposure to cytotoxic chemotherapy and providing a potential rationale for CDK4/6 inhibitors in the supportive care of patients receiving conventional chemotherapy [123]. Continued exploration of the effects of CDK4/6 inhibitors on other cell types, including adaptive and innate immune cells, endothelial cells, and other mesenchymal cells represents an important area of investigation.
Concluding Remarks
Pharmacologic inhibitors of CDKs 4 and 6 have changed the treatment landscape for breast cancer, and it is likely that their clinical indications will expand in the years to come. Although they were developed to inhibit tumor cell proliferation, their effects on cancer cells and other cells in the tumor microenvironment are clearly more extensive. Of particular importance, recent reports have highlighted that these agents can modulate intracellular kinase signaling, induce a senescence-like state, and enhance tumor cell immunogenicity. There are still many gaps in our understanding of how CDK4/6 inhibitors affect tumor cells, and why their impact differs in different cancer types (see Outstanding Questions). The key challenge now is to build upon this new information, much of it unexpected, and to then successfully integrate it into the design of new clinical trials. Only then will our patients stand the greatest chance of reaping the benefits of many decades of cell cycle research.
Outstanding Questions.
Which cancer types should be prioritized in future CDK4/6 inhibitor trials? Answering this question will require a deeper understanding of the molecular determinants of cancer cell sensitivity to these agents.
To what extent are the phenotypes induced by CDK4/6 inhibitors in vitro (e.g. senescence, immunogenicity, autophagy) observed in human tumors? This can only be addressed by analyzing tumor biopsies obtained from patients that are receiving these agents.
How do CDK4/6 inhibitors modify kinase signaling in cancer cells controlled by different driving oncogenes (e.g. PI3K pathway vs RAS pathway)?
By what mechanisms do CDK4/6 inhibitors impact the functions of T cells and other components of the adaptive immune system, and can they be used to enhance the effects of cancer immunotherapy?
What are the mechanisms by which tumor cells acquire resistance to CDK4/6 inhibitors?
Highlights.
Selective pharmacologic inhibitors of cyclin-dependent kinases 4 and 6 (CDK4/6) show immense promise as treatment for a wide variety of cancers
The primary mechanism of CDK4/6 inhibitor activity is suppression of RB phosphorylation, enforcing G1 cell cycle arrest, thus inhibiting proliferation.
CDK4/6 inhibitors can also impact other aspects of cancer cell behavior by inducing a senescence-like state, enhancing immunogenicity, and modulating kinase signaling
CDK4/6 inhibitors may also exert their activity through a direct effect on other cell types within tumors, including immune cells
Understanding the complex biological phenotypes induced by CDK4/6 inhibitors will ultimately allow for the development of new therapeutic combinations to further benefit patients
Abbreviations:
- AR
androgen receptor
- ER
estrogen receptor
- GFR
growth factor receptor
- GSK3β
glycogen synthase kinase-3 beta
- mTORC
mammalian target of rapamycin complex
- PI3K
phosphoinositide 3-kinase
- TSC
tuberous sclerosis complex
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5), 646–74. [DOI] [PubMed] [Google Scholar]
- 2.Fry DW et al. (2004) Specific inhibition of cyclin-dependent kinase 4/6 by PD 0332991 and associated antitumor activity in human tumor xenografts. Mol Cancer Ther 3 (11), 1427–38. [PubMed] [Google Scholar]
- 3.Kim S et al. (2014) LEE011: An orally bioavailable, selective small molecule inhibitor of CDK4/6-Reactivating Rb in cancer. Molecular Cancer Therapeutics 12 (PR02–PR02). [Google Scholar]
- 4.Gelbert LM et al. (2014) Preclinical characterization of the CDK4/6 inhibitor LY2835219: in-vivo cell cycle-dependent/independent anti-tumor activities alone/in combination with gemcitabine. Invest New Drugs 32 (5), 825–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Finn RS et al. (2009) PD 0332991, a selective cyclin D kinase 4/6 inhibitor, preferentially inhibits proliferation of luminal estrogen receptor-positive human breast cancer cell lines in vitro. Breast Cancer Res 11 (5), R77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Leary B et al. (2016) Treating cancer with selective CDK4/6 inhibitors. Nat Rev Clin Oncol 13 (7), 417–30. [DOI] [PubMed] [Google Scholar]
- 7.Malumbres M and Barbacid M (2009) Cell cycle, CDKs and cancer: a changing paradigm. Nat Rev Cancer 9 (3), 153–66. [DOI] [PubMed] [Google Scholar]
- 8.Narasimha AM et al. (2014) Cyclin D activates the Rb tumor suppressor by monophosphorylation. Elife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato J et al. (1993) Direct binding of cyclin D to the retinoblastoma gene product (pRb) and pRb phosphorylation by the cyclin D-dependent kinase CDK4. Genes Dev 7 (3), 331–42. [DOI] [PubMed] [Google Scholar]
- 10.Baldin V et al. (1993) Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev 7 (5), 812–21. [DOI] [PubMed] [Google Scholar]
- 11.Weintraub SJ et al. (1995) Mechanism of active transcriptional repression by the retinoblastoma protein. Nature 375 (6534), 812–5. [DOI] [PubMed] [Google Scholar]
- 12.Hiebert SW et al. (1992) The interaction of RB with E2F coincides with an inhibition of the transcriptional activity of E2F. Genes Dev 6 (2), 177–85. [DOI] [PubMed] [Google Scholar]
- 13.Harbour JW et al. (1999) Cdk phosphorylation triggers sequential intramolecular interactions that progressively block Rb functions as cells move through G1. Cell 98 (6), 859–69. [DOI] [PubMed] [Google Scholar]
- 14.Talluri S et al. (2010) A G1 checkpoint mediated by the retinoblastoma protein that is dispensable in terminal differentiation but essential for senescence. Mol Cell Biol 30 (4), 948–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chicas A et al. (2012) H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci U S A 109 (23), 8971–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rubin SM (2013) Deciphering the retinoblastoma protein phosphorylation code. Trends Biochem Sci 38 (1), 12–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sherr CJ and Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13 (12), 1501–12. [DOI] [PubMed] [Google Scholar]
- 18.Harper JW et al. (1993) The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 75 (4), 805–16. [DOI] [PubMed] [Google Scholar]
- 19.Blain SW et al. (1997) Differential interaction of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 with cyclin A-Cdk2 and cyclin D2-Cdk4. J Biol Chem 272 (41), 25863–72. [DOI] [PubMed] [Google Scholar]
- 20.LaBaer J et al. (1997) New functional activities for the p21 family of CDK inhibitors. Genes Dev 11 (7), 847–62. [DOI] [PubMed] [Google Scholar]
- 21.Sicinski P et al. (1995) Cyclin D1 provides a link between development and oncogenesis in the retina and breast. Cell 82 (4), 621–30. [DOI] [PubMed] [Google Scholar]
- 22.Malumbres M et al. (2004) Mammalian Cells Cycle without the D-Type Cyclin-Dependent Kinases Cdk4 and Cdk6. Cell 118, 493–504. [DOI] [PubMed] [Google Scholar]
- 23.Kozar K et al. (2004) Mouse development and cell proliferation in the absence of D-cyclins. Cell 118 (4), 477–91. [DOI] [PubMed] [Google Scholar]
- 24.Santamaria D et al. (2007) Cdk1 is sufficient to drive the mammalian cell cycle. Nature 448 (7155), 811–5. [DOI] [PubMed] [Google Scholar]
- 25.Sherr CJ et al. (2016) Targeting CDK4 and CDK6: From Discovery to Therapy. Cancer Discov 6 (4), 353–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gong X et al. (2017) Genomic Aberrations that Activate D-type Cyclins Are Associated with Enhanced Sensitivity to the CDK4 and CDK6 Inhibitor Abemaciclib. Cancer Cell 32 (6), 761–776 e6. [DOI] [PubMed] [Google Scholar]
- 27.Cerami E et al. (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2 (5), 401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gao J et al. (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signaling 6 (269), 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lundgren K et al. (2012) Effects of cyclin D1 gene amplification and protein expression on time to recurrence in postmenopausal breast cancer patients treated with anastrozole or tamoxifen: a TransATAC study. Breast Cancer Res 14 (2), R57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Erikson J et al. (1984) The chromosome 14 breakpoint in neoplastic B cells with the t(11;14) translocation involves the immunoglobulin heavy chain locus. Proc Natl Acad Sci U S A 81 (13), 4144–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Filmus J et al. (1994) Induction of cyclin D1 overexpression by activated ras. Oncogene 9 (12), 3627–33. [PubMed] [Google Scholar]
- 32.Roberts PJ and Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26 (22), 3291–310. [DOI] [PubMed] [Google Scholar]
- 33.Diehl JA et al. (1998) Glycogen synthase kinase-3beta regulates cyclin D1 proteolysis and subcellular localization. Genes Dev 12 (22), 3499–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Albers MW et al. (1993) FKBP-rapamycin inhibits a cyclin-dependent kinase activity and a cyclin D1-Cdk association in early G1 of an osteosarcoma cell line. J Biol Chem 268 (30), 22825–9. [PubMed] [Google Scholar]
- 35.Rosenwald IB et al. (1995) Eukaryotic translation initiation factor 4E regulates expression of cyclin D1 at transcriptional and post-transcriptional levels. J Biol Chem 270 (36), 21176–80. [DOI] [PubMed] [Google Scholar]
- 36.Yu Q et al. (2001) Specific protection against breast cancers by cyclin D1 ablation. Nature 411 (6841), 1017–21. [DOI] [PubMed] [Google Scholar]
- 37.Finn RS et al. (2016) Palbociclib and Letrozole in Advanced Breast Cancer. N Engl J Med 375 (20), 1925–1936. [DOI] [PubMed] [Google Scholar]
- 38.Hortobagyi GN et al. (2016) Ribociclib as First-Line Therapy for HR-Positive, Advanced Breast Cancer. N Engl J Med. [DOI] [PubMed] [Google Scholar]
- 39.Goetz MP et al. (2017) MONARCH 3: Abemaciclib As Initial Therapy for Advanced Breast Cancer. J Clin Oncol 35 (32), 3638–3646. [DOI] [PubMed] [Google Scholar]
- 40.Dickler MN et al. (2017) MONARCH 1, A Phase II Study of Abemaciclib, a CDK4 and CDK6 Inhibitor, as a Single Agent, in Patients with Refractory HR(+)/HER2(−) Metastatic Breast Cancer. Clin Cancer Res 23 (17), 5218–5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu Q et al. (2006) Requirement for CDK4 kinase function in breast cancer. Cancer Cell 9 (1), 23–32. [DOI] [PubMed] [Google Scholar]
- 42.Jeselsohn R et al. (2010) Cyclin D1 kinase activity is required for the self-renewal of mammary stem and progenitor cells that are targets of MMTV-ErbB2 tumorigenesis. Cancer Cell 17 (1), 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parker JS et al. (2009) Supervised risk predictor of breast cancer based on intrinsic subtypes. J Clin Oncol 27 (8), 1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Leonard JP et al. (2012) Selective CDK4/6 inhibition with tumor responses by PD0332991 in patients with mantle cell lymphoma. Blood 119 (20), 4597–607. [DOI] [PubMed] [Google Scholar]
- 45.Dickson MA et al. (2016) Progression-Free Survival Among Patients With Well-Differentiated or Dedifferentiated Liposarcoma Treated With CDK4 Inhibitor Palbociclib: A Phase 2 Clinical Trial. JAMA Oncol 2 (7), 937–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Patnaik A et al. (2016) Efficacy and Safety of Abemaciclib, an Inhibitor of CDK4 and CDK6, for Patients with Breast Cancer, Non-Small Cell Lung Cancer, and Other Solid Tumors. Cancer Discov 6 (7), 740–53. [DOI] [PubMed] [Google Scholar]
- 47.Geoerger B et al. (2017) A Phase I Study of the CDK4/6 Inhibitor Ribociclib (LEE011) in Pediatric Patients with Malignant Rhabdoid Tumors, Neuroblastoma, and Other Solid Tumors. Clin Cancer Res 23 (10), 2433–2441. [DOI] [PubMed] [Google Scholar]
- 48.DeMichele A et al. (2015) CDK 4/6 inhibitor palbociclib (PD0332991) in Rb+ advanced breast cancer: phase II activity, safety, and predictive biomarker assessment. Clin Cancer Res 21 (5), 995–1001. [DOI] [PubMed] [Google Scholar]
- 49.Shapiro GI et al. (2017) Phase 1 dose-escalation study of the CDK inhibitor dinaciclib in combination with the PARP inhibitor veliparib in patients with advanced solid tumors. Proc Am Assoc Cancer Res, C0147 [abstract]. [Google Scholar]
- 50.Garrido-Castro AC and Goel S (2017) CDK4/6 Inhibition in Breast Cancer: Mechanisms of Response and Treatment Failure. Curr Breast Cancer Rep 9 (1), 26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Finn R et al. (2016) Biomarker analyses from the phase 3 PALOMA-2 trial of palbociclib with letrozole compared with placebo plus letrozole in postmenopausal women with ER+/HER2-advanced breast cancer. Annals of Oncology 27 (7), 1–36. [Google Scholar]
- 52.Finn RS et al. , Comprehensive gene expression biomarker analysis of CDK 4/6 and endocrine pathways from the PALOMA-2 study, San Antonio Breast Cancer Symposium, San Antonio, TX, 2017. [Google Scholar]
- 53.Finn RS et al. (2015) The cyclin-dependent kinase 4/6 inhibitor palbociclib in combination with letrozole versus letrozole alone as first-line treatment of oestrogen receptor-positive, HER2-negative, advanced breast cancer (PALOMA-1/TRIO-18): a randomised phase 2 study. Lancet Oncol 16 (1), 25–35. [DOI] [PubMed] [Google Scholar]
- 54.Choi YJ et al. (2012) The requirement for cyclin D function in tumor maintenance. Cancer Cell 22 (4), 438–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sawai CM et al. (2012) Therapeutic targeting of the cyclin D3:CDK4/6 complex in T cell leukemia. Cancer Cell 22 (4), 452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dean JL et al. (2012) Therapeutic response to CDK4/6 inhibition in breast cancer defined by ex vivo analyses of human tumors. Cell Cycle 11 (14), 2756–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Condorelli R et al. (2017) Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Ann Oncol. [DOI] [PubMed] [Google Scholar]
- 58.Herschkowitz JI et al. (2008) The functional loss of the retinoblastoma tumour suppressor is a common event in basal-like and luminal B breast carcinomas. Breast Cancer Res 10 (5), R75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shapiro GI et al. (1995) Reciprocal Rb inactivation and p16INK4 expression in primary lung cancers and cell lines. Cancer Res 55 (3), 505–9. [PubMed] [Google Scholar]
- 60.Shapiro GI et al. (1995) Multiple mechanisms of p16INK4A inactivation in non-small cell lung cancer cell lines. Cancer Res 55 (24), 6200–9. [PubMed] [Google Scholar]
- 61.Turner NC et al. , Cyclin El (CCNE1) expression associates with benefit from palbociclib in metastatic breast cancer (MBC) in the PALOMA3 trial, AACR Annual Meeting, Chicago, IL, 2018, pp. Abstr CT-039. [Google Scholar]
- 62.Hortobagyi GN et al. , First-line ribociclib + letrozole in hormone receptor-positive, HER2-negative advanced breast cancer: Efficacy by baseline circulating tumor DNA alterations in MONALEESA-2, San Antonio Breast Cancer Symposium, San Antonio, TX, 2017, pp. PD4–06. [Google Scholar]
- 63.Wardell SE et al. (2015) Efficacy of SERD/SERM Hybrid-CDK4/6 Inhibitor Combinations in Models of Endocrine Therapy-Resistant Breast Cancer. Clin Cancer Res 21 (22), 5121–5130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Malorni L et al. (2017) A phase II trial of the CDK4/6 inhibitor palbociclib (P) as single agent or in combination with the same endocrine therapy (ET) received prior to disease progression, in patients (pts) with hormone receptor positive (HR+) HER2 negative (HER2-) metastatic breast cancer (mBC) (TREnd trial). Journal of Clinical Oncology 35 (15_suppl), 1002–1002.28297625 [Google Scholar]
- 65.Watts CK et al. (1994) Antiestrogen regulation of cell cycle progression and cyclin D1 gene expression in MCF-7 human breast cancer cells. Breast Cancer Res Treat 31 (1), 95–105. [DOI] [PubMed] [Google Scholar]
- 66.Miller TW et al. (2011) ERalpha-dependent E2F transcription can mediate resistance to estrogen deprivation in human breast cancer. Cancer Discov 1 (4), 338–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stice JP et al. (2017) CDK4/6 Therapeutic Intervention and Viable Alternative to Taxanes in CRPC. Mol Cancer Res 15 (6), 660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zacharek SJ et al. (2005) Negative regulation of TSC1-TSC2 by mammalian D-type cyclins. Cancer Res 65 (24), 11354–60. [DOI] [PubMed] [Google Scholar]
- 69.Goel S et al. (2016) Overcoming Therapeutic Resistance in HER2-Positive Breast Cancers with CDK4/6 Inhibitors. Cancer Cell 29 (3), 255–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Witkiewicz AK et al. (2014) CDK4/6 inhibition provides a potent adjunct to Her2-targeted therapies in preclinical breast cancer models. Genes Cancer 5 (7–8), 261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vora SR et al. (2014) CDK 4/6 inhibitors sensitize PIK3 CA mutant breast cancer to PI3K inhibitors. Cancer Cell 26 (1), 136–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Teo ZL et al. (2017) Combined CDK4/6 and PI3Kalpha Inhibition Is Synergistic and Immunogenic in Triple-Negative Breast Cancer. Cancer Res 77 (22), 6340–6352. [DOI] [PubMed] [Google Scholar]
- 73.Bonelli MA et al. (2017) Combined Inhibition of CDK4/6 and PI3K/AKT/mTOR Pathways Induces a Synergistic Anti-Tumor Effect in Malignant Pleural Mesothelioma Cells. Neoplasia 19 (8), 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ku BM et al. (2016) The CDK4/6 inhibitor LY2835219 has potent activity in combination with mTOR inhibitor in head and neck squamous cell carcinoma. Oncotarget 7 (12), 14803–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Michaloglou C et al. (2018) Combined inhibition of mTOR and CDK4/6 is required for optimal blockade of E2F function and long term growth inhibition in estrogen receptor positive breast cancer. Mol Cancer Ther. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Heilmann AM et al. (2014) CDK4/6 and IGF1 receptor inhibitors synergize to suppress the growth of p16INK4A-deficient pancreatic cancers. Cancer Res 74 (14), 3947–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wood AC et al. (2017) Dual ALK and CDK4/6 Inhibition Demonstrates Synergy against Neuroblastoma. Clin Cancer Res 23 (11), 2856–2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Romero-Pozuelo J et al. (2017) CycD/Cdk4 and Discontinuities in Dpp Signaling Activate TORC1 in the Drosophila Wing Disc. Dev Cell 42 (4), 376–387 e5. [DOI] [PubMed] [Google Scholar]
- 79.Inoki K et al. (2002) TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 4 (9), 648–57. [DOI] [PubMed] [Google Scholar]
- 80.Olmez I et al. (2017) Combined CDK4/6 and mTOR Inhibition Is Synergistic against Glioblastoma via Multiple Mechanisms. Clin Cancer Res 23 (22), 6958–6968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Fingar DC et al. (2004) mTOR controls cell cycle progression through its cell growth effectors S6K1 and 4E-BP1/eukaryotic translation initiation factor 4E. Mol Cell Biol 24 (1), 200–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Imperial R et al. (2017) Comprehensive pancancer genomic analysis reveals (RTK)-RAS-RAF-MEK as a key dysregulated pathway in cancer: Its clinical implications. Semin Cancer Biol. [DOI] [PubMed] [Google Scholar]
- 83.Chen SH et al. (2018) RAF inhibitor LY3009120 sensitizes RAS or BRAF mutant cancer to CDK4/6 inhibition by abemaciclib via superior inhibition of phospho-RB and suppression of cyclin D1. Oncogene 37 (6), 821–832. [DOI] [PubMed] [Google Scholar]
- 84.Ziemke EK et al. (2016) Sensitivity of KRAS-Mutant Colorectal Cancers to Combination Therapy That Cotargets MEK and CDK4/6. Clin Cancer Res 22 (2), 405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yadav V et al. (2014) The CDK4/6 inhibitor LY2835219 overcomes vemurafenib resistance resulting from MAPK reactivation and cyclin D1 upregulation. Mol Cancer Ther 13 (10), 2253–63. [DOI] [PubMed] [Google Scholar]
- 86.Tao Z et al. (2016) Coadministration of Trametinib and Palbociclib Radiosensitizes KRAS-Mutant Non-Small Cell Lung Cancers In Vitro and In Vivo. Clin Cancer Res 22 (1), 122–33. [DOI] [PubMed] [Google Scholar]
- 87.Hart LS et al. (2017) Preclinical Therapeutic Synergy of MEK½ and CDK4/6 Inhibition in Neuroblastoma. Clin Cancer Res 23 (7), 1785–1796. [DOI] [PubMed] [Google Scholar]
- 88.Kim HS et al. (2017) Oncogenic BRAF fusions in mucosal melanomas activate the MAPK pathway and are sensitive to MEK/PI3K inhibition or MEK/CDK4/6 inhibition. Oncogene 36 (23), 3334–3345. [DOI] [PubMed] [Google Scholar]
- 89.Pek M et al. (2017) Oncogenic KRAS-associated gene signature defines co-targeting of CDK4/6 and MEK as a viable therapeutic strategy in colorectal cancer. Oncogene 36 (35), 4975–4986. [DOI] [PubMed] [Google Scholar]
- 90.Zhou J et al. (2017) Palbociclib, a selective CDK4/6 inhibitor, enhances the effect of selumetinib in RAS-driven non-small cell lung cancer. Cancer Lett 408, 130–137. [DOI] [PubMed] [Google Scholar]
- 91.Kwong LN et al. (2012) Oncogenic NRAS signaling differentially regulates survival and proliferation in melanoma. Nat Med 18 (10), 1503–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roux PP et al. (2004) Tumor-promoting phorbol esters and activated Ras inactivate the tuberous sclerosis tumor suppressor complex via p90 ribosomal S6 kinase. Proc Natl Acad Sci U S A 101 (37), 13489–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ma L et al. (2005) Phosphorylation and functional inactivation of TSC2 by Erk implications for tuberous sclerosis and cancer pathogenesis. Cell 121 (2), 179–93. [DOI] [PubMed] [Google Scholar]
- 94.Lee MS et al. (2016) Efficacy of the combination of MEK and CDK4/6 inhibitors in vitro and in vivo in KRAS mutant colorectal cancer models. Oncotarget 7 (26), 39595–39608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Teh JLF et al. (2018) In vivo E2F reporting reveals efficacious schedules of MEK½-CDK4/6 targeting and mTOR-S6 resistance mechanisms. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Romano G et al. (2018) A pre-existing rare PIK3CAE545K subpopulation confers clinical resistance to MEK plus CDK4/6 inhibition in NRAS melanoma and is dependent on S6K1 signaling. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Narita M et al. (2003) Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 113 (6), 703–16. [DOI] [PubMed] [Google Scholar]
- 98.Shay JW et al. (1991) A role for both RB and p53 in the regulation of human cellular senescence. Exp Cell Res 196 (1), 33–9. [DOI] [PubMed] [Google Scholar]
- 99.Goel S et al. (2017) CDK4/6 inhibition triggers anti-tumour immunity. Nature 548 (7668), 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yoshida A et al. (2016) Induction of Therapeutic Senescence in Vemurafenib-Resistant Melanoma by Extended Inhibition of CDK4/6. Cancer Res 76 (10), 2990–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hernandez-Segura A et al. (2018) Hallmarks of Cellular Senescence. Trends Cell Biol. [DOI] [PubMed] [Google Scholar]
- 102.Chang J et al. (2016) Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat Med 22 (1), 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vijayaraghavan S et al. (2017) CDK4/6 and autophagy inhibitors synergistically induce senescence in Rb positive cytoplasmic cyclin E negative cancers. Nat Commun 8, 15916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Acevedo M et al. (2016) A CDK4/6-Dependent Epigenetic Mechanism Protects Cancer Cells from PML-induced Senescence. Cancer Res 76 (11), 3252–64. [DOI] [PubMed] [Google Scholar]
- 105.Valenzuela CA et al. (2017) Palbociclib-induced autophagy and senescence in gastric cancer cells. Exp Cell Res 360 (2), 390–396. [DOI] [PubMed] [Google Scholar]
- 106.Brown NE et al. (2012) Cyclin D1 activity regulates autophagy and senescence in the mammary epithelium. Cancer Res 72 (24), 6477–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Schaer DA et al. (2018) The CDK4/6 Inhibitor Abemaciclib Induces a T Cell Inflamed Tumor Microenvironment and Enhances the Efficacy of PD-L1 Checkpoint Blockade. Cell Rep 22 (11), 2978–2994. [DOI] [PubMed] [Google Scholar]
- 108.Kim YM et al. (2013) Implications of time-series gene expression profiles of replicative senescence. Aging Cell 12 (4), 622–34. [DOI] [PubMed] [Google Scholar]
- 109.Lu Y et al. (1996) Apoptosis-independent retinoblastoma protein rescue of HLA class II messenger RNA IFN-gamma inducibility in non-small cell lung carcinoma cells. Lack of surface class II expression associated with a specific defect in HLA-DRA induction. J Immunol 156 (7), 2495–502. [PubMed] [Google Scholar]
- 110.Lu Y et al. (1994) Evidence for retinoblastoma protein (RB) dependent and independent IFN-gamma responses: RB coordinately rescues IFN-gamma induction of MHC class II gene transcription in noninducible breast carcinoma cells. Oncogene 9 (4), 1015–9. [PubMed] [Google Scholar]
- 111.Rugo HS et al. , A phase 1b study of abemaciclib plus pembrolizumab for patients with hormone receptor-positive (HR+), human epidermal growth factor receptor 2-negative (HER2-) metastatic breast cancer (MBC), San Antonio Breast Cancer Symposium, San Antonio, TX., 2017, pp. P1-09-01. [Google Scholar]
- 112.Deng J et al. (2018) CDK4/6 Inhibition Augments Antitumor Immunity by Enhancing T-cell Activation. Cancer Discov 8 (2), 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zhang J et al. (2018) Cyclin D-CDK4 kinase destabilizes PD-L1 via cullin 3-SPOP to control cancer immune surveillance. Nature 553 (7686), 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Jirawatnotai S et al. (2014) The cyclin D1-CDK4 oncogenic interactome enables identification of potential novel oncogenes and clinical prognosis. Cell Cycle 13 (18), 2889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Anders L et al. (2011) A systematic screen for CDK4/6 substrates links FOXM1 phosphorylation to senescence suppression in cancer cells. Cancer Cell 20 (5), 620–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Wang H et al. (2017) The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature 546 (7658), 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lagarrigue S et al. (2016) CDK4 is an essential insulin effector in adipocytes. J Clin Invest 126 (1), 335–48. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 118.Liu T et al. (2017) CDK4/6-dependent activation of DUB3 regulates cancer metastasis through SNAIL1. Nat Commun 8, 13923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cheng M et al. (1999) The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J 18 (6), 1571–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Guan X et al. (2017) Stromal Senescence By Prolonged CDK4/6 Inhibition Potentiates Tumor Growth. Mol Cancer Res 15 (3), 237–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rowell EA et al. (2014) Regulation of T cell differentiation and alloimmunity by the cyclin-dependent kinase inhibitor p18ink4c. PLoS One 9 (3), e91587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Alhaja E et al. (2004) Anti-migratory and anti-angiogenic effect of p16: a novel localization at membrane ruffles and lamellipodia in endothelial cells. Angiogenesis 7 (4), 323–33. [DOI] [PubMed] [Google Scholar]
- 123.He S et al. (2017) Transient CDK4/6 inhibition protects hematopoietic stem cells from chemotherapy-induced exhaustion. Sci Transl Med 9 (387). [DOI] [PMC free article] [PubMed] [Google Scholar]