

Abstract
Epilepsy is often associated with altered expression or function of ion channels. One example of such a channelopathy is the reduction of A-type potassium currents in the hippocampal CA1 region. The underlying mechanisms of reduced A-type channel function in epilepsy are unclear. Here, we show that inhibiting a single microRNA, miR-324-5p, which targets the pore-forming A-type potassium channel subunit Kv4.2, selectively increased A-type potassium currents in hippocampal CA1 pyramidal neurons in mice. Resting membrane potential, input resistance and other potassium currents were not altered. In a mouse model of acquired chronic epilepsy, inhibition of miR-324-5p reduced the frequency of spontaneous seizures and interictal epileptiform spikes supporting the physiological relevance of miR-324-5p-mediated control of A-type currents in regulating neuronal excitability. Mechanistic analyses demonstrated that microRNA-induced silencing of Kv4.2 mRNA is increased in epileptic mice leading to reduced Kv4.2 protein levels, which is mitigated by miR-324-5p inhibition. By contrast, other targets of miR-324-5p were unchanged. These results suggest a selective miR-324-5p-dependent mechanism in epilepsy regulating potassium channel function, hyperexcitability and seizures.
Keywords: microRNA, Kv4.2, mir-324-5p, epilepsy, A-type potassium currents, antagomir, seizures, RNA-induced silencing complex, RISC, epileptiform spikes
Graphical Abstract
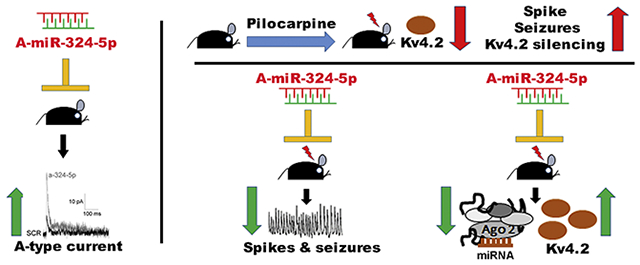
Introduction
Altered expression or function of ion channels, summarized under the term channelopathies, can lead to various neurological disorders including epilepsy. Seizures, the phenotypic determinants of epilepsy, are defined by periods of abnormally synchronized electrical activity in the brain due to neuronal hyperexcitability. Potassium channels are key regulators of neuronal excitability and are altered in genetic and acquired forms of epilepsy (Brenner and Wilcox, 2012; Villa and Combi, 2016). Members of all classes of potassium channels have been shown to be mutated or altered in epilepsy, with voltage-gated potassium channels comprising the largest group of affected channels (Köhling and Wolfart, 2016). For example, mutations in KCNA1 (coding for Kv1.1) were found in episodic ataxia type 1 and partial epilepsy (Zuberi et al., 1999), mutations in KCNQ1 (coding for Kv7.1) were linked to SUDEP (sudden unexplained death in epilepsy) (Goldman et al., 2009), and a growing number of mutations in KCNQ2 and KCNQ3 (coding for the M-type channels Kv7.2 and Kv7.3) have been associated with epilepsy (Köhling and Wolfart, 2016). Another example of a voltage-gated potassium channel altered in epilepsy is Kv4.2 (KCND2, Shal related subfamily D, member 2), the focus of this study.
Kv4.2 mediates somatodendritic A-type potassium currents in the hippocampus and cortex and is the predominant pore-forming A-type potassium channel subunit controlling neuronal excitability in the hippocampal CA1 region (Jerng and Pfaffinger, 2014; Jerng et al., 2004). Although mutations in KCND2 have been identified in epilepsy patients (Lee et al., 2014; Singh et al., 2006), it is not a bona fide epilepsy susceptibility gene. By contrast, there is strong evidence that impaired Kv4.2 function, independent of mutations in the gene, is associated with epilepsy: reduced Kv4.2 expression and function have been observed in several rodent models of acquired epilepsy, including pilocarpine-induced temporal lobe epilepsy (Bernard et al., 2004; Monaghan et al., 2008), traumatic brain injury (Lei et al., 2012), ischemic insult (Lei et al., 2014) and in acute seizure models (Francis et al., 1997; Tsaur et al., 1992). A causal relationship between reduced Kv4.2 function and neuronal hyperexcitability is corroborated by studies showing increased dendritic excitability and/or seizure susceptibility in mice lacking Kv4.2 or its auxiliary subunits DPP6 (Dipeptidyl-peptidase-like protein 6) and KChIP2 (potassium channel interacting protein 2) (Barnwell et al., 2009; Chen et al., 2006; Sun et al., 2011; Wang et al., 2013). Increased Kv4.2 protein levels in the brain correlate with prolonged latency to kainic acid-induced seizure (Gross et al., 2016). Together, these studies suggest that molecular mechanisms regulating Kv4.2 expression play an important role in the development and progression of epilepsy.
MicroRNAs regulate posttranscriptional gene expression by reducing the stability or translation of target mRNAs via the RNA-induced silencing complex (RISC) (Bartel, 2018). In recent years, many studies in human patients and animal models for epilepsy have suggested a role of microRNA-induced silencing in epilepsy (Henshall et al., 2016). The first microRNA shown to regulate the susceptibility to seizures was miR-134, which, when inhibited with antisense oligonucleotides before a seizure-inducing treatment, had an anticonvulsant effect (Jimenez-Mateos et al., 2012). In the following years, other microRNAs, for example miR-132, miR-146a, miR-34a, miR-128, miR-324-5p and miR-124 have also been implicated as positive or negative regulators of neuronal hyperexcitability (Tiwari et al., 2018). These findings suggest an important function of microRNA-induced silencing in epilepsy, but knowledge about the underlying mechanisms is limited. We have recently shown that the microRNA miR-324-5p reduces Kv4.2 expression. Inhibiting miR-324-5p is neuroprotective and increases the latency to seizure onset in mouse models of status epilepticus (Gross et al., 2016); however, it is unknown whether miR-324-5p-induced silencing regulates the functional expression of A-type currents in the hippocampus or contributes to seizure occurrence in established epilepsy.
The present study provides mechanistic insight into how microRNA-mediated control of neuronal excitability is dysregulated in epilepsy and may contribute to epilepsy-associated channelopathies. We show that inhibiting microRNA-induced silencing of Kv4.2 in mice with intracerebroventricular (ICV) injection of an antagomir to miR-324-5p reduces neuronal excitability by selectively increasing A-type potassium currents in hippocampal CA1 pyramidal neurons. MicroRNA-mediated silencing of Kv4.2 is enhanced in epileptic mice, whereas inhibition of miR-324-5p in epileptic mice impairs microRNA-induced silencing of Kv4.2, increases Kv4.2 protein levels, and reduces spontaneous recurrent seizures and interictal electrographic spike frequency. Overall, this study reveals a microRNA-mediated mechanism regulating A-type potassium currents that controls neuronal hyperexcitability in a mouse model of temporal lobe epilepsy.
Material and Methods
Animals
All experiments were conducted in accordance with the Institutional Animal Care and Use Committees of Cincinnati Children’s Hospital Medical Center (CCHMC) and University of Texas at Austin (UT Austin) and followed National Institute of Health Guidelines for the Care and Use of Laboratory Animals. All mice were housed in standard cages on a 14/10 (CCHMC) or 12/12 (UT Austin) light/dark cycle with access to food and water ad libitum. For all pilocarpine treatment experiments and associated controls, mice were obtained from the F1 offspring of male C57BL/6J (IMSR Cat# JAX:000664, RRID: IMSR_JAX:000664) and female FVB/NJ (IMSR Cat# JAX:001800, RRID: IMSR_JAX:001800) breeders at CCHMC and were used at 8-10 weeks of age. Offspring from these mixed breeding pairs require the same dose of pilocarpine to induce status epilepticus and have similar patterns of spontaneous recurrent seizures and mossy fiber sprouting as C57BL/6J mice but have shown better survivability to pilocarpine treatment compared to C57BL/6J mice (Hosford et al., 2016; Hosford et al., 2017). C57BL/6J mice (directly procured from Jackson Laboratory) were used at 6-8 weeks of age for patch clamp experiments. Kv4.2 knockout (KO) mice (in C57BL/6J background) were a generous gift from Dr. Jeanne Nerbonne (Guo et al., 2005) and were bred at CCHMC. Mice were housed in groups of up to four per cage. After surgeries (stereotaxic injections or EEG electrode and cannula implanting), mice were single-housed. Only male mice were used in the study. In all experiments, animals were assigned randomly to experimental groups and conditions. The number of animals and cohorts used for each experiment are indicated in the respective experimental sections.
Drugs, antibodies, antagomirs, and primers
Pilocarpine (cat# 0694/100) was obtained from Tocris (Minneapolis, MN); scopolamine methyl nitrate (cat# S2250) and diazepam (cat# 1185008) were obtained from Sigma-Aldrich (St. Louis, MO). Antagomirs were the same as described in Gross et al. (2016) and were LNA™-modified and custom-made by Qiagen (Hilden, Germany; formerly Exiqon). The following antibodies were used: rabbit polyclonal anti-Kv4.2 (Proteintech Group, Rosemont, IL Cat# 21298-1-AP, RRID:AB_10733102; used for all Kv4.2 western blots shown), rabbit polyclonal anti-Tubulin-β3 (BioLegend, San Diego, CA Cat# 802001, RRID:AB_2564645), rabbit monoclonal anti-Akt (Cell Signaling Technology, Danvers, MA Cat# 4691, RRID:AB_915783), mouse monoclonal anti-Akt (Cell Signaling Technology Cat# 2920, RRID:AB_1147620), mouse monoclonal anti-Kv4.2 (UC Davis/NIH NeuroMab Facility Cat# 75-016, RRID:AB_2131945), mouse monoclonal anti-Gli1 (Cell Signaling Technology Cat# 2643, RRID:AB_2294746), and mouse monoclonal anti-Ago2/eIFC2 (Abnova Corporation, Walnut, CA Cat# H00027161-M01, RRID:AB_565459). The following qRT-PCR primers were used: Kv4.2for: GCTTTGAGACACAGCACCAC; Kv4.2rev: TGTTCATCGACAAACTCATGG; β-tubfor: TCGTGGAATGGATCCCCAAC; β-tubrev: TCCATCTCGTCCATGCCCT; Gli1for: TGGAGGTCTGCGTGGTAGA; Gli1rev: TTGAACATGGCGTCTCAGG; Smofor: GCAAGCTCGTGCTCTGGT; Smorev: GGGCATGTAGACAGCACACA; miR-324-5p: CGCATCCCCTAGGGCATTGGTGT; RU19: GAGATCGTGTTACACTGTTGG.
Intracerebroventricular antagomir injection for slice electrophysiology
Six- to eight-week-old C57BL/6J mice received a unilateral ICV injection of miR-324-5p antagomir or scrambled control (0.5 nmol in 2 μl sterile artificial cerebrospinal fluid (ACSF)) as described previously (Gross et al., 2016). Briefly, one burr hole was drilled at the following stereotaxic coordinates from bregma, AP =−0.3 mm; L = ± 1.0 mm and V = 2.0 mm. Using a 5 μl Hamilton syringe, the antagomir was slowly administered over a 5-minute duration. The needle was left in place for 10 minutes to allow diffusion of the injected liquid followed by a slow, 5-minute retraction. Post injection, the open skin incision was closed using surgical sutures (Covidien, Medtronic, Minneapolis, MN), and GLUture (Zoetis Inc., Kalamazoo, MI). An antibiotic ointment (RARO, Hawthorne, NY) was applied to the top of the sutures. Mice were administered with 1 ml of sterile Ringer’s saline intraperitoneally (i.p.), placed on a heating pad and monitored until recovery. Two days post injection the mice were shipped to UT Austin for electrophysiological analyses. Four cohorts of mice sent in four separate shipments with three mice per condition (scrambled and miR-324-5p antagomir) were used for these experiments (24 mice total).
Acute hippocampal slices
Mice were allowed to recover for at least 2-3 days after arriving at UT Austin. Acute hippocampal slices were prepared as described (Brager et al., 2012). Briefly, mice were anesthetized with ketamine and xylazine and perfused intracardially using ice cold modified ACSF of the following composition (in mM): 210 sucrose, 2.5 KCl, 1.2 NaH2PO4, 25 NaHCO3, 0.5 CaCl2, 7.0 MgCl2, and 7.0 dextrose, bubbled with 95% O2/5% CO2. The brain was extracted and cut into 300 μm hippocampal sections from the middle hippocampus using a vibratome. The sections were placed in a holding chamber with ACSF of the following composition (mM): 125 NaCl, 2.5 KCl, 1.25 NaHPO4, 25 NaHCO3, 2.0 CaCl2, 2.0 MgCl2 and 21 dextrose, pH 7.4, bubbled with 95% O2/5% CO2 at 35°C for 45-60 mins and then at room temperature.
Hippocampal slices were individually placed into a submerged recording chamber and were continuously perfused with oxygenated extracellular saline of the following composition (mM): 125 NaCl, 3.0 KCl, 1.25 NaHPO4, 25 NaHCO3, 2.0 CaCl2, 2.0 MgCl2 and 21 dextrose, pH 7.4, bubbled with 95% O2/5% CO2 at 32 – 34°C. The slices were viewed using a Zeiss AxioExaminer D microscope fitted with 60X water-immersion objective and Dodt gradient contrast optics.
Electrophysiology
For cell-attached recordings, patch pipettes were pulled from borosilicate glass, wrapped with Parafilm to reduce capacitance, and filled with the following solution (in mM): 125 NaCl, 3 KCl, 10 HEPES, 2.0 CaCl2, 1.0 MgCl2 and 0.001 TTX, pH 7.3 with KOH. Membrane currents were recorded using an Axopatch 200B amplifier (Molecular Devices, San Jose, CA) and were sampled at 10 kHz, analog filtered at 2 kHz, and digitized by an ITC-18 interface connected to a computer running Axograph X. Isolation of the 3 different potassium currents, rapidly inactivating (IKA), slowly inactivating (IKslow) and sustained (IKsust), was performed as described previously (Kalmbach et al., 2015; Ordemann et al., 2019; Routh et al., 2013). Briefly, total potassium current IK was recorded from a hyperpolarizing holding potential of −90 mV. In a second protocol, a 100-ms pre-step to −20 mV was used to inactivate the IKA component. A third protocol was used to isolate the sustained component (IKsust) by measuring potassium currents from a more depolarized holding potential of −30mV. IKA, and when present IKslow, were calculated by offline subtraction. Activation curves were generated by depolarizing voltage commands (−90 to 50 mV in 10 mV steps) to activate IKA starting from a holding potential of −100 mV. Activation data were fit to single Boltzmann functions using a least-squares program. Linear leakage and capacitive currents were digitally subtracted by scaling traces at smaller command voltages in which no voltage-dependent current was activated.
For current clamp recordings, the pipette solution contained (in mM): 120 K-gluconate, 16 KCl, 10 Hepes, 8 NaCl, 7 K2-phosphocreatine, 0.3 Na-GTP, 4 Mg-ATP (pH 7.3 with KOH). Data were acquired using a Dagan BVC-700 amplifier (Dagan Corp., Minneapolis, MN, USA) and AxoGraph X (AxoGraph Scientific, Sydney, Australia) data acquisition software. Data were acquired at 10–50 kHz, filtered at 5–10 kHz and digitized by an ITC-18 (HEKA Instruments Inc., Holliston, MA, USA) interface. Pipette capacitance was compensated, and the bridge was balanced during each recording. Series resistance was monitored and compensated throughout each experiment and was 10–25 MΩ. Voltages were not corrected for the liquid-junction potential (estimated as ~8 mV). Data were analyzed using AxoGraph X. Input resistance was calculated from the linear portion of the current–voltage relationship in response to a family of 1-second current injections (−150 to +50 pA, 20 pA steps).
Epilepsy model (pilocarpine treatment)
Eight- to ten-week-old mice received i.p. injections of scopolamine methyl nitrate (1mg/kg) dissolved in sterile Ringer’s solution. After 15 minutes, the mice were injected i.p. with pilocarpine (320 mg/kg, in saline) or an equal volume of saline and were monitored for seizure activity and onset of status epilepticus (SE). The onset of SE was determined behaviorally as occurrence of multiple tonic-clonic seizures (class V), followed by which mice experienced continuous seizure activity (class II and III or higher) that lasted through the entire 3-hour observation period (Racine, 1972). Throughout the procedure the mice were housed in a standard cage without food, bedding, wired lids and water bottles to prevent injury and accidental chocking during SE. After 3 hours of continuous SE, seizure activity was terminated by i.p. injection of diazepam (10mg/kg). The mice were returned to their normal housing environment and kept on a heating pad, with soft food (diet gel) and water ad libitum. Sterile 0.9% saline solution was injected i.p. twice daily until the mice returned to their normal grooming behavior. Mice failing to enter SE within 1 hour of pilocarpine treatment were excluded from the study. All EEG-monitored mice exhibited spontaneous seizures within 2-3 weeks after pilocarpine injection. The overall survival rate after pilocarpine-induced SE in these experiments was approximately 51 percent 3 weeks after SE.
EEG transmitter and cannula implantation and antagomir injection
Three weeks post pilocarpine treatment (11 weeks of age), the mice were implanted with cortical electrodes for EEG monitoring (Castro et al., 2012; Gross et al., 2016; Hosford et al., 2016) and a cannula for future ICV treatment with antagomir or scrambled control. Mice were first anesthetized by placing them in a chamber supplied with 4% isoflurane in 1.5% oxygen. After anesthesia was established, the mice were placed on a stereotaxic frame and supplied with 1-1.5% isoflurane to maintain anesthesia throughout the procedure. Three burr holes were drilled into the skull with one for ICV cannula placement (Bregma; AP = −0.3 mm, L = ± 1.0 mm and V = −2.0mm), and two for the placement of EEG electrodes (Bregma; AP = −2.5 mm, L = ± 2.0mm (Tse et al., 2014)). The transmitter (TA11ETAF-10, Data Sciences International, St. Paul, MN) was placed subcutaneously behind the neck and the leads of the transmitter were placed at each burr hole for cortical surface EEG recordings. Electrodes were placed on the dura mater and care was taken not to penetrate it. A guide cannula with an outer closed cap (RWD Life Sciences, San Diego, CA; Plastics One, Roanoke, VA) was placed into the hole at the ICV coordinates and secured in place using bone wax. A stainless-steel screw (Plastics One, Roanoke, VA) was attached to the base of the skull and dental cement was applied to secure the setup in place. The incision was then closed with surgical suture, and GLUture and antibiotic ointment was applied on the top of the sutures. After continuous EEG/video-recording of spontaneous recurrent seizures for 2-3 weeks, mice were lightly anaesthetized with isoflurane and injected with 0.5 nmol scrambled or miR-324-5p-specific antagomir (in 2 μl ACSF) through the canula. EEG/video-monitoring continued for up to 4 additional weeks, after which mice were euthanized (9-10 weeks post SE). A total of 41 animals from 13 separate cohorts were injected with pilocarpine, of which 24 mice survived and were implanted with electrodes and a cannula 3 weeks after SE. One mouse was removed from the study because of insufficient EEG signal, and 5 mice died during the initial recording period. The remaining 18 mice were injected with antagomir and monitored for up to 4 weeks (10 miR-324-5p, 8 scrambled, randomly assigned). Most mice died before the end of the 4-week recording period post antagomir injection (8 in the miR-324-5p group, 6 in the scrambled group, also see Fig. S4). To ensure sufficient mouse numbers per group, spontaneous seizures were analyzed for 10 days post injection. Using these criteria, one miR-324-5p-injected mouse was excluded from the analysis due to premature death (8 days after injection). In addition, one scrambled-injected mouse was excluded due to technical issues with cannula implantation.
Continuous video-EEG recording
Mice were housed in individual static cages placed on wireless receiver plates (RPC-1, Data Sciences International (DSI™), St. Paul, MN). DATAQUEST A.R.T software was used for recording video EEG data (Axis 221, Axis communication, Lund, Sweden). Mice were continuously video-EEG recorded for the entire experiment (up to 7 weeks). Video and EEG data were analyzed using NeuroScore™ version 3.2.1 (DSI™). Spontaneous recurrent seizures were identified by behavioral changes evident in video analyses (Racine scale) and electrographic changes in the EEG. A seizure was defined as an abrupt increase in frequency and amplitude of the EEG signal (at least 2× baseline) with a minimum duration of 10 seconds. Seizure cessation was indicated by return of EEG signal to or below baseline and cessation of seizure behavior. For analysis of seizure frequency and seizure duration, continuous data from 10 days before and after antagomir injection were quantified.
Electrographic spike analysis
Interictal electrographic spikes were detected using the spike detector module in NeuroScore™ version 3.2.1. A spike was defined as having a duration of 1 – 70 ms and an absolute amplitude value of 150 μV – 1500 μV (Puttachary et al., 2015). Spike trains were defined as having a minimum duration of 5 ms with a minimum of 4 spikes spaced 80 – 5000 ms apart (Losing et al., 2017; Puttachary et al., 2015). EEG artifacts were manually removed. Mice with EKG signal or highly noisy EEG were excluded from the analysis (2 scrambled, 4 a-miR-324-5p). Data was analyzed during 2-hour periods between 10:00 am – 7:00 pm (light phase) daily for 3 days before and 3 days after injection of either miR-324-5p antagomir or scrambled control. Analysis periods were selected to be at least 1 hour before and after a seizure to avoid any pre-ictal or post-ictal spikes.
Generation of tissue samples from epileptic mice for biochemical analysis
Epileptic mice were generated by pilocarpine injection as described above. Hippocampal tissue was collected 3 and 5 weeks post pilocarpine treatment. These time points were chosen to analyze the expression levels of Kv4.2 and Gli1 protein, as well as RISC association and levels of Kv4.2, Gli1 and Smo mRNA and miR-324-5p at the corresponding time points used for electrode placement surgery and antagomir injection, respectively. The 3-week mice (8 saline, 8 pilocarpine) and 5-week mice (8 saline, 9 pilocarpine) were each generated from 6 different cohorts with 1-2 mice per condition. In another set of experiments, mice were ICV-injected with miR-324-5p-specific antagomir or scrambled control as described above 5 weeks post pilocarpine treatment. Ten days later, hippocampal tissue was collected for analysis. These mice were generated from 7 cohorts with 1-3 mice per condition (15 scrambled, 14 miR-324-5p). Mice were not implanted with electrodes or cannula to avoid confounding effects and reduce mortality.
Ago2 immunoprecipitation and mRNA quantification
Ago2 immunoprecipitations (IPs) were performed as described in Gross et al., 2016 with a few modifications. Tissue lysates were prepared in lysis buffer (50 mM Tris pH7.5, 40 mM NaCl, 1 mM EDTA, 0.5% Triton X-100, 50 mM NaF, 10 mM sodium pyrophosphate, 10 mM sodium β-glycerol phosphate, containing proteinase and RNase inhibitors). 200 μg of hippocampal lysate was incubated with 4 μg of Ago2 antibody or mouse normal IgG as control for 4 hours at 4°C on an end-over-end rotator, followed by incubation with Protein G-coupled agarose beads (Sigma-Aldrich, St. Louis, MO) for 2 hours. Precipitates were washed 6 times with wash buffer (20 mM Tris pH7.5, 150 mM NaCl, 5 mM MgCl2, 1% NP40, containing proteinase and RNase inhibitors), and RNA was extracted using Trizol® (Life Technologies, Carlsbad, CA). 20 μg of the lysate was kept as input sample at 4°C during the IP, and RNA was likewise extracted using Trizol®. Precipitated RNA from the IP was dissolved in 18 μl water and precipitated RNA from the input samples was dissolved in 36 μl water. cDNA was generated using the High Capacity RNA-to-cDNA kit (Applied Biosystems, Foster City, CA) for mRNA with 9 μl of the RNA solution, and the qScript™ microRNA cDNA synthesis kit (Quanta BioSciences, Gaithersburg, MD) for microRNA with 7 μl of the RNA solution, respectively. Specific mRNAs or microRNAs were quantified with SYBR green quantitative real-time PCR (qRT-PCR) (Bio-Rad Laboratories, Hercules, CA) using the QuantStudio 3 Real-Time PCR System (Applied Biosystems, Foster City, CA). Relative changes in expression were determined using the comparative cycle threshold method (2−ΔCT).
Western blotting
SDS-PAGE and Western blots were performed as described previously (Muddashetty et al., 2007) and quantified using NIH ImageJ software by experimenters blind to the condition. Samples were always loaded in duplicates, and signal intensities of Kv4.2 and Gli1 were normalized to βTubulin or Akt signal on the same blot. The different experimental conditions (saline/pilocarpine or miR-324-5p/scrambled antagomir-injected) were run on the same gels and equal protein amounts were loaded. For Kv4.2-specific western blots, hippocampal lysate from Kv4.2 KO mice was run on each gel to identify the Kv4.2-specific band.
Image processing and quantification
Figures were prepared using Adobe Photoshop CS6. To enhance visibility, brightness and contrast of western blot images were adjusted equally across conditions using the “level” tool in Adobe Photoshop software without changing the midtone. Densitometric quantification of western blots was performed on raw images using the gel analysis tool in ImageJ (NIH).
Experimental Design and Statistical Analysis
The goal of this study was to assess miR-324-5p-mediated silencing of Kv4.2 expression and function as a molecular mechanism contributing to hyperexcitability in epilepsy. At least 7 mice per condition were used and experimental groups were randomly assigned. Potassium currents and other membrane conductance properties were assessed in 4 separate cohorts with 3 mice per condition each. Mice used for all other experiments were obtained from at least 6 different litters to avoid litter bias. Slice electrophysiology, western blot, qRT-PCR and Ago-IP analyses were performed by experimenters blind to the conditions. Data were analyzed using GraphPad Prism7. Data sets were tested for normality using the Shapiro-Wilk test, and the appropriate parametric (unpaired t-test) or non-parametric tests (unpaired Mann-Whitney test) were used. Two-way ANOVA was used for analyses of data with two factors (Fig. 1C and S1B). Significance level was set to a<0.05, and p<0.05 was considered significant. Exclusion criteria for mice used for analyses of seizures and spikes in long-term EEG recordings are indicated in the respective method sections. Data are expressed in scatter plots and as means +/− SEM. Statistical tests used and their results as well as sample sizes are indicated in the figure legends.
Figure 1: Antagonizing miR-324-5p increases hippocampal A-type potassium currents in mice.

(A) Timeline depicting ICV antagomir injection, shipping and patch clamp recording for potassium current measurement. (B) Representative traces showing A-type potassium current recorded from a CA1 pyramidal neuron from mice injected with either scrambled or miR-324-5p antagomir. (C) Summary plot showing that the peak IKA was significantly greater at the most depolarized test voltages in mice treated with the antagomir compared to scrambled (SCR: n=9 from 3 mice; a-324-5p: n=7 from 3 mice; 2-way RM ANOVA with Sidak’s multiple comparison post hoc testing, interaction antagomir X membrane potential: F(16,224)=3.194, p<0.0001; *p=0.004 at VM = 50, and *p=0.005 at VM = 60 mV). (D) The maximum IKA current density was significantly greater in mice treated with antagomir compared to scrambled (SCR: n=20 from 9 mice; a-324-5p: n=16 from 9 mice; unpaired two-tailed Mann-Whitney test, *p=0.036). (E) There was no difference in the voltage-dependence of activation of IKA between scrambled and miR-324-5p antagomir-injected mice (n=8 from 3 mice each). (F,G) There was no significant difference in either the slowly inactivating (F: SCR: n=8 from 9 mice; a-324-5p: n=7 from 9 mice; unpaired two-tailed t-test, t(13)=1.032, p=0.321) or sustained (G: SCR: n=20 from 9 mice; a-324-5p: n=15 from 9 mice; two-tailed Mann-Whitney test, p=0.382) potassium currents between neurons from miR-324-5p antagomir- and scrambled antagomir-injected mice. Error bars represent SEM. Additional analyses shown in Fig. S1.
Contact for reagent and resource sharing:
The corresponding author Dr. Christina Gross (christina.gross@cchmc.org) can be contacted for further information and requests on reagent and resource sharing.
Results
In vivo inhibition of miR-324-5p increases A-type potassium current in CA1 pyramidal neurons
To assess if manipulation of the Kv4.2-targeting microRNA miR-324-5p modulates the functional expression of A-type potassium currents (IKA), we ICV-injected mice with miR-324-5p antagomirs and measured perisomatic IKA from CA1 pyramidal neurons 7-14 days later (timeline shown in Fig. 1A). Perisomatic potassium currents could be divided into multiple components based on kinetics and voltage-dependence (Kalmbach et al., 2015; Routh et al., 2013). Consistent with previous results (Hoffman et al., 1997; Routh et al., 2013) we found that the perisomatic expression of IKA in control mice (injected with scrambled antagomir) was relatively small. Treatment with miR-324-5p antagomirs significantly increased the maximum transient fast-inactivating potassium current (Fig. 1B–D). The area of the patch of membrane from which IKA was measured was not significantly different between scrambled and antagomir groups (scrambled: 1.4±0.2 μm2 (n=19) vs. miR-324-5p antagomir: 1.2±0.2 μm2 (n=16); unpaired t-test, t(33)=0.566, p=0.575). The increase in IKA was not due to a change in the voltage-dependence of activation between scrambled and antagomir treatment (Fig. 1E). A slowly inactivating potassium current was present in a small subset of cell-attached patches, consistent with previous reports from CA1 pyramidal neurons (Hoffman et al., 1997; Routh et al., 2013) (Fig. 1F). There was no significant difference in either the slowly inactivating or sustained potassium currents (Fig. 1F and G). Current clamp recordings revealed that resting membrane potential and input resistance were also not significantly different between the groups (Fig. S1A–C). These results suggest that miR-324-5p inhibition selectively increases the maximum perisomatic A-type potassium current in CA1 pyramidal neurons without affecting other perisomatic potassium currents.
miR-324-5p inhibition reduces seizure frequency in epileptic mice
We next tested if increasing A-type potassium currents by miR-324-5p inhibition affected the frequency of spontaneous seizures in a mouse model of temporal lobe epilepsy. Previous research has shown that A-type potassium channel function is decreased in acquired epilepsy (Bernard et al., 2004; Poolos and Johnston, 2012). A-type potassium currents reduce neuronal excitability suggesting that their impaired function may contribute to the occurrence of spontaneous recurrent seizures in epilepsy. Increasing A-type potassium currents by inhibition of miR-324-5p may thus reduce the seizure phenotype in acquired epilepsy. To test this hypothesis, we induced epilepsy in mice through pilocarpine-evoked SE (timeline shown in Fig. 2A). Three weeks after SE, mice were implanted with cortical surface electrodes and a capped ICV cannula for later injections. Mice were continuously video-EEG monitored for 2 to 3 weeks to confirm the occurrence of spontaneous recurrent seizures, after which they were ICV-injected through the cannula with either scrambled or miR-324-5p-specific antagomirs and video-EEG monitored for another 10-28 days (example EEG traces shown in Fig. 2B).
Figure 2: Antagonizing miR-324-5p in vivo reduces seizure frequency in a pilocarpine mouse model of epilepsy.

(A) Timeline depicting age of mice during pilocarpine injection and antagomir treatment. (B) Representative EEG traces of spontaneous seizures in the pilocarpine model following scrambled or miR-324-5p antagomir treatment. Seizures are indicated with lines above the EEG. (C) No significant difference was observed in seizure frequency in mice before injection of scrambled or miR-324-5p-specific antagomirs (unpaired two-tailed t-test, t(14)=0.898, p=0.384). (D) ICV injection of a miR-324-5p-specific antagomir reduces seizure frequency on average by half compared to the scrambled control (unpaired two-tailed t-test, t(14)=2.221, *p=0.043). (E) No significant effect between the two treatment groups was observed on the mean seizure duration between scrambled or miR-324-5p antagomir-injected mice during the pre-treatment period (two-tailed Mann-Whitney test, p=0.142). (F) Similarly, no difference in seizure duration after miR-324-5p antagomir treatment was observed (two-tailed Mann-Whitney test, p=0.142). N was 7 for scrambled, and 9 for miR-324-5p antagomir-treated mice. Error bars represent SEM.
No significant difference in mean seizure frequency was observed between groups during the pre-treatment period (Fig. 2C). Following miR-324-5p antagomir injection, however, treated animals displayed a significant reduction in seizure frequency compared to scrambled controls (Fig. 2D). Seizure frequency was, on average, reduced to 50% compared to scrambled antagomir-treated mice, although individual results varied. No significant difference in the duration of individual seizures between the two groups was detected before (Fig. 2E) or after the antagomir treatment (Fig 2F). There was also no difference in survival rates after treatment during a 4-week observation period (Fig. S4). Experiments using a fluorescently labeled antagomir confirmed previous findings (Jimenez-Mateos et al., 2012) and showed that the antagomir was stable over the 10-day monitoring period (Fig. S5). Overall, these findings suggest that inhibiting miR-324-5p reduces the occurrence but not duration of spontaneous recurrent seizures in epileptic mice.
miR-324-5p inhibition reduces the number of spikes and spike trains during interictal phases in epileptic mice
Neuronal network hyperexcitability in epilepsy is often characterized by irregular and increased numbers of electrographic spikes or spike trains during interictal periods (Chabrol et al., 2010; Puttachary et al., 2015) (examples shown in Fig. 3A, extended trace shown in Fig. S6). No difference in the number of spikes and spike trains was observed between groups during the pre-treatment period (Fig. 3B and 3D); however, a significant reduction in the number of spikes and spike trains was observed post treatment with miR-324-5p-specific antagomirs compared to scrambled controls (Fig. 3C and 3E). These results further corroborate the suppressive effects of miR-324-5p inhibition on hyperexcitability in epileptic mice.
Figure 3: Antagonizing miR-324-5p in vivo reduces the number of interictal spikes and spike trains in epileptic mice.

(A) Representative EEG traces of interictal spikes in scrambled or miR-324-5p antagomir-injected mice. (B,D) Before treatment, no significant differences were observed in total spike count (B, unpaired two-tailed t-test, t(8)=0.590, p=0.572) and the number of spike trains (D, unpaired two-tailed t-test, t(8)=1.195, p=0.266) between scrambled and miR-324-5p mice (2-hour period over 3 days). (C,E) After treatment, total spike number (C, unpaired two-tailed t-test, t(8)=2.676, *p=0.028) and total spike trains (E, unpaired two-tailed t-test, t(8)=2.51, *p=0.036) were significantly lower in miR-324-5p-specific antagomir-injected mice compared to the scrambled control (2-hour period over 3 days). Error bars represent SEM, n=5 for both conditions. Also see extended EEG trace in Fig. S6.
Antagonizing miR-324-5p in chronically seizing mice reduces microRNA-induced silencing of Kv4.2
We next tested the hypothesis that miR-324-5p inhibition reduces microRNA-induced silencing of Kv4.2 in the pilocarpine mouse model of epilepsy. In a separate experiment, mice were ICV-injected with a miR-324-5p antagomir or a scrambled antagomir 5 weeks post pilocarpine treatment, and hippocampal tissue was collected 10 days later (timeline shown in Fig. 4A). The timeline for these experiments paralleled the timeline used for seizure and spike analyses. RISC association of Kv4.2 mRNA was assessed as a surrogate for its microRNA-mediated silencing through RNA co-immunoprecipitation with an antibody specific to the RISC component Ago2 (Argonaute 2) followed by qRT-PCR (schematic shown in Fig. 4B) (Choe et al., 2010; Jimenez-Mateos et al., 2012). These experiments confirmed our hypothesis and showed that ICV injection with the miR-324-5p antagomir resulted in reduced association of Kv4.2 mRNA with Ago2 (Fig. 4C) and in elevated Kv4.2 protein levels (Fig. 4D and E) in the hippocampus of spontaneously seizing mice. Kv4.2 mRNA levels were not significantly changed (Fig. 4F). These results suggest that miR-324-5p inhibition reduces microRNA-induced silencing of Kv4.2 in epileptic mice leading to increased Kv4.2 protein levels.
Figure 4: miR-324-5p antagomir reduces microRNA-induced silencing of Kv4.2 in the pilocarpine model.

(A) Timeline depicting age of the mice during pilocarpine injection, antagomir treatment and tissue collection. (B) Schematic of Ago2-specific immunoprecipitation to assess Kv4.2 mRNA levels in the RISC. (C) Reduced association of Kv4.2 mRNA with Ago2 was observed after miR-324-5p antagomir treatment compared to scrambled control (C: SCR: n=15, a-324-5p: n=14, unpaired one-tailed t-test, t(27)=1.940, *p=0.031). mRNA levels in Ago2-IPs were quantified by qRT-PCR and normalized to input levels. (D,E) Western blot analyses of hippocampal lysates show significantly increased Kv4.2 expression in chronically seizing mice 10 days after miR-324-5p antagomir treatment compared to scrambled control (SCR: n=15, a-324-5p: n=14, unpaired one-tailed Mann-Whitney test, *p=0.047). Example blot of 2 different mice per condition (#1-4, loaded in duplicates) shown in D, cumulative quantification shown in E. Kv4.2-specific signal was normalized to loading control (Akt or βTubulin) on the same blot. Asterisk indicates Kv4.2-specific band that is absent in lysates from Kv4.2 KO mice. (F) Kv4.2 mRNA levels (normalized to βtubulin) were not significantly changed after antagomir treatment (SCR: n=15, a-324-5p: n=14, unpaired one-tailed t-test, t(27)=0.916, p=0.184). Error bars represent SEM.
MicroRNAs target more than one mRNA, and it is thus likely that additional mRNA targets, apart from Kv4.2, contribute to the seizure-suppressing effect of miR-324-5p. Only a few other mRNAs have been experimentally validated to be targeted by miR-324-5p. Two of them, Gli1 and Smoothened (Smo), are strong candidates because they have been shown to be targets of miR-324-5p in independent studies (Ferretti et al., 2008; Xu et al., 2014). Moreover, Gli1 and Smo are components of the Sonic Hedgehog signaling pathway, which is implicated in epilepsy (Banerjee et al., 2005; Fang et al., 2011; Feng et al., 2016). We therefore tested if microRNA-induced silencing of Gli1 and Smo was reduced by miR-324-5p inhibition in epileptic mice, similarly, as observed for Kv4.2. We observed no significant changes in Gli1 or Smo mRNA association with the RISC in epileptic mice following miR-324-5p inhibition compared to treatment with a scrambled antagomir (Fig. 5A and B). Likewise, Gli1 protein levels, and Smo and Gli1 mRNA levels were unchanged (Fig. 5C–E). We were unable to analyze Smo protein levels due to lack of a reliable antibody. These results suggest that the suppressive effects of the miR-324-5p antagomir on hyperexcitability in epilepsy are mediated through inhibition of microRNA-induced silencing of Kv4.2 but not Gli1 or Smo.
Figure 5: Gli1 and Smo expression are not affected by miR-324-5p inhibition in epileptic mice.

(A,B) In contrast to Kv4.2 mRNA, association of Gli1 (A) and Smo (B) mRNA with Ago2 is not significantly changed in epileptic mice after miR-324-5p inhibition (SCR: n=8, a-324-5p: n=7, unpaired one-tailed t-test, A: t(13)=1.087, p=0.148; B: t(13)=0.836, p=0.209). (C-E) Likewise, neither Gli1 protein (C) nor Gli1 (D) or Smo (E) mRNA are affected by the in vivo antagomir treatment (C: n=9, unpaired one-tailed t-test, t(16)=0.450, p=0.330; D: SCR: n=9, a-324-5p: n=7, unpaired one-tailed t-test, t(14)=0.039, p=0.485; E: SCR: n=9, a-324-5p: n=7, one-tailed Mann-Whitney test, p=0.105). Example western blot of two mice per condition (#1-4, loaded in duplicates) for C shown at top. Ago-IPs were normalized to input. Error bars represent SEM.
MicroRNA-induced silencing of Kv4.2 is increased in epileptic mice
To assess if miR-324-5p inhibition exerts its effect through targeting an epilepsy-associated mechanism (as opposed to merely reducing neuronal excitability by increasing A-type potassium currents independent of underlying mechanisms), we next tested if microRNA-induced silencing of Kv4.2 mRNA is increased in the chronic phase of the pilocarpine model of epilepsy (Fig. 6). In line with previous results (Bernard et al., 2004) we observed that in the chronic phase of the pilocarpine model of epilepsy, 3 or 5 weeks after SE, respectively, Kv4.2 mRNA was reduced (Fig. 6C and F, timeline shown in Fig. 6A). By contrast, Kv4.2 protein levels were significantly decreased only after 5 weeks (Fig. 6E) but not after 3 weeks (Fig. 6B). As an estimate of the level of microRNA-induced silencing of Kv4.2 in epileptic mice, we then quantified RISC association using Ago2-specific immunoprecipitation as above. No significant change in RISC-association of Kv4.2 mRNA was observed 3 weeks after pilocarpine treatment (Fig. 6D); however, association of Kv4.2 mRNA with the RISC was significantly increased 5 weeks after pilocarpine-induced SE compared to saline control suggesting that microRNA-induced silencing of Kv4.2 is elevated in mice with frequent spontaneous seizures (Fig. 6G). These results suggest that microRNA-mediated silencing of Kv4.2 is increased in epileptic mice which may contribute to reduced Kv4.2 protein levels and function. miR-324-5p levels or association with the RISC were not significantly changed 3 and 5 weeks after SE (Fig. S2). We also did not detect significant changes in the expression of either Gli1 mRNA and protein, or Smo mRNA 3 and 5 weeks after SE (Fig. S3A–F). In line with these findings, no significant changes in RISC association of Gli or Smo mRNA were detected 5 weeks after SE, although both showed a trend towards increased association (Fig. S3G and S3H). Together with the lack of an effect of the miR-324-5p antagomir on Gli1 and Smo in epileptic mice (Fig. 5), these results further support the hypothesis that miR-324-5p reduces seizure and interictal spike frequency in epilepsy through regulating Kv4.2 but not Gli1 or Smo.
Figure 6: MicroRNA-induced silencing of Kv4.2 is increased in epileptic mice.

(A) Timeline depicting age of mice during pilocarpine injection and tissue collection. (B-D) Three weeks after pilocarpine injection, when mice start to develop spontaneous seizures, no significant reduction in Kv4.2 protein levels (B: n=8, unpaired two-tailed t-test, t(14)=1.899, p=0.078) or Kv4.2 mRNA association with Ago2 (i.e. the RISC) (D: n=8, unpaired two-tailed t-test, t(14)=1.168, p=0.262), but significantly reduced Kv4.2 mRNA (C: n=8, unpaired two-tailed t-test, t(14)=4.470, *p<0.001) was observed. (E-G) Five weeks following pilocarpine treatment, when mice have frequent spontaneous seizures, significantly reduced Kv4.2 protein (E: n=8, unpaired two-tailed t-test, t(14)=3.513, *p=0.003) and mRNA levels (F: saline: n=8, pilo: n=9; unpaired two-tailed t-test, t(15)=4.218, *p<0.001), and significantly increased association of Kv4.2 mRNA with Ago2 (G: saline: n=7, pilocarpine: n=9, two-tailed Mann-Whitney test, *p=0.008) compared to saline control was observed. Example western blots for B and E are shown at top (samples loaded in duplicates), Kv4.2-specific signal was normalized to Akt on the same blot. Asterisk indicates Kv4.2-specific band that is absent in lysates from Kv4.2 KO mice. Total mRNA levels in C and F were quantified by qRT-PCR and normalized to βtubulin. mRNA levels in Ago2-IPs in D and G were quantified by qRT-PCR and normalized to input levels. Error bars represent SEM.
Discussion
Discovering the molecular mechanisms that contribute to neuronal hyperexcitability in epilepsy is crucial to understand how seizures develop and epileptogenesis progresses, and may help to identify novel treatment strategies. Hippocampal A-type potassium currents are reduced in models of acquired and genetic epilepsy suggesting a mechanism of increased neuronal excitability; however, the underlying molecular processes are largely unknown. The present study contributes to the understanding of these mechanisms by reporting a microRNA-mediated mechanism regulating neuronal excitability in a mouse model of epilepsy through specific modulation of hippocampal A-type potassium currents. Our results show that inhibition of a single microRNA, miR-324-5p, in epileptic mice reduces epilepsy-associated downregulation of Kv4.2, the major channel responsible for A-type potassium currents in the hippocampal CA1 region. This leads to overall reduced neuronal excitability as evident by decreased seizure frequency and decreased numbers of interictal epileptiform spikes. These results have several important implications. First, they show that a single microRNA can regulate the intrinsic excitability of neurons by modulating A-type potassium currents. Second, they show that microRNA-induced silencing of select mRNAs is increased in epilepsy, suggesting a mechanism contributing to neuronal hyperexcitability. Third, they corroborate the important role of Kv4.2 in regulating excitability in chronically seizing mice. Last but not least, they also hint at the potential therapeutic value of antagomirs in established epilepsy – so far, the majority of studies has tested the effect of microRNA inhibition before or shortly after SE on the development of spontaneous seizures.
Several previous studies, including our own, have shown that manipulation of select microRNAs can alter the response to proconvulsive stimuli or reduce the development of spontaneous seizures following SE (Gross et al., 2016; Iori et al., 2017; Jimenez-Mateos et al., 2012); however, it was previously unknown if any of these microRNAs directly regulate the intrinsic excitability of neurons by modulating specific ion channels. Here, we show that ICV injection of an antagomir to miR-324-5p selectively increased A-type potassium currents in CA1 pyramidal neurons (Fig. 1). The maximum IKA in CA1 pyramidal neurons was increased in mice treated with the miR-324-5p antagomir. By contrast, the miR-324-5p antagomir did not alter the slowly inactivating or sustained potassium currents and did not change resting membrane potential or input resistance. These observations suggest that miR-324-5p may be a useful tool to specifically modulate A-type potassium currents in the brain in vivo, while leaving other potassium currents unaltered. We cannot exclude that the miR-324-5p antagomir affects other ion channels that were not tested here. Nevertheless, the observed selectivity towards A-type potassium currents could alleviate concerns about the application of antagomirs as therapeutics due to their assumed widespread effect on many targets. Endogenous Kv4.2 channels are expressed somatodendritically with increasing density towards distal parts of the dendrite. Here, we show that perisomatic A-type currents are elevated by the miR-324-5p antagomir, suggesting that an ectopic increase of Kv4.2 close to the soma can contribute to the reduction of neuronal hyperexcitability in epilepsy.
We observed increased association of Kv4.2 mRNA with the RISC in chronically seizing mice, which was reduced by inhibition of miR-324-5p and correlated with increased Kv4.2 protein levels (Fig. 4 and 6). These results suggest that the miR-324-5p antagomir directly targets an underlying disease mechanism that contributes to the reduction of Kv4.2 expression and function in epileptic mice, namely increased microRNA-induced silencing of Kv4.2. Notably, we only observed significantly reduced Kv4.2 protein at 5 weeks, but not 3 weeks after SE. The pilocarpine model of epilepsy is progressive, and mice usually have less spontaneous recurrent seizures initially (3 weeks) compared to later stages of the disease (5 weeks). In our experience, many mice just start to develop spontaneous recurrent seizures around 3 weeks after SE. Our results thus suggest that a longer period of spontaneous seizures or a more severe seizure phenotype are required to consistently reduce Kv4.2 protein levels. The mechanisms of how Kv4.2 mRNA is recruited to the RISC in epilepsy are unclear. miR-324-5p was not increased in the RISC (Fig. S2), and we therefore speculate that other mechanisms, e.g. phosphorylation of protein components of the RISC, may be involved. Of note, in a previous study by Song et al. miR-324-5p was shown to be upregulated 60 days following Lithium-pilocarpine-induced SE in rats (Song et al., 2011). In our investigation, we observed a non-significant upregulation of miR-324-5p expression in hippocampal tissue at 3 and 5 weeks after pilocarpine-induced SE, supporting this previous study. As we report relative association of miR-324-5p and Kv4.2 mRNA with the RISC (i.e. the ratio of IP versus total levels), the total amount of miR-324-5p in the RISC may thus still be increased in epileptic mice. Interestingly, another study has reported that miR-324-5p is downregulated 24 hours post pilocarpine-induced SE (Kretschmann et al., 2015). This suggests an A-type potassium current-upregulating, compensatory effect during early epileptogenesis, and is supported by another study showing increased Kv4.2 levels within hours after SE (Su et al., 2008). The role of microRNA-induced silencing in regulation of Kv4.2 during these earlier time points is unclear and requires further investigation.
An important finding of this study is that antagomir-mediated inhibition of miR-324-5p reduced the occurrence of interictal epileptiform spikes (Fig. 3), in addition to decreasing overall seizure frequency (Fig. 2). Previous studies have shown that single microRNAs can alter seizure susceptibility and frequency in rodent models of epilepsy (Tiwari et al., 2018). Our results add to the impact single microRNAs have on epilepsy phenotypes by demonstrating that they can also alter neuronal excitability during interictal phases, which may contribute to their anticonvulsant effects. We did not detect significant changes in duration of single seizures between scrambled and miR-324-5p antagomir-treated mice (Fig. 2F). This suggests that the mechanisms which regulate seizure duration are not affected by miR-324-5p inhibition.
We observed some variability in the effect of miR-324-5p-inhibition on seizure frequency in individual mice. The pilocarpine model of epilepsy is progressive over time and variable in development and severity of spontaneous seizures (Arida et al., 1999; Hosford et al., 2017; Lim et al., 2018), which may have contributed to the fluctuations in effectiveness of antagomir treatment in this and a previous study (Iori et al., 2017). Other factors that might have influenced seizure severity in this model are the repeated exposure to isoflurane, which was shown to affect seizure susceptibility and occurrence of spontaneous seizures (Bar-Klein et al., 2016), and the potential injury or trauma caused by the ICV cannula. Even if these variables factored into seizure frequency, they would have not influenced the overall result that miR-324-5p inhibition was seizure-suppressive, because mice in both groups were treated equally and would have been affected similarly.
Future work is needed to evaluate miR-324-5p inhibition in less severe models of epilepsy. Related to this, it is unknown if the observed mechanism of increased microRNA-induced silencing of Kv4.2 is present in epilepsy caused by other factors, such as traumatic brain injury or gene mutations. Addressing this question will be important to assess the potential utility of miR-324-5p inhibition as a treatment strategy. In this study, we showed that miR-324-5p inhibition reduces spontaneous recurrent seizures and increases Kv4.2 protein expression in epileptic mice within 10 days post treatment but it is unclear if this effect is temporary or permanent. It will be vital to investigate if a transient inhibition of miR-324-5p leads to lasting changes that alter the long-term progression of the disease.
Our results suggest that inhibition of miR-324-5p in epileptic mice does not affect the previously reported miR-324-5p targets Gli1 and Smo; however, we only analyzed one dose and time point. MicroRNA-induced silencing of mRNAs strongly depends on the relative amounts of microRNA, their mRNA targets and competing endogenous RNAs within a cell (Chan and Tay, 2018); therefore, the effect of inhibiting a microRNA can vary between its targets. A recent study suggests that miR-324-5p targets Gli1 only in human but not murine cells, which could also have contributed to a lack of an effect in our experiments (Woods et al., 2018). Gli1 and Smo were chosen for analysis because they have been experimentally confirmed as miR-324-5p targets in two different studies (Ferretti et al., 2008; Xu et al., 2014), and because the Sonic Hedgehog signaling pathway has been implicated in epilepsy (Banerjee et al., 2005; Fang et al., 2011; Feng et al., 2016). We have not assessed other potential targets of miR-324-5p. While our electrophysiological analyses support a specific effect on CA1 A-type potassium currents, we speculate that modulation of other miR-324-5p-targeted mRNAs, for example in different brain regions, contributes to the anticonvulsant effects of its inhibition. The relatively subtle effect of miR-324-5p inhibition on Kv4.2 protein levels and RNA-induced silencing in epileptic mice and the fact that the antagomir can be taken up by neurons and glial cells further supports the hypothesis that other targets in other cell types, in addition to the neuronal Kv4.2, may be involved. Notably, microRNAs can have opposing effects on seizure development through targeting multiple mRNAs and cellular mechanisms (Brennan et al., 2016). It therefore will be important to reveal a more complete picture of the mRNAs regulated by miR-324-5p in epilepsy to fully understand its function.
In conclusion, our study illustrates the potential of miR-324-5p inhibition to reduce seizures and network excitability in epileptic mice by reducing microRNA-induced silencing of the A-type potassium channel Kv4.2. Our results support the hypothesis that miR-324-5p inhibition targets a disease-contributing mechanism. However, due to the relatively short timeline of analyses in this study, it is unclear whether or not miR-324-5p inhibition has a long-term disease-modifying effect. Future investigation is needed to fully understand the underlying mechanisms and mRNA targets involved; nonetheless, this study further corroborates the possible use of specific antagomirs in the treatment of epilepsy (Henshall et al., 2016), a strategy that is already being actively pursued in clinical trials for other diseases, for example Hepatitis C (Qiu and Dai, 2014; van der Ree et al., 2014; van der Ree et al., 2016).
Supplementary Material
Highlights.
miR-324-5p regulates intrinsic excitability by modulating A-type potassium currents.
miR-324-5p inhibition reduces frequency of seizures in a mouse model of epilepsy.
miR-324-5p inhibition reduces interictal epileptiform spikes in epileptic mice.
miR-324-5p inhibition specifically reduces silencing of Kv4.2 in epileptic mice.
MicroRNA-induced silencing of Kv4.2 is increased in epileptic mice.
Acknowledgements
This research was supported by the National Institutes of Health [grant numbers R01NS092705 (to C.G.) and R01MH100510 (to D.H.B.)], a postdoctoral fellowship from the American Epilepsy Society, Chicago, IL [grant number 506835 (to D.T.)], and a Trustee Award from the Cincinnati Children’s Research Foundation, Cincinnati, OH (to C.G.). We would like to thank Dr. J. Nerbonne for the Kv4.2 KO mice, and all members of the Gross and Danzer labs for helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
C.G. is co-inventor on US patent 9,932,585 B2. All other authors declare no competing financial interests.
References
- Arida RM, Scorza FA, de Araujo Peres C, Cavalheiro EA, 1999. The course of untreated seizures in the pilocarpine model of epilepsy. Epilepsy Res 34, 99–107. 10.1016/S0920-1211(98)00092-8 [DOI] [PubMed] [Google Scholar]
- Banerjee SB, Rajendran R, Dias BG, Ladiwala U, Tole S, Vaidya VA, 2005. Recruitment of the Sonic hedgehog signalling cascade in electroconvulsive seizure-mediated regulation of adult rat hippocampal neurogenesis. Eur J Neurosci 22, 1570–1580. 10.1111/j.1460-9568.2005.04317.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Klein G, Klee R, Brandt C, Bankstahl M, Bascunana P, Tollner K, Dalipaj H, Bankstahl JP, Friedman A, Loscher W, 2016. Isoflurane prevents acquired epilepsy in rat models of temporal lobe epilepsy. Ann Neurol 80, 896–908. 10.1002/ana.24804 [DOI] [PubMed] [Google Scholar]
- Barnwell LFS, Lugo JN, Lee WL, Willis SE, Gertz SJ, Hrachovy RA, Anderson AE, 2009. Kv4.2 knockout mice demonstrate increased susceptibility to convulsant stimulation. Epilepsia. 50, 1741–1751. 10.1111/j.1528-1167.2009.02086.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartel DP, 2018. Metazoan MicroRNAs. Cell. 173, 20–51. 10.1016/j.cell.2018.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard C, Anderson A, Becker A, Poolos NP, Beck H, Johnston D, 2004. Acquired dendritic channelopathy in temporal lobe epilepsy. Science. 305, 532–5. 10.1126/science.1097065 [DOI] [PubMed] [Google Scholar]
- Brager DH, Akhavan AR, Johnston D, 2012. Impaired dendritic expression and plasticity of h-channels in the fmr1(-/y) mouse model of fragile X syndrome. Cell Rep 1,225–33. 10.1016/j.celrep.2012.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan GP, Dey D, Chen Y, Patterson KP, Magnetta EJ, Hall AM, Dube CM, Mei Y-T, Baram TZ, 2016. Dual and opposing roles of microRNA-124 in epilepsy are mediated through inflammatory and NRSF-dependent gene networks. Cell Rep 14, 2402–2412. 10.1016/j.celrep.2016.02.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner R, Wilcox KS, Potassium Channelopathies of Epilepsy In: Noebels JL, et al. , (Eds.), Jasper’s Basic Mechanisms of the Epilepsies. National Center for Biotechnology Information (US), Bethesda (MD), 2012. [PubMed] [Google Scholar]
- Castro OW, Santos VR, Pun RY, McKlveen JM, Batie M, Holland KD, Gardner M, Garcia-Cairasco N, Herman JP, Danzer SC, 2012. Impact of corticosterone treatment on spontaneous seizure frequency and epileptiform activity in mice with chronic epilepsy. PLoS One. 7, e46044 10.1371/journal.pone.0046044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabrol E, Navarro V, Provenzano G, Cohen I, Dinocourt C, Rivaud-Péchoux S, Fricker D, Baulac M, Miles R, LeGuern E, Baulac S, 2010. Electroclinical characterization of epileptic seizures in leucine-rich, glioma-inactivated 1-deficient mice. Brain. 133, 2749–2762. 10.1093/brain/awq171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JJ, Tay Y, 2018. Noncoding RNA:RNA Regulatory Networks in Cancer. Int J Mol Sci 19, pii: E1310. 10.3390/ijms19051310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Yuan LL, Zhao C, Birnbaum SG, Frick A, Jung WE, Schwarz TL, Sweatt JD, Johnston D, 2006. Deletion of Kv4.2 gene eliminates dendritic A-type K+ current and enhances induction of long-term potentiation in hippocampal CA1 pyramidal neurons. J Neurosci 26, 12143–51. 10.1523/jneurosci.2667-06.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe J, Cho H, Lee HC, Kim YK, 2010. microRNA/Argonaute 2 regulates nonsense-mediated messenger RNA decay. EMBO Rep 11,380–6. 10.1038/embor.2010.44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang M, Lu Y, Chen GJ, Shen L, Pan YM, Wang XF, 2011. Increased expression of Sonic hedgehog in temporal lobe epileptic foci in humans and experimental rats. Neuroscience. 182, 62–70. 10.1016/j.neuroscience.2011.02.060 [DOI] [PubMed] [Google Scholar]
- Feng S, Ma S, Jia C, Su Y, Yang S, Zhou K, Liu Y, Cheng J, Lu D, Fan L, Wang Y, 2016. Sonic hedgehog is a regulator of extracellular glutamate levels and epilepsy. EMBO Rep 17, 682–94. 10.15252/embr.201541569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferretti E, De Smaele E, Miele E, Laneve P, Po A, Pelloni M, Paganelli A, Di Marcotullio L, Caffarelli E, Screpanti I, Bozzoni I, Gulino A, 2008. Concerted microRNA control of Hedgehog signalling in cerebellar neuronal progenitor and tumour cells. EMBO J. 27, 2616–27. 10.1038/emboj.2008.172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis J, Jugloff DG, Mingo NS, Wallace MC, Jones OT, Burnham WM, Eubanks JH, 1997. Kainic acid-induced generalized seizures alter the regional hippocampal expression of the rat Kv4.2 potassium channel gene. Neurosci Lett 232, 91–4. 10.1016/S0304-3940(97)00593-4 [DOI] [PubMed] [Google Scholar]
- Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL, 2009. Arrhythmia in Heart and Brain: KCNQ1 Mutations Link Epilepsy and Sudden Unexplained Death. Sci Transl Med 1,2ra6–2ra6. 10.1126/scitranslmed.3000289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, Yao X, Engel T, Tiwari D, Xing L, Rowley S, Danielson SW, Thomas KT, Jimenez-Mateos EM, Schroeder LM, Pun RY, Danzer SC, Henshall DC, Bassell GJ, 2016. MicroRNA-Mediated Downregulation of the Potassium Channel Kv4.2 Contributes to Seizure Onset. Cell Rep 17, 37–45. 10.1016/j.celrep.2016.08.074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Jung WE, Marionneau C, Aimond F, Xu H, Yamada KA, Schwarz TL, Demolombe S, Nerbonne JM, 2005. Targeted deletion of Kv4.2 eliminates I(to,f) and results in electrical and molecular remodeling, with no evidence of ventricular hypertrophy or myocardial dysfunction. Circ Res 97, 1342–50. 10.1161/01.RES.0000196559.63223.aa [DOI] [PubMed] [Google Scholar]
- Henshall DC, Hamer HM, Pasterkamp RJ, Goldstein DB, Kjems J, Prehn JHM, Schorge S, Lamottke K, Rosenow F, 2016. MicroRNAs in epilepsy: pathophysiology and clinical utility. Lancet Neurol 15, 1368–1376. 10.1016/S1474-4422(16)30246-0 [DOI] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D, 1997. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 387, 869–75. 10.1038/43119 [DOI] [PubMed] [Google Scholar]
- Hosford BE, Liska JP, Danzer SC, 2016. Ablation of Newly Generated Hippocampal Granule Cells Has Disease-Modifying Effects in Epilepsy. J Neurosci 36, 11013–11023. 10.1523/jneurosci.1371-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosford BE, Rowley S, Liska JP, Danzer SC, 2017. Ablation of peri-insult generated granule cells after epilepsy onset halts disease progression. Sci Rep 7, 18015 10.1038/s41598-017-18237-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iori V, Iyer AM, Ravizza T, Beltrame L, Paracchini L, Marchini S, Cerovic M, Hill C, Ferrari M, Zucchetti M, Molteni M, Rossetti C, Brambilla R, Steve White H, D’lncalci M, Aronica E, Vezzani A, 2017. Blockade of the IL-1R1/TLR4 pathway mediates disease-modification therapeutic effects in a model of acquired epilepsy. Neurobiol Dis 99, 12–23. 10.1016/j.nbd.2016.12.007 [DOI] [PubMed] [Google Scholar]
- Jerng HH, Pfaffinger PJ, 2014. Modulatory mechanisms and multiple functions of somatodendritic A-type K (+) channel auxiliary subunits. Front Cell Neurosci 8, 82 10.3389/fncel.2014.00082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerng HH, Pfaffinger PJ, Covarrubias M, 2004. Molecular physiology and modulation of somatodendritic A-type potassium channels. Mol Cell Neurosci 27, 343–369. 10.1016/j.mcn.2004.06.011 [DOI] [PubMed] [Google Scholar]
- Jimenez-Mateos EM, Engel T, Merino-Serrais P, McKiernan RC, Tanaka K, Mouri G, Sano T, O’Tuathaigh C, Waddington JL, Prenter S, Delanty N, Farrell MA, O’Brien DF, Conroy RM, Stallings RL, Defelipe J, Henshall DC, 2012. Silencing microRNA-134 produces neuroprotective and prolonged seizure-suppressive effects. Nat Med 18, 1087–94. 10.1038/nm.2834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalmbach BE, Johnston D, Brager DH, 2015. Cell-Type Specific Channelopathies in the Prefrontal Cortex of the fmr1-/y Mouse Model of Fragile X Syndrome. eNeuro. 2, ENEURO.0114-15.2015. 10.1523/eneuro.0114-15.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhling R, Wolfart J, 2016. Potassium Channels in Epilepsy. Cold Spring Harbor Perspect Med. 6 10.1101/cshperspect.a022871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmann A, Danis B, Andonovic L, Abnaof K, van Rikxoort M, Siegel F, Mazzuferi M, Godard P, Hanon E, Frohlich H, Kaminski RM, Foerch P, Pfeifer A, 2015. Different MicroRNA Profiles in Chronic Epilepsy Versus Acute Seizure Mouse Models. J Mol Neurosci 55, 466–479. 10.1007/s12031-014-0368-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Lin MC, Kornblum HI, Papazian DM, Nelson SF, 2014. Exome sequencing identifies de novo gain of function missense mutation in KCND2 in identical twins with autism and seizures that slows potassium channel inactivation. Hum Mol Genet 23, 3481–9. 10.1093/hmg/ddu056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Z, Deng P, Li J, Xu ZC, 2012. Alterations of A-type potassium channels in hippocampal neurons after traumatic brain injury. J Neurotrauma. 29, 235–45. 10.1089/neu.2010.1537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei Z, Zhang H, Liang Y, Cui Q, Xu Z, Xu ZC, 2014. Reduced expression of I channels is associated with postischemic seizures in hyperglycemic rats. J Neurosci Res 92, 1775–84. 10.1002/jnr.23445 [DOI] [PubMed] [Google Scholar]
- Lim J-A, Moon J, Kim T-J, Jun J-S, Park B, Byun J-I, Sunwoo J-S, Park K-I, Lee S-T, Jung K-H, Jung K-Y, Kim M, Jeon D, Chu K, Lee SK, 2018. Clustering of spontaneous recurrent seizures separated by long seizure-free periods: An extended video-EEG monitoring study of a pilocarpine mouse model. PLOS ONE. 13, e0194552 10.1371/journal.pone.0194552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losing P, Niturad CE, Harrer M, Reckendorf CMZ, Schatz T, Sinske D, Lerche H, Maljevic S, Knoll B, 2017. SRF modulates seizure occurrence, activity induced gene transcription and hippocampal circuit reorganization in the mouse pilocarpine epilepsy model. Mol Brain. 10, 30 10.1186/s13041-017-0310-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaghan MM, Menegola M, Vacher H, Rhodes KJ, Trimmer JS, 2008. Altered expression and localization of hippocampal A-type potassium channel subunits in the pilocarpine-induced model of temporal lobe epilepsy. Neuroscience. 156, 550–62. 10.1016/j.neuroscience.2008.07.057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muddashetty RS, Kelic S, Gross C, Xu M, Bassell GJ, 2007. Dysregulated metabotropic glutamate receptor-dependent translation of AMPA receptor and postsynaptic density-95 mRNAs at synapses in a mouse model of fragile X syndrome. J Neurosci 27, 5338–48. 10.1523/jneurosci.0937-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordemann GJ, Apgar CJ, Brager DH, 2019. D-type potassium channels normalize action potential firing between dorsal and ventral CA1 neurons of the mouse hippocampus. J Neurophysiol 121, 983–995. 10.1152/jn.00737.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poolos NP, Johnston D, 2012. Dendritic ion channelopathy in acquired epilepsy. Epilepsia. 53, 32–40. 10.1111/epi.12033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puttachary S, Sharma S, Tse K, Beamer E, Sexton A, Crutison J, Thippeswamy T, 2015. Immediate Epileptogenesis after Kainate-Induced Status Epilepticus in C57BL/6J Mice: Evidence from Long Term Continuous Video-EEG Telemetry. PLoS ONE. 10, e0131705 10.1371/journal.pone.0131705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu Z, Dai Y, 2014. Roadmap of miR-122-related clinical application from bench to bedside. Expert Opin Investig Drugs. 23, 347–55. 10.1517/13543784.2014.867327 [DOI] [PubMed] [Google Scholar]
- Racine RJ, 1972. Modification of seizure activity by electrical stimulation. II. Motor seizure. Electroencephalogr Clin Neurophysiol 32, 281–94. [DOI] [PubMed] [Google Scholar]
- Routh BN, Johnston D, Brager DH, 2013. Loss of functional A-type potassium channels in the dendrites of CA1 pyramidal neurons from a mouse model of fragile X syndrome. J Neurosci 33, 19442–50. 10.1523/JNEUROSCI.3256-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh B, Ogiwara I, Kaneda M, Tokonami N, Mazaki E, Baba K, Matsuda K, Inoue Y, Yamakawa K, 2006. A Kv4.2 truncation mutation in a patient with temporal lobe epilepsy. Neurobiol Dis 24, 245–53. 10.1016/j.nbd.2006.07.001 [DOI] [PubMed] [Google Scholar]
- Song YJ, Tian XB, Zhang S, Zhang YX, Li X, Li D, Cheng Y, Zhang JN, Kang CS, Zhao W, 2011. Temporal lobe epilepsy induces differential expression of hippocampal miRNAs including let-7e and miR-23a/b. Brain Res 1387, 134–40. 10.1016/j.brainres.2011.02.073 [DOI] [PubMed] [Google Scholar]
- Su T, Cong WD, Long YS, Luo AH, Sun WW, Deng WY, Liao WP, 2008. Altered expression of voltage-gated potassium channel 4.2 and voltage-gated potassium channel 4-interacting protein, and changes in intracellular calcium levels following lithium-pilocarpine-induced status epilepticus. Neuroscience. 157, 566–76. 10.1016/j.neuroscience.2008.09.027 [DOI] [PubMed] [Google Scholar]
- Sun W, Maffie JK, Lin L, Petralia RS, Rudy B, Hoffman DA, 2011. DPP6 establishes the A-type K(+) current gradient critical for the regulation of dendritic excitability in CA1 hippocampal neurons. Neuron. 71, 1102–15. 10.1016/j.neuron.2011.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiwari D, Peariso K, Gross C, 2018. MicroRNA-induced silencing in epilepsy: Opportunities and challenges for clinical application. Dev Dyn 247, 94–110. 10.1002/dvdy.24582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaur ML, Sheng M, Lowenstein DH, Jan YN, Jan LY, 1992. Differential expression of K+ channel mRNAs in the rat brain and down-regulation in the hippocampus following seizures. Neuron. 8, 1055–67. 0896-6273(92)90127-Y [DOI] [PubMed] [Google Scholar]
- van der Ree MH, van der Meer AJ, de Bruijne J, Maan R, van Vliet A, Welzel TM, Zeuzem S, Lawitz EJ, Rodriguez-Torres M, Kupcova V, Wiercinska-Drapalo A, Hodges MR, Janssen HLA, Reesink HW, 2014. Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antiviral Res 111,53–59. 10.1016/j.antiviral.2014.08.015 [DOI] [PubMed] [Google Scholar]
- van der Ree MH, van der Meer AJ, van Nuenen AC, de Bruijne J, Ottosen S, Janssen HL, Kootstra NA, Reesink HW, 2016. Miravirsen dosing in chronic hepatitis C patients results in decreased microRNA-122 levels without affecting other microRNAs in plasma. Aliment Pharmacol Ther 43, 102–13. 10.1111/apt.13432 [DOI] [PubMed] [Google Scholar]
- Villa C, Combi R, 2016. Potassium Channels and Human Epileptic Phenotypes: An Updated Overview. Front Cell Neurosci 10, 81 10.3389/fncel.2016.00081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HG, He XP, Li Q, Madison RD, Moore SD, McNamara JO, Pitt GS, 2013. The auxiliary subunit KChIP2 is an essential regulator of homeostatic excitability. J Biol Chem 288, 13258–68. 10.1074/jbc.M112.434548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods S, Barter MJ, Elliott HR, McGillivray CM, Birch MA, Clark IM, Young DA, 2018. miR-324-5p is up regulated in end-stage osteoarthritis and regulates Indian Hedgehog signalling by differing mechanisms in human and mouse. Matrix Biol 77, 87–100. 10.1016/j.matbio.2018.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HS, Zong HL, Shang M, Ming X, Zhao JP, Ma C, Cao L, 2014. miR-324-5p inhibits proliferation of glioma by target regulation of GLI1. Eur Rev Med Pharmacol Sci 18, 828–32. [PubMed] [Google Scholar]
- Zuberi SM, Eunson LH, Spauschus A, De Silva R, Tolmie J, Wood NW, McWilliam RC, Stephenson JB, Kullmann DM, Hanna MG, 1999. A novel mutation in the human voltage-gated potassium channel gene (Kv1.1) associates with episodic ataxia type 1 and sometimes with partial epilepsy. Brain. 122 (Pt 5), 817–25. 10.1093/brain/122.5.817 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.