

Abstract
Although β-amyloid plaques are a well-recognized hallmark of Alzheimer’s disease (AD) neuropathology, no drugs reducing amyloid burden have shown efficacy in clinical trials, suggesting that once AD symptoms emerge, disease progression becomes independent of Aβ production. Reactive astrocytes are another neuropathological feature of AD, where there is an emergence of neurotoxic (A1) reactive astrocytes. We find that serine racemase (SR), the neuronal enzyme that produces the N-methyl-D-aspartate receptor (NMDAR) co-agonist D-serine, is robustly expressed in A1-reactive neurotoxic astrocytes in the hippocampus and entorhinal cortex of AD subjects and an AD rat model. Furthermore, we observe intracellular signaling changes consistent with increased extra-synaptic NMDAR activation, excitotoxicity and decreased neuronal survival. Thus, reducing neurotoxic D-serine release from A1 inflammatory astrocytes could have therapeutic benefit for mild to advanced AD, when anti-amyloid strategies are ineffective.
Keywords: glial fibrillary acidic protein, complement C3, N-methyl-D-asparate receptor, extrasynaptic, hippocampus, entorhinal cortex, Tg-F344AD, death associated protein kinase
Introduction
Millions of people are living with Alzheimer’s disease (AD), the most common cause of late-onset dementia. From a neuropathological perspective, AD is characterized by the presence of extracellular amyloid-beta (Aβ) plaques and intraneuronal neurofibrillary tangles composed of misfolded, hyperphosphorylated tau protein in the brain. Therefore, the predominant strategy for developing therapies to treat AD has focused on mechanisms that reduce amyloid burden. However, this approach has failed to significantly improve cognitive and functional outcomes in large-scale Phase III clinical trials (Amanatkar et al., 2017; Egan et al., 2018). These failures suggest that once clinical AD symptoms emerge, disease progression becomes independent of Aβ production (Hardy and De Strooper, 2017). In addition to plaques and tangles, reactive glial cells, including astrocytes and microglia, are neuropathological hallmarks of AD that were first identified as components of amyloid plaques in AD patients several decades ago (Brendel et al., 2016; Perez-Nievas and Serrano-Pozo, 2018). Recent genome-wide association studies indicate that inflammatory factors play an important role in AD pathophysiology (Villegas-Llerena et al., 2016). Activated microglia can convert quiescent astrocytes into neurotoxic A1 inflammatory astrocytes in neurodegenerative disorders, including AD (Liddelow et al., 2017; Yun et al., 2018).
We recently demonstrated with in vitro (Li et al., 2018) studies and in a mouse model of traumatic brain injury (TBI) (Perez et al., 2017) that reactive (inflammatory) astrocytes express serine racemase (SR), the enzyme that converts L-serine to D-serine. D-serine, rather than glycine, is the predominant co-agonist at synaptic N-methyl-D-aspartate receptor (NMDARs) in the forebrain (Curcio et al., 2013; Papouin et al., 2012). Importantly, conditionally knocking-out SR only from glial fibrillary acidic protein (GFAP)-expressing astrocytes protects the mice from the damaging effects of TBI on hippocampal synaptic plasticity and memory, demonstrating that D-serine released from reactive astrocytes into the extra-synaptic space was detrimental to synaptic function (Perez et al., 2017). In contrast, astrocytic SR elimination in naive mice did not impair hippocampal synaptic plasticity (Benneyworth et al., 2012; Perez et al., 2017). Hence, the current study sought to determine whether neurotoxic, reactive astrocytes express D-serine synthesizing SR in AD, a disorder with pronounced neuroinflammation.
Materials and Methods
Human postmortem immunofluorescence labeling
Blocks of paraffin embedded midbody hippocampi and entorhinal cortices were obtained from the Neuropathology Core of the Massachusetts Alzheimer’s Disease Research Center (ADRC) at the MassGeneral Institute for Neurodegenerative Disease. Cases were assessed by a neuropathologist and were matched for age and gender. “Control” cases (n=5) were selected that had received Braak staging scores of 0/I/II, had sparse plaque pathology, and were rated as having low probability of AD by National Institute on Aging and the Alzheimer’s Association (NIA-AA) criteria. AD cases were selected that had received Braak staging scores of III-IV (n=6) and V-VI (n=7), had frequent plaques and were rated as having intermediate or high probability of AD. See Supplemental Table 1 for details all of the subjects’ Braak stage, age, post-mortem interval (PMI), sex, hemisphere, ABC burden (derives from three separate four-point scales: Aβ/amyloid plaques (A) by the method of Thal phases, neurofibrillary tangles stage by the method of Braak (B), and neuritic plaque score by the method of CERAD (C). The combination of A, B, and C scores receive a descriptor of “Not”, “Low”, Intermediate” or “High” AD neuropathologic change (Hyman et al., 2012), exposure to AD medications categorized as acetylcholinesterase inhibitors, NMDAR antagonists, or no exposure, and other neuropathological comments. Control subjects for which no records of AD medication exposure were available, and did not show neuropathological changes consistent with AD were categorized as having no exposure to AD medications.
Blocks were cut at 10μm and sections placed on positively charged slides. Two sections from each case were used for each experiment of two immunofluorescent studies: SR/GFAP and SR/GFAP/C3. Paraffin was removed from the sections in xylene (2x, 10 min each), after which they were serially dehydrated in ethanol, and then placed in water for 5 min. Antigen unmasking was performed by placing the sections in tubes containing 80°C 0.1M citric acid buffer (pH 6) that were then placed in an 80°C water bath for 20 minutes. All of the following incubations occurred at room temperature. Sections were allowed to cool for several minutes before washing in 0.01M PBS/0.5% Triton-X 100 (3x, 10 min each). Sections were then incubated for 30 min in wash buffer containing 10% methanol and 0.3% hydrogen peroxide. Sections were then washed (3x, 5 minutes each) and incubated in blocking buffer (2% bovine serum albumin; Cell Signal Technologies) for 1 h. After blocking, sections were incubated with blocking buffer containing primary antibodies (experiment 1: rabbit anti-SR and mouse anti-GFAP; experiment 2: rabbit anti-SR, mouse anti-GFAP, and goat anti-C3) in a humidifying chamber overnight. See Supplemental Table 2 for all antibody concentrations. The sections were then washed, incubated in blocking buffer (1% BSA) containing biotinylated goat anti-rabbit secondary antibody for 2 h, washed, and then incubated with blocking buffer (1% BSA) containing streptavidin Alexa488 and goat-anti mouse Alex647 (experiment 1) or streptavidin Alexa488, donkey-anti mouse Alex568, and donkey-anti mouse Alex647 (experiment 2) for 4 h. Sections were then incubated for 10 min with copper sulfate (0.05M ammonium acetate, 0.001M copper sulfate, pH 5) to quench lipofucsin autofluorescence, followed by two washes in 0.1M phosphate buffer. Sections were allowed to completely dry and were cover-slipped using Vectashield liquid mounting media (Vector Labs). The anti-SR antibody was validated as specific for SR by using SR−/− mice as histological controls (Wolosker et al., 2016) (Supplemental Figure 4). The C3 antibody was previously validated in C3KO mice (Schafer et al., 2016) and the GFAP antibody is widely used in the literature and shows no cross-reactivity with vimentin (Millipore data sheet).
Stereology-based quantification of immunoreactive glia in human tissue
All samples were coded so that researchers involved in all studies were blind to diagnosis. Stereology-based sampling methods were used for quantification of postmortem samples as described in previous studies (Berretta et al., 2007; Coggeshall and Lekan, 1996; Dorph-Petersen and Lewis, 2011; Guillery and Herrup, 1997; Gundersen et al., 1999; Hyman et al., 1998; Pantazopoulos et al., 2007). In order to generate an accurate sampling strategy, we first manually counted all GFAP+ and SR+/GFAP+ glial cells in the full extent of each hippocampal subfield and entorhinal cortex divisions of one tissue section each from a representative control, AD (Braak IV), and AD (Braak VI) subject. This allowed us to obtain a complete number of immunoreactive cells per section to use as a baseline for establishing optimal stereological sampling parameters to use for the complete cohort quantification. With this information, we optimized sampling parameters (counting frame [300μm2 for all brain regions], grid size) for each sub-region in order to obtain stereological estimates that were within ~10% of the actual counts in each section for each subregion across diagnoses. This resulted in an average of 25 sampling sites per section for sector CA4, 20 for sector CA3, 14 for sector CA2, 28 for sector CA1, 26 for the superficial layers of the ECs, and 27 for the deep layers of the entorhinal cortex. We then stereologically estimated (optical fractionator: Stereoinvestigator software; MBF Biosciences, VT), the number of glial cells that were GFAP+ and GFAP+/SR+ (experiment 1) or GFAP+/C3+ and GFAP+/C3+/SR+ (experiment 2) in hippocampus (dentate gyrus, CA4, CA3, CA2, CA1) and entorhinal cortex (superficial [I-III] and deep [IV-VI] layers) using our defined sampling strategy from two slides per subject. The total number of estimated cells was calculated using the following equation: Cell estimate = # of counted cells x [(grid area2 / counting frame area2)](Berretta et al., 2007; Pantazopoulos et al., 2007). Cell number estimates were then normalized to the area of each region (mm3). Z-axis measurements of paraffin embedded sections following immunofluorescence processing revealed no measurable thickness collapse, as the average measured thickness was 10.5 μm. This thickness is comparable to the z-axis sampling thickness used for stereological sampling in our previous studies following z-axis collapse in free floating PFA fixed sections (Markota et al., 2014; Pantazopoulos et al., 2007; Pantazopoulos et al., 2015).
Animals
TgF344-AD rats heterozygous for an APPsw/PS1ΔE9 transgene and wild-type littermates were group housed and maintained in the animal facility at Emory University. No sex differences in Alzheimer’s disease-like pathology or behavior have been reported for these rats (Cohen et al., 2013; Rorabaugh et al., 2017), and thus roughly equal numbers of males and females were used for all studies. Rats were pair-housed on a 12-h light/dark cycle and given ad libitum access to food and chow.
TgF344-AD rat immunofluorescence labeling
At 6 or 16 months, rats were anaesthetized with isoflurane (Henry Schein) and perfused with ice cold K-PBS followed by 4% paraformaldehyde. Brains were removed, immersed in 30% sucrose (6 months: WT: 2 female; TgF344-AD 2 male; 16 months: WT: 3 female; TgF344-AD 3 male), and sectioned at 30μm at the level of hippocampus. Triple-antigen immunofluorescence was performed on free-floating sections. For SR/GFAP/C3 staining, sections were blocked with 10% NDS/1% BSA and incubated with primary antibodies (mouse anti-GFAP: 1:10,000; rabbit anti-SR (Kartvelishvily et al., 2006): 1:1500; goat anti-C3: 1:150) overnight at 4°C, followed by incubation with donkey anti-rabbit AlexaFluor647 IgG (H+L) (1:500) and donkey anti-mouse AlexaFluor488 IgG (H+L), and donkey anti-goat AlexaFluor568 IgG (H+L).
Stereology-based estimation of immunoreactive astrocytes in aged rat hippocampus
The number of neurotoxic, reactive astrocytes (GFAP/C3) and SR-expressing reactive astrocytes (GFAP/C3/SR) in the hippocampus of 16-month old WT (n=3) and 16-month old TgAD (n=3) rats was estimated using systematic random sampling (SRS) with a standardized Optical Fractionator workflow (StereoInvesitgator; MBF Bioscience). Six hippocampal sections from each rat (section interval = 6) matched for rostral-caudal position and spanning approximately −3.14mm though −5.6mm bregma (anterior/posterior; Paxinos, George, and Watson) were used for analysis. Cells were counted blinded to genotype at 20x using exposure and image settings that were kept consistent across all sections. Each of the hippocampal subfields were analyzed separately (CA1, CA2/3, DG [dentate gyrus; molecular layer]) and then summed together to a total hippocampal cell estimate. The average volumes of each of the analyzed subfields did not significantly differ between genotypes (Supplemental Table 3). A counting frame size of 120μm x 120μm and a sampling grid size of 450μm x 450μm were used for CA1 and CA2/3, while a counting frame size of 150μm x 150μm and a sampling grid size of 400μm x 400μm were used for DG. These parameters resulted in a minimum of 300 GFAP/C3 counts per subfield and highly accurate cell population estimates across genotypes and subfields (coefficient of error values < 0.1; Supplemental Table 3)
Western Blot Analysis
SDS-PAGE and immunoblotting were performed using hippocampi from WT and TgF344-AD rats collected at either 6mo (WT: 4 female, 2 male; TgF344-AD: 2 female, 4 male) or 16mo (WT: 2 female, 4 male; TgF344-AD: 3 female, 3 male) of age. A single hippocampal lobe from each rat was bisected longitudinally, along its dorsal-ventral axis. Protein for Western blotting and amino acids for HPLC were extracted from the same half of hippocampus. Nitrocellulose membranes were incubated overnight at 4°C with primary antibody (see Supplemental Table 2). Membranes were then incubated with species-appropriate horseradish peroxidase-conjugated secondary antibodies (1:5,000). Chemiluminescent values of the protein of interest were divided by its corresponding β-actin chemiluminescent values (Chemidoc XRS+ Imager equipped with ImageLab 6.0 analysis software; Bio-Rad). The ratio of each experimental sample was divided by the average of all the control sample values in each gel and multiplied by 100. The average of the normalized control values from each gel was 100% ± SEM. The normalized values were then averaged and used for statistical analysis.
HPLC analysis of amino acid
L-serine, D-serine, and glutamate levels in the hippocampus were determined by HPLC analysis as previously described (Hashimoto et al., 1992). Briefly, the samples were de-proteinized by precipitation with TCA 5% and centrifugation. After extraction of TCA with water-saturated ether, the samples were analyzed in a Merck-Hitachi LaChrom liquid chromatograph equipped with an autosampler (L-7250), a quaternary gradient pump (L-7100), degasser unit (L-7614), a fluorescent detector (FL-7485), and a column oven adjusted to 30°C (L-7350). After derivatization with o-phtalodialdehyde and Boc-L-cysteine (Radzishevsky and Wolosker, 2012), the enantiomers were separated using a Spheri-5C18 column (220 mm 4.6 mm i.d., 5mm particle size) from Alltech (Deerfield), fitted with precolumn NewGuard RP-18, 7 mm, 15 mm, 3.2 mm by Grace (Deerfield).
Statistical Analyses .
Statistical analyses were performed using SPSS (human) and Graphpad Prism 7.0 (La Jolla, CA; rat) software, and JMP Pro v. 14 (SAS Institute, Cary, NC) for analysis of covariance of human postmortem data. For statistical analyses of human data, we applied classical and robust linear regression of main outcome measures on diagnostic groups (Control, Braak stage III-IV, Braak stage V-VI), illness-related and demographic covariates (age, sex, postmortem interval, hemisphere, anterior/posterior position of tissue section, ABC burden, exposure to acetylcholinesterase inhibitors or NMDAR antagonists, and date range of dissection). Differences between groups relative to the main outcome measures were assessed for statistical significance using stepwise linear regression (ANCOVA). Potential confounds were tested systematically for their effects on main outcome measures and included in the model if they significantly improved goodness-of-fit. Effects of co-variates are reported whenever significant effects were detected, together with main effects of diagnostic groups (Supplemental Table 4). Unpaired Student’s t-test was used to compared astrocyte population estimates between aged WT and TgAD rats. Two-way ANOVA was used to examine the effect of age and genotype for all Western blot and high pressure liquid chromatography (HPLC) experiments, with Holm-Sidak or Fisher’s LSD post-hoc tests used when there was a significant interaction between age and genotype. Values of P < 0.05 were considered statistically significant.
Study approval.
All studies involving rats were approved by the Emory University Institutional Animal Care and Use Committee in accordance with the NIH Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011).
Results
Serine Racemase is Expressed in Neurotoxic Reactive Astrocytes in the Entorhinal Cortex and Hippocampus of Subjects with AD
When quiescent astrocytes transition to a reactive state, they become hypertrophic and overexpress GFAP (Perez-Nievas and Serrano-Pozo, 2018). Thus, we performed dual-antigen immunofluorescence (SR, GFAP) on human post-mortem entorhinal cortex samples from age-matched, non-demented controls and on subjects with a clinical diagnosis of AD (Supplemental Table 1). The entorhinal cortex was chosen as it is one of the first brain regions affected in AD (Braak et al., 2006). Control subjects on average had a negligible amount of GFAP+/SR+ astrocytes in this region. In AD, we found a robust increase in the number of GFAP+ and GFAP+/SR+ reactive astrocytes, with a higher density of reactive astrocytes in superficial as compared to deep layers (Figure 1A–G; Supplemental Figure 1A–C). Exposure to AD medication had a significant effect on the density of both SR and GFAP astrocytes, resulting in a loss of significance of GFAP increases in the overall entorhinal cortex, as well as the superficial and deep layers (Fig.1A–G; Supplemental Figure 1A–C). In contrast, a significant effect of exposure to AD medication resulted in a larger increase in significant differences in the density of SR+ astrocytes in the whole entorhinal cortex, as well as in the superficial and deep layers (Fig.1A–G; Supplemental Figure 1A–C).
Figure 1. SR is expressed by reactive astrocytes in the entorhinal cortex of AD subjects.
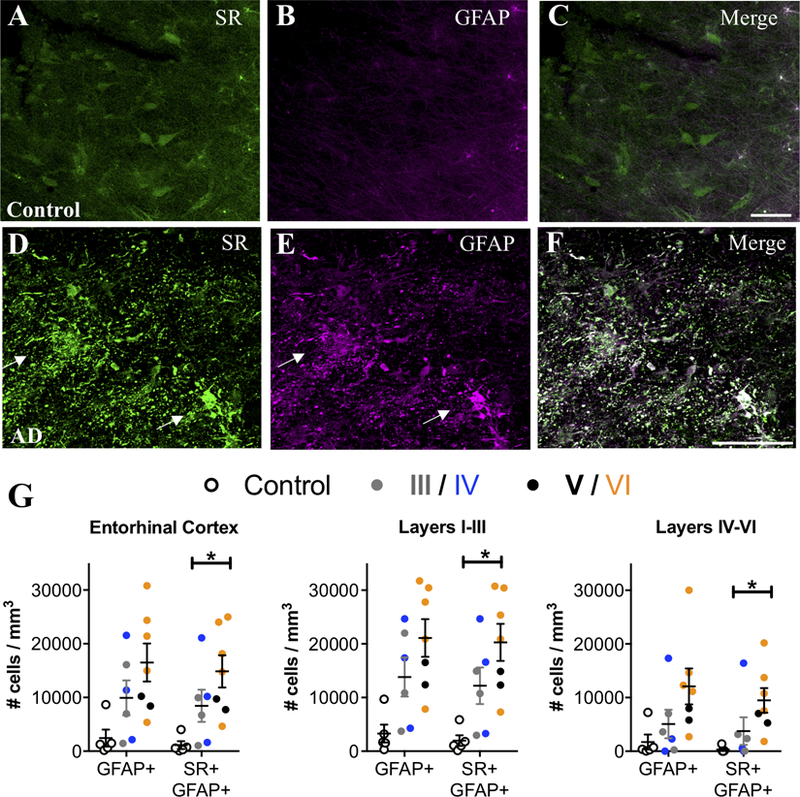
Representative images from layer II entorhinal cortex showing serine racemase (SR; green) and glial fibrillary associated protein (GFAP; magenta) immunostaining of control (A–C) and AD subjects (D–F). White arrows show cells that express both markers (white). Scale bars=25μm. (G) The number of GFAP+/ SR+ cells/mm3 was increased in AD subjects (filled circles; Braak III: gray, IV: blue; V: black, VI: orange) compared to controls (open circles) in the entire entorhinal cortex (GFAP/SR: F2,15=8.72, P=0.03), as well as in layers I-III (GFAP/SR: F2,15=7.37, P=0.0035) and layers IV-VI (GFAP/SR: F2,15=22.29, P=0.005). All values are adjusted for significant effects of exposure to AD medication.
Neuroinflammation induces type A1 reactive astrocytes that dramatically up-regulates many classical complement cascade genes that are known to be destructive to synapses (Zamanian et al., 2012). These neurotoxic A1 reactive astrocytes, identified by complement component 3 (C3) expression, are induced by activated microglia and are increased in AD and other neurodegenerative disorders (Liddelow et al., 2017; Yun et al., 2018). We found that AD subjects had a several fold increase of C3+ reactive astrocytes compared to matched controls in the entorhinal cortex, particularly in layers I-III, with the majority of these C3+ astrocytes co-expressing SR (Figure 2 A–I). Furthermore, a significant effect of exposure to AD medication further enhanced the significance of the increase in C3+ astrocytes, particularly in layers I-III (Figure 2 A–I).
Figure 2. SR-expressing reactive astrocytes in the entorhinal cortex of AD subjects are neurotoxic.
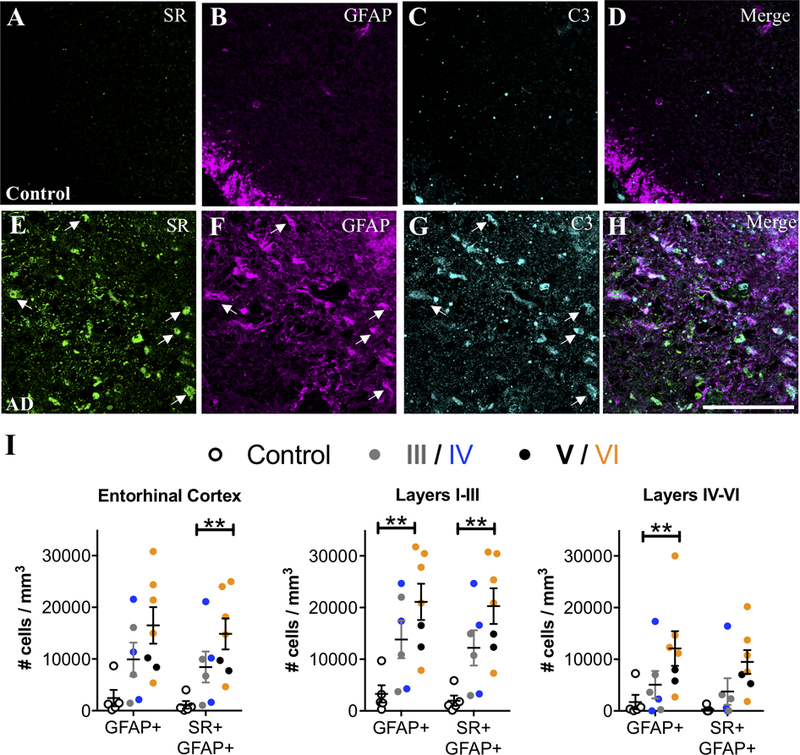
Representative images showing SR (green), GFAP (magenta), and C3 (turquoise) immunostaining in layer II entorhinal cortex from control (A–D) and AD subjects (E–H). White arrows show cells that express all three markers. Scale bars=25μm. (I) The number of GFAP+/C3+ and GFAP+/C3+/SR+ cells/mm3 was increased in AD subjects (filled circles; Braak III: gray, IV: blue; V: black, VI: orange) compared to controls (open circles) in the entire entorhinal cortex (GFAP/C3: F2,15=10.88**, P=0.22**; GFAP/C3/SR: F2,15=7.03**, P=0.04**). Numbers of GFAP+/C3+ and GFAP+/C3+/SR+ cells/mm3 were increased in layers I-III (GFAP/C3: F2,15=5.19**, P=0.02**; GFAP/C3/SR: F2,15=17.79**, P=0.008**) and layers IV-VI (GFAP/C3: F2,15=7.3**, P=0.04**; GFAP/C3/SR: F2,15=1.39, P=0.27) were also analyzed separately. Values represent the mean ± SEM. ** indicates values that are adjusted for significant effect of exposure to AD medication.
We then examined the hippocampus, a region that also exhibits neuroinflammation in AD (Braak et al., 2006). Similar to the entorhinal cortex, controls had very few SR+/GFAP+ reactive astrocytes, whereas SR+/GFAP+ reactive astrocytes were markedly more abundant in subjects with AD (Figure 3A–G). A significant effect of exposure to AD medication was observed in the hippocampus, which eliminated the significantly increased density of GFAP astrocytes, but enhanced the significant increase in density of GFAP-SR astrocytes (Fig. 3A–G). When we analyzed individual hippocampal subfields, we found significant increases in reactive astrocytes in CA2 and CA1, which was enhanced when accounting for a significant effect of exposure to AD medication, (Figure 3G), while there was a greater variability among AD cases in the density of reactive astrocytes (± SR) in DG, CA4, and CA3. In CA3-CA1, reactive astrocytes were concentrated primarily in stratum oriens and lacunosum moleculare (Supplemental Figure 1D–F). A significant effect of ABC burden was observed in the DG, which resulted in a significant increase in the density of GFAP astrocytes when included in the ANCOVA model. AD subjects had significantly more C3+ reactive astrocytes than controls in the hippocampus, particularly in CA1, with a majority of the C3+ reactive astrocytes co-expressing SR (Figure 4A–I). Significant effects of exposure to AD medication were only detected in the DG and in CA2, which enhanced the increased density of SR-GFAP-C3 astrocytes when included in the ANCOVA models. Finally, when control subjects were compared to late stage (Braak V-VI) subjects, the statistical differences and effect sizes were more robust and observed across more subdivisions of the entorhinal cortex and hippocampus (Supplemental Table 5), further highlighting the progressive nature of the inflammatory astrocyte pathology.
Figure 3. SR is expressed by reactive astrocytes in hippocampus of AD subjects.
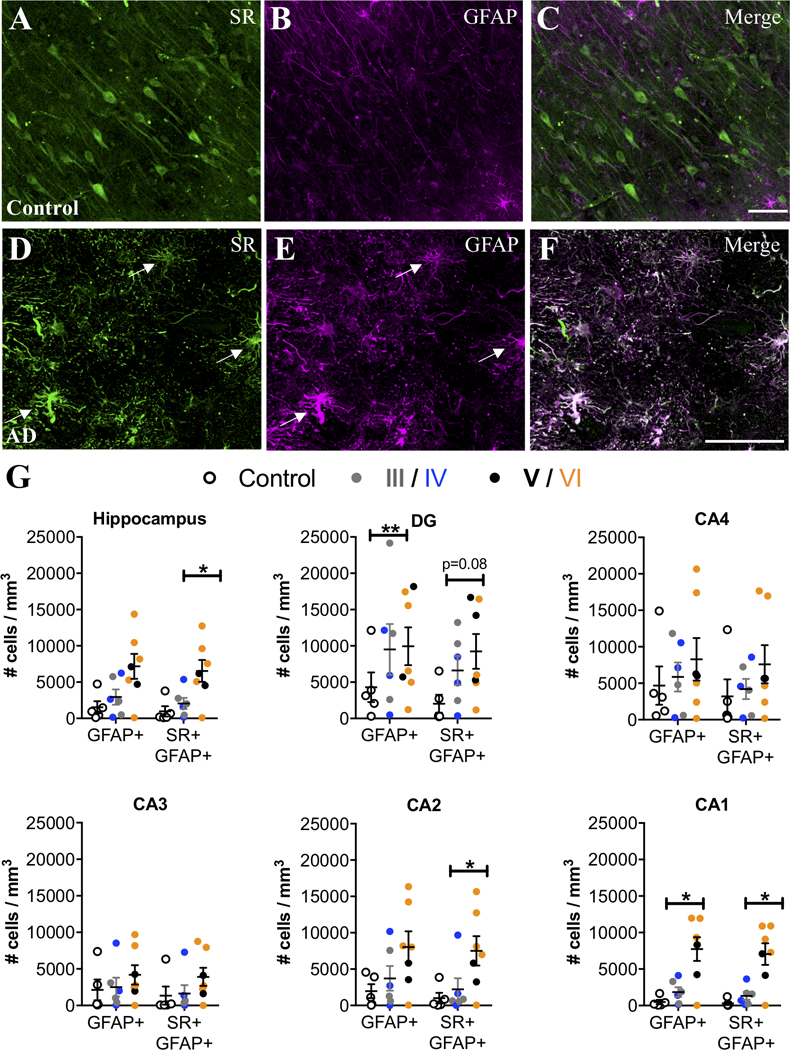
Representative images from the anterior hippocampus (pyramidal cell layer/stratum oriens) showing SR (green) and GFAP (magenta) immunostaining of control (A–C) and AD subjects (D–F). White arrows show cells that express both markers (white). The number of GFAP+ and GFAP+/ SR+ cells/mm3 was increased in AD subjects (filled circles; Braak III: gray, IV: blue; V: black, VI: orange) compared to controls (open circles) in the entire hippocampus (GFAP: F2,15=2.54*, P=0.17*; GFAP/SR: F2,15=8.93*, P=0.03*). We also analyzed hippocampal subfields separately: DG (GFAP: F2,15=6.76**, P=0.01**.; GFAP/SR: F2,15=2.94, P=0.08), CA4 (GFAP: F2,15=0.5, P=0.61.; GFAP/SR: F2,15=1.1, P=0.35), CA3 (GFAP: F2,15=0.69, P=0.51; GFAP/SR: F2,15=1.35, P=0.28), CA2 (GFAP: F2,15=1.43*, P=0.28*; GFAP/SR: F2,15=10.79*, P=0.02*), CA1 (GFAP: F2,15=18.69*, P=0.008*; GFAP/SR: F2,15=24.96*, P=0.0007*). * indicates values are adjusted for significant effect of exposure to AD medication. ** indicates values are adjusted for significant effect of ABC burden. Values represent the mean ± SEM.
Figure 4. SR-expressing reactive astrocytes in the hippocampus of AD subjects are neurotoxic.
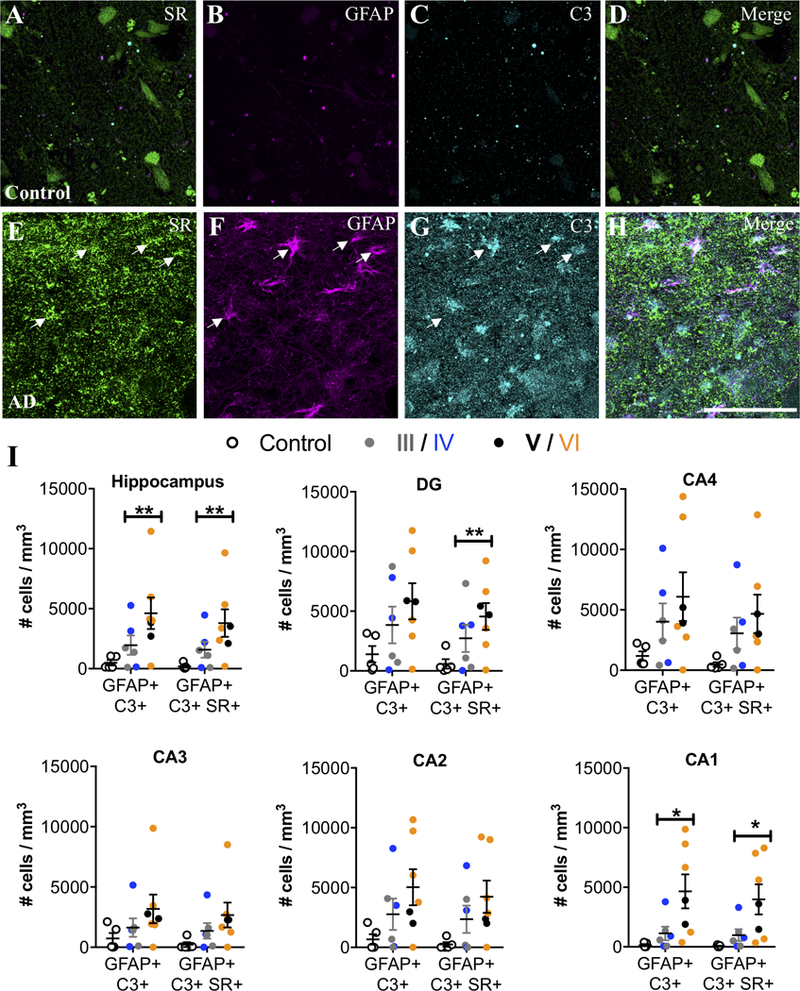
Representative images showing SR (green), GFAP (magenta), and C3 (turquoise) immunostaining of control (A–D) and AD subjects (E–H). White arrows show cells that express all three markers. Scale bars = 25μm. (I) The number of GFAP+/C3+ and GFAP+/C3+/SR+ cells/mm3 was increased in AD subjects (filled circles; Braak III: gray, IV: blue; V: black, VI: orange) compared to controls (open circles) in the entire hippocampus (GFAP/C3: F2,15=16.84**, P=0.0009**; GFAP/C3/SR: F2,15=8.68**, P=0.03**). Hippocampal subfields were also analyzed separately: DG (GFAP/C3: F2,15=2.45, P=0.12; GFAP/C3/SR: F2,15=7.14**, P=0.04**), CA4 (GFAP/C3: F2,15=2.16, P=0.15; GFAP/C3/SR: F2,15=2.48, P=0.18), CA3 (GFAP/C3: F2,15=1.71, P=0.21; GFAP/C3/SR: F2,15=2.36, P=0.13), CA2 (GFAP/C3: F2,15=0.35**, P=0.58**; GFAP/C3/SR: F2,15=0.58**, P=0.48**), and CA1 (GFAP/C3: F2,15 = 5.49, P = 0.01; GFAP/C3/SR: F2,15=5.32, P=0.01). * indicates significant differences between control and AD. ** indicates values are adjusted for significant effect of exposure to AD medication. Values represent the mean ± SEM.
Expression of Serine Racemase in Neurotoxic Reactive Astrocytes in TgF344-AD Rats is Associated with Elevated Extrasynaptic NMDAR Signaling
We next determined whether a similar phenomenon exists in a transgenic AD rat model (TgF344-AD) that expresses the disease-causing mutant human amyloid precursor protein (APPsw) and presenilin 1 (PS1ΔE9) (Cohen et al., 2013; Rorabaugh et al., 2017). TgF344-AD rats display an age-dependent phenotype that includes amyloid plaques, cognitive impairment, endogenous tau hyperphosphorylation, gliosis, and apoptotic neuronal loss (Cohen et al., 2013; Rorabaugh et al., 2017). Similar to what we found in human post-mortem tissue, we observed robust SR expression in C3+-reactive astrocytes in the hippocampus (Figure 5A–P) and entorhinal cortex (Figure S2) of aged (16mo) TgF344-AD rats, with some astrocytic SR expression detected in 16mo WT and 6mo TgF344-AD rats. We used unbiased, stereological sampling to estimate the number of neurotoxic, reactive astrocytes that express SR in the hippocampus of 16mo WT and TgF344-AD rats. We found that aged TgF344-AD rats had both significantly more neurotoxic astrocytes (GFAP/C3) and more SR-expressing neurotoxic astrocytes than aged WT rats in the hippocampus (Figure 5Q). Similar to the human post-mortem data, the CA1 region had the most robust increase in GFAP/C3/SR astrocytes (Figure 5Q).
Figure 5. Neurotoxic reactive astrocytes express SR in the hippocampus of TgF344-AD rats.
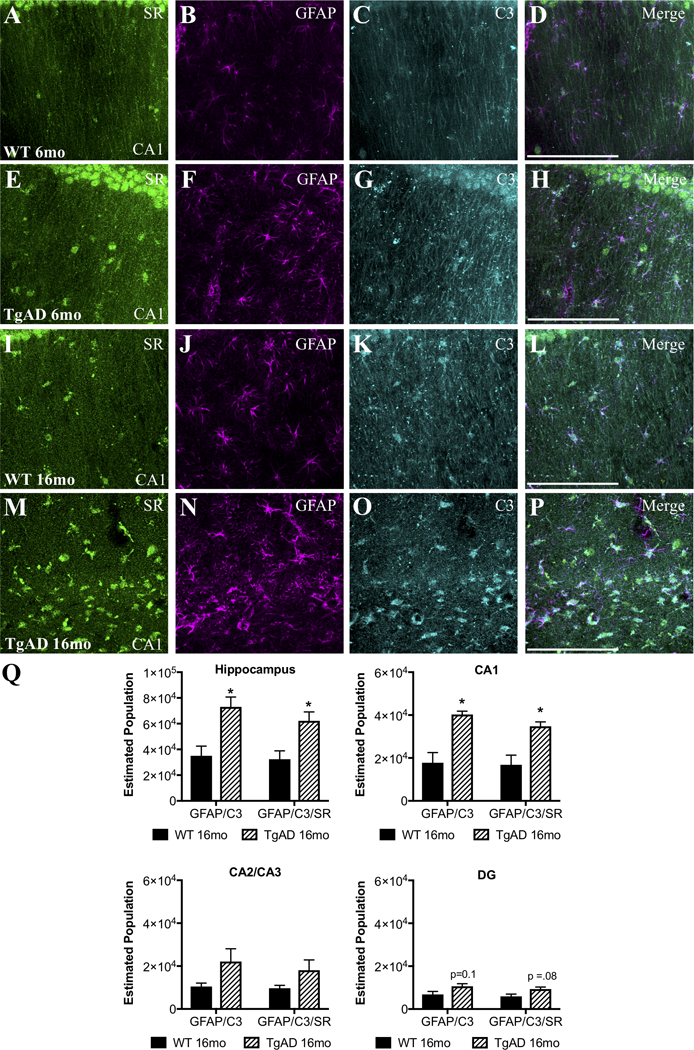
Representative images from the dorsal hippocampus (CA1 region) of wild-type (WT: 6-months: A–D, 16-months: I–L) and TgF344-AD (6-months: E–H, 16-months: M–P) rats showing SR (green), glial fibrillary associated protein (GFAP; magenta), and C3 (turquoise) immunostaining. Scale bars = 25μm. (Q) The total estimated number of GFAP+/C3+ and GFAP+/C3+/SR+ cells was increased in 16mo TgF344AD subjects (striped bars) compared to controls (black bars) in the entire hippocampus (GFAP/C3: t(4) = 3.5, P = 0.02; GFAP/C3/SR: t(4) = 3.1, P = 0.03). Hippocampal subfields were also analyzed separately: CA1: GFAP/C3: t(4) = 4.5, P = 0.01; GFAP/C3?SR: t(4) = 3.7, P = 0.02; CA2/3: GFAP/C3: t(4) = 1.9, P = 0.13; GFAP/C3/SR: t(4) = 1.7, P = 0.16; DG: GFAP/C3: t(4) = 2.1, P = 0.10; GFAP/C3/SR: t(4) = 2.4, P = 0.08. * indicates significant differences between WT and TgF344AD. Values represent the mean ± SEM.
We found increased hippocampal protein levels of GFAP (Figure 6A; age: F(1, 20) = 3.00, P =0.098; genotype: F(1,20)=8.31, P=0.01; interaction: F (1, 20) = 6.790, P=0.017) and ionized calcium binding adaptor molecule-1 (Iba-1) in 16-month, but not 6-month old, TgF344-AD rats (Supplemental Figure 3A), consistent with prior research showing increased numbers of astrocytes and microglia in aged TgF344-AD rats (Cohen et al., 2013; Rorabaugh et al., 2017). Interestingly, we did not detect a difference between WT and TgF344-AD rats in hippocampal SR protein levels at any age (Supplemental Figure 3B). This suggests that the loss of SR expression by neurons due to neuronal excitotoxic damage and degeneration in TgF344-AD rats (Cohen et al., 2013) is offset by an increase in astrocytic SR production, similar to what we demonstrated in mice following TBI (Perez et al., 2017). TgF344-AD rats had increased tissue content of L-serine (Figure 6B; age: F(1, 19) = 3.22, P=0.09; genotype: F(1,19)=6.45, P=0.02; interaction: F(1, 20) = 0.06, P=0.80) in the hippocampus compared to WT littermates, accompanied by a trend for increased D-serine content (Supplemental Figure 3C). As astrocytes are the primary site of brain L-serine synthesis (Yang et al., 2010), elevated L-serine is consistent with the increased number of astrocytes in TgF344-AD rats. Excessive extracellular glutamate causes excitotoxicity and cell death (Olney, 1969) via NMDAR activation (Coyle and Puttfarcken, 1993; Rothman and Olney, 1995). In aged TgF344-AD rats, there was also a robust increase in hippocampal glutamate levels (Figure 6C; age: F(1, 19) = 4.56, P=0.046; genotype: F(1,19) = 5.10, P=0.036; interaction: F(1, 20) = 3.13, P=0.09).
Figure 6. Increased extra-synaptic NMDAR signaling in the hippocampus of TgF344-AD rats.
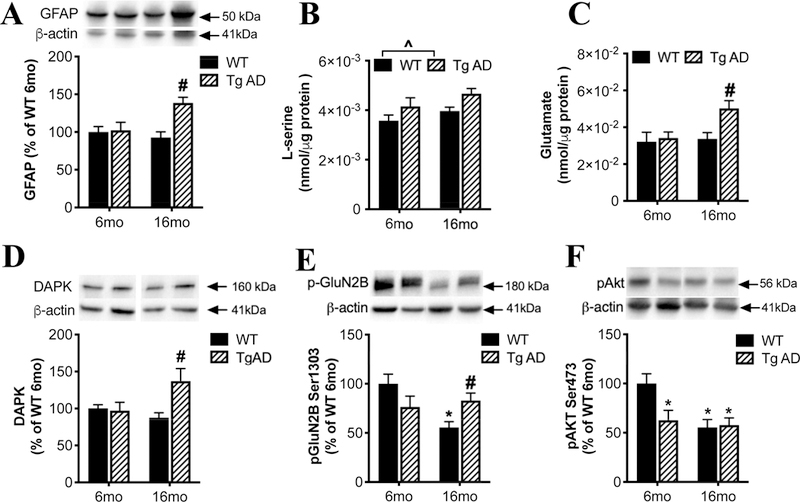
In the hippocampus of 6 or 16 months old wild-type (WT; black bars) and transgenic AD (Tg AD; striped bars) rats, we measured: (A) protein levels of GFAP, tissue levels of (B) L-serine and (C) glutamate, protein levels of (D) death associated protein kinase (DAPK), (E) phospho-GluN2B Ser1303, and (F) phospho-Akt-Ser473. Asterisk (*) and # indicates significant difference from 6-months and 16-months old WT, respectively (P < 0.05). ^ indicated significant difference between genotypes. All values represent the mean ± SEM.
Evidence suggests that activation of extra-synaptic NMDARs by excess glutamate spillover is responsible for the deleterious effects of glutamate on plasticity and neuronal survival (Bading, 2017). The cellular consequences of synaptic versus extra-synaptic NMDAR stimulation are very different. While synaptic NMDARs activate signaling pathways critical for neuronal survival, extrasynaptic NMDARs, which preferentially contain the GluN2B subunit, trigger cell death pathways (Bading, 2017). Phosphorylation of GluN2B at Ser1303 by Death-associated protein kinase (DAPK) is linked to cell death pathways via extrasynaptic NMDAR activation (Tu et al., 2010). DAPK (Figure 6D; age: F(1, 19) = 1.60, P=0.22; genotype: F(1,19) = 4.61, P=0.047; interaction: F(1, 20) = 5.97, P=0.03) and P-GluN2BSer1303 (Figure 6E; age: F(1, 19) = 4.62, P=0.04; genotype: F(1,19) = 0.04, P=0.83; interaction: F(1, 20) = 8.32, P=0.009) were significantly increased in the 16-month TgF344-AD rats compared to 16-month WT rats. We also found reduced pAkt Ser473 in 6-month TgF344-AD rats that remained low at 16months-old and did not differ from 16 month WT rats (Figure 6F; age: F(1, 19) = 7.72, P=0.01; genotype: F(1,19) = 3.86, P=0.06; interaction: F(1, 20) = 4.98, P=0.037). The changes in phosphorylation were not associated with changes in total levels of GluN2B or Akt (Supplemental Figure 3D and E). These findings are consistent with the hypothesis that increased extrasynaptic NMDAR activation leads to decreased cell survival via phosphorylated GluN2BSer1303 (Parsons and Raymond, 2014; Tu et al., 2010).
Discussion
We demonstrate a robust up-regulation of SR in reactive astrocytes in the hippocampus and entorhinal cortex of subjects with AD that increased with disease progression. The majority of these SR-expressing astrocytes were also neurotoxic, as they co-expressed C3. We observed a similar phenomenon in the AD rat model, whereby neurotoxic reactive astrocytes expressing SR increased with age. Finally, we observed an increased activation of signaling pathways associated with extrasynaptic NMDAR activation in the hippocampus of aged TgF344-AD rats.
Healthy astrocytes perform numerous homeostatic functions, such as maintaining and modulating neuronal communication and synaptic physiology. When astrocytes undergo a pronounced transformation called ‘reactive astrogliosis,’ it interferes with their ability to perform their normal functions. These reactive astrocytes are not able to effectively modulate neuronal glutamatergic synaptic transmission. For example, the microenvironment surrounding plaques in an AD mouse model has chronically elevated glutamate concentrations that correlate with a reduced expression of the glutamate transporter-1 (GLT-1) in the reactive astrocytes surrounding plaques, while soluble Aβ oligomers reduce the expression of GLT-1 in astrocyte cultures (Abdul et al., 2009; Scimemi et al., 2013). Thus, one mechanism by which reactive astrocytes contribute to neuronal loss is via glutamate excitotoxicity through excessive activation of extrasynaptic NMDARs. The production and release of D-serine by reactive astrocytes would promote excitotoxicity because D-serine is the co-agonist required for the excess agonist (glutamate) to activate extrasynaptic NMDARs. Recent research has identified a subtype of reactive astrocytes, termed A1, that highly express C3 and are abundant in several human neurodegenerative diseases, including AD, which lose their ability to sustain neuronal survival and synapse formation, and are neurotoxic by undefined mechanisms (Liddelow et al., 2017). Our results suggest that D-serine released from A1 reactive astrocytes could be contributing to their neurotoxic phenotype. Future studies should identify the molecular mechanism(s) that account for the up-regulation of SR transcription and/or translation, as well as D-serine uptake and release in reactive astrocytes.
Evidence suggests that activation of extra-synaptic NMDARs, which preferentially contain the GluN2B subunit, triggers cell death pathways and is responsible for the deleterious effects of glutamate on plasticity and neuronal survival (Bading, 2017). In particular, phosphorylation of GluN2B at Ser1303 by DAPK is linked to cell death pathways via extrasynaptic NMDAR activation (Tu et al., 2010), with selective activation of DAPK in excitatory pyramidal neurons in the entorhinal cortical layer II contributing to the synapse and memory loss in Tg2576-APPswe mice (Shu et al., 2016). In agreement with this, we found increased protein levels of both DAPK and GluN2BSer1303 in the hippocampus of 16 month-old TgF344-AD rats. Contrary to extra-synaptic receptors, synaptic NMDARs activate signaling pathways critical for neuronal survival (Bading, 2017). For example, synaptic NMDAR activity promotes sustained activation of the Akt pathway that can inhibit the activation of pro-death transcription factors (Hardingham and Bading, 2010). We found that p-Akt (active form) was reduced in 6 month-old TgF344-AD rats and remained at that level at 16-months old. Interestingly, p-Akt decreased with age in WT rats to a level not different from age-matched TgF344-AD rats. This suggests that neuronal and synaptic loss in the hippocampus of aged TgF344-AD rats is due, in part, to elevated GluN2BSer1303 signaling. As these downstream signaling pathways are very complex and can be influenced by numerous upstream effectors, future work will aim to tease apart the contributions of synaptic versus extrasynaptic NMDAR signaling and glial-mediated D-serine release.
Our human post-mortem data showed that the highest density of SR-expressing reactive astrocytes occurred in the CA1 region of the hippocampus and superficial layers of the entorhinal cortex, areas in which neurons are particularly vulnerable to degeneration in early AD (Fu et al., 2018). In other areas where neurons are less vulnerable, such as deep layers of the entorhinal cortex and CA3 of the hippocampus (Fu et al., 2018), the density of SR-expressing reactive astrocytes was higher only in late Braak stages or not significantly increased at any stage. Thus, the brain regions with the highest density of SR-expressing reactive astrocytes observed here align well with brain regions known to be particularly susceptible to neurodegeneration in AD. The higher number of GFAP+ astrocytes, which we detected in AD, is unlikely due to an increase in proliferation, but rather increased expression of GFAP in pre-existing astrocytes; GFAP expression in quiescent astrocytes is often below the detection level of immunohistochemistry (Perez-Nievas and Serrano-Pozo, 2018). It should also be noted that the neuronal SR expression was reduced in AD in both entorhinal cortex and hippocampus. Future studies are needed to delineate whether this was due to neuronal loss, a down-regulation of SR expression in remaining neurons, or a combination of both. We previously observed a reduction in neuronal SR expression without neuronal loss in a mouse model of TBI (Perez et al., 2017). Finally, we included AD medication as a co-variant in our ANCOVA model that affected the significance of some of our findings. The statistical interaction between GFAP density and exposure to AD medication is consistent with current evidence indicating that acetylcholinesterase inhibitors and memantine reduce neuroinflammation and glial activation (Folch et al., 2018; Matsuda and Hisatsune, 2017; Rajasekar et al., 2016). This covariant inclusion does have two confounds: 1) we were able to obtain AD-related medication history for only five AD subjects and 2) we only know that the AD subjects had been prescribed these medications, but do not know what the drug levels were at the time of death. However, with the available data, our results suggest that increased density of SR and C3 immunoreactive astrocytes is not due to an effect of AD medication.
In sum, our current AD and previous TBI findings (Perez et al., 2017) support a model whereby D-serine released from reactive, neurotoxic astrocytes binds to extrasynaptic GluN2B-containing NMDARs, thereby driving excitotoxic signaling and neuronal damage and death. These results highlight the “Janus-like” role of D-serine as both the gate-keeper for neuroplasticity (Balu et al., 2012; Balu and Coyle, 2012; Balu et al., 2013; Basu et al., 2009; Benneyworth et al., 2012; Li et al., 2013; Lin et al., 2016) and a toxic mediator of reactive astrocytes. Our findings have important therapeutic implications for patients with mild to advanced AD when anti-amyloid strategies will likely be ineffective. Thus, our results highlight astrocytic SR as a potential target for blocking NMDAR excitoxicity in AD and other diseases associated with SR-expressing reactive astrocytes, by limiting D-serine availability.
Supplementary Material
Acknowledgements
The research described in this manuscript was supported by 5R00MH099252-04 and a subcontract of R01NS098740-02 to DTB, Program for Advancing Strategic International Networks to Accelerate the Circulation of Talented Researchers (S2702) of the Japan Society for the Promotion of Science to YU, RF1AG047667 to DW, R01MH104488 and R01MH105608 to SB, and R01MH51290-18 to JTC.
Abbreviations
- AD
Alzheimer’s disease
- C3
complement C3
- DAPK
death associated protein kinase
- GFAP
glial fibrillary acid protein
- NMDAR
N-methyl-D-aspartate receptor
- SR
serine racemase
Footnotes
Conflicts of interest: DTB served as a consultant for LifeSci Capital, JTC has served as a consultant for Concert Pharmaceuticals, and DW has served as a consultant for Longitude Capital. JTC is also an inventor on US Patent Number 7,704,978 and four other related US patents, that are assigned to The General Hospital Corporation, Boston, Massachusetts, USA. The remaining authors declare no competing interests.
References
- Abdul HM, et al. , 2009. Cognitive decline in Alzheimer’s disease is associated with selective changes in calcineurin/NFAT signaling. J Neurosci 29, 12957–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amanatkar HR, et al. , 2017. Analysis of recent failures of disease modifying therapies in Alzheimer’s disease suggesting a new methodology for future studies. Expert Rev Neurother 17, 7–16. [DOI] [PubMed] [Google Scholar]
- Bading H, 2017. Therapeutic targeting of the pathological triad of extrasynaptic NMDA receptor signaling in neurodegenerations. J Exp Med 214, 569–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, et al. , 2012. The NMDA receptor co-agonists, d-serine and glycine, regulate neuronal dendritic architecture in the somatosensory cortex. Neurobiology of disease 45, 671–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, Coyle JT, 2012. Neuronal d-serine regulates dendritic architecture in the somatosensory cortex. Neuroscience letters 517, 77–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balu DT, et al. , 2013. Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc Natl Acad Sci U S A 110, E2400–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu AC, et al. , 2009. Targeted disruption of serine racemase affects glutamatergic neurotransmission and behavior. Mol Psychiatry 14, 719–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benneyworth MA, et al. , 2012. Cell Selective Conditional Null Mutations of Serine Racemase Demonstrate a Predominate Localization in Cortical Glutamatergic Neurons. Cellular and molecular neurobiology [DOI] [PMC free article] [PubMed]
- Berretta S, et al. , 2007. Neuron numbers and volume of the amygdala in subjects diagnosed with bipolar disorder or schizophrenia. Biol Psychiatry 62, 884–93. [DOI] [PubMed] [Google Scholar]
- Braak H, et al. , 2006. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 112, 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendel M, et al. , 2016. Glial Activation and Glucose Metabolism in a Transgenic Amyloid Mouse Model: A Triple-Tracer PET Study. J Nucl Med 57, 954–60. [DOI] [PubMed] [Google Scholar]
- Coggeshall RE, Lekan HA, 1996. Methods for determining numbers of cells and synapses: a case for more uniform standards of review. J Comp Neurol 364, 6–15. [DOI] [PubMed] [Google Scholar]
- Cohen RM, et al. , 2013. A transgenic Alzheimer rat with plaques, tau pathology, behavioral impairment, oligomeric abeta, and frank neuronal loss. J Neurosci 33, 6245–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P, 1993. Oxidative stress, glutamate, and neurodegenerative disorders. Science 262, 689–95. [DOI] [PubMed] [Google Scholar]
- Curcio L, et al. , 2013. Reduced D-serine levels in the nucleus accumbens of cocaine-treated rats hinder the induction of NMDA receptor-dependent synaptic plasticity. Brain 136, 1216–30. [DOI] [PubMed] [Google Scholar]
- Dorph-Petersen KA, Lewis DA, 2011. Stereological approaches to identifying neuropathology in psychosis. Biol Psychiatry 69, 113–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan MF, et al. , 2018. Randomized Trial of Verubecestat for Mild-to-Moderate Alzheimer’s Disease. N Engl J Med 378, 1691–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folch J, et al. , 2018. Memantine for the Treatment of Dementia: A Review on its Current and Future Applications. J Alzheimers Dis 62, 1223–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu H, et al. , 2018. Selective vulnerability in neurodegenerative diseases. Nat Neurosci 21, 1350–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillery RW, Herrup K, 1997. Quantification without pontification: choosing a method for counting objects in sectioned tissues. J Comp Neurol 386, 2–7. [DOI] [PubMed] [Google Scholar]
- Gundersen HJ, et al. , 1999. The efficiency of systematic sampling in stereology--reconsidered. J Microsc 193, 199–211. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Bading H, 2010. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci 11, 682–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, De Strooper B, 2017. Alzheimer’s disease: where next for anti-amyloid therapies? Brain 140, 853–855. [DOI] [PubMed] [Google Scholar]
- Hashimoto A, et al. , 1992. Determination of free amino acid enantiomers in rat brain and serum by high-performance liquid chromatography after derivatization with N-tert.-butyloxycarbonyl-L-cysteine and o-phthaldialdehyde. J Chromatogr 582, 41–8. [DOI] [PubMed] [Google Scholar]
- Hyman BT, et al. , 1998. Stereology: a practical primer for neuropathology. J Neuropathol Exp Neurol 57, 305–10. [DOI] [PubMed] [Google Scholar]
- Hyman BT, et al. , 2012. National Institute on Aging-Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement 8, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kartvelishvily E, et al. , 2006. Neuron-derived D-serine release provides a novel means to activate N-methyl-D-aspartate receptors. The Journal of biological chemistry 281, 14151–62. [DOI] [PubMed] [Google Scholar]
- Li S, et al. , 2018. Astrocytes in primary cultures express serine racemase, synthesize d-serine and acquire A1 reactive astrocyte features. Biochem Pharmacol [DOI] [PMC free article] [PubMed]
- Li Y, et al. , 2013. Identity of endogenous NMDAR glycine site agonist in amygdala is determined by synaptic activity level. Nat Commun 4, 1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddelow SA, et al. , 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin H, et al. , 2016. D-Serine and Serine Racemase Are Associated with PSD-95 and Glutamatergic Synapse Stability. Front Cell Neurosci 10, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markota M, et al. , 2014. Reduced dopamine transporter expression in the amygdala of subjects diagnosed with schizophrenia. Schizophr Bull 40, 984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda T, Hisatsune T, 2017. Cholinergic Modification of Neurogenesis and Gliosis Improves the Memory of AbetaPPswe/PSEN1dE9 Alzheimer’s Disease Model Mice Fed a High-Fat Diet. J Alzheimers Dis 56, 1–23. [DOI] [PubMed] [Google Scholar]
- Olney JW, 1969. Brain lesions, obesity, and other disturbances in mice treated with monosodium glutamate. Science 164, 719–21. [DOI] [PubMed] [Google Scholar]
- Pantazopoulos H, et al. , 2007. Parvalbumin neurons in the entorhinal cortex of subjects diagnosed with bipolar disorder or schizophrenia. Biol Psychiatry 61, 640–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantazopoulos H, et al. , 2015. Aggrecan and chondroitin-6-sulfate abnormalities in schizophrenia and bipolar disorder: a postmortem study on the amygdala. Transl Psychiatry 5, e496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papouin T, et al. , 2012. Synaptic and extrasynaptic NMDA receptors are gated by different endogenous coagonists. Cell 150, 633–46. [DOI] [PubMed] [Google Scholar]
- Parsons MP, Raymond LA, 2014. Extrasynaptic NMDA receptor involvement in central nervous system disorders. Neuron 82, 279–93. [DOI] [PubMed] [Google Scholar]
- Perez EJ, et al. , 2017. Enhanced astrocytic d-serine underlies synaptic damage after traumatic brain injury. J Clin Invest 127, 3114–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Nievas BG, Serrano-Pozo A, 2018. Deciphering the Astrocyte Reaction in Alzheimer’s Disease. Front Aging Neurosci 10, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzishevsky I, Wolosker H, 2012. An enzymatic-HPLC assay to monitor endogenous D-serine release from neuronal cultures. Methods Mol Biol 794, 291–7. [DOI] [PubMed] [Google Scholar]
- Rajasekar N, et al. , 2016. Inhibitory Effect of Memantine on Streptozotocin-Induced Insulin Receptor Dysfunction, Neuroinflammation, Amyloidogenesis, and Neurotrophic Factor Decline in Astrocytes. Mol Neurobiol 53, 6730–6744. [DOI] [PubMed] [Google Scholar]
- Rorabaugh JM, et al. , 2017. Chemogenetic locus coeruleus activation restores reversal learning in a rat model of Alzheimer’s disease. Brain 140, 3023–3038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothman SM, Olney JW, 1995. Excitotoxicity and the NMDA receptor--still lethal after eight years. Trends Neurosci 18, 57–8. [DOI] [PubMed] [Google Scholar]
- Schafer DP, et al. , 2016. Microglia contribute to circuit defects in Mecp2 null mice independent of microglia-specific loss of Mecp2 expression. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scimemi A, et al. , 2013. Amyloid-beta1–42 slows clearance of synaptically released glutamate by mislocalizing astrocytic GLT-1. J Neurosci 33, 5312–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu S, et al. , 2016. Selective Degeneration of Entorhinal-CA1 Synapses in Alzheimer’s Disease via Activation of DAPK1. J Neurosci 36, 10843–10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu W, et al. , 2010. DAPK1 interaction with NMDA receptor NR2B subunits mediates brain damage in stroke. Cell 140, 222–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villegas-Llerena C, et al. , 2016. Microglial genes regulating neuroinflammation in the progression of Alzheimer’s disease. Curr Opin Neurobiol 36, 74–81. [DOI] [PubMed] [Google Scholar]
- Wolosker H, et al. , 2016. The Rise and Fall of the d-Serine-Mediated Gliotransmission Hypothesis. Trends Neurosci 39, 712–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JH, et al. , 2010. Brain-specific Phgdh deletion reveals a pivotal role for L-serine biosynthesis in controlling the level of D-serine, an N-methyl-D-aspartate receptor co-agonist, in adult brain. The Journal of biological chemistry 285, 41380–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun SP, et al. , 2018. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med 24, 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian JL, et al. , 2012. Genomic analysis of reactive astrogliosis. J Neurosci 32, 6391–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.