

Abstract
Objective
Pathogenic variants in SCN8A, encoding the voltage‐gated sodium (Na+) channel α subunit Nav1.6, is a known cause of epilepsy. Here, we describe clinical and genetic features of all patients with SCN8A epilepsy evaluated at a single‐tertiary care center, with biophysical data on identified Nav1.6 variants and pharmacological response to selected Na+ channel blockers.
Methods
SCN8A variants were identified via an exome‐based panel of epilepsy‐associated genes for next generation sequencing (NGS), or via exome sequencing. Biophysical characterization was performed using voltage‐clamp recordings of ionic currents in heterologous cells.
Results
We observed a range in age of onset and severity of epilepsy and associated developmental delay/intellectual disability. Na+ channel blockers were highly or partially effective in most patients. Nav1.6 variants exhibited one or more biophysical defects largely consistent with gain of channel function. We found that clinical severity was correlated with the presence of multiple observed biophysical defects and the extent to which pathological Na+ channel activity could be normalized pharmacologically. For variants not previously reported, functional studies enhanced the evidence of pathogenicity.
Interpretation
We present a comprehensive single‐center dataset for SCN8A epilepsy that includes clinical, genetic, electrophysiologic, and pharmacologic data. We confirm a spectrum of severity and a variety of biophysical defects of Nav1.6 variants consistent with gain of channel function. Na+ channel blockers in the treatment of SCN8A epilepsy may correlate with the effect of such agents on pathological Na+ current observed in heterologous systems.
Introduction
Voltage‐gated sodium (Na+) channels (VGSCs) mediate the generation and propagation of electrical signals in excitable cells.1, 2, 3 Pathogenic variants in or deletion of the predominant brain‐expressed Na+ channel genes SCN1A, 2A, 3A, and 8A 1 are known causes of epilepsy.2
De novo pathogenic missense variants in SCN8A are associated with a spectrum of epilepsy severity, from benign familial infantile seizures (BFIS), infantile convulsions, and paroxysmal choreoathetosis (ICCA), to early infantile epileptic encephalopathy (EIEE)/developmental and epileptic encephalopathy (DEE).4, 5, 6, 7, 8, 9 In general, variants associated with severe childhood‐onset epilepsy are de novo missense variants, and electrophysiological studies of such variants typically reveal “gain of (channel) function” consistent with enhanced Na+ current.7, 10, 11 Mice harboring missense mutations associated with EIEE in humans exhibit severe, early onset epilepsy and increased epilepsy‐associated mortality.12, 13 Electrophysiological recordings of acutely dissociated cells and acute brain slices prepared from mice harboring an Scn8a‐p.Asn1768Asp amino acid substitution (Scn8a N1768D/+ mice), modeling a recurrent SCN8A pathogenic variant in humans,14 demonstrate neuronal hyperexcitability in selected subsets of neurons.15, 16
Consistent with this gain of channel function, patients with SCN8A epilepsy may exhibit clinical response to Na+ channel blockers.17, 18 The Na+ channel modulator GS967, which exhibits more selective block of persistent relative to peak transient current, has been shown to be an effective treatment for seizures in Scn8a N1768D/+ and Scn8a R1872W/+ mice,12, 19 both of which harbor variants shown previously to exhibit increased slowly inactivating/“persistent” current.
We characterized all patients with SCN8A epilepsy seen and evaluated at a single center and determined the biophysical effects of the corresponding identified pathogenic variants on electrophysiological function. We then tested Na+ channel modulators on selected epilepsy‐associated pathogenic variants in heterologous systems.
Patients and Methods
Study subjects
Patients included in the study (Tables 1 and 2) were seen and evaluated at The Children’s Hospital of Philadelphia, Philadelphia, PA, USA, and this study was approved by the Institutional Review board.
Table 1.
Clinical features of patients with SCN8A epilepsy.
| Patient | Variant | Epilepsy features | DD? | MRI | ||
|---|---|---|---|---|---|---|
| Onset (age) | Epilepsy Type | Epilepsy treatment | ||||
| 1 | p.Gly1475Arg | 4 months | Focal | −LVT; ++OXC; −ZON; +LAC | Mild | Macrocephaly; mild atrophy |
| 2 | p.Arg1872Leu | 6 weeks | Focal | −LVT; −LMG; −TPM; −CLB; −VPA; +OXC; +PHT | Severe | Normal |
| 3 | p.Ala1491Val | 3 weeks | Focal, Tonic, Tonic‐clonic | −PHB; −PHT; −LVT; −LAC; −OXC; −TPM; −CBD | Profound | Normal |
| 4 | p.Asn374Lys | 7 years | Focal, Tonic‐clonic | −OXC; −VPA; −LVT; −ZNS; −CLB; −LAC; ++PHT | Mild | PVNH |
| 5 | p.Leu483Phe | 23 months | Atypical absence | +ZAR; −LVT | No | Normal |
| p.Val1758Ala | ||||||
| 6 | p.Met139Ile | 9 years | Tonic‐clonic | −LVT; ++LMG | Mild | Normal |
| 7 | p.Arg1872Trp | 2 months | Focal, Tonic‐clonic | −LVT; −OXC; +PHT | Moderate | Normal |
LVT, levetiracetam; OXC, oxcarbazepine; ZON, zonisamide; LAC, lacosamide; LMG, lamotrigine; TPM, topiramate; CLB, clobazam; CBD, cannabidiol; DD, developmental delay; SE, status epilepticus; FS, febrile seizure(s).
Note that "−" indicates no response; "+" positive response; "++" strong positive response.
Table 2.
Genetic features of patients with SCN8A epilepsy.
| Patient | Variant | gnomAD | CADD score | Inheritance pattern | |
|---|---|---|---|---|---|
| Nucleotide | Protein change | ||||
| WT | – | ||||
| 1 | c.4423G>A | p.Gly1475Arg | 0 | 43 | De novo |
| 2 | c.5615G>T | p.Arg1872Leu | 0 | 32 | De novo |
| 3 | c.4472C>T | p.Ala1491Val | 0 | 27 | De novo |
| 4 | c.1122C>G | p.Asn374Lys | 0 | 21.6 | De novo |
| 5 | c.1447C>T | p.Leu483Phe | 1/230,832 | 24.5 | Paternal |
| c.5372T>C | p.Val1758Ala | 0 | 22.6 | De novo | |
| 6 | c.417G>A | p.Met139Ile | 0 | 23.2 | De novo |
| 7 | c.5614C>T | p.Arg1872Trp | 0 | 26.4 | De novo |
Patient 1 was a male with epilepsy onset at 4 months of age. Seizures were stabilized on supratherapeutic doses of oxcarbazepine, with infrequent breakthrough seizures described as transient focal onset with impaired awareness or nonmotor autonomic onset, with occasional progression to bilateral tonic‐clonic seizures. Epilepsy was accompanied by mild global developmental delay as well as attention deficit/hyperactivity disorder and features of autism spectrum disorder. Magnetic resonance imaging (MRI) of the brain was initially normal and later revealed mild atrophy. Interictal electroencephalogram (EEG) remained normal. At last follow‐up, the patient remained on oxcarbazepine and low‐dose clobazam, and was in a mainstream school with additional assistance, and continued to have 1–3 brief seizures per year.
Patient 2 was a female with first recognized seizure at 6 weeks of age. Seizures were of focal onset with impaired awareness accompanied by automatisms, tonic movements, or autonomic phenomena, with progression to bilateral tonic‐clonic seizures and frequent episodes of status epilepticus. Epilepsy was accompanied by profound developmental delay/intellectual disability as well as choreoathetoid limb movements.
Patient 3 was a male with epilepsy onset at 3 weeks of age progressing to treatment‐resistant epilepsy with recurrent status epilepticus, profound global developmental delay, and status‐post gastrostomy tube and trachestomy placement, hypotonia and central visual impairment. EEG showed multifocal epileptiform discharges while MRI showed mild underopercularization. The patient died of sudden unexpected death (SUDEP) at 2 years of age.
Patient 4 was a female with focal onset epilepsy with first seizure at 7 years of age, accompanied by moderate intellectual disability and behavioral disorder. At last follow‐up at 13 years of age, the patient was ambulatory and conversant in full sentences. Epilepsy was controlled using high‐dose phenytoin, with periods of seizure freedom up to 1 year in duration.
Patient 5 was a female with febrile seizures prior to 2 years of age, then the onset of absence epilepsy at 2 years of age with frequent, brief episodes of arrest of activity, eye fluttering, and staring correlated with bursts of rhythmic delta in the bilateral occipital regions on EEG. Interictal EEG showed irregularly generalized spike‐wave discharges. MRI of the brain was normal. The patient was seizure‐free on ethosuxamide. Epilepsy panel revealed two heterozygous variants in SCN8A, one of which was determined to be paternally inherited while the other was de novo.
Patient 6 was a male with febrile seizures prior to 2 years of age. First unprovoked seizure was at 9 years of age. At last follow‐up at 12 years of age, the patient had approximately 10 total seizures and remained seizure‐free for a period of 2 years on moderate‐dose lamotrigine. EEG showed occasional independent bilateral spike and wave discharges without clinical signs. MRI was normal.
Patient 7 was a female with first seizure of life at 2 months of age progressing to DEE with recurrent status epilepticus and global developmental delay with central hypotonia. MRI was normal. EEG showed disorganization of the background.
Genetic testing
Testing was performed either through commercial laboratories or in‐house at The Children's Hospital of Philadelphia. For in‐house testing, we performed a comprehensive exome‐based sequencing and copy number analysis of 100 epilepsy‐associated genes. Genomic DNA was extracted from peripheral blood to obtain DNA material for sequencing and an in‐house bioinformatics pipeline was used for analysis.20, 21 After filtering, variants were classified using published guidelines from the American College of Medical Genetics and Genomics.22
Plasmid preparation
A plasmid encoding human SCN8A was used (Reference Sequence NM_014191.3), and variants were introduced by site‐directed mutagenesis. All plasmids were sequenced prior to transfection.
Cell culture and transfection
HEK293T cells (ATCC, CRL‐3216) expressing either wild‐type or epilepsy‐associated SCN8A variants were grown under standard conditions.23 Auxiliary subunits β1 (hβ1‐V5‐2A‐dsRed) and β2 (pGFP‐IRES‐ hβ2) were co‐transfected with pcDNA3.1‐SCN8A constructs as described in detail in previous studies.23
Electrophysiology
Whole‐cell patch clamp biophysical experiments were performed at room temperature using a MultiClamp 700B amplifier (Molecular Devices, Sunnyvale, CA) in an extracellular solution consisting of the following: 109 mmol/L NaCl, 36 mmol/L choline chloride, 4 mmol/L KCl, 1.8 mmol/L CaCl2, 1.8 mmol/L MgCl2, 10 mmol/L HEPES, and 10 mmol/L glucose; pH was adjusted to 7.35 with NaOH while osmolarity was adjusted to 305 mOsm/L with sucrose.23 Intracellular solution contained, in mmol/L: CsF, 110; NaF, 10; CsCl, 20; EGTA, 2.0; HEPES, 10. pH was adjusted to 7.35 with CsOH and osmolarity to 300 mOsm/L with sucrose.
Recording pipettes were fashioned from thin‐walled borosilicate glass (Sutter Instruments, Novato, CA), fire‐polished, and wrapped with parafilm. Cells with access resistance of 2–4 MΩ were considered for recording; those with an increase in access resistance by 20% were excluded from analysis. Recording was initiated 10 min after achieving the whole‐cell configuration, after which recorded currents were found to be stable for the duration of the recording period. Currents were corrected for capacitive and leak currents and voltage errors were reduced via series resistance compensation up to 80%. Voltage‐clamp pulses were generated using Clampex 10.6, acquired at 10 kHz, and filtered at 5 kHz. Standard protocols for the determination of activation properties, steady state inactivation, and recovery from inactivation were described previously.23 Persistent currents were measured as the average value of the currents in response in the last 10 ms of a 200 ms test pulse to − 10 mV. Ramp currents were obtained using a voltage ramp at 0.8 mV/ms from holding potential to +40 mV. Total charge (area under the curve; in Coulombs/pF) were calculated. Kinetics of recovery from channel inactivation was determined as described previously.23
All recordings and data analysis (below) were performed blind to experimental group.
Pharmacology
Pharmacologic experiments were performed via bath perfusion. Oxcarbazepine was purchased from Tocris, GS967 was purchased from Cayman Chemical, dissolved in DMSO, and stored as 10‐ and 0.5‐mmol/L stock solution respectively prior to 1:1000 dilution in ACSF.
Data analysis
Data for standard electrophysiological parameters were obtained from at least n = 20 cells from multiple separate transfections for each experiment. Data were analyzed using custom Matlab scripts or Clampfit 10.6 (Molecular Devices). Results are presented as the mean ± standard error of the mean (SEM) and statistical significance was established using the P value calculated from a one‐way analysis of variance (ANOVA) with post hoc Bonferroni test for correction of multiple comparisons, or, for pharmacological experiments (pre/post), using a paired Student’s t‐test, with the P‐value reported exactly. Normality of the data was tested using the D’Agostino and Pearson normality test with P < 0.05.
Results
A single‐center cohort of patients with SCN8A epilepsy
We identified a cohort of seven patients with epilepsy seen and evaluated over the course of 6 years who were determined to harbor de novo heterozygous pathogenic variants in SCN8A (Tables 1 and 2; Figure 1; see Patients and Methods). Age at epilepsy onset was variable, ranging from 3 weeks to 9 years. Seizures were generalized tonic‐clonic, focal, and focal with evolution to a bilateral tonic‐clonic, in all patients except for one; a single patient had atypical childhood absence epilepsy. Developmental delay was present in 6 of 7 patients (86%) and ranged in severity from mild to profound. Magnetic resonance imaging (MRI) of the brain was performed in all cases and was normal in 5 of 7 patients (71%); it was abnormal in 2 of 7 patients (29%), showing mild atrophy in one case and periventricular nodular heterotopiae in a second case. Choreoathetosis was seen in one patient. SUDEP, which is known to occur at a relatively high rate in patients with SCN8A encephalopathy,5, 24 occurred in one patient in the cohort (14%).
Figure 1.
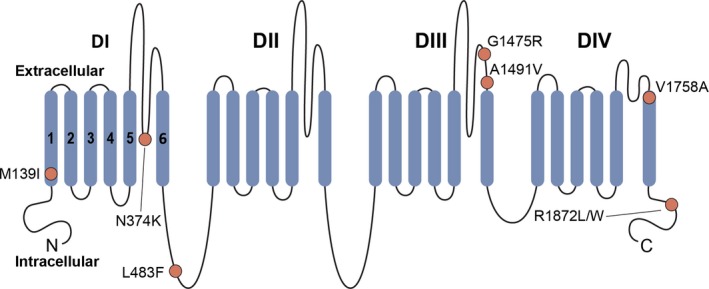
Schematic of Nav1.6. Na+ channels are comprised of a single polypeptide that is composed of four repeated domains (domains (D) I–IV), with each domain formed by six transmembrane segments (S1–S6); S1–S4 represent the voltage‐sensing region, while S5‐S6 forms the conducting pore. Shown is a schematic of the Nav1.6 protein, with DI‐IV and S1–S6 as well as the Nav1.6 variants included in this study (circles).
Identified pathogenic variants in SCN8A lead to a variety of biophysical Na+ channel defects
To determine the functional effects of the identified variants, we performed whole‐cell voltage‐clamp electrophysiological recordings of wild‐type and variant hNav1.6. We included the inherited variant SCN8A‐p.Leu483Phe found in Patient 5 as a negative control.
Representative families of currents are shown for wild‐type Nav1.6 and each variant, along with the G‐V curves (Figure 2).
Figure 2.
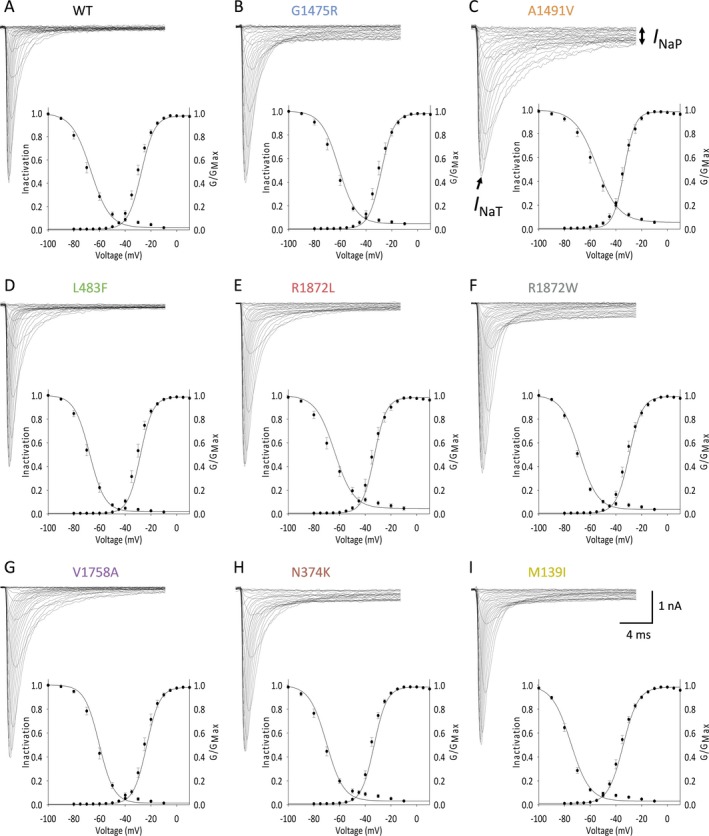
Selected epilepsy‐associated Nav1.6 variants lead to changes in the voltage dependence of channel availability. A–I, Representative traces (top) showing families of sodium current elicited by 20 ms depolarizing voltage steps from −80 mV to +50 mV in 5 mV increments, for wild‐type (WT, n = 28) and NaV1.6 variants Gly1475Arg (n = 25), Ala1491Val (n = 26), Leu483Phe (n = 22), Arg1872Leu (n = 27), Arg1872Trp (n = 25), Val1758Ala (n = 23) Asp374Lys (n = 23), and Met139Ile (n = 25). Included are the corresponding G‐V curves for the population data (bottom) illustrating the voltage dependence of steady‐state activation and inactivation, presented as mean ± SEM, with lines representing the Boltzman fit to the data points.
Peak current density was not different between groups (Table 3).
Table 3.
Biophysical properties of wild‐type and Nav1.6 mutant channels.
| Variant | PeakI Na density | Voltage dependence of activation | Persistent current | Decay | n | ||||
|---|---|---|---|---|---|---|---|---|---|
| pA/pF | n | V 1/2 | k | n | % of peak | n | τ (ms) | ||
| Wild‐type | 360.4 ± 16.6 | 28 | −27.5 ± 0.7 | 5.2 ± 0.1 | 28 | 1.7 ± 0.2 | 31 | 0.8 ± 0.03 | 31 |
| G1475R | 371.8 ± 28.3 | 25 | −28.1 ± 1.2 | 4.9 ± 0.3 | 25 | 7.2 ± 0.7*** | 28 | 1.1 ± 0.06 | 28 |
| A1491V | 397.6 ± 24.9 | 26 | −33.6 ± 0.9*** | 4.2 ± 0.3 | 26 | 7.4 ± 0.8*** | 26 | 2.4 ± 0.15*** | 26 |
| L483F | 425.6 ± 24.6 | 22 | −28.3 ± 0.8 | 4.8 ± 0.2 | 21 | 1.6 ± 0.2 | 23 | 0.8 ± 0.05 | 23 |
| R1872L | 434.1 ± 34.9 | 27 | −32.9 ± 1.0*** | 4.7 ± 0.2 | 27 | 2.9 ± 0.4* | 25 | 1.3 ± 0.11** | 25 |
| R1872W | 461.8 ± 23.3 | 25 | −29.7 ± 0.9 | 5.1 ± 0.2 | 25 | 5.8 ± 0.6*** | 25 | 1.7 ± 0.12*** | 25 |
| V1758A | 427.9 ± 28.1 | 23 | −23.4 ± 1.1† | 5.1 ± 0.2 | 23 | 1.1 ± 0.2 | 23 | 0.8 ± 0.07 | 23 |
| N374K | 447.9 ± 23.7 | 25 | −33.2 ± 0.7*** | 4.9 ± 0.3 | 25 | 3.5 ± 0.3*** | 25 | 0.9 ± 0.06 | 25 |
| M139I | 385.2 ± 27.1 | 25 | −34.4 ± 0.7*** | 5.9 ± 0.2 | 25 | 4.9 ± 0.5*** | 26 | 1.2 ± 0.12* | 26 |
† P = 0.052; *P < 0.05; **P < 0.01; ***P < 0.001; vs. wild‐type via one‐way ANOVA (with Bonferroni correction for multiple comparisons).
*P < 0.05; **P < 0.01; ***P < 0.001 vs. wild‐type (noncorrected).
The voltage dependence of channel availability, quantified as the voltage at half‐maximal activation (V 1/2 of activation), was − 27.5 ± 0.7 mV (mean ± standard error of the mean (SEM); n = 28), similar to previously reported values.25 We observed a left (hyperpolarizing) shift in the voltage dependence of channel availability of the Nav1.6‐p.Ala1491Val (−33.2 ± 1.0 mV; n = 26; P < 0.0001 vs. wild‐type via one‐way ANOVA with post hoc correction for multiple comparisons with Bonferroni test), Arg1872Leu (−32.9 ± 1.0 mV; n = 27; P = 0.0007 vs. wild‐type), Asn374Lys (−33.2 ± 0.7 mV; n = 25; P = 0.0004 vs. wild‐type), and Met139Ile variants (−29.7 ± 0.7 mV; n = 25; P < 0.0001 vs. wild‐type). The V 1/2 of activation of Nav1.6‐p.Val1758Ala was − 23.4 ± 1.1 mV (n = 23; P = 0.0518 via one‐way ANOVA with post hoc correction for multiple comparisons; P = 0.0014 via one‐way ANOVA without correction).
There were prominent differences in the slowly inactivating/persistent component of the Na+ current (I NaP) (Figure 3A–B). I NaP was 1.7 ± 0.2% (n = 31) of peak transient current (I NaT) for wild‐type channels, but was 7.2 ± 0.7% for Nav1.6‐p.Gly1475Arg (n = 28; P < 0.0001 vs. wild‐type via one‐way ANOVA with post hoc correction for multiple comparisons via Bonferroni test), 7.4 ± 0.8% for Ala1491Val (n = 26; P < 0.0001 vs. wild‐type), 5.8 ± 0.6% for Arg1872Trp (n = 25; P < 0.0001 vs. wild‐type), and 4.9 ± 0.5% for Met139Ile (n = 26; P < 0.0001 vs. wild‐type). The ratio of persistent to transient current for Nav1.6‐p.Asn374Lys was 3.5 ± 0.3% (n = 25; P = 0.2842 via one‐way ANOVA with post hoc correction for multiple comparisons; P = 0.0079 via one‐way ANOVA without correction).
Figure 3.
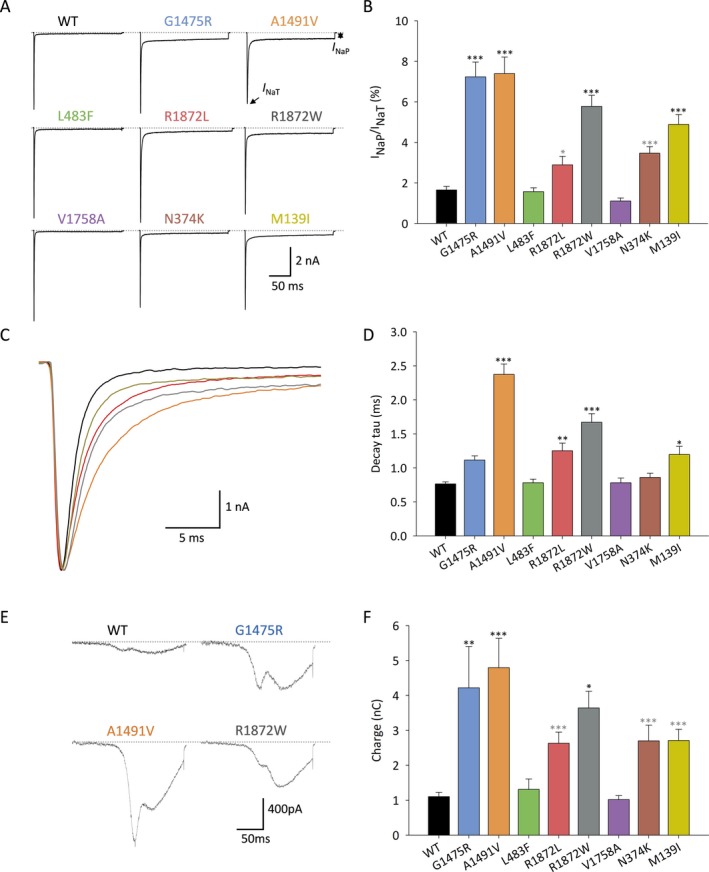
Selected epilepsy‐associated Nav1.6 variants result in enhanced persistent sodium current and/or impaired fast inactivation. A–B, Increased persistent current. A, Representative current traces showing transient (I NaT) and persistent (I NaP) Na+ current for channels composed of wild‐type (WT) and variant Nav1.6 subunits in response to a voltage steps from − 120 mV to − 10 mV for 200 ms. B, Bar graph showing I NaP: I NaT ratio for WT (n = 31) and variant Nav1.6 subunits Gly1475Arg (n = 28), Ala1491Val (n = 26), Leu483Phe (n = 23), Arg1872Leu (n = 25), Arg1872Trp (n = 25), Val1758Ala (n = 23), Asn374Lys (n = 25), and Met139Ile (n = 26). C–D, Impaired fast inactivation. C, Representative single traces showing I NaT for wild‐type and variant Nav1.6 illustrating fast inactivation. Note that traces were scaled to the peak. D, Quantification of decay tau of I NaT in wild‐type (WT) and variant Nav1.6 channels. E‐F, Ramp currents are increased in Na+ channels composed of variant NaV1.6 subunits. E, Shown is a representative current traces of wild‐type (WT) and variant Nav1.6 in response to slow depolarizing voltage ramps evoked by linear increases from −120 mV to +40 mV at 0.8 mV/ms. F, Quantitative analysis of total charge (in nanocoulombs; nC) for WT (n = 27) versus Nav1.6 variants Gly1475Arg (n = 22), Ala1491Val (n = 26), Leu483Phe (n = 22), Arg1872Leu (n = 24), Arg1872Trp (n = 21), Val1758Ala (n = 22), Asp374Lys (n = 23), and Met139Ile (n = 23). Data are presented as mean ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001; via one‐way ANOVA with Bonferonni correction for multiple comparisons. Light gray **P < 0.01; light gray ***P < 0.001, via one‐way ANOVA without correction.
A subset of epilepsy‐associated pathogenic variants exhibited impaired fast inactivation (Figure 3C‐D). For wild‐type Nav1.6, decay of the fast, transient, inward current was well fit with a single exponential (τ1) of 0.77 ± 0.03 ms (mean ± SEM; n = 18). In comparison, the decay tau was 1.11 ± 0.06 ms for Nav1.6‐p.Gly1475Arg (n = 28; P = 0.22 vs. wild‐type via one‐way ANOVA with Bonferroni correction for multiple comparisons), 2.37 ± 0.15 ms for Nav1.6‐p.Ala1491Val (n = 26; P < 0.0001), 0.78 ± 0.05 for Nav1.6‐p.Leu483Phe (n = 23; P> 0.9999), 1.25 ± 0.11 for Nav1.6‐p.Arg1872Leu (n = 25; P = 0.0086), 1.67 ± 0.13 for Nav1.6‐p.Arg1872Trp (n = 25; P < 0.0001), 0.78 ± 0.07 for Nav1.6‐p.Val1758Ala (n = 23; P> 0.9999), 0.86 ± 0.06 for Nav1.6‐p.Asn374Lys (n = 25; P> 0.9999), and 1.20 ± 0.12 for Nav1.6‐p.Met139Ile (n = 26; P = 0.0335).
We found that pathogenic epilepsy‐associated Nav1.6 variants that displayed increased I NaP also showed increased currents in response to slow ramp depolarizations (Figure 3E–F). Total charge (in nanocoulombs) was 1.10 ± 0.12 nC (n = 27) for wild‐type Nav1.6, 4.23 ± 0.12 nC for Nav1.6‐p.Gly1475Arg (n = 26; P = 0.003 vs. wild‐type via one‐way ANOVA with post hoc correction for multiple comparisons using Bonferroni test), 4.80 ± 0.84 for Nav1.6‐p.Ala1491Val (n = 26; P < 0.0001), 1.31 ± 0.30 nC for Nav1.6‐p.Leu483Phe (n = 22; P> 0.9999), 2.63 ± 0.32 nC for Nav1.6‐p.Arg1872Leu (n = 24; P> 0.9999), 3.64 ± 0.48 nC for Nav1.6‐p.Arg1872Trp (n = 21; P = 0.053), 2.70 ± 0.45 nC for Nav1.6‐p.Asn374Lys (n = 23; P> 0.9999), and 2.70 ± 0.32 for Nav1.6‐p.Met139Ile (n = 23; P> 0.9999).
Modulation of pathological Nav1.6 subunit‐containing sodium channels with compounds that target persistent sodium current
Na+ channel modulators that selectively block I NaP over I NaT have been shown previously to reduce seizures in mouse models of SCN8A epilepsy.12, 19 We found that both oxcarbazepine and the novel Na+ channel modulator GS967 were highly specific for I NaP over I NaT, as reported previously (Figure 4A–D).23, 26 Oxcarbazepine produced a 71.9 ± 11.4% block of wild‐type Nav1.6 (n = 5; P = 0.04 vs. pretreatment control via paired Student’s t‐test) (Figure 4A–B). Oxcarbazepine was highly effective in seizure reduction in Patient 1 (Table 1) and produced a 62.7 ± 8.8% block of Nav1.6‐p.Gly1475Arg (n = 10; P = 0.001 vs. control) to 2.69 ± 0.57% (P = 1.00 for posttreatment Nav1.6‐p.Gly1475Arg vs. pretreatment wild‐type Nav1.6). Oxcarbazepine yielded a 67.4 ± 2.1% block of Nav1.6‐p.Ala1491Val (n = 8; P = 0.001 vs. control) and 68.4 ± 4.4% block of Nav1.6‐p.Arg1872Trp (n = 9; P = 0.001 vs. control) (Figure 4A–B).
Figure 4.
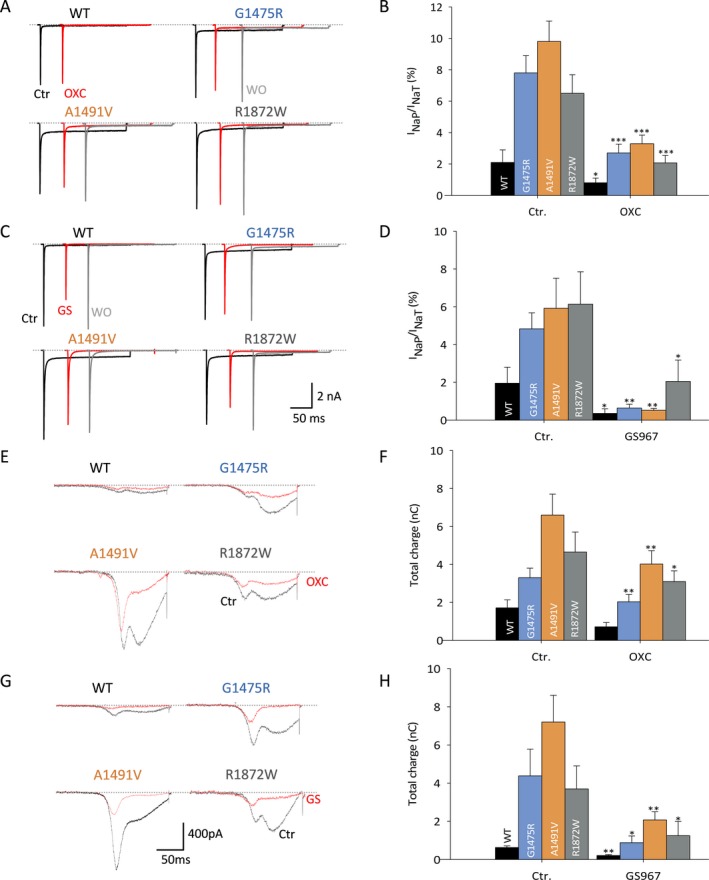
Anti‐seizure medications block pathological INaP and slow ramp currents. A–D, Block of persistent currents. A, C, Traces showing I NaT and I NaP of WT and variant Nav1.6 before (black) and after (red) the application of 10 μmol/L oxcarbazepine (A) and 0.5 μmol/L GS967 (C), with washout (gray). B, D, Bar graphs showing I NaP: I NaT ratio before (Ctr.) and after the application of oxcarbazepine (OXC; B) or GS‐967 (D). B, control Nav1.6 (Ctr.), n = 10; Nav1.6‐p.Gly1475Arg, n = 10; Ala1491Val, n = 8; Arg1872Trp, n = 9). D, GS967, control Nav1.6 (Ctr.), n = 5; Gly1475Arg, n = 5; Ala1491Val, n = 5; Arg1872Trp, n = 4). E–H, Ramp currents are increased in Na+ channels composed of variant NaV1.6 subunits. E, G, Example traces showing ramp currents for WT and variant Nav1.6 channels before (Ctr., black) and after (red traces) application of 10 μmol/L oxcarbazepine (OXC; E) and 0.5 μmol/L GS967 (G). F, H, Bar graph showing % total charge before and after the application of oxcarbazepine (WT, n = 5; Gly1475Arg, n = 10; Ala1491Val, n = 8 and Arg1872Trp, n = 9) and GS967 (WT, n = 4; Gly1475Arg, n = 5; Ala1491Val, n = 5 and Arg1872Trp, n = 4). Data are presented as mean ± SEM; *P < 0.05; **P < 0.01; ***P < 0.001 (Ctr. vs. drug via two‐tailed paired Student’s t‐test).
GS967 produced a 86.8 ± 4.1% block of I NaP in the Nav1.6‐p.Gly1475Arg variant identified in Patient 1 (n = 5; P = 0.006 vs. pretreatment control) (Figure 4C–D). Residual I NaP of 0.64 ± 0.2% was not different than pretreatment wild‐type (P = 0.142). GS967 also normalized the pathological epilepsy‐associated I NaP observed in Nav1.6‐p.Ala1491Val (87.0 ± 5.5% block; n = 5; P = 0.029 vs. pretreatment control) and p.Arg1872Trp variants (73.5 ± 8.9% block; n = 4; P = 0.007 vs. pretreatment control), but had no effect on the hyperpolarizing shift in the voltage dependence of activation or impaired fast inactivation observed in the Nav1.6‐p.Ala1491Val or p.Arg1872Trp variants.
Discussion
A single‐center cohort of patient with SCN8A epilepsy
Here we report the clinical, genetic, and electrophysiological properties of all pathogenic variants in SCN8A identified in children with epilepsy at a single center. We found a spectrum of clinical phenotypes, with a broad range of severity in terms of epilepsy and developmental delay/intellectual disability as well as of underlying biophysical Na+ channel dysfunction.
Identified variants were found to be de novo (identified in the patient/proband, and not directly inherited from either parent). Such variants were absent from control databases of human genetic variation. Functional studies in heterologous expression systems generally demonstrated biophysical defects consistent with gain of channel function via a diversity of mechanisms, consistent with prior reports.7, 27 For the variants that had not previously reported, functional studies increased the evidence of pathogenicity from likely pathogenic (based on one strong and one moderate criteria) to pathogenic (two strong criteria).22
Clinically, we encountered a variety of phenotypes.4, 5 The severity of developmental delay/intellectual disability ranged from none (Patient 5; SCN8A‐p.Val1758Ala) to profound (Patient 3; SCN8A‐p.Ala1491Val). Epilepsy age of onset ranged from 3 weeks to 9 years of age. Epilepsy was focal/multifocal in most (6 of 7) cases.
One variant (SCN8A‐p.Val1758Ala identified in Patient 5) exhibited an apparent loss of channel function, with a rightward/depolarizing shift in the voltage dependence of channel availability. Interestingly, this patient presented with atypical absence epilepsy.
De novo pathogenic variants in SCN8A causing EIEE are highly penetrant. However, clinical severity does not seem to strictly correlate with biophysical abnormalities identified experimentally via voltage‐clamp recordings of Na+ channel currents in heterologous systems. In fact, it has been reported that patients with the same missense variant in SCN8A can apparently have different clinical phenotypes.5 How to rationalize these observations? It could be the case that genetic background or inheritance of a complement of other rare variants in ion channels and/or other regulatory genes might have a modifying effect, as has been hypothesized for SCN9A acting as a modifier of the phenotype associated with pathogenic variants in SCN1A. 28 Interestingly, we identified a second, inherited variant in SCN8A in Patient 5 (p.Leu483Phe), although we do not know if the two identified variants exist in cis or trans and did not test the functional impact of a p.Leu483Phe variant on the biophysical properties of Nav1.6‐p.Val1758Ala.
In our dataset, channel dysfunction appeared to loosely correlate with clinical severity. Three of the four Nav1.6 variants identified in more mildly affected patients either showed an increased persistent current alone (Patient 1) or a shift in the voltage dependence of activation (Patients 4 and 5). In contrast, variants identified in patients with intractable epilepsy and moderately severe to profound global developmental delay (Patients 2, 3, and 7) exhibited more complex channel dysfunction, with two or more identified abnormalities. One variant, SCN8A‐p.Met139Ile, did not follow this correlation, being identified in a patient with mild intellectual disability yet exhibiting a left‐shift in the voltage dependence of channel availability, increased persistent current, and slightly impaired fast channel inactivation. How the variants characterized here might affect the function of neurons can only be inferred. A recent study found that the biophysical properties of Nav1.6 variants in heterologous expression systems did not necessarily predict the effect of overexpression of such variants in cultured hippocampal neurons.27
The identification of a gene‐specific diagnosis is important as it may inform prognosis, including, importantly, risk of SUDEP,5, 7, 24 as well as treatment. Epilepsy in some patients with SCN8A encephalopathy may respond well to Na+ channel antagonists such as phenytoin.17, 29 We found that 5 of 7 patients (71%) had a positive clinical response to anti‐seizure drugs that have a prominent mechanism of action of Na+ channel blockade, including 3 (Patients 1, 4, and 6) who had seizure‐free periods lasting over 1 year. For one patient (Patient 1) with a dramatic clinical response to oxcarbazepine, we tested the effect of this anti‐seizure medication on Na+ channels containing the associated Nav1.6 variant, and found that it produced normalization of pathological epilepsy‐associated increased persistent current. However, oxcarbazepine did not alter fast inactivation or the voltage dependence of channel availability, which may at least partially explain why oxcarbazepine and other Na+ channel blockers were without appreciable clinical effect in Patients 2 and 3.
Future work will aim to fully define the clinical‐genetic landscape of SCN8A epilepsy, the physiological properties of identified pathogenic variants, and the pharmacological response of such variants to existing and candidate anti‐seizure compounds targeting Na+ channels. Advanced model systems such as induced pluripotent stem cell‐derived neurons and mouse models will facilitate a link across levels of analysis to provide insight into how genetic variants in ion channels lead to seizures and epilepsy, which may inform development of novel treatments for SCN8A epilepsy and related ion channelopathies.
Author Contributions
TZ, AAT, and EMG contributed to the conception and design of the study; TZ, AAT, and EMG contributed to the acquisition and analysis of data; TZ and EMG contributed to preparing the figures and EMG wrote the manuscript.
Conflict of Interest
None declared.
Acknowledgments
We thank Dr. Xiaohong Zhang for expert technical assistance. We thank Lori L. Isom for the gift of a β‐1 cDNA clone and Alfred L. George for the gift of a β‐2 cDNA clone. This work was supported by NIH NINDS K08 NS097633 and U54 NS108874 and the Burroughs Wellcome Fund Career Award for Medical Scientists to EMG.
Funding Information
This work was supported by National Institutes of Health (NIH) NINDS grant K08 NS097633 and a Burroughs Wellcome Fund Career Award for Medical Scientists to E.M.G., as well as by NIH NINDS U54 NS108874.
Funding Statement
This work was funded by National Institute of Neurological Disorders and Stroke grants K08 NS097633 and U54 NS108874; Burroughs Wellcome Fund grant Career Award for Medical Scientists.
References
- 1. Catterall WA. From ionic currents to molecular mechanisms: the structure and function of voltage‐gated sodium channels. Neuron 2000;26:13–25. [DOI] [PubMed] [Google Scholar]
- 2. Catterall WA. Voltage‐gated sodium channels at 60: structure, function and pathophysiology. J Physiol 2012;590:2577–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. O’Malley HA, Isom LL. Sodium channel β subunits: emerging targets in channelopathies. Annu Rev Physiol 2015;77:481–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Larsen J, Carvill GL, Gardella E, et al. The phenotypic spectrum of SCN8A encephalopathy. Neurology 2015;84:480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gardella E, Marini C, Trivisano M, et al. The phenotype of SCN8A developmental and epileptic encephalopathy. Neurology 2018;91:e1112–e1124. [DOI] [PubMed] [Google Scholar]
- 6. Gardella E, Becker F, Møller RS, et al. Benign infantile seizures and paroxysmal dyskinesia caused by an SCN8A mutation. Ann Neurol 2016;79:428–436. [DOI] [PubMed] [Google Scholar]
- 7. Veeramah KR, O’Brien JE, Meisler MH, et al. De novo pathogenic SCN8A mutation identified by whole‐genome sequencing of a family quartet affected by infantile epileptic encephalopathy and SUDEP. Am J Hum Genet 2012;90:502–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Johannesen KM, Gardella E, Encinas AC, et al. The spectrum of intermediate SCN8A‐related epilepsy. Epilepsia 2019;60:830–844. [DOI] [PubMed] [Google Scholar]
- 9. Denis J, Villeneuve N, Cacciagli P, et al. Clinical study of 19 patients with SCN 8A ‐related epilepsy: two modes of onset regarding EEG and seizures. Epilepsia 2019;60:845–856. [DOI] [PubMed] [Google Scholar]
- 10. Patel RR, Barbosa C, Brustovetsky T, et al. Aberrant epilepsy‐associated mutant Nav1.6 sodium channel activity can be targeted with cannabidiol. Brain 2016;139(Pt 8):2164–2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wagnon JL, Barker BS, Hounshell JA, et al. Pathogenic mechanism of recurrent mutations of SCN8A in epileptic encephalopathy. Ann Clin Transl Neurol 2016;3:114–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bunton‐Stasyshyn RKA, Wagnon JL, Wengert ER, et al. Prominent role of forebrain excitatory neurons in SCN8A encephalopathy. Brain 2019;142:362–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wagnon JL, Korn MJ, Parent R, et al. Convulsive seizures and SUDEP in a mouse model of SCN8A epileptic encephalopathy. Hum Mol Genet 2015;24:506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wagnon JL, Meisler MH. Recurrent and non‐recurrent mutations of SCN8A in epileptic encephalopathy. Front Neurol 2015;6:104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lopez‐Santiago LF, Yuan Y, Wagnon JL, et al. Neuronal hyperexcitability in a mouse model of SCN8A epileptic encephalopathy. Proc Natl Acad Sci 2017;114:2383–2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ottolini M, Barker BS, Gaykema RP, et al. Aberrant sodium channel currents and hyperexcitability of medial entorhinal cortex neurons in a mouse model of SCN8A encephalopathy. J Neurosci 2017;37:7643–7655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Boerma RS, Braun KP, van de Broek MPH, et al. Remarkable phenytoin sensitivity in 4 children with SCN8A‐related epilepsy: a molecular neuropharmacological approach. Neurotherapeutics 2016;13:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Møller RS, Johannesen KM. Precision medicine: SCN8A encephalopathy treated with sodium channel blockers. Neurotherapeutics 2016;13:190–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Baker EM, Thompson CH, Hawkins NA, et al. The novel sodium channel modulator GS‐458967 (GS967) is an effective treatment in a mouse model of SCN8A encephalopathy. Epilepsia 2018;59:1166–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Niazi R, Gonzalez MA, Balciuniene J, et al. The development and validation of clinical exome‐based panels using exomeslicer. J Mol Diagnostics 2018;20:643–652. [DOI] [PubMed] [Google Scholar]
- 21. Balciuniene J, DeChene ET, Akgumus G, et al. Use of a dynamic genetic testing approach for childhood‐onset epilepsy. JAMA Netw Open 2019;2:e192129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zaman T, Helbig I, Božović IB, et al. Mutations in SCN3A cause early infantile epileptic encephalopathy. Neurol Ann 2018;83:703–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johannesen KM, Gardella E, Scheffer I, et al. Early mortality in SCN8A ‐related epilepsies. Epilepsy Res 2018;143:79–81. [DOI] [PubMed] [Google Scholar]
- 25. Burbidge SA, Dale TJ, Powell AJ, et al. Molecular cloning, distribution and functional analysis of the NA(V)1.6. Voltage‐gated sodium channel from human brain. Brain Res Mol Brain Res 2002;103:80–90. [DOI] [PubMed] [Google Scholar]
- 26. Anderson LL, Thompson CH, Hawkins NA, et al. Antiepileptic activity of preferential inhibitors of persistent sodium current. Epilepsia 2014;55:1274–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu Y, Schubert J, Sonnenberg L, et al. Neuronal mechanisms of mutations in SCN8A causing epilepsy or intellectual disability. Brain 2019;142:376–390. [DOI] [PubMed] [Google Scholar]
- 28. Mulley JC, Hodgson B, McMahon JM, et al. Role of the sodium channel SCN9A in genetic epilepsy with febrile seizures plus and Dravet syndrome. Epilepsia 2013;54:e122–e126. [DOI] [PubMed] [Google Scholar]
- 29. Wolff M, Johannesen KM, Hedrich UBS, et al. Genetic and phenotypic heterogeneity suggest therapeutic implications in SCN2A‐related disorders. Brain 2017;140:1316–1336. [DOI] [PubMed] [Google Scholar]