

Abstract
Mutations in the MAPT gene cause frontotemporal dementia with tau deposits. We report the novel p.P397S MAPT variant in eight subjects from five apparently nonrelated families suffering from frontotemporal dementia with autosomal dominant pattern of inheritance. In silico analysis reported conflicting evidence of pathogenicity. The segregation analysis support that this variant is likely pathogenic. The mean age at onset (61.4 years) and mean disease duration (13.9 years) of these subjects and their affected relatives were significantly higher compared with our series of p.P301L MAPT mutation carriers. These findings suggest that p.P397S variant could be a new MAPT mutation associated with a less aggressive phenotype than other MAPT mutations.
Introduction
Mutations in the microtubule‐associated protein tau (MAPT) gene in chromosome 17 were described in 1998 as a cause of frontotemporal lobar degeneration (FTLD) with tau pathology.1, 2, 3 To date, nearly 50 pathogenic MAPT mutations have been reported.4 Clinically, FTLD related to MAPT mutations may affect behavior, language, memory, and executive functions. Parkinsonism has also been reported in some cases. Most MAPT mutations are characterized by early onset (mean 47.9 years) and a rapidly progressive clinical deterioration with a mean age at death of 58.7 years.5
We describe the clinical phenotype of a novel exon 13 MAPT variant – a Proline to Serine substitution at codon 397 (p.P397S) – in eight subjects with FTLD with an autosomal dominant pattern of inheritance from five apparently nonrelated families. We hypothesize that this variant could be causative of FTLD. In addition, we compare the clinical phenotype of these cases with our series of the most frequent (p.P301L) MAPT mutation.6
Methods
Subjects were recruited from the Genetic counseling program for familial dementias (PICOGEN program) at the Hospital Clínic de Barcelona.7 A detailed clinical history and neurological evaluation of all subjects were performed. We reviewed the information available from all the affected cases in the pedigrees. The diagnosis of behavioral variant frontotemporal dementia (bvFTD) was performed following the current diagnostic criteria.8 Cerebrospinal fluid (CSF) analysis for Alzheimer's disease (AD) biomarkers was measured with INNOTEST ELISAs following manufacturer's instructions (Fujirebio, Ghent, Belgium). Our laboratory normal reference values for amyloid beta 42 (AB42), total tau (t‐tau), and phosphorylated tau at threonine 181 (p‐tau) are >660, <385, and >65 pg/mL, respectively.
Ethics
This research was performed according to the guidelines of the Declaration of Helsinki. The participants provided written informed consent for genetic testing and publication of relevant findings. The study was approved by the Hospital Clínic Ethics committee.
Genetic analysis
Genetic screening for MAPT (exons 1 and 9–13) and progranulin (GRN) (exons 1–13) or serum progranulin levels was performed in the eight subjects as previously described.9 The MAPT haplotype (H1/H2) was determined studying the SNP rs1800547 and APOE genotype with two genotyping assays (rs429358 and rs7412), using TaqMan genotyping technologies (Life Technologies, Carlsbad, CA). C9ORF72 GGGGCC hexanucleotide repeat expansion was tested with a repeat‐primed PCR. Subject II.II.VIII was examined by exome sequencing using MedExome (Roche, Basel, Switzerland) in an Illumina NextSeq500.
We searched for functional information at ENSEMBL database (https://www.ensembl.org), where the variant was described as rs1295855402, and with algorithms that predict whether an amino acid substitution affects the protein function such as SIFT, Polyphen‐2, REVEL, Mutation Assessor, CADD, and MetaLR.
Statistical analysis
Mean age at onset, disease duration, and age at death were calculated considering together subjects with proven variant and their affected relatives. Results in p.P397S carriers were compared with our series of p.P301L using Fisher's exact test. Disease duration was analyzed using Kaplan–Meier estimator and compared with Log‐rank test. The analyses were performed using the SPSS Statistics Version 20.0 IBM Corp, Chicago, IL), and the level of significance was established at a P level of 0.05 (two‐sided).
Results
Clinical phenotype
We include here the description of several representative cases.
Family I
Subject I.III.IV developed behavioral disinhibition, sweet food preference, deficits in executive tasks, and semantic language impairment at the age of 60 meeting criteria for bvFTD. MRI showed severe medial, lateral, and polar bitemporal atrophy. Currently, after 17 years of disease, she is in a stage of moderately severe dementia with severe semantic impairment scoring 5/30 at Boston Naming Test (BNT), but preserved motor functions. She had an autosomal dominant family history of dementia on her mother, aunt, grandmother, and three siblings (Fig. 1). All of them developed cognitive and behavioral dysfunction starting from 50 to 77 years old and showing slow disease progression. Due to this family history of dementia genetic exam was performed in subject I.III.IV and one of her brothers (subject I.III.III) revealing the presence of the p.P397S MAPT variant in both.
Figure 1.
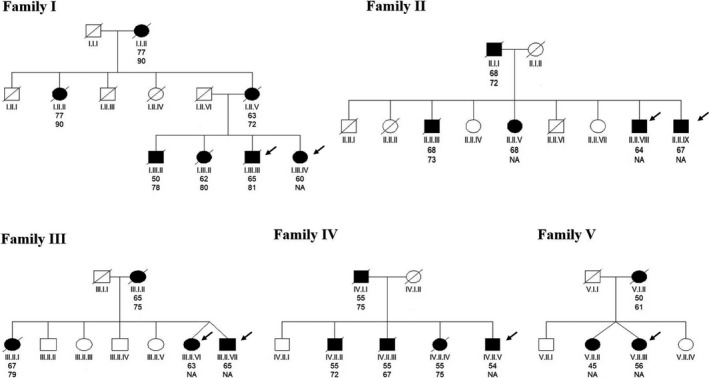
Pedigree of the five reported families. Arrows indicate the probands described in the main text. Upper ages beneath each symbol are age at onset. Lower ages beneath each symbol are age at death.
Family II
Subject II.II.IX developed socially inappropriate behavior at the age of 64. Neuropsychological evaluation revealed short‐term memory and semantic language impairment (BNT 35/60). The MRI demonstrated moderate bitemporal atrophy (Fig. 2). The CSF core biomarkers analysis showed normal levels of AB42 (1308 pg/mL), but increased levels of t‐tau (414 pg/mL) and p‐tau (87 pg/mL). He had a family history of late‐onset dementia on his father, and three siblings (Fig. 1). The genetic study demonstrated the presence of the p.P397S variant in the MAPT gene. Currently, after 8 years of disease duration, he still is in a moderate stage of dementia, scoring 24 at the MMSE and 102 at the Revised Cambridge Behavioral Inventory (CBI‐R).
Figure 2.
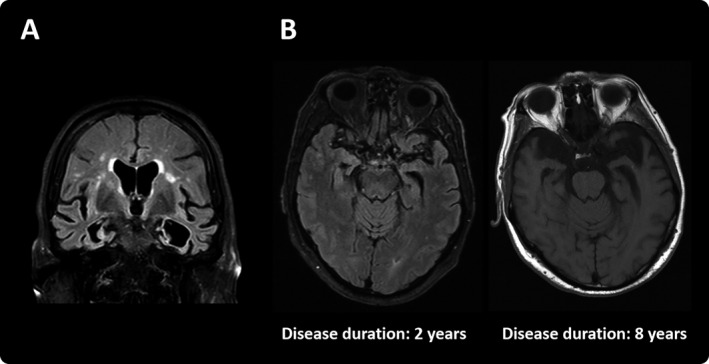
MRI of subject III.II.VI showing important medial and polar bitemporal atrophy (A). Longitudinal MRI examinations of subject II.II.IX (B).
Subject II.II.VIII was a sibling of subject II.II.IX. He initially developed episodic and semantic memory loss and behavioral disinhibition at the age of 67. The CT scan showed bitemporal atrophy and the CSF exam presented decreased levels of AB42 (437 pg/mL) and increased t‐tau (543 pg/mL) and p‐tau (80 pg/mL), so AD diagnosis was established. During the follow‐up, the patient developed severe apathy, loss of empathy, hyperorality, and executive dysfunction, highly suggestive of bvFTD. He also presented mild rigid‐akinetic parkinsonism. Currently, after 8 years of disease progression, he scores 25 at the Mini Mental State Examination (MMSE) and he is independent for basic activities of daily living and most of the instrumental activities. The genetic test confirmed the presence of the p.P397S variant, consequently the final diagnosis of genetic bvFTD with possible concomitant AD was established.
The p.P397S MAPT variant was not found in subject II.II.VII, an asymptomatic 68‐year‐old sibling.
Family III
Subjects III.II.VI and III.II.VII were dizygotic twins. Their mother developed dementia at the age of 65. Another sibling presented clinically with behavioral impairment dysfunction strongly suggestive of bvFTD (Fig. 1). Subject III.II.VI presented with disinhibition, apathy, and ritualistic behaviors at the age of 63. Neuropsychological evaluation revealed impairment in memory and naming. MMSE score was 19 at the age of 69. The MRI showed severe medial bitemporal atrophy (Fig. 2) and the amyloid PET scan (IMM Flutemetamol‐F18) was negative. Subject III.II.VII became apathetic, exhibit diminished social interest, poor hygiene, and significantly increased smoking and alcohol intake at the age of 64. MRI demonstrated severe bitemporal atrophy. The genetic study revealed the p.P397S variant in both twins.
Summary of the clinical phenotype of p.P397S carriers
Table 1 summarizes the demographic and clinical features of the eight subjects who are confirmed carriers of the p.P397S MAPT variant.
Table 1.
Demographic and clinical features of subjects with the p.P397S MAPT variant.
| Subject | Gender | Ethnicity | Age at onset (y) | Age at death (y) | Disease duration (y) | Current dementia stage | Current MMSE | Boston Naming Test | CBI‐R | Parkinsonism | Clinical Diagnosis | MAPT haplotype | APOE | AD biomarkers | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AB42 | t‐tau | p‐tau | ||||||||||||||
| I.III.IV | Female | Caucasian | 60 | Alive | 16 | Moderately severe | 9 | 5/30 | 94 | Yes | bvFTD | 1/2 | 3/3 | NA | NA | NA |
| I.III.III | Male | Caucasian | 64 | 81 | 17 | Dead | – | NA | NA | No | AD | 1/2 | 3/3 | NA | NA | NA |
| II.II.IX | Male | Caucasian | 64 | Alive | 8 | Moderate | 24 | 30/60 | 102 | No | bvFTD | 1/2 | 3/3 | 1308 | 414 | 87 |
| II.II.VIII | Male | Caucasian | 67 | Alive | 8 | Moderate | 25 | 35/60 | NA | Yes | bvFTD | 1/2 | 3/3 | 437 | 543 | 80 |
| III.II.VI | Female | Caucasian | 63 | Alive | 6 | Moderate | 19 | NA | NA | No | bvFTD | 1/1 | 2/3 | NA | NA | NA |
| III.II.VII | Male | Caucasian | 65 | Alive | 3 | Moderate | 25 | NA | NA | No | bvFTD | NA | NA | NA | NA | NA |
| IV.II.V | Male | Caucasian | 54 | Alive | 18 | Severe | 0 | NA | NA | Yes | bvFTD | NA | NA | NA | NA | NA |
| V.II.III | Female | Caucasian | 56 | Alive | 3 | Mild | 27 | 31/60 | 22 | No | bvFTD | 1/2 | 2/3 | 1423 | 850 | 91 |
AD, Alzheimer's Disease; bvFTD, behavioral variant frontotemporal dementia; CBI‐R, Revised Cambridge Behavioral Inventory; NA, not available; y, years.
All of them developed behavior problems and semantic language impairment. Three of them developed mild rigid‐akinetic parkinsonism. None of the subjects presented oculomotor impairment. All subjects presented bitemporal atrophy with relative frontal lobe preservation (Fig. 2). The frequency of these clinical symptoms does not differ from our published series of p.P301L mutation carriers.6
Subjects with the p.P397S MAPT variant presented a significantly older age at onset than the patients with the p.P301L MAPT mutation (61.3 years vs. 53.5 years; P = 0.016). In addition, p.P397S carriers showed a significantly older age at death (76.3 years vs. 60.3 years; P = 0.009) and longer disease duration (14.0 years vs. 6.9 years; P = 0.002) (Fig. 3).
Figure 3.
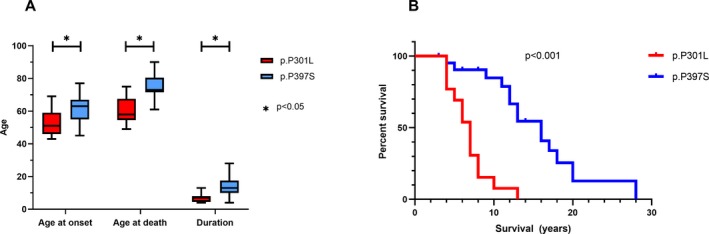
Distribution of onset ages, disease durations, and ages at death associated with p.P397S and P301L MAPT carriers (A). Survival curves of p.P397S and P301L MAPT carriers (B).
Genetic results
The eight patients studied carried the exon 13 MAPT p.P397S variant. Six of them were probands (subjects I.III.IV, II.II.IX, III.II.VI, III.II.VII, IV.II.V and V.II.III) and the other two (I.III.III and II.II.VIII) were tested after the results found in their relatives. None of them present c9ORF72 expansion. GRN mutations were excluded by direct sequencing in three p.P397S carriers (subjects I.III.IV, II.II.VIII, and IV.II.V) and the rest presented normal serum progranulin levels. In addition, one subject was studied by whole‐exome sequencing (II.II.VIII) excluding any other variants in known genes involved in neurodegeneration. This novel variant was predicted as likely pathogenic according to the American College of Medical Genetics and Genomics and the Association for Molecular Pathology guidelines and as definitively pathogenic by Guerreiro et. al. algorithm for mutation's pathogenicity.10, 11 In silico analysis produced different results, being predicted pathogenic by SIFT bioinformatics algorithm and probably damaging by Polyphen‐2. However, other algorithms scores prediction classified this variant as likely benign (CADD and REVEL algorithms), tolerated (MetaLR), or without a functional consequence (Mutation Assessor). This variant was not described in gnomAD database (http://gnomad.broadinstitute.org/) and it was present at ENSEMBL database as rs1295855402.
Discussion
In this study, we report a new MAPT variant (p.P397S) in eight subjects from five families suffering from FTLD with autosomal dominant pattern of inheritance. Even if the eight probands belong to five apparently nonrelated families, we could track a common geographical origin in the southeast of Spain, suggesting a possible founder effect of the p.P397S variant in this area.
We hypothesize that this new MAPT variant might be causative of FTLD. According to segregation analysis, their presence in eight affected bvFTD patients and their absence in one unaffected relative is predicted to be likely pathogenic in segregation analysis. In silico analysis produced discrepant results. The p.P397S variant is predicted to destroy a Proline/Serine phosphorylation site at the S396 position. Previous experimental data suggest that phosphorylation at S396 site is necessary to promote the long‐term depression at the hippocampus. In this sense, the p.P397S variant potentially will modify the physiological function of tau.12, 13
All patients with the p.P397S variant presented clinical features consistent with the diagnosis of bvFTD. Most patients also developed semantic language impairment. Three patients developed mild rigid‐akinetic parkinsonism. These clinical features do not differ from those of patients with the p.P301L mutation. However, patients with the p.P397S MAPT variant showed a significantly older age at disease onset and slower disease progression compared with p.P301L mutation carriers. Neuroimaging of all patients revealed bitemporal atrophy in concordance with the typical atrophy pattern described in FTLD due to MAPT mutations, but with relative preservation of other areas of the brain including frontal areas.14, 15
Mutations in the MAPT gene produce FTLD characterized by early onset dementia, with an age of onset between the third and fifth decade of life and 8–10 years of disease duration.5 However, previous studies reported that different MAPT mutations may show considerable phenotypic variations. This variability might be explained by the microtubule binding properties of the mutant protein.16 In spite of this, some mutations show an important variability in their age at onset or progression suggesting that other genetic or environmental factors can play an important role in the phenotypic expression.17 Only a few cases of MAPT mutation carriers have been reported to date presenting after the sixth decade.18, 19, 20 Interestingly, other mutations located at the exon 13 are also characterized by a slow rate of disease progression. The p.R406W mutation has a median age of onset of 55 (IQR 51.25–61.75) years and a median disease duration of 14 (IQR 9–26 years).21 As in our series, memory impairment was a marked symptom in p.R406W carriers. The p.T427M MAPT mutation, described only in one family, also seems to have a delayed age at onset (range 60–71 years) and death (67–79 years).22 However, the p.G389R mutation, also located in exon 13, has been related to an early onset presentation.23
The CSF AD biomarkers were tested in three of the p.P397S carriers (subjects II.II.IX, II.II.VIII, and V.II.III). Surprisingly, levels of p‐tau ant t‐tau were increased in all three, with decreased levels of Aβ42 only in one of them. Several previous reports have described diminished CSF Aβ levels in sporadic or/and genetic FTLD.24, 25, 26, 27 It is discussed whether this finding is related to an increased deposition of Aβ spices or to a reduction of Aβ production, as it is associated with a reduction in soluble APP levels. However, in the absence of neuropathological studies in our patients, we cannot rule out the presence of concomitant AD pathology. Of note, the presence of increased p‐tau and t‐tau at CSF, in addition with the temporal lobe atrophy, some of these patients could be misdiagnosed as suffering from AD. Of course, it is not possible to discard that the presence of AD concomitant pathology was the cause of these results.
The main limitation of this study is the absence of neuropathological and direct functional data. In the absence of functional evidence, we cannot ultimately rule out the possibility that this MAPT variant is just a rare polymorphism. Future functional assays are needed to confirm this variant as a pathogenic mutation.
In conclusion, we report a novel MAPT variant, consisting in the substitution of a Proline for a Serine in the codon 397, in eight subjects from five apparently nonrelated families suffering from frontotemporal dementia with autosomal dominant pattern of inheritance. We hypothesize that this new MAPT variant might be causative of a less aggressive FTLD than other MAPT mutations. In silico analysis using several prediction software produce different results about the potential pathogenicity of this novel variant and segregation analysis predicted it to be likely pathogenic. Thus, in the absence of neuropathology or functional data, we cannot confirm this variant is definitively pathogenic. Future studies are needed in order to determine the pathogenicity of this variant.
Author Contributions
Conception and design of the study: S. B.‐E., A. L., R. S.‐V. Acquisition of the data: S. B.‐E., I. P., C. P. B., M. T. A.‐V., J. O., N. F. Analysis of the data: S. B.‐E. Genetic and laboratory analysis: A. A., J. A. P.‐B. All authors contributed to the critical revision of the manuscript. Study supervision: R. S.‐V.
Conflict of Interest
None of the authors have conflict of interest to be disclosed.
Acknowledgments
Dr. Sergi Borrego‐Écija is the recipient of the Rio Hortega post‐residency research grant from the Instituto de Salud Carlos III, Spain (CM18/00028). This study was partially funded by Fundació Marató de TV3 (grant no. 20143810 to RSV) and Spanish Ministry of Economy and Competitiveness‐Instituto de Salud Carlos III and Fondo Europeo de Desarrollo Regional (FEDER), Unión Europea, “Una manera de hacer Europa” (PI14/00282 to Dr. A. Lladó). A. Antonell received funding from Departament de Salut de la Generalitat de Catalunya (PERIS 2016‐2020 SLT002/16/00329). The authors thank Dr. M Goedert and Dr. J Ávila for the information and references provided about previous experimental data.
Funding Information
Dr. Sergi Borrego‐Écija is the recipient of the Rio Hortega post‐residency research grant from the Instituto de Salud Carlos III, Spain (CM18/00028). This study was partially funded by Fundació la Marató de TV3 (grant no. 20143810 to R. S.‐V.) and Spanish Ministry of Economy and Competitiveness‐Instituto de Salud Carlos III and Fondo Europeo de Desarrollo Regional (FEDER), Unión Europea, “Una manera de hacer Europa” (PI14/00282 to Dr. A. Lladó). A. Antonell received funding from Departament de Salut de la Generalitat de Catalunya (PERIS 2016‐2020 SLT002/16/00329).
Funding Statement
This work was funded by Hospital Clínic de Barcelona grant ; Fundació la Marató de TV3 grant 20143810; Spanish Ministry of Economy and Competitiveness‐Instituto de Salud Carlos III grant ; Fondo Europeo de Desarrollo Regional grant ; Unión Europea grant PI14/00282; Departament de Salut de la Generalitat de Catalunya grant SLT002/16/00329.
References
- 1. Foster NL, Wilhelmsen K, Sima AA, et al. Frontotemporal dementia and parkinsonism linked to chromosome 17: a consensus conference. Conference Participants. Ann Neurol 1997;41:706–715. [DOI] [PubMed] [Google Scholar]
- 2. Poorkaj P, Bird TD, Wijsman E, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol 1998;43:815–825. [DOI] [PubMed] [Google Scholar]
- 3. Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5'‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature 1998;393:702–705. [DOI] [PubMed] [Google Scholar]
- 4. Cruts M, Theuns J, Van Broeckhoven C. Locus‐specific mutation databases for neurodegenerative brain diseases. Hum Mutat 2012;33:1340–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cruts M, Van Broeckhoven C. Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet 2008;24:186–194. [DOI] [PubMed] [Google Scholar]
- 6. Borrego‐Écija S, Morgado J, Palencia‐Madrid L, et al. Frontotemporal dementia caused by the P301L mutation in the MAPT gene: clinicopathological features of 13 cases from the same geographical origin in Barcelona, Spain. Dement Geriatr Cogn Disord 2017;44:213–221. [DOI] [PubMed] [Google Scholar]
- 7. Fortea J, Lladó A, Clarimón J, et al. PICOGEN: five years experience with a genetic counselling program for dementia. Neurologia 2011;26:143–149. [DOI] [PubMed] [Google Scholar]
- 8. Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain 2011;134:2456–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Antonell A, Gil S, Sánchez‐Valle R, et al. Serum progranulin levels in patients with frontotemporal lobar degeneration and Alzheimer's disease: detection of GRN mutations in a Spanish cohort. J Alzheimers Dis 2012;31:581–591. [DOI] [PubMed] [Google Scholar]
- 10. Guerreiro RJ, Baquero M, Blesa R, et al. Genetic screening of Alzheimer's disease genes in Iberian and African samples yields novel mutations in presenilins and APP. Neurobiol Aging 2010;31:725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Richards S, Aziz N, Bale S, et al; Laboratory Quality Assurance Committee . Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Iqbal K, Liu F, Gong CX. Tau and neurodegenerative disease: the story so far. Nat Rev Neurol 2016;12:15–27. [DOI] [PubMed] [Google Scholar]
- 13. Regan P, Piers T, Yi J‐H, et al. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J Neurosci 2015;35:4804–4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Whitwell JL, Jack CR, Boeve BF, et al. Atrophy patterns in IVS10+16, IVS10+3, N279K, S305N, P301L, and V337M MAPT mutations. Neurology 2009;73:1058–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fumagalli GG, Basilico P, Arighi A, et al. Distinct patterns of brain atrophy in Genetic Frontotemporal Dementia Initiative (GENFI) cohort revealed by visual rating scales. Alzheimers Res Ther 2018;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. van Swieten JC, Stevens M, Rosso SM, et al. Phenotypic variation in hereditary frontotemporal dementia with tau mutations. Ann Neurol 1999;46:617–626. [DOI] [PubMed] [Google Scholar]
- 17. van Herpen E, Rosso SM, Serverijnen LA, et al. Variable phenotypic expression and extensive tau pathology in two families with the novel tau mutation L315R. Ann Neurol 2003;54:573–581. [DOI] [PubMed] [Google Scholar]
- 18. Reed LA, Grabowski TJ, Schmidt ML, et al. Autosomal dominant dementia with widespread neurofibrillary tangles. Ann Neurol 1997;42:564–572. [DOI] [PubMed] [Google Scholar]
- 19. Hayashi S, Toyoshima Y, Hasegawa M, et al. Late‐onset frontotemporal dementia with a novel exon 1 (Arg5His) tau gene mutation. Ann Neurol 2002;51:525–530. [DOI] [PubMed] [Google Scholar]
- 20. Poorkaj P, Muma NA, Zhukareva V, et al. AnR5Lτ mutation in a subject with a progressive supranuclear palsy phenotype. Ann Neurol 2002;52:511–516. [DOI] [PubMed] [Google Scholar]
- 21. Ygland E, van Westen D, Englund E, et al. Slowly progressive dementia caused by MAPT R406W mutations: longitudinal report on a new kindred and systematic review. Alzheimers Res Ther 2018;10:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Giaccone G, Rossi G, Farina L, et al. Familial frontotemporal dementia associated with the novel MAPT mutation T427M [4]. J Neurol 2005;252:1543–1545. [DOI] [PubMed] [Google Scholar]
- 23. Chaunua MP, Deramecourt V, Buée‐Scherrer V, et al. Juvenile frontotemporal dementia with parkinsonism associated with tau mutation G389R. J Alzheimers Dis 2013;37:769–776. [DOI] [PubMed] [Google Scholar]
- 24. Blauwendraat C, Wilke C, Simón‐Sánchez J, et al. The wide genetic landscape of clinical frontotemporal dementia: systematic combined sequencing of 121 consecutive subjects. Genet Med 2018;20:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Alcolea D, Vilaplana E, Suárez‐Calvet M, et al. CSF sAPPβ, YKL‐40, and neurofilament light in frontotemporal lobar degeneration. Neurology 2017;89:178–188. [DOI] [PubMed] [Google Scholar]
- 26. Alcolea D, Irwin DJ, Illán‐Gala I, et al. Elevated YKL‐40 and low sAPPβ:YKL‐40 ratio in antemortem cerebrospinal fluid of patients with pathologically confirmed FTLD. J Neurol Neurosurg Psychiatry 2019;90:180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Paterson RW, Slattery CF, Poole T, et al. Cerebrospinal fluid in the differential diagnosis of Alzheimer's disease: clinical utility of an extended panel of biomarkers in a specialist cognitive clinic. Alzheimers Res Ther 2018;10:32. [DOI] [PMC free article] [PubMed] [Google Scholar]