

Abstract
Objective
To characterize the molecular and clinical phenotypic basis of developmental and epileptic encephalopathies caused by rare biallelic variants in CACNA2D2.
Methods
Two affected individuals from a family with clinical features of early onset epileptic encephalopathy were recruited for exome sequencing at the Centers for Mendelian Genomics to identify their molecular diagnosis. GeneMatcher facilitated identification of a second family with a shared candidate disease gene identified through clinical gene panel‐based testing.
Results
Rare biallelic CACNA2D2 variants have been previously reported in three families with developmental and epileptic encephalopathy, and one family with congenital ataxia. We identified three individuals in two unrelated families with novel homozygous rare variants in CACNA2D2 with clinical features of developmental and epileptic encephalopathy and cerebellar atrophy. Family 1 includes two affected siblings with a likely damaging homozygous rare missense variant c.1778G>C; p.(Arg593Pro) in CACNA2D2. Family 2 includes a proband with a homozygous rare nonsense variant c.485_486del; p.(Tyr162Ter) in CACNA2D2. We compared clinical and molecular findings from all nine individuals reported to date and note that cerebellar atrophy is shared among all.
Interpretation
Our study supports the candidacy of CACNA2D2 as a disease gene associated with a phenotypic spectrum of neurological disease that include features of developmental and epileptic encephalopathy, ataxia, and cerebellar atrophy. Age at presentation may affect apparent penetrance of neurogenetic trait manifestations and of a particular clinical neurological endophenotype, for example, seizures or ataxia.
Introduction
Developmental and epileptic encephalopathies (DEEs) encompass a heterogenous group of early‐onset clinically severe disorders characterized by intractable seizures, abundant epileptiform activity on EEG, and developmental impairment or regression.1
A substantial number of DEE patients have been shown to have an underlying genetic etiology, with de novo heterozygous variants playing an important role.2, 3, 4, 5 With rapid advancements in molecular biology and genetic techniques, >100 genes have been associated with DEEs in the last 20 years, revolutionizing our understanding of their molecular etiology.6 These genes have been linked to pathways involved in synaptic transmission, ion channels, transcriptional regulation, DNA damage repair, chromatin remodeling, and metabolism.3 Pathogenic variants in genes involved in ion channel function represent perhaps one of the most common causes of DEEs, also called channelopathies, and their identification and molecular diagnostic specification in individuals with EE can have important therapeutic implications.7 Pathogenic variants in genes encoding subunits of voltage‐gated calcium channels (VGCCs), that is, CACNA1A, CACNA2D2,8, 9, 10, 11 and CACNA2D1 12 have been linked to epileptic encephalopathy (EE) and/or ataxia phenotypes [MIM#617106, 108500]. CACNA2D2 encodes the α2δ‐2 auxiliary subunit of high VGCCs, is highly expressed in the cerebellar cortex, and is reportedly a receptor for the antiepileptic drugs pregabalin and gabapentin.13 Auxiliary α2δ subunits support calcium channels in their assembly and trafficking to form complexes, increase channel density, and modulate channel kinetic properties.14 Biallelic pathogenic variants in CACNA2D2 have been reported in three families with DEEs and one family with congenital ataxia.8, 9, 10, 11 In this study, we present an additional two families with novel homozygous variants in CACNA2D2 causing DEE and cerebellar atrophy. We also compared the molecular and clinical features of all previously reported cases, thus providing further evidence to support CACNA2D2 as a disease‐causing gene for DEE and a syndromic neurological trait that includes ataxia as a manifestation of the neurobiology of cerebellar dysfunction.
Patients and Methods
Participants and ethical approval
Informed consent was obtained from all family members in Family 1 in accordance with the Baylor‐Hopkins Center for Mendelian Genomics (BHCMG) research protocol (Baylor College of Medicine IRB protocol number: H‐29697). A second family, Family 2, was identified and recruited via GeneMatcher following informed consent.
Sequencing, variant interpretation, and bioinformatics
Exome sequencing (ES) was performed on two affected siblings (BH9685‐1 and BH9685‐4) of Turkish descent who were born to consanguineous parents in Family 1 using our standard sequencing and variant prioritization workflow that has been previously described, including BafCalculator for determination of absence of heterozygosity (AOH), potentially reflecting identity‐by‐descent (IBD), from exome variant data.15 The proband in Family 2 had commercial panel testing from Invitae (San Francisco, California), the Early Infantile Epileptic Encephalopathy Panel (gene content enumerated alphabetically in Table S1), which includes CACNA2D2. The CACNA2D2 variants in Family 1 and 2 have been added to ClinVar under the accession numbers SCV000920875 and SCV000551913.2, respectively. Both CACNA2D2 variants are absent from the public variant databases gnomAD, ExAC, ARIC, and ESP. Bioinformatic analyses were used to predict potential variant effects on protein function (SIFT, PolyPhen2, MutationTaster, Phylop, CADD) and assess predicted (NMDEscPredictor) premature termination codons (PTCs).
Variant confirmation
Potential disease‐causing variants in CACNA2D2 were examined for segregation in all family members with available DNA for both families using standard PCR and Sanger sequencing techniques.
Clinical evaluation
Family 1
BH9685‐1 and BH9685‐4 are affected siblings born to consanguineous Turkish parents with no known clinical history of seizures, and have an unaffected elder female sibling (Fig. 1A). BH9685‐1 is currently a 29‐year‐old male who was born at term via C‐section after an uncomplicated pregnancy. His seizures started at 1 month of life and quickly became refractory to several antiepileptic drugs (AEDs) including levetiracetam, oxcarbazepine, topiramate, and clonazepam. His most prominent clinical seizure type is tonic‐clonic. He had global developmental delay (GDD) and currently has intellectual disability (ID). At present, he is unable to walk unassisted, has a limited vocabulary of a few words and some behavioral issues including restlessness and sleep disturbance. He has subtle dysmorphic craniofacial features including thick eyebrows and a prominent nose not observed in his parents. Brain MRI at age 26 years showed marked cerebellar and mild cerebral atrophy (Fig1C).
Figure 1.
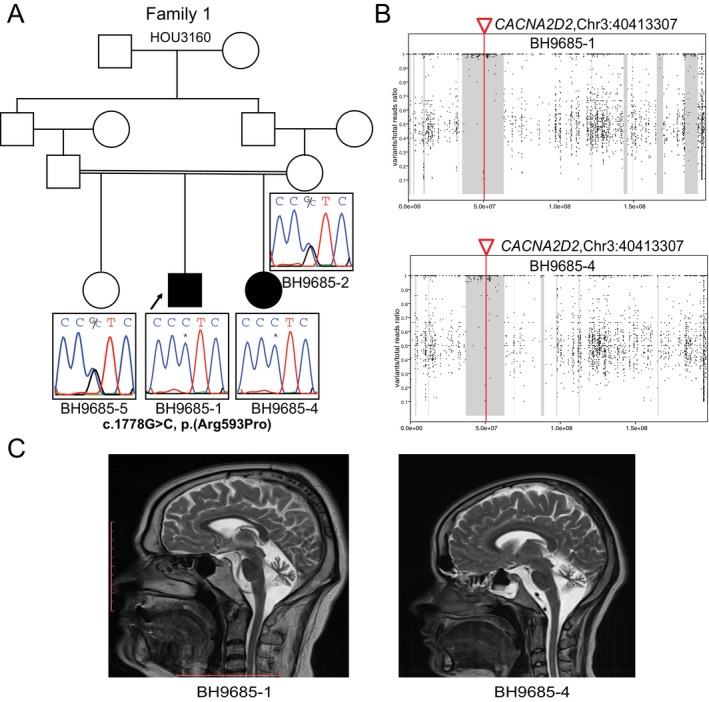
Biallelic variant in CACNA2D2 in two siblings with developmental and epileptic encephalopathy (Family 1). (A) Pedigree diagram for Family 1 shows family structure and Sanger sequencing traces for the c.1778G>C, p.(Arg593Pro) CACNA2D2 variant in family members with available DNA. The variant is homozygous in both affected siblings (BH9685‐1 and BH9685‐4) and heterozygous in the unaffected mother (BH9685‐2) and unaffected sibling (BH9685‐5) conforming to Mendelian expectations for an autosomal recessive (AR) disease trait. DNA from the unaffected father was not available for segregation studies. (B) Absence of heterozygosity (AOH) plots for Family 1 show that the CACNA2D2 variant lies in a region of AOH in chromosome 3 that is represented by the gray shaded area for both affected siblings (BH9685‐1 and BH9685‐4). AOH regions are calculated using B‐allele frequency data from exome data.15 (C) T2‐weighted brain MRI images for affected siblings BH9685‐1 and BH9685‐4 show cerebellar atrophy.
BH9685‐4 is BH9685‐1’s 24‐year‐old sister. She was delivered by C‐section at term, following an uncomplicated pregnancy. Her seizures started at 2 months of life, and quickly became refractory to multiple AEDs. Her seizures spontaneously resolved following puberty, and currently she is in remission (seizure‐free). Additional clinical features are similar in severity and character to those of her affected brother. Brain MRI at age 21 years showed marked cerebellar and mild cerebral atrophy similar to her affected brother.
Family 2
The proband of Family 2 was a 3‐year‐old male born premature at 31 weeks of corrected gestational age to first cousin consanguineous parents of Afghani descent (Fig. 2A). Parents did not report any personal history of seizures. He was noted to have GDD, was able to sit with support but had an unsteady posture, showed dysmetria, axial hypotonia, and ataxia. He showed pincer grasp and reciprocal smile. He produced a few simple words and had not yet walked. His brain MRI demonstrated global cerebral and cerebellar atrophy (Fig. 2B). He was first noted to have seizures at 7 months, and these became refractory to several antiseizure medications including amantadine, ethosuximide, valproic acid, levetiracetam, and clobazam. He had several seizure types including typical absence, generalized tonic‐clonic, generalized myoclonic and focal sensory seizures with decreased level of awareness. A routine EEG performed at 13 months of age captured two typical absence seizures associated with 3Hz spike and slow wave activity lasting for 8 seconds (Fig. 2C). A 48‐hour EEG at 3 years of age showed background slowing, focal and generalized epileptiform discharges, and captured a single 12‐second typical absence seizure. He eventually underwent placement of a vagal nerve stimulator (VNS) passing away less than 48 hours after insertion due to broncho‐aspiration.
Figure 2.
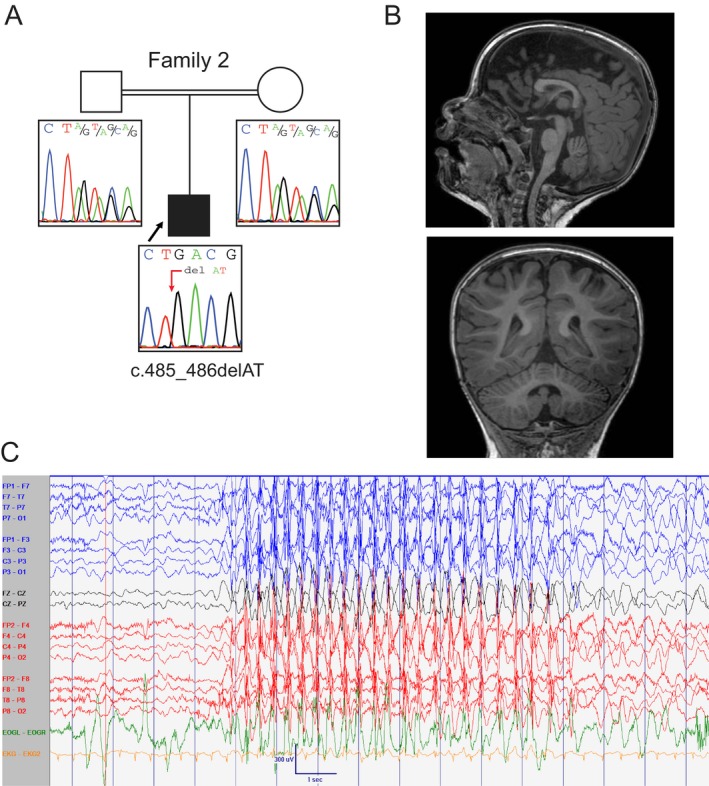
Biallelic variant in CACNA2D2 in proband with epileptic encephalopathy (Family 2). (A) Pedigree diagram and Sanger sequencing validation in Family 2 for CACNA2D2 variant c.485_486del, p.(Tyr162Ter) show heterozygosity for the variant in both parents, and homozygosity for the variant in the affected proband, consistent with an AR disease pattern. (B) T1‐weighted brain MRI images for affected proband show cerebral and cerebellar atrophy. (C) Routine EEG recording at 13 months of age captured an episode of unresponsiveness and eye‐blinking lasting for 8 seconds associated with 3 Hz generalized spike and slow wave activity consistent with early‐onset absence seizure [Settings: Low frequency filter at 1 Hz; high frequency filter at 70 Hz; sensitivity at 15 μV/mm; paperspeed 30 mm/sec].
Results
Genetic analyses
In Family 1, ES analyses showed a homozygous rare missense variant c.1778G>C, p.(Arg593Pro) in CACNA2D2 (NM_006030.3) in both affected siblings. This variant is predicted to be disease causing (SIFT, Polyphen‐2, MutationTaster), conserved (PhyloP), and has a CADD score of 34. In Family 2, an epilepsy gene panel test showed a homozygous rare nonsense variant c.485_486del, p.(Tyr162Ter) in CACNA2D2 (NM_006030.3) in the proband. This variant is predicted to result in a truncated protein by producing a stopgain at amino acid residue position 162 in exon 5 of this 38‐exon gene. Sanger sequencing confirmed the variants and segregation in accordance with Mendelian expectations for an autosomal recessive (AR) disease trait, showing heterozygosity for the identified CACNA2D2 variant in the mother and unaffected older sister in Family 1 and heterozygosity for the CACNA2D2 variant in both parents in Family 2 (Figs. 1A and 2A).
Analysis of absence of heterozygosity (AOH) from exome variant data in Family 1 demonstrated overlapping regions of AOH in both siblings, measuring ~29.9 Mb and ~25.8 Mb in size (Fig. 1B). Notably, these regions of AOH were not the largest AOH interval in either sibling (~37 Mb and ~30.6 Mb), and the siblings demonstrated a total AOH of ~380 Mb and ~393 Mb consistent with the reported history of parental consanguinity. The rare likely damaging variant in CACNA2D2 found in the ~30 Mb and ~26 Mb regions of AOH was the only such variant found in any AOH interval. For Family 2, a SNP array was carried out and it demonstrated that the rare CACNA2D2 variant is in ~27.1 Mb region of AOH, which also happens to be the largest AOH interval.
CACNA2D2 has a high probability of being loss‐of function intolerant, that is, pLI (probability of being loss‐of‐function intolerant) score of 1, and analysis using the NMDEscPredictor tool indicates that it has a noninformative transcript for predicting escape of nonsense‐mediated decay (NMD).16
Comparison of clinical and molecular features
The principal molecular and clinical features of all individuals reported including the two families (three individuals) in our study and four families reported earlier are shown in Tables 1 and 2 and Table S2. Affected individuals in all five DEE families show different seizure types and clinically severe epilepsy, as demonstrated by drug resistance to AEDs, in 7/8 individuals. Typical absence seizures with early onset (before 4 years of age) were observed in three individuals (Family 2, Pippucci et al., Butler et al.), and treatment with ethosuximide (ESM) improved absence seizure frequency in one individual (Pippucci et al.). The individual reported with congenital ataxia (Valence et al.) has only had one seizure, interpreted as a febrile seizure, and no features of intellectual disability (ID) or developmental delay (DD), distinct from the DEE individuals for whom recurrent seizure activity and severe DD were both present. Available MRI images of all individuals were compared by a pediatric neuroradiologist (JVH). All individuals (9/9) show cerebellar atrophy by MRI. Of note is the remarkable consistency of the MRI patterns from the affected siblings of Family 1 (Fig1C). Three individuals with DEE also showed moderate or truncal ataxia. Although ataxia was not mentioned in the clinical features for the other two families, these individuals showed intermittent choreiform movements (Edvardson et al.) and dyskinesia (Pippucci et al.). The positions of the biallelic variants within the transcript and protein domains have been mapped and are shown in Figure 3.
Table 1.
Molecular features of individuals with rare biallelic CACNA2D2 variants.
| Family 1 | Family 2 | Edvardson et al. | Pippucci et al. | Valence et al. | Butler et al. | ||||
|---|---|---|---|---|---|---|---|---|---|
| Ethnicity | Turkish | Afghani | Arab‐Palestinian | Italian | Portuguese | Unknown | |||
| Gender | M | F | M | M | M | F | M | M | M |
| Age at last evaluation | 29 years | 24 years | 5 years | 7 years | 4 years | – | 10 years | 20 years | 5 years |
| Parental consanguinity | Yes | Yes | Yes | Yes | unknown | No | |||
| Zygosity | Hom | Hom | Hom | Hom | Hom | Compound het | |||
| cDNA (NM_006030.3) | c.1778G>C | c.485_486delAT | c.3119A>G | c.1295delA | c.2971G>A | c.782C>T, c.3137T>C | |||
| Exon (of 38) | Exon 20 | Exon 5 | Exon 36 | Exon 13 | Exon 34 | Exon7, Exon 36 | |||
| Protein | p.(Arg593Pro) | p.(Tyr162Ter) | p.(Leu1040Pro) | p.(Asn432fs) | p.(Asp991Asn) | p.(Pro261Leu), p.(Leu1046Pro) | |||
| Variant information | Missense | Nonsense/potential LoF | Missense | Frameshift/potential LoF | Missense | Missense + missense | |||
| Testing | ES | ES | Epilepsy panel | ES | ES | ES | ES | ||
Abbreviations: ES, exome sequencing; F, female; het, heterozygous; hom, homozygous; LoF, loss of function; M, male.
Table 2.
Clinical features of individuals with rare biallelic CACNA2D2 variants.
| Family 1 | Family 2 | Edvardson et al. | Pippucci et al. | Valence et al. | Butler et al. | ||
|---|---|---|---|---|---|---|---|
| Age at onset | 1 month | 2 months | 7 months | 1–2 months | 5 months | ‐ | 7 months |
| Seizure types | Tonic‐clonic | Tonic‐clonic | Tonic‐clonic, typical absence, myoclonic, focal sensory | Atonic, clonic, tonic‐clonic | Clonic, clonic‐tonic, typical absence | Single febrile seizure at 1 year of age | Typical absence, atonic, tonic, tonic‐clonic |
| Epilepsy | + | + | + | + | + | ‐ | + |
| DR | DR | DR | DR | DR | ‐ | Controlled | |
| AED treatment & response | LEV, OCBZ, TPM, CLN | Amantadine, ESM, VPA, CLB; VNS placement | VGB, CLN, TPM, CLB PB, VPA | PB, BZD, VPA, LEV, LTG, ESM (effective) | n/a | VPA, ESM, CLN (effective), ketogenic diet | |
| ID/GDD | ID, GDD | Severe GDD | Severe GDD | Severe GDD | ‐ | GDD | |
| Brain MRI findings | Mild supratentorial WM loss, thinning of CC, marked; diffuse CA (at ages 21 years, 26 years) | Normal WM, foreshortened CC, mild CA (at 1.1 years) | Normal WM, borderline lower to normal CC, mild‐moderate; diffuse CA (at 8 years) | Prominent ventral WM loss, thinning and slight foreshortening of CC, moderate‐marked; diffuse CA (at 7 years) | Mild WM loss, slight foreshortening of CC, moderate CA; localized to superior vermis (at 18 years) | Prominent ventral WM loss, normal CC, marked; diffuse CA (at 2.4 years) | |
| Ataxia | Truncal ataxia | Axial ataxia | ‐ | ‐ | Congenital ataxia | Moderate ataxia | |
| Other neurological features | Mild behavioral problems, wheelchair dependent, speech limited to a few words | Nonambulatory, absent speech, axial hypotonia, dysmetria | Intermittent choreiform movements, axial hypotonia, absent speech | Dyskinesia, tremors, myoclonic jerks, hypertonia, axial hypotonia, oculomotor apraxia, recurrent SE | Motor delay, dysmetria, dysarthria | Nonambulatory, absent speech, hypotonia, recurrent SE | |
Abbreviations: AED, antiepileptic drugs; BZD, benzodiazepine; CA, cerebellar atrophy; CC, corpus callosum; CLB, clobazam; CLN, clonazepam; DR, drug resistant; EEG, electroencephalogram; ESM, Ethosuximide; GDD, global developmental delay; GSSW, generalized spike and slow waves; ID, intellectual disability; LEV, levetiracetam; LTG, lamotrigine; OCBZ, oxcarbazepine; PB, phenobarbital; SE, status epilepticus; TPM, topiramate; VNS, vagal nerve stimulator; VGB, vigabatrin; VPA, valproic acid; WM, white matter.
Figure 3.
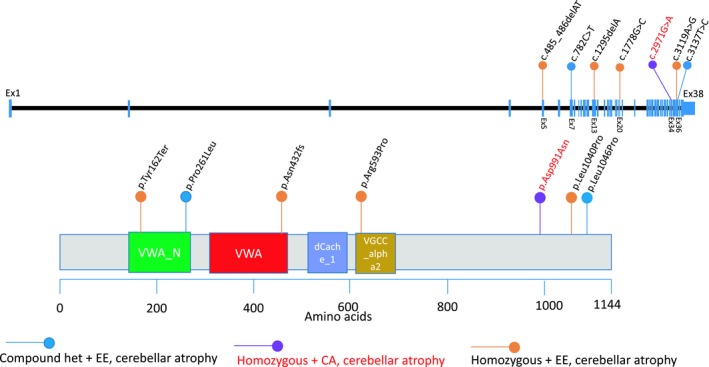
Exonic and protein map positions of CACNA2D2 variant alleles showing potential protein domains mutated. Lollipop diagram showing biallelic variants reported in this study (2 families) and reported previously in the literature (4 families) mapped across the transcript and protein domain structures of CACNA2D2. The orange lollipops indicate the positions of homozygous variants in CACNA2D2 from four families (Edvardson et al.8, orange, Pippucci et al.9, orange, Family 1, orange, and 2, orange, in this report) with developmental and epileptic encephalopathy (DEE) and cerebellar atrophy. The blue lollipops show the positions of reported compound heterozygous variants inherited in trans in a patient with DEE and cerebellar atrophy (Butler et al.11, blue). The purple lollipop and red text indicate the position of a homozygous variant reported to cause a milder phenotype of congenital ataxia (CA) and cerebellar atrophy (Valence et al.10, purple). The genomic structure of CACNA2D2 represented here is based on information from the UCSC genome browser (https://genome.ucsc.edu) using transcript NM_006030; note the CACNA2D2 transcript is encoded by the minus strand and has been flipped here for representational purposes. The protein domain structures were visualized and variants mapped using the Mutation Mapper portal (http://www.cbioportal.org/mutation_mapper). Protein domains: VWA_N: VWA N‐terminal (141–265), VWA: von Willebrand factor type A domain (291–454), dCache_1: Cache domain (485–573), VGCC_alpha2: Neuronal voltage‐dependent calcium channel alpha 2acd (580–662).
Discussion
Inherited channelopathies represent a heterogenous group of disorders caused by mutations in genes encoding ion channel subunits or their regulators, and account for a substantial proportion of Mendelian epilepsy syndromes.17 Genes encoding voltage‐gated calcium channels (VGCCs) have been linked to both epilepsy and/or ataxia neurological phenotypes (Table 3). Gene expression pattern of these five genes (CACNA1A, CACNA1G, CACNB4, CACNA2D1, and CACNA2D2) reveals high expression in the brain with a pattern of higher expression in the cerebellum in genes that present with symptoms of ataxia or cerebellar atrophy/hypoplasia on brain MRI, that is, CACNA1A, CACNA1G, CACNB4, and CACNA2D2 (Fig. 4). Cerebellar Purkinje cell impairment is a pathological hallmark of ataxia and may result in a wide spectrum of motor symptoms.18 CACNA2D1 has been proposed as a candidate gene for intellectual disability and epilepsy, and affected individuals have been reported to show features of cortical atrophy without any cerebellar involvement, concordant with its expression pattern in the brain (Fig. 4).12
Table 3.
Genes encoding voltage‐gated Calcium channels that are linked to epilepsy and/or ataxia phenotypes.
| Gene | Disease | Phenotype MIM# | Inheritance | Gene expression |
|---|---|---|---|---|
| CACNA1A | Epileptic encephalopathy, early infantile, 42 | 617106 | AD | Cerebellum, caudate, brain, other |
| Episodic ataxia, type 2 | 108500 | AD | ||
| Migraine, familial hemiplegic, 1 | 141500 | AD | ||
| Migraine, familial hemiplegic, 1, with progressive cerebellar ataxia | 141500 | AD | ||
| Spinocerebellar ataxia 6 | 183086 | AD | ||
| CACNA1G | Spinocerebellar ataxia 42 | 616795 | AD | Cerebellum, caudate, brain, retina, ovary, uterine, other |
| Spinocerebellar ataxia 42, early‐onset, severe, with neurodevelopmental deficits | 618087 | AD | ||
| CACNB4 | Episodic ataxia, type 5 | 613855 | AD | Cerebellum, caudate, brain, hippocampus, other |
| {Epilepsy, idiopathic generalized, susceptibility to, 9} | 607682 | AD | ||
| {Epilepsy, juvenile myoclonic, susceptibility to, 6} | 607682 | AD | ||
| CACNA2D1 | Intellectual disability and epilepsy (Vergult et al., 2015)12 | – | AD | Skeletal muscle, hippocampus, brain, caudate, smooth muscle, thyroid, pituitary gland, other |
| CACNA2D2 | Early‐onset epileptic encephalopathy (Edvardson et al.8, Pippucci et al.9, Butler et al.11) | – | AR | Lung, cerebellum, retina, caudate, other |
| Congenital ataxia (Valence et al. 10) | – | AR |
Abbreviations: AD, Autosomal dominant; AR, Autosomal recessive. Note that gene expression in major tissues (>5% total expression in all tissues) from humans was incorporated from Seitter et al.28 and the GTEx database.
Figure 4.
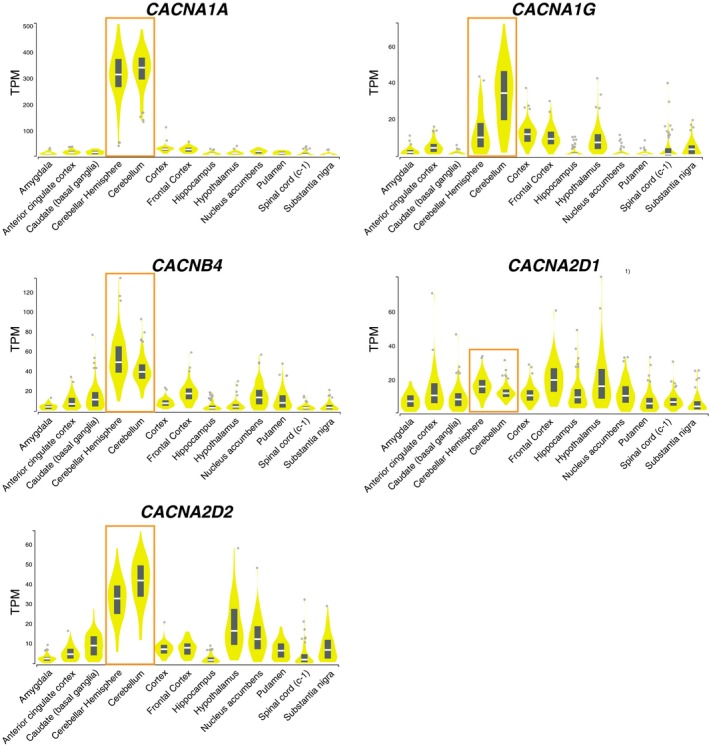
Gene expression in brain tissues of genes encoding voltage‐gated calcium channels linked to epilepsy and/or ataxia. Gene expression pattern in all brain tissues from GTEx portal for the genes encoding voltage‐gated calcium channels CACNA1A, CACNA1G, CACNB4, CACNA2D1, and CACNA2D2. The orange box highlights expression in the specific brain regions of cerebellar hemisphere and cerebellum. TPM: Transcripts Per Million, Data Source: GTEx Analysis Release V7 (dbGaP Accession phs000424.v7.p2), accessed on 30th March, 2019.
CACNA2D2 was first described as a candidate DEE gene in 20138, 9, however, its role as a disease gene remained ambiguous as both studies also reported rare homozygous variants (rs149614835, rs587777163) in CELSR3 that segregated with disease in both families. Recently, there have been two additional reports describing biallelic variants in CACNA2D2, rare homozygous variants in CELSR3 were absent in both individuals reported. 10, 11 Valence et al. describe an individual with congenital ataxia and cerebellar atrophy, this clinically milder neurologic phenotype was attributed to the nature of the rare homozygous splice variant observed that was shown by RT‐PCR to produce a leaky splicing defect, impacting only a proportion of the transcripts.10 Butler et al. reported an individual with compound heterozygous variants in CACNA2D2 and a phenotype of DEE with cerebellar atrophy.11 Rare variants in CELSR3 are absent in Family 1 in this report (Family 2 had gene panel testing that did not include CELSR3), providing further evidence for CACNA2D2 as an independent disease‐causing gene.
Figure 3 represents all biallelic variants mapped to the transcript and protein domain structures of CACNA2D2 showing that the variants are not limited to a specific protein domain. Of the two missense variants that are shown to occur in protein domains, the homozygous missense variant p.Arg593Pro in Family 1 lies in the neuronal voltage‐dependent calcium channel alpha‐2 domain that is thought to play a role in calcium channel subunit assembly.19 The heterozygous missense variant reported in Butler et al. (compound heterozygous variants) is predicted to be in the von Willebrand factor type A, N‐terminus (VWA_N) domain which may be involved in the trafficking and function of VDCC subunits.20 The other missense variants are not associated with known protein domains and thus their functional impact is uncertain; however, Edvardson et al. performed functional studies in Xenopus laevis oocytes and observed that the CACNA2D2 p.(Leu1040Pro) variant resulted in the reduction of current density and slower inactivation of calcium channels compared to wildtype.8 The identification of two frameshifting variants predicted to undergo nonsense‐mediated decay (NMD) suggests a loss‐of‐function mechanism for the association between rare variation in CACNA2D2 and DEE.
Additional support for CACNA2D2 as a disease gene is seen in its mouse models. The spontaneous autosomal recessive (AR) mouse mutant ducky (du) is a model for absence epilepsy, and is characterized by spike‐wave seizures and cerebellar ataxia.21 Mutations in Cacna2d2 were identified in the original du strain and a new allele du2j, both resulted in loss of the full‐length protein and were associated with decreased calcium channel current in cerebellar Purkinje cells.22, 23 Du mice also showed an ataxic wide‐based gait, dyskinesia, reduced size and did not survive beyond 35 days; neuropathology showed dysgenesis of the cerebellum, medulla, and spinal cord.24
Another spontaneous AR mouse model “entla” with mutations in Cacna2d2 was observed with neurological features of epilepsy and ataxia. Entla (ent) mice result from an in‐frame duplication in Cacna2d2 and resemble the phenotype of ducky mice except that entla mice do not show neuroanatomical or central nervous system abnormalities.25 Ligand‐binding assays using gabapentin on cerebellar Purkinje cells isolated from entla mice showed >60% reduced binding to the membrane compared to wildtype and heterozygous mice demonstrating that α2δ‐2 contributes to gabapentin binding.
Targeted knockout of Cacna2d2 resulted in Cacna2d2tmINCIF mice with ataxic gait, growth retardation, seizure susceptibility, and cardiac abnormalities.13 Cerebellar degeneration was observed in Cacna2d2tmINCIF mice with initial loss of the granule cell layer followed by gradual depletion of Purkinje cells. The cerebellar pathology observed was less severe than the ducky mice and the authors hypothesized this may be due to elimination of longer segments of the α2δ‐2 protein in ducky mutants.
Overall, the studies in mice display a robust link between Cacna2d2 and neurological disorders with features including epilepsy and ataxia. Pippucci et al. noted that the individual with a biallelic, loss‐of‐function (LoF) CACNA2D2 variant in their study more closely resembled the absence epilepsy phenotype of the LoF ducky mouse compared to individuals with biallelic, missense variants.9 The proband in family 2 with a biallelic, potential LoF CACNA2D2 variant also shows absence seizures indicating that biallelic LoF variants may result in a slightly different phenotype.
Antiepileptic drugs gabapentin and pregabalin have been observed to bind to α2δ‐1 and α2δ‐2, high VGCCs encoded by CACNA2D1 and CACNA2D2, respectively, with similar affinity.14, 24 Ataxia is reported as a side effect of gabapentin that may potentially be due to α2δ‐2 binding in the cerebellum.26 Notably, individuals with CACNA2D2‐associated DEE and drug‐resistant seizures have not been treated with gabapentin. Although treatment with gabapentin in these cases has not been objectively explored, it is possible that rare pathogenic variation in CACNA2D2 may impact the efficacy of gabapentin as an AED.
To date, biallelic rare variants in CACNA2D2 have been reported in a total of three unrelated families with DEE, notably, one additional individual has been described with a milder phenotype of congenital ataxia and cerebellar atrophy by MRI, which lies within the phenotypic spectrum of clinical features associated with CACNA2D2. Genotype–phenotype associations in another VGCC encoding gene, CACNA1A, have indicated that functional effects (gain‐of‐function, loss‐of‐function, hypomorphic) can impact phenotypic expression of disease, and may have important clinical consequences, including the response to therapeutic treatment by AED.27 Detailed studies of additional individuals with CACNA2D2 rare biallelic variants will help illuminate the penetrance of neurological endophenotypes at this locus. The potential effectiveness of gabapentin as an AED of choice in treating DEE due to CACNA2D2 pathogenic biallelic variants remains to be explored. CACNA2D2 should be considered as part of the differential for early onset absence seizure conditions such as GLUT1 deficiency syndrome (MIM#606777). Rapid advances in medical genetics and clinical genomics are predicated on establishing robust genotype/phenotype correlations; constructing allelic series at every locus will be a prerequisite for such analyses. Progress in the field of epilepsy continues to be made by employing a gene/variant‐based model to inform epilepsy classification and therapeutics, driving the way for epilepsies to serve as a model for future precision‐based medicine.
Author Contributions
Study concept and design: JEP, RAG, JRL; Acquisition of data: JP, EK, AG, DP, SNJ, REL, JPA, AMI; Data analyses and interpretation: All authors; Drafting and revision of manuscript: All authors; Study supervision: JEP, JRL.
Study funding: RAG, JRL.
Conflict of Interest
JVH receives royalties for a chapter on pediatric neuroimaging in UpToDate. JRL has stock ownership in 23andMe, is a paid consultant for Regeneron, and is a coinventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting.
Supporting information
Table S1. List of genes tested using the commercial Early Infantile Epileptic Encephalopathy Panel for proband of Family 2.
Table S2. Detailed clinical features of individuals with rare biallelic CACNA2D2 variants.
Acknowledgments
We thank the patients and families for participating in this study. AMI and REL would like to acknowledge Linda MacLaren and Melanie Keller for clinical and laboratory support of the workup of this patient and to the family for their kind participation in this study.
Funding information
This work was supported in part by the National Institutes of Health, National Institute of Neurologic Disorders and Stroke [R35 NS105078] and a jointly funded National Human Genome Research Institute (NHGRI) and National Heart, Lung, and Blood Institute (NHLBI) grant to the Baylor‐Hopkins Center for Mendelian Genomics [UM1 HG006542] to JRL. JP is supported by the Muscular Dystrophy Association, grant #512848 awarded to JRL. JEP is supported by NHGRI [K08 HG008986]. DP is supported by the Clinical Research Training Scholarship in Neuromuscular Disease partnered by American Brain Foundation (ABF) and Muscle Study Group (MSG), and NIH – Brain Disorders and Development Training Grant (T32 NS043124‐17). DM is supported by a training grant through the National Institutes of Health [T32 GM007526].
Funding Statement
This work was funded by Muscular Dystrophy Association grant #512848 ; National Institute of General Medical Sciences grant T32 GM007526; National Human Genome Research Institute grants K08 HG008986 and UM1 HG006542; National Institute of Neurological Disorders and Stroke grants R35 NS105078 and T32 NS043124-17; National Heart, Lung, and Blood Institute grant ; Baylor‐Hopkins Center for Mendelian Genomics grant UM1 HG006542; American Brain Foundation grant ; National Institutes of Health grant .
Contributor Information
Jennifer E. Posey, Email: Jennifer.Posey@bcm.edu.
James R. Lupski, Email: jlupski@bcm.edu
References
- 1. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia 2017;58:512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Thomas RH, Berkovic SF. The hidden genetics of epilepsy‐a clinically important new paradigm. Nat Rev Neurol 2014;10:283–292. [DOI] [PubMed] [Google Scholar]
- 3. Epi4K Consortium ; Epilepsy Phenome/Genome Project , Allen AS, et al. De novo mutations in epileptic encephalopathies. Nature 2013;501:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Helbig KL, Farwell Hagman KD, Shinde DN, et al. Diagnostic exome sequencing provides a molecular diagnosis for a significant proportion of patients with epilepsy. Genet Med 2016;18:898–905. [DOI] [PubMed] [Google Scholar]
- 5. Papuc SM, Abela L, Steindl K, et al. The role of recessive inheritance in early‐onset epileptic encephalopathies: a combined whole‐exome sequencing and copy number study. Eur J Hum Genet 2019;27:408–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. McTague A, Howell KB, Cross JH, et al. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol 2016;15:304–316. [DOI] [PubMed] [Google Scholar]
- 7. Maljevic S, Reid CA, Petrou S. Models for discovery of targeted therapy in genetic epileptic encephalopathies. J Neurochem 2017;143:30–48. [DOI] [PubMed] [Google Scholar]
- 8. Edvardson S, Oz S, Abulhijaa FA, et al. Early infantile epileptic encephalopathy associated with a high voltage gated calcium channelopathy. J Med Genet 2013;50:118–123. [DOI] [PubMed] [Google Scholar]
- 9. Pippucci T, Parmeggiani A, Palombo F, et al. A novel null homozygous mutation confirms CACNA2D2 as a gene mutated in epileptic encephalopathy. PLoS ONE 2013;8:e82154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Valence S, Cochet E, Rougeot C, et al. Exome sequencing in congenital ataxia identifies two new candidate genes and highlights a pathophysiological link between some congenital ataxias and early infantile epileptic encephalopathies. Genet Med 2019;21:553–563. [DOI] [PubMed] [Google Scholar]
- 11. Butler KM, Holt PJ, Milla SS, et al. Epileptic encephalopathy and cerebellar atrophy resulting from compound heterozygous CACNA2D2 variants. Case Rep Genet 2018;2018:6308283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vergult S, Dheedene A, Meurs A, et al. Genomic aberrations of the CACNA2D1 gene in three patients with epilepsy and intellectual disability. Eur J Hum Genet 2015;23:628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gong HC, Hang J, Kohler W, et al. Tissue‐specific expression and gabapentin‐binding properties of calcium channel alpha2delta subunit subtypes. J Membr Biol 2001;184:35–43. [DOI] [PubMed] [Google Scholar]
- 14. Zamponi GW, Striessnig J, Koschak A, Dolphin AC. The physiology, pathology, and pharmacology of voltage‐gated calcium channels and their future therapeutic potential. Pharmacol Rev 2015;67:821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karaca E, Posey JE, Coban Akdemir Z, et al. Phenotypic expansion illuminates multilocus pathogenic variation. Genet Med 2018;20:1528–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Coban‐Akdemir Z, White JJ, Song X, et al. Identifying genes whose mutant transcripts cause dominant disease traits by potential gain‐of‐function alleles. Am J Hum Genet 2018;103:171–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Spillane J, Kullmann DM, Hanna MG. Genetic neurological channelopathies: molecular genetics and clinical phenotypes. J Neurol Neurosurg Psychiatry 2016;87:37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hoxha E, Balbo I, Miniaci MC, Tempia F. Purkinje cell signaling deficits in animal models of ataxia. Front Synaptic Neurosci 2018;10:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Snell GD. Ducky, a new second chromosome mutation in the mouse. J Hered 1955;46:27–29. [Google Scholar]
- 20. Brust PF, Simerson S, McCue AF, et al. Human neuronal voltage‐dependent calcium channels: studies on subunit structure and role in channel assembly. Neuropharmacology 1993;32:1089–1102. [DOI] [PubMed] [Google Scholar]
- 21. Barclay J, Balaguero N, Mione M, et al. Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J Neurosci 2001;21:6095–6104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brodbeck J, Davies A, Courtney JM, et al. The ducky mutation in Cacna2d2 results in altered Purkinje cell morphology and is associated with the expression of a truncated alpha 2 delta‐2 protein with abnormal function. J Biol Chem 2002;277:7684–93. [DOI] [PubMed] [Google Scholar]
- 23. Meier H. The neuropathology of ducky, a neurological mutation of the mouse. A pathological and preliminary histochemical study. Acta Neuropathol 1968;11:15–28. [DOI] [PubMed] [Google Scholar]
- 24. Brill J, Klocke R, Paul D, et al. entla, a novel epileptic and ataxic Cacna2d2 mutant of the mouse. J Biol Chem 2004;279:7322–7330. [DOI] [PubMed] [Google Scholar]
- 25. Ivanov SV, Ward JM, Tessarollo L, et al. Cerebellar ataxia, seizures, premature death, and cardiac abnormalities in mice with targeted disruption of the Cacna2d2 gene. Am J Pathol 2004;165:1007–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Field MJ, Li Z, Schwarz JB. Ca2+ channel alpha2‐delta ligands for the treatment of neuropathic pain. J Med Chem 2007;50:2569–2575. [DOI] [PubMed] [Google Scholar]
- 27. Luo X, Rosenfeld JA, Yamamoto S, et al. Clinically severe CACNA1A alleles affect synaptic function and neurodegeneration differentially. PLoS Genet 2017;13:e1006905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Seitter H, Koschak A. Relevance of tissue specific subunit expression in channelopathies. Neuropharmacology 2018;132:58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. List of genes tested using the commercial Early Infantile Epileptic Encephalopathy Panel for proband of Family 2.
Table S2. Detailed clinical features of individuals with rare biallelic CACNA2D2 variants.