

Abstract
Objectives
Recent evidence showed that myotonic dystrophy type I (DM1) patients are at increased risk of certain cancers, but the risk of benign tumors is unknown. We compared the risk of benign tumors in DM1 patients with matched DM1‐free individuals and assessed the association between benign tumors and subsequent cancers.
Methods
We identified 927 DM1 patients and 13,085 DM1‐free individuals matched on gender, birth‐year, clinic, and clinic‐registration year from the UK Clinical Practice Research Datalink, a primary care records database. We used Cox regression models for statistical analyses.
Results
DM1 patients had elevated risks of thyroid nodules (Hazard Ratio [HR] = 10.4; 95% Confidence Interval [CI] = 3.91–27.52; P < 0.001), benign tumors of the brain or nervous system (HR = 8.4; 95% CI = 2.48–28.47; P < 0.001), colorectal polyps (HR = 4.3; 95% CI = 1.76–10.41; P = 0.001), and possibly uterine fibroids (HR = 2.7; 95% CI = 1.22–5.88; P = 0.01). Pilomatricomas and salivary gland adenomas occurred almost exclusively in DM1 patients (Fisher's exact P < 0.001). The HR for colorectal polyps was elevated in DM1 males but not in females (HR = 8.2 vs. 1.3, respectively; P‐heterogeneity < 0.001), whereas endocrine and brain tumors occurred exclusively in females. The data suggested an association between benign tumors and subsequent cancer in classic DM1 patients (HR = 2.7; 95% CI = 0.93–7.59; P = 0.07).
Interpretation
Our study showed a similar site‐specific benign tumor profile to that previously reported for DM1‐associated cancers. The possible association between benign tumors and subsequent cancer in classic DM1 patients warrants further investigation as it may guide identifying patients at elevated risk of cancer. Our findings underscore the importance of following population‐based screening recommendations in DM1 patients, for example, for colorectal cancer.
Introduction
Myotonic dystrophy type I (Dystrophia Myotonica I; DM1; Steinert's disease) is an autosomal dominant multisystem disorder, primarily presenting with progressive muscle weakness and myotonia.1, 2 It is one of the most common adult‐onset muscular dystrophies, with an estimated prevalence ranging from 5 to 20 per 100,000 worldwide.3 DM1 results from a CTG tri‐nucleotide repeat expansion in the 3' untranslated region of the dystrophia myotonica protein kinase (DMPK) gene on chromosome 19q13.3.4, 5, 6 Clinically, the DM1 phenotype varies widely and is classified into three major subtypes: congenital/childhood (most severe), classic, and late‐onset (mild).2 Recent quantitative studies have shown that DM1 patients are at increased risk of cancers arising in the skin, thyroid, uterus, and possibly colon, testis, and brain.7, 8, 9, 10, 11, 12, 13 We have previously shown that the excess risk of cancer in DM1 may be restricted to patients with the classic subtype (diagnosed between ages 11–40 years).14
Case reports as early as 1965 suggested an association between certain benign tumors and DM1, most commonly pilomatricoma, a rare calcifying cutaneous neoplasm.15 Other reported benign tumors included those arising in the parotid, parathyroid, thyroid, and pituitary glands, as well as thymoma, meningioma, uterine fibroids, and insulinoma (reviewed by Mueller et al. 15). Cross‐sectional studies of 255 Italian16 and 231 United Kingdom (UK)17 DM1 patients reported overall benign tumor frequencies of approximately 21% and 12%, respectively, with female reproductive tumors most commonly reported. Another Italian study in which dermatological examinations were conducted in 90 DM1 patients and 103 age‐ and gender‐matched controls found that DM1 patients were more likely to have dysplastic nevi and pilomatricoma.18 Finally, a Spanish study found an increased risk of pilomatricoma in 102 DM1 patients versus age‐ and gender‐matched controls.19
In this study, we aimed to evaluate organ site‐specific benign tumor risk in a large cohort of DM1 patients compared with matched DM1‐free individuals (control cohort). We also evaluated the association between benign tumors and subsequent cancer in both cohorts.
Methods
The study design was previously described.12, 14 Briefly, we utilized the UK Clinical Practice Research Datalink (CPRD), a primary care physician database linkable to other data sources and identified 927 patients with a record of DM1 (defined using Read codes “F392011: Steinert's disease”, and “F392000: Dystrophia myotonica [Steinert's disease]”) from the October 2016 CPRD release, and 13,085 DM1‐free controls matched on year of birth (±2 years), gender, clinic, and clinic registration year (±1 year). All participants were cancer‐free prior to the start of follow‐up. With the exception of cancer, all study variables were identified from CPRD using Read codes (available upon request). Cancer cases were identified from CPRD (n = 525), linkable inpatient records from the national Hospital Episode Statistics database (HES; n = 213), or cause of death data from the Office of National Statistics (ONS; n = 14), as previously described.14
Statistical analysis
Cox proportional hazards regression models were used to assess the association between developing benign tumors (overall and by anatomic site) and DM1 status (DM1‐affected vs. DM1‐free matched controls). Age was the time scale used for all analyses. Follow‐up started at the latest of either age at first DM1‐diagnosis/DM1‐free selection, clinic registration, or study start date (January 1st, 1988; after the start of CPRD). Patients were followed to the earliest of either age at first benign tumor under study, death, transfer out of the CPRD‐participating clinic, last data collection in CPRD, or end of study (February 29, 2016). To accommodate the matched design, baseline hazards were stratified on the matched sets. Potential effect measure modification was assessed by fitting separate Cox models to strata defined by gender and DM1 subtype categories (using age at DM1 diagnosis as a proxy; age 0–10 years = congenital/childhood, 11–40 years = classic, and >40 years = late‐onset). Heterogeneity of estimates across strata was tested using the Wald test. The proportional hazards assumption was assessed using Schoenfeld residuals; no significant violations were observed. When Cox models failed due to zero or sparse events, we used Fisher's exact test to examine intergroup differences.
To assess the robustness of our findings, we conducted several sensitivity analyses using more stringent definitions of DM1. We repeated the analysis using only: (1) DM1 patients diagnosed after their clinic's “up‐to‐standard” date (a CPRD practice‐level data quality metric);20 (2) DM1 patients diagnosed in 1995 or later (after DM1 gene discovery in 1992 and subsequent implementation of DM1 genetic testing in the UK);21 and (3) DM1 patients who had a DM1 record in at least two of the three data sources (CPRD primary care database, HES, and ONS). Additionally, we conducted a sensitivity analysis ending follow‐up at the maximum of three years (median follow‐up in the DM1‐free control group) for all patients, to assess whether differential follow‐up time for the DM1 and DM1‐free cohorts affected our results. Finally, to address the possibility of detection bias, we adjusted all models for the number of healthcare encounters and indicators of health seeking behavior. The number of healthcare encounters were ascertained from CPRD and/or HES (maximum of one per day) as a continuous, time‐varying covariate with a one‐year time lag, and were counted in 12‐month intervals between start of follow‐up and censor or first benign tumor dates. Indicators of health‐seeking behavior in this study included participation in cancer screening or influenza vaccination identified from CPRD records. In all analyses, we only used the corresponding matched DM1‐free controls for included DM1 patients.
Cox proportional hazards regression models were also used to evaluate the association between benign tumors and subsequent cancer risk in DM1 patients and the DM1‐free cohort, separately. Benign tumor was considered a time‐dependent variable, in which study participants belonged to the benign tumor‐free group until they developed their first benign tumor. Final models were adjusted for gender and number of healthcare care encounters as described above. To adjust for cancer ascertainment from multiple databases for patients eligible for linkage, the regression models were additionally adjusted for linkage status as a time‐varying covariate.
We conducted all analyses using SAS version 9.4 (Cary, NC) and R version 3.3.1 (R Foundation for Statistical Computing, Vienna, Austria). For site‐specific benign tumor risk analyses, we defined statistical significance as P‐value < 0.01 to minimize the possibility of false discovery; for all others, P‐value < 0.05 was used.
This study was approved by the CPRD Independent Scientific Advisory Committee (Protocol # 16_005RA2); see Acknowledgments for details on data sources. The use of the CPRD database was exempted from full Institutional Review Board (IRB) review by the National Institutes of Health Office of Human Subject Research and the University of Maryland's IRB, because of the anonymized nature of the data.
Role of the funding source
The funding source had no role in the design, conduct, or reporting of the study.
Results
Table 1 summarizes the study cohort characteristics. During 80,177 person‐years of follow‐up, we observed 138 benign tumors in 132 (14%) DM1 patients and 844 benign tumors in 800 (6%) DM1‐free controls. Skin tumors comprised the most frequent diagnosis in both cohorts (DM1 = 85/132 [64%]; DM1‐free = 623/844 [78%]), followed by female genital tumors (DM1 = 11%; DM1‐free = 10%), digestive tract tumors (DM1 = 8%; DM1‐free = 5%), endocrine tumors (DM1 = 7%, DM1‐free = 2%), and brain or nervous system tumors (including cerebral meningiomas, neuromas, neurofibromas, and other unspecified benign neoplasms; DM1 = 5%; DM1‐free = 1%). In DM1, 89% of endocrine tumors were thyroid nodules or cysts, 79% of female genital tumors were uterine fibroids, all digestive tumors were colorectal polyps, and all salivary gland tumors were pleomorphic adenomas (see Table 2 footnote for details of tumor subtypes).
Table 1.
Characteristics of the myotonic dystrophy type I (DM1)‐affected patients and their matched DM1‐free controls.
| Characteristics | DM1 (n = 927) | DM1‐free (n = 13,085) |
|---|---|---|
| Age at DM1 diagnosis, <i>n</i> (%) | ||
| Congenital/childhood (0–10 years) | 132 (14%) | – |
| Classic (11–40 years) | 500 (54%) | – |
| Late‐onset (>40 years) | 295 (32%) | – |
| Mean age at diagnosis, mean (SD) | 31.2 (17.8) | – |
| Gender, n (%) | ||
| Male | 455 (49%) | 6,474 (49%) |
| Female | 472 (51%) | 6,611 (51%) |
| Benign tumor | ||
| All sites combined, n (%) | 132 (14%) | 800 (6%) |
| Age at first tumor, mean (SD) | 43.8 (14.6) | 42.0 (15.0) |
| Tumor status | ||
| None | 762 (82%) | 11,660 (89%) |
| Benign only | 124 (13%) | 717 (5%) |
| Malignant only | 33 (4%) | 625 (5%) |
| Benign and malignant | 8 (1%) | 83 (1%) |
| Follow‐up time | ||
| Median (range) | 5.6 (0‐28.2) | 3.7 (0‐28.2) |
| Total, Person‐years | 7,100.7 | 73,076.5 |
Table 2.
Adjusted hazard ratios and 95% confidence intervals of selected benign tumors comparing myotonic dystrophy type I (DM1)‐affected with matched DM1‐free controls.
| Tumor/Site | Tumor frequency1 n (%) | HR2 | 95% CI | P‐value | ||
|---|---|---|---|---|---|---|
| DM1 | DM1‐free | |||||
| Skin | 85 (9.2%) | 623 (4.8%) | ||||
| Lipoma | 0.9 | 0.45 | 1.86 | 0.81 | ||
| Other skin tumors3 | 1.4 | 1.08 | 1.95 | 0.01 | ||
| Pilomatricoma | – | – | – | <0.0016 | ||
| Female genital | 14 (1.5%) | 82 (0.6%) | ||||
| Uterine fibroids | 2.7 | 1.22 | 5.88 | 0.01 | ||
| Uterine polyps | 9.6 | 1.20 | 77.49 | 0.03 | ||
| Cervical polyps | 0.4 | 0.05 | 2.96 | 0.36 | ||
| Endocrine | 9 (1.0%) | 19 (0.2%) | ||||
| Pituitary tumors4 | 11.0 | 0.49 | 250.14 | 0.13 | ||
| Thyroid nodules/cysts | 10.4 | 3.91 | 27.52 | <0.001 | ||
| Digestive | 10 (1.1%) | 42 (0.3%) | ||||
| Colorectal polyps | 4.3 | 1.76 | 10.41 | 0.001 | ||
| Other digestive tumors | – | – | – | >0.996 | ||
| Brain & nervous system5 | 7 (0.8%) | 11 (0.1%) | 8.4 | 2.48 | 28.47 | <0.001 |
| Other tumors | 13 (1.4%) | 67 (0.5%) | ||||
| Salivary glands | – | – | – | <0.0016 | ||
| Breast fibroadenoma | – | – | – | >0.996 | ||
| Other/Unknown7 | 1.4 | 0.67 | 3.09 | 0.35 | ||
In accordance with CPRD policy which prohibits the reporting of cells with fewer than five events, frequencies were reported by system, where applicable.
Baseline hazards were stratified on the matched sets and models were adjusted for number of healthcare care encounters.
Skin tumors included nevi, dermatofibromas, papillomas, moles, and other benign neoplasm of the skin.
In DM1, all pituitary tumors were adenomas; in controls, pituitary tumors included adenomas and craniopharyngiomas.
Brain & nervous system tumors included meningiomas, neuromas, neurofibromas, and other benign neoplasms of the brain & nervous system.
Obtained from Fisher's Exact test due to sparse or zero events in at least one group.
Other/unknown tumors include tumors of the bone & connective tissue, lip/oral cavity/pharynx, lymphatic/hematopoietic, respiratory, and unspecified sites (total n = 75).
The age at first benign tumor diagnosis was similar in DM1 patients (median = 43 years, range = 14–81) and DM1‐free controls (mean = 42 years, range = 3–90) (P = 0.27). However, site‐specific differences were noted. Specifically, DM1 patients developed colorectal polyps at a younger age compared with DM1‐free controls (median age at first polyp: DM1 = 50 years [range = 14–81] vs. DM1‐free = 65 years [range = 33–78], P = 0.04). Similarly, age differences were observed for tumors of the brain and nervous system (median age DM1 = 48 years, range = 16–59 vs. DM1‐free = 59 years, range = 42–63; P = 0.05) and thyroid nodules (median age DM1 = 50 years, range = 28–70 vs. DM1‐free = 63, range = 30–83, P = 0.16), although these differences were not statistically significant. No differences were noted for uterine fibroids (median age DM1 = 44 years, range = 37–53 vs. DM1‐free = 45, range = 29–65, P = 0.87), or other sites (Fig. 1).
Figure 1.
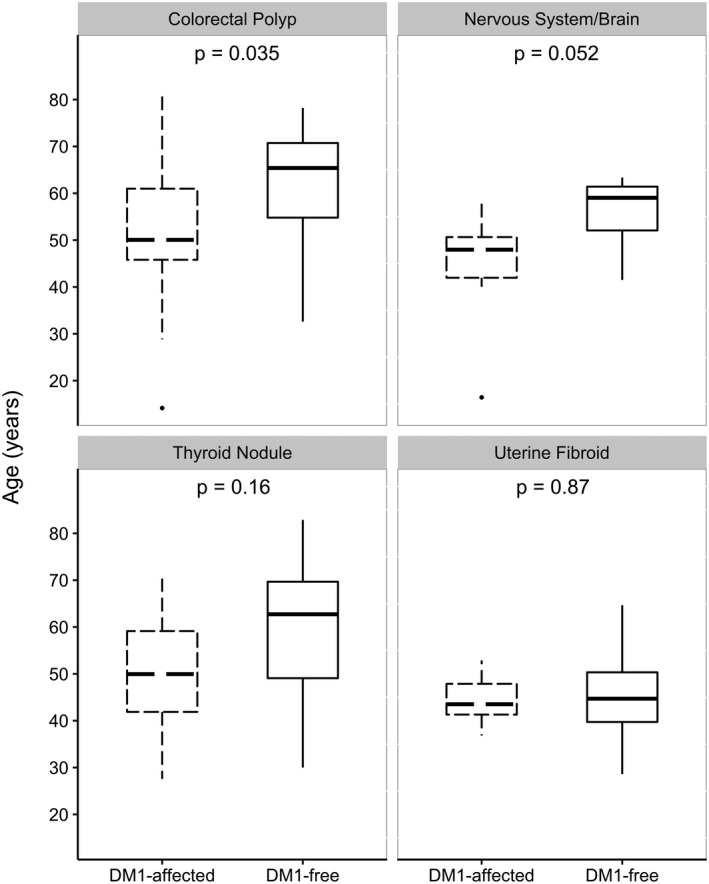
Distribution of age at diagnosis for selected benign tumors comparing DM1‐affected patients with DM1‐free controls.
Compared with matched DM1‐free controls, DM1 patients were at elevated risk of developing benign tumors (HR for all sites combined = 1.5; 95% CI = 1.23–1.88; P < 0.001). The observed excess in risk was driven by thyroid nodules (HR = 10.4; 95% CI = 3.91–27.52; P < 0.001), benign tumors of the brain or nervous system (HR = 8.4; 95% CI = 2.48–28.47; P < 0.001), colorectal polyps (HR = 4.3; 95% CI = 1.76–10.41; P = 0.001), and possibly uterine fibroids (HR = 2.7; 95% CI = 1.22–5.88; P = 0.01) and skin tumors other than lipoma or pilomatricoma (HR = 1.4; 95% CI = 1.08–1.94; 0.01). Pilomatricomas and salivary gland adenomas were found almost exclusively in DM1 patients, with Fisher's exact P < 0.001 for both (Table 2).
We found no significant heterogeneity by gender for all sites combined (P‐heterogeneity = 0.93); however, some site‐specific gender differences were observed. An elevated relative risk of colorectal polyps was noted in males (HR for DM1 vs. DM1‐free = 8.2; 95% CI = 2.70–24.75; P < 0.001; but not females (HR = 1.3; 95% CI = 0.24–7.28; P = 0.75; P‐heterogeneity < 0.001). In DM1, all benign endocrine and brain tumors occurred exclusively in females vs. only 74%, and 50%, respectively, occurred in female DM1‐free controls. Pilomatricomas in DM1 occurred primarily in males (83%). No statistically significant heterogeneity in the relative risk of benign tumors was noted between strata defined by DM1 subtype (congenital/childhood, classic, and late‐onset) (P‐heterogeneity = 0.75). Results from sensitivity analyses showed similar or higher HRs when restricting to DM1 patients diagnosed after their clinic's up‐to‐standard date (HR = 1.65, P = 0.001), diagnosed on or after 1995 (HR = 1.55, P = 0.004), or when ending follow‐up at a maximum of 3 years (HR = 1.98, P < 0.001). In analyses restricted to patients with DM1 records in at least 2 data sources, the HR was slightly attenuated and did not reach statistical significance (HR = 1.25; P = 0.19), likely due to the significant reduction in sample size and/or over‐representation of severe cases (with hospitalization or death record) (Table 3).
Table 3.
Sensitivity analyses of benign tumor risk estimates (all sites combined) comparing myotonic dystrophy type I (DM1)‐affected patients with DM1‐free controls.
| Sensitivity analysis1 | Number of patients with tumor/Total | HR | 95% CI | P‐value | ||
|---|---|---|---|---|---|---|
| DM1 | DM1‐free | |||||
| 1 | 71/454 | 362/5936 | 1.65 | 1.22 | 2.23 | 0.001 |
| 2 | 55/376 | 332/5541 | 1.25 | 0.89 | 1.75 | 0.19 |
| 3 | 72/530 | 383/6988 | 1.55 | 1.16 | 2.08 | 0.004 |
| 4 | 42/927 | 364/13,085 | 1.98 | 1.35 | 2.89 | <0.001 |
Analyses: (1) Restricted to DM1 patients diagnosed after their clinic's “up to standard” date and their matched cohort. (2) Restricted to DM1 patients with DM1 records in at least two data sources and their matched cohort. (3) Restricted to DM1 patients diagnosed in 1995 or later and their matched cohort. (4) Ending follow‐up at a maximum of 3 years for both cohorts.
Among DM1 patients, a suggested association between benign tumors and risk of subsequent cancer (all sites combined) was only observed in those with the classic subtype (age at onset = 11–40 years) (HR comparing patients with benign tumors with those who were tumor‐free = 2.7; 95% CI = 0.93–7.59; P = 0.07 vs. HR in late‐onset DM1 = 0.7; 95% CI = 0.15–3.38; P = 0.67) (Table 4). No cancers were observed subsequent to benign tumors in the congenital/childhood DM1 cohort or their matched controls.
Table 4.
Adjusted hazard ratios of cancer risk (all sites combined) subsequent to benign tumors (all sites combined) in the myotonic dystrophy type I (DM1)‐affected and DM1‐free cohorts, stratified by age at DM1 diagnosis.
| Cohort | Classic DM1 (age at diagnosis = 11–40 years) | Late‐onset DM1 (age at diagnosis >40 years) | ||||||
|---|---|---|---|---|---|---|---|---|
| HR1 | 95% CI | P‐value | HR1 | 95% CI | P‐value | |||
| DM1‐affected | 2.7 | 0.93 | 7.59 | 0.07 | 0.7 | 0.15 | 3.38 | 0.67 |
| Matched DM1‐free controls | 1.2 | 0.70 | 2.08 | 0.50 | 2.0 | 1.47 | 2.63 | <0.001 |
| P‐heterogeneity = 0.01 | P‐heterogeneity = 0.12 | |||||||
No cancers were observed subsequent to benign tumors in congenital/childhood DM1 patients or their matched controls.
Hazard ratios of subsequent cancer comparing patients with benign tumors to those who are tumor‐free; models were adjusted for gender and number of healthcare care encounters.
Discussion
In this large study, we demonstrated that DM1 patients were at elevated risk of developing certain benign tumors, including pilomatricoma, colorectal polyps, thyroid nodules, salivary gland adenomas, tumors of the brain and nervous system, and possibly uterine fibroids. We also found that classic DM1 patients with benign tumors may be more likely to develop a subsequent cancer, compared with those who were tumor‐free.
Previous studies found DM1 patients to be at elevated risk of developing specific cancers including melanoma and basal cell carcinoma of the skin, as well as cancers of the thyroid, uterus, ovary, and possibly colon, testis, and brain.7, 8, 9, 10, 11, 12, 14, 22 Here, we identified a similar organ profile for benign tumors, suggesting that DM1 pathogenesis could include a selective proliferative cellular growth advantage in those same organs. Of possible clinical importance, we found that DM1 patients are at high risk of developing colorectal polyps at a younger age compared with DM1‐free control (median age in years = 50 vs. 65, respectively, P = 0.035). The younger age at colorectal polyp in DM1 patients may suggest that this observation is affected by a detection bias related to the high prevalence of gastrointestinal abnormalities in DM1 patients. Yet, our observation of a male predominance of risk argues against detection bias since gastrointestinal symptoms are more prevalent in female DM1 patients.23 If validated, this suggests that earlier colorectal cancer screening may warrant consideration for DM1 patients. Colorectal polypectomy has been proven to reduce colon cancer risk and mortality in the general population.24, 25 Cancer studies in DM were inconclusive in relation to the risk of colon cancer compared with the general population (excess risk in DM patients was suggested in five of the six published studies, but only one reached statistical significance7, 9, 10, 11, 14). This inconsistency may be the consequence of differences in colorectal cancer screening practices between countries. In this study population, approximately 1% of the DM1 patients developed a colorectal cancer, comprising 17% of all DM1 cancers; a suggested increase in cancer risk was observed among patients with classic DM1 (HR = 1.82, 95% CI = 0.32–10.31) compared with matched controls (cancer risk findings published previously14). The histopathological classification of detected polyps in the current study was unavailable; however, recent data from the English National Health Service bowel cancer screening program showed that adenomas represented 67% of all polyps.26 Nonetheless, it is advisable that population‐based colorectal cancer screening guidelines be part of the routine, ongoing care of DM1 patients.
In contrast with our previous study which showed significant differences in cancer relative risk by DM1 disease subtype (excess cancer risk was restricted to patients with classic DM1),14 here we observed no heterogeneity in the overall risk of benign tumors by disease subtype (HR for benign tumors in DM1 vs. DM1 free control = 1.53 in congenital/childhood, 1.92 in classic, and 1.87 in late‐onset, P‐heterogeneity = 0.62). However, our analysis exploring the possible association between benign tumors and the risk of subsequent cancer in DM1, showed that classic DM1 patients with benign tumors may be at higher risk of developing cancer compared with those who were tumor‐free, an association that was not observed in late‐onset DM1. Although this finding was not statistically significant, likely due to the small number of cancer events, it could suggest that benign tumors in patients with classic DM1 may aid in identifying patients with a higher genetic predisposition to tumorigenesis, in general. Due to the relatively small number of cancer events, we were unable to evaluate site‐specific risks of cancer subsequent to benign tumor. Larger studies are warranted to investigate these associations.
Consistent with previous cross‐sectional studies of DM1‐associated tumors,16, 17, 27 benign tumors were more frequently observed in female DM1‐patients (18%) versus males (10%), with endocrine, brain, and salivary tumors occurring exclusively in females. In contrast, most pilomatricomas occurred in male DM1 patients, a finding consistent with a previous cross‐sectional study of 255 Italian DM1 patients,16 but not with the female predominance as reported in published literature of pilomatricomas in the general population.28, 29, 30, 31, 32, 33 This inconsistency may be explained, at least in part, by differential healthcare‐seeking behavior between males and females; females, in general, are more likely to visit their primary care provider,34, 35, 36, 37, 38 possibly resulting in higher detection of pilomatricoma in the general population. In DM1, we observed a higher frequency in males despite a similar pattern (average number of healthcare encounters per year in DM1 females: median = 9.5 vs. males = 7.8; P < 0.001). Gender‐specific phenotypic variations in DM1 patients have been previously described, but the biological mechanisms behind such differences are not known.37 In a small number of DM1 patients, downregulation of the microRNA 200c/141 tumor suppressor family was reported in women, but a slight elevated expression in men when compared with healthy controls.11, 23
The molecular mechanisms underlying DM1‐related tumorigenesis remain unknown. It has been previously hypothesized that abnormal accumulation of ß‐catenin through the Wnt/ß‐catenin signaling pathway may play a role in DM‐related tumorigenesis.15 Mutations in the ß‐catenin (CTNNB1) gene were previously associated with pilomatricomas.15, 39, 40 Other proposed hypotheses include modified expression of downstream oncogenes or tumor suppressor genes as a result of abnormal RNA‐splicing, and/or altered protein‐coding mechanisms.41, 42, 43, 44 Further studies to investigate molecular mechanisms underlying DM1‐related tumorigenesis are needed to better understand tumor etiology as well as guide clinical management and therapeutic interventions in those patients.
To our knowledge, this study is the first to comprehensively quantify the risk of developing benign tumors in DM1 patients. The use of electronic primary care medical records enabled us to capture the full spectrum of DM1 patients, minimizing selection bias resulting from ascertaining patients through tertiary care facilities, or voluntary registries. Additionally, primary care records captured data on benign tumors, which are not routinely collected in inpatient hospitalization records or cancer registries. Our large sample size, longitudinal design, and the use of a matched comparison group represent some of the major strengths of this study. Nonetheless, several limitations existed, most notably lack of information on genetic testing or repeat expansion size, which may have led to misclassification of DM1 status. To minimize this possibility, we only included patients with diagnostic codes specific to DM1 (described above). Additionally, we conducted several sensitivity analyses with more stringent definitions of DM1, and our conclusions remained unchanged, suggesting that diagnostic misclassification did not have a large impact and demonstrating the robustness of our findings. It is also possible that the higher frequency of detected benign tumors in DM1 patients compared with DM1‐free controls may be the result of the relatively intensive medical care required by DM1 patients. To address this possibility, all models were adjusted for the number of healthcare care encounters, and indicators of health seeking behaviors. Due to incomplete procedure records, we were unable to conduct analyses restricted to patients undergoing diagnostic procedures such as colonoscopy for colon polyps, or imaging studies for thyroid or brain tumor detection; therefore, validation of these results in a population with complete procedure data is important. Notably, the low risks of other benign tumors equally likely to be affected by detection bias, such as lipoma and cervical polyps in those patients provide reassurance that our results are not likely driven by detection bias. We also observed the occurrence of pilomatricomas (anecdotally associated with DM1 in previous literature15) in several patients before their first DM1 record. Finally, the lack of histopathological data limited our ability to assess if detected tumors were premalignant and the relatively small number of cancer events restricted our ability to evaluate site‐specific risks of cancer subsequent to benign tumor.
The current study suggests a similar organ profile for benign and malignant tumors in DM1 patients, and that benign tumors, in patients with the classic subtype of DM1, may identify those at higher future risk of developing cancer. These findings, if confirmed, may guide future studies aiming at understanding mechanisms of DM1‐related tumorigenesis and inform clinical management of DM1 patients. Our findings highlight the importance of following population‐based screening guidelines as part of the routine care for DM1 patients.
Author Contributions
Conception and design: RA, DMSG, RMP, MHG, SMG. Data acquisition, analysis, and interpretation: RA, MZ, RMP, YW, LAA, ZL, JK, AJB, KRW, SA, SMG. Drafting manuscript or figures: RA, SMG. Manuscript critical review: All.
Conflicts of Interest
Nothing to report.
Acknowledgments
The authors thank Ms. Emily Carver, BS, and Mr. David Ruggieri, BS, both from the Information Management Services Inc. (Calverton, MD, USA) for their important contributions to database management. This study is based on data from the CPRD GOLD database October 2016 release, obtained from the UK Medicines and Healthcare Products Regulatory Agency, Hospital Episode Statistics (HES) database (Copyright © (2016)), and Office of National Statistics (ONS) database (Copyright © (2016)) reused with the permission of The Health & Social Care Information Centre. All rights reserved. The interpretation and conclusions contained in this study are those of the authors alone.
Funding Information
The Intramural Research Program of the Division of Cancer Epidemiology and Genetics, National Cancer Institute, National Institutes of Health.
Funding Statement
This work was funded by Division of Cancer Epidemiology and Genetics, National Cancer Institute grant ; National Institutes of Health grant .
References
- 1. Bird TD. Myotonic dystrophy type 1 In: R. A. Pagon, M. P. Adam, H. H. Ardinger et al., eds. GeneReviews(R). Seattle (WA): University of Washington, 1993. [Google Scholar]
- 2. Thornton CA. Myotonic dystrophy. Neurol Clin 2014;32:705–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Johnson NE, Heatwole CR. Myotonic dystrophy: from bench to bedside. Semin Neurol 2012;32:246–254. [DOI] [PubMed] [Google Scholar]
- 4. Brook JD, McCurrach ME, Harley HG, et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3' end of a transcript encoding a protein kinase family member. Cell 1992;68:799–808. [DOI] [PubMed] [Google Scholar]
- 5. Fu YH, Pizzuti A, Fenwick RG Jr, et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992;255:1256–1258. [DOI] [PubMed] [Google Scholar]
- 6. Mahadevan M, Tsilfidis C, Sabourin L, et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3' untranslated region of the gene. Science 1992;255:1253–1255. [DOI] [PubMed] [Google Scholar]
- 7. Gadalla SM, Lund M, Pfeiffer RM, et al. Cancer risk among patients with myotonic muscular dystrophy. JAMA 2011;306:2480–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Win AK, Perattur PG, Pulido JS, et al. Increased cancer risks in myotonic dystrophy. Mayo Clin Proc 2012;87:130–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mohamed S, Pruna L, Kaminsky P. Increasing risk of tumors in myotonic dystrophy type 1. Presse Med 2013;42(9 Pt 1):e281–e284. [DOI] [PubMed] [Google Scholar]
- 10. Abbott D, Johnson NE, Cannon‐Albright LA. A population‐based survey of risk for cancer in individuals diagnosed with myotonic dystrophy. Muscle Nerve 2016;54:783–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fernandez‐Torron R, Garcia‐Puga M, Emparanza JI, et al. Cancer risk in DM1 is sex‐related and linked to miRNA‐200/141 downregulation. Neurology 2016;87:1250–1257. [DOI] [PubMed] [Google Scholar]
- 12. Wang Y, Pfeiffer RM, Alsaggaf R, et al. Risk of skin cancer among patients with myotonic dystrophy type 1 based on primary care physician data from the U.K. Clinical Practice Research Datalink. Int J Cancer 2018;142:1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Emparanza JI, Lopez de Munain A, Greene MH, et al. Cancer phenotype in myotonic dystrophy patients: results from a meta‐analysis. Muscle Nerve 2018;58:517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Alsaggaf R, St George DM, Zhan M, et al. Cancer risk in myotonic dystrophy type i: evidence of a role for disease severity. JNCI Cancer Spectrum 2018;2:pky052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mueller CM, Hilbert JE, Martens W, et al. Hypothesis: neoplasms in myotonic dystrophy. Cancer Causes Control 2009;20:2009–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bianchi ML, Leoncini E, Masciullo M, et al. Increased risk of tumor in DM1 is not related to exposure to common lifestyle risk factors. J Neurol 2016;263:492–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Alsaggaf R, Wang Y, Marini‐Bettolo C, et al. Benign and malignant tumors in the UK myotonic dystrophy patient registry. Muscle Nerve 2018;57:316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zampetti A, Silvestri G, Manco S, et al. Dysplastic nevi, cutaneous melanoma, and other skin neoplasms in patients with myotonic dystrophy type 1: a cross‐sectional study. J Am Acad Dermatol 2015;72:85–91. [DOI] [PubMed] [Google Scholar]
- 19. Marcoval J, Olive M, Bonfill‐Orti M, et al. Cutaneous neoplasms in myotonic dystrophy type 1. Dermatology 2016;232:700–703. [DOI] [PubMed] [Google Scholar]
- 20. Herrett E, Thomas SL, Schoonen WM, et al. Validation and validity of diagnoses in the General Practice Research Database: a systematic review. Br J Clin Pharmacol 2010;69:4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wood L, Cordts I, Atalaia A, et al. The UK Myotonic Dystrophy Patient Registry: facilitating and accelerating clinical research. J Neurol 2017;264:979–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Emparanza JI, de Munain AL, Greene MH, et al. Cancer phenotype in myotonic dystrophy patients: results from a meta‐analysis. Muscle Nerve 2018;58:517–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dogan C, De Antonio M, Hamroun D, et al. Gender as a modifying factor influencing myotonic dystrophy type 1 phenotype severity and mortality: a nationwide multiple databases cross‐sectional observational study. PLoS ONE 2016;11:e0148264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Winawer SJ, Zauber AG, Ho MN, et al. Prevention of colorectal cancer by colonoscopic polypectomy. The National Polyp Study Workgroup. N Engl J Med 1993;329:1977–1981. [DOI] [PubMed] [Google Scholar]
- 25. Zauber AG, Winawer SJ, O'Brien MJ, et al. Colonoscopic polypectomy and long‐term prevention of colorectal‐cancer deaths. N Engl J Med 2012;366:687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Majumdar D, Patnick J, Nickerson C, Rutter M. OC‐156 analysis of colorectal polyps detected in the English NHS bowel cancer screening programme with emphasis on advanced adenoma and polyp cancer detected. Gut 2012;61:A67. [Google Scholar]
- 27. Das M, Moxley RT 3rd, Hilbert JE, et al. Correlates of tumor development in patients with myotonic dystrophy. J Neurol 2012;259:2161–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lan MY, Lan MC, Ho CY, et al. Pilomatricoma of the head and neck: a retrospective review of 179 cases. Arch Otolaryngol Head Neck Surg 2003;129:1327–1330. [DOI] [PubMed] [Google Scholar]
- 29. Yencha MW. Head and neck pilomatricoma in the pediatric age group: a retrospective study and literature review. Int J Pediatr Otorhinolaryngol 2001;57:123–128. [DOI] [PubMed] [Google Scholar]
- 30. Danielson‐Cohen A, Lin SJ, Hughes CA, et al. Head and neck pilomatrixoma in children. Arch Otolaryngol Head Neck Surg 2001;127:1481–1483. [DOI] [PubMed] [Google Scholar]
- 31. Duflo S, Nicollas R, Roman S, et al. Pilomatrixoma of the head and neck in children: a study of 38 cases and a review of the literature. Arch Otolaryngol Head Neck Surg 1998;124:1239–1242. [DOI] [PubMed] [Google Scholar]
- 32. Julian CG, Bowers PW. A clinical review of 209 pilomatricomas. J Am Acad Dermatol 1998;39(2 Pt 1):191–195. [DOI] [PubMed] [Google Scholar]
- 33. Thomas RW, Perkins JA. Ruegemer JL, Munaretto JA. Surgical excision of pilomatrixoma of the head and neck: a retrospective review of 26 cases. Ear Nose Throat J 1999;78;541, 544–546, 548. [PubMed] [Google Scholar]
- 34. Thompson AE, Anisimowicz Y, Miedema B, et al. The influence of gender and other patient characteristics on health care‐seeking behaviour: a QUALICOPC study. BMC Fam Pract 2016;17:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nabalamba A, Millar WJ. Going to the doctor. Health Rep 2007;18(1):23–35. [PubMed] [Google Scholar]
- 36. Carriere G. Consultations with doctors and nurses. Health Rep 2005;16:45–48. [PubMed] [Google Scholar]
- 37. Deveugele M, Derese A, van den Brink‐Muinen A, et al. Consultation length in general practice: cross sectional study in six European countries. BMJ 2002;325:472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Cleary PD, Mechanic D, Greenley JR. Sex differences in medical care utilization: an empirical investigation. J Health Soc Behav 1982;23:106–119. [PubMed] [Google Scholar]
- 39. Kajino Y, Yamaguchi A, Hashimoto N, et al. beta‐Catenin gene mutation in human hair follicle‐related tumors. Pathol Int 2001;51:543–548. [DOI] [PubMed] [Google Scholar]
- 40. Chan E, Gat U, McNiff JM, Fuchs E. A common human skin tumour is caused by activating mutations in β‐catenin. Nat Genet 1999;21:410. [DOI] [PubMed] [Google Scholar]
- 41. Du H, Cline MS, Osborne RJ, et al. Aberrant alternative splicing and extracellular matrix gene expression in mouse models of myotonic dystrophy. Nat Struct Mol Biol 2010;17:187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Osborne RJ, Lin X, Welle S, et al. Transcriptional and post‐transcriptional impact of toxic RNA in myotonic dystrophy. Hum Mol Genet 2009;18:1471–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ranum LP, Day JW. Myotonic dystrophy: clinical and molecular parallels between myotonic dystrophy type 1 and type 2. Curr Neurol Neurosci Rep 2002;2:465–470. [DOI] [PubMed] [Google Scholar]
- 44. Udd B, Krahe R. The myotonic dystrophies: molecular, clinical, and therapeutic challenges. Lancet Neurol 2012;11:891–905. [DOI] [PubMed] [Google Scholar]