

Abstract
Objective
Increased translocator protein (TSPO), previously known as the peripheral benzodiazepine receptor (PBR), in glial cells of the brain has been used as a neuroinflammation marker in the early and middle stages of neurodegenerative diseases, such as Alzheimer’s disease (AD) and Dementia with Lewy Bodies (DLB). In this study, we investigated the changes in TSPO density with respect to late stage AD and DLB.
Methods
TSPO density was measured in multiple regions of postmortem human brains in 20 different cases: seven late stage AD cases (Braak amyloid average: C; Braak tangle average: VI; Aged 74–88, mean: 83 ± 5 years), five DLB cases (Braak amyloid average: C; Braak tangle average: V; Aged 79–91, mean: 84 ± 4 years), and eight age‐matched normal control cases (3 males, 5 females: aged 77–92 years; mean: 87 ± 6 years). Measurements were taken by quantitative autoradiography using [3H]PK11195 and [3H]PBR28.
Results
No significant changes were found in TSPO density of the frontal cortex, striatum, thalamus, or red nucleus of the AD and DLB brains. A significant reduction in TSPO density was found in the substantia nigra (SN) of the AD and DLB brains compared to that of age‐matched healthy controls.
Interpretation
This distinct pattern of TSPO density change in late stage AD and DLB cases may imply the occurrence of microglia dystrophy in late stage neurodegeneration. Furthermore, TSPO may not only be a microglia activation marker in early stage AD and DLB, but TSPO may also be used to monitor microglia dysfunction in the late stage of these diseases.
Introduction
Translocator protein 18 kDa (TSPO), previously known as the peripheral benzodiazepine receptor (PBR), is located on the outer membrane of mitochondria and is a part of mitochondrial permeability transition pore.1 Outside of the central nervous system, it is ubiquitously expressed in the kidneys, heart, adrenal gland, and reproductive tract2, 3; in the brain it is expressed at a low level in the resting glial cells.2 TSPO is believed to be involved in the synthesis of steroids and the translocation of cholesterol from the outer to the inner mitochondria membrane.4 TSPO is also involved in the regulation of cell death,1 cell proliferation, and inflammation.5 TSPO density has been found to be increased in many neurological diseases such as ischemic stroke, multiple sclerosis, amyotrophic lateral sclerosis, encephalitis, and frontotemporal dementia.6 The upregulation of TSPO is often accompanied by microglial changes from resting to activated morphology and the secretion of cytokines which can induce tissue inflammatory response; hence TSPO is currently accepted as a neuroinflammation marker.
TSPO density in the human AD and DLB brain has been extensively explored by PET studies using [11C]PK11195,7, 8, 9 [11C]DAA1106,10 [11C]vinpocetine,11 and [11C]PBR28;12, 13, 14, 15 and by autoradiography studies using [3H]PK11195,16, 17 [3H] DAA1106,17 and [125I]DAA1106.18 In these studies early stage patients with mild to moderate cognitive impairment (clinical dementia rate (CDR) 1 and 2) were usually recruited and increases in TSPO density were often found in multiple brain regions.7, 10, 15 Two studies showed no significant change in TSPO density between aged AD (CDR unknown) and the age‐matched control brain.9, 11 Because it is a marker of microglia activation, increases in TSPO density in early stage AD may reflect the involvement of microglia‐mediated neuroinflammation in the pathogenesis of AD,19 although the enhanced phagocytosis by activated microglia has been reported to have a protective effect.20, 21
Microglial dystrophy has been recently observed in the AD brain; this is thought to play a key role in the pathogenesis of AD.22, 23, 24, 25, 26, 27, 28 Both Tau pathology and amyloid beta pathology have been reported to promote microglial dystrophy,29, 30 which may occur via telomere shortening.29, 31, 32 Although microglial dystrophy is closely related to AD stage, TSPO density in late stage AD and DLB has not been well documented until now. In this communication, we hypothesize that DLB and AD patients have more microglial dystrophy than healthy controls and TSPO PET may be useful for measuring late stage microglia dystrophy in DLB and AD patients. To explore the relationship between TSPO density change and microglial dystrophy, we systematically studied a group of late stage AD and DLB cases and measured TSPO density in multiple brain regions using quantitative autoradiography with two radioligands [3H]PK11195 and [3H]PBR28.
Materials and Methods
Ethics statement
After death, the written consent of the next of kin was obtained for brain removal per local Ethical Committee procedures (Washington University Institutional Review Board, Washington University School of Medicine). Use of this tissue for the postmortem receptor autoradiography study was approved by the Charles F. and Joanne Knight Alzheimer's Disease Research Center (Knight ADRC) Leadership Committee.
Chemicals and radioligands
Chemical reagents and the standard compounds were purchased from Sigma‐Aldrich (St. Louis, MO) and Tocris (Ellisville, MO). [3H]PK11195 (69.9 Ci/mmol) was purchased from Perkin Elmer Life Sciences (Boston, MA) and [3H]PBR28 (80 Ci/mmol) was purchased from American Radiolabeled Chemicals (St Louis, MO).
Postmortem human brain cases
Clinically and neuropathologically well‐characterized human brain tissues were obtained from the Knight ADRC at Washington University School of Medicine including: seven AD (1 male, 6 females) aged 74–88 (mean: 83 ± 5) years at death, five DLB (2 males, 3 females) aged 79–91 (mean: 84 ± 4) years at death, and eight age‐matched normal control cases (3 males, 5 females) aged 77–92 (mean: 87 ± 6) years at death. Dementia level was evaluated by CDR. Alzheimer disease pathological changes were assessed using Braak staging.33, 34 Stages of amyloid beta deposition refer to initial deposits in the basal neocortex (A), deposits extended into the association areas of the neocortex (B), and heavy deposition throughout the entire cortex (C). Stages of neurofibrillary pathology correspond to transentorhinal (I–II), limbic (III–IV), and neocortical (V and VI). There is no significant difference between the average age and post mortem interval time among these three groups; whereas all the AD and DLB cases show a heavy burden of amyloid beta (Average: C) and neurofibrillary tangles (NFTs) (Average: VI for AD; V for DLB) and are significantly different from that of age‐matched control cases (Average amyloid beta: A; NFTs: II). There exist significant differences in the brain weight and the CDR between the control cases and the AD or DLB cases. Detailed information on the clinical and pathological features is summarized in Table 1.
Table 1.
Demographic details, clinical, and pathological features of cases.
| Case # | Age (years) | Gender | Brain weight (g) | PMI (h) | Braak tangle | Braak amyloid | Exp CDR | Diagnosis |
|---|---|---|---|---|---|---|---|---|
| 1 | 77 | F | 1410 | 10 | 1 | A | 0 | Con |
| 2 | 91.6 | F | 1310 | 16 | 2 | 0 | 0 | Con |
| 3 | 92.1 | F | 1120 | 6 | 0 | 0 | 0 | Con |
| 4 | 91.6 | F | 1220 | 16 | 2 | A | 0 | Con |
| 5 | 79 | F | 1100 | 25 | 1 | A | 0 | Con |
| 6 | 90 | M | 1150 | 10 | 4 | A | 0 | Con |
| 7 | 84 | M | 1010 | 5.5 | 1 | B | 0 | Con |
| 8 | 91 | M | 1170 | 8.5 | 1 | A | 0 | Con |
| 9 | 84 | M | 1180 | 21 | 5 | C | 3 | DLB |
| 10 | 82 | M | 1200 | 16.25 | 5 | C | 3 | DLB |
| 11 | 84 | F | 950 | 5 | 3 | A | 1 | DLB |
| 12 | 79 | F | 940 | 21.25 | 6 | C | 3 | DLB |
| 13 | 91 | F | 940 | 18 | 6 | C | 3 | DLB |
| 14 | 80 | F | 1130 | 7 | 6 | C | 3 | AD |
| 15 | 83 | F | 950 | 6.45 | 6 | C | 3 | AD |
| 16 | 74 | M | 1120 | 4 | 6 | C | 3 | AD |
| 17 | 88 | F | 1050 | 6 | 6 | C | 3 | AD |
| 18 | 88 | F | 785 | 21 6 | C | 3 | AD | |
| 19 | 86 | F | 1250 | 7 | 5 | C | 1 | AD |
| 20 | 81 | F | 1050 | 8.5 | 4 | C | 3 | AD |
AD, Alzheimer’s disease; Con, Control; DLB, Dementia with Lewy Bodies; PMI, Post‐mortem interval; Exp CDR, Estimated clinical dementia rating at expiration.
Tissue collection
Brains were removed at the time of autopsy and the right hemisphere was coronally sectioned and snap‐frozen by contact with Teflon‐coated aluminum plates cooled in liquid nitrogen vapor. Tissue blocks were subsequently placed in airtight zip‐lock plastic bags and stored at − 80°C until used. Microscopic examination to establish neuropathology was performed using established rating scales. For autoradiography studies, frozen coronal sections (20 µm) were cut with a Microm cryotome and mounted on Superfrost Plus glass slides (Fisher Scientific, Pittsburgh, PA) from the following brain regions: the striatal regions, containing the caudate and putamen, of the frontal cortex, and the middle brain containing the substantia nigra (SN), the red nucleus (RN), and the thalamus (Th). Data from 2–4 sections were averaged to determine total binding. Nonspecific binding was defined by the average of two adjacent sections for the two radioligands.
Quantitative autoradiography protocol
Sections on slides were incubated in an open staining jar with the respective radiotracer for 30 min as detailed below; slides were then rinsed five times at 1 min intervals with ice‐cold buffer, then air dried. This protocol results in a free radioligand concentration loss < 5%, as described previously.35, 36
TSPO binding
TSPO was labeled with [3H]PK11195 and [3H]PBR28. Briefly, adjacent brain sections were incubated for 30 min in buffer solution at room temperature with the addition of 2 nmol/L [3H]PK11195 and [3H]PBR28, respectively. Nonspecific binding was determined from incubating the slides in the presence of 1 µmol/L PBR28 for [3H]PK11195, and 1 µmol/L PK11195 for [3H]PBR28.
Quantification of total radioactivity
Slides were made conductive by coating the free side with a copper foil tape. Slides were then placed in the sample holder and loaded into a gas chamber containing a mixture of argon and triethylamine (Sigma‐Aldrich) as part of a gaseous detector system, the Beta Imager 2000Z Digital Beta Imaging System (Biospace, France). After the gas was well mixed and a homogenous state was reached, exposure of the slides for 20 h yielded high‐quality images. A [3H]Microscale (American Radiolabeled Chemicals, St Louis, Missouri) was counted simultaneously as a reference for total radioactivity quantitative analysis. Quantitative analysis was performed using the program Beta‐Vision Plus (BioSpace, France) for anatomical regions of interest.
Statistical analysis
The receptor‐bound radioligand binding apparent densities were calculated as described previously using the specific activity of each radioligand expressed as fmol/mg tissue.35, 36 The experimenter was blinded to all conditions during the analysis. Comparison of receptor densities was analyzed using an unpaired Student’s t test. The correlation between receptor binding densities across different brain regions was assessed using Pearson product‐moment correlation coefficient.
Results
Quantitative analysis of TSPO binding
The sensitivity limit of the Beta Imager 2000Z Digital Beta Imaging System is 0.07 dpm/mm2. A tritium standard was counted with each set of slides, the [3H]Microscale contains multiple strips with a known amount of radioactivity (ranging from 0 to 36.3 nCi/mg) and was used to create a standard curve. In each case the standard curve had a correlation coefficient (R) greater than 0.99. The binding density of TSPO was determined by quantitative autoradiography using 2 nmol/L [3H]PK11195 and [3H]PBR28 (Fig. 1). The nonspecific binding was determined by using the high affinity cold compound and was much higher with [3H]PK11195 than [3H]PBR28 (Figs. 2, 3, 4C and D). TSPO receptor density values in different brain regions are summarized in Table 2.
Figure 1.
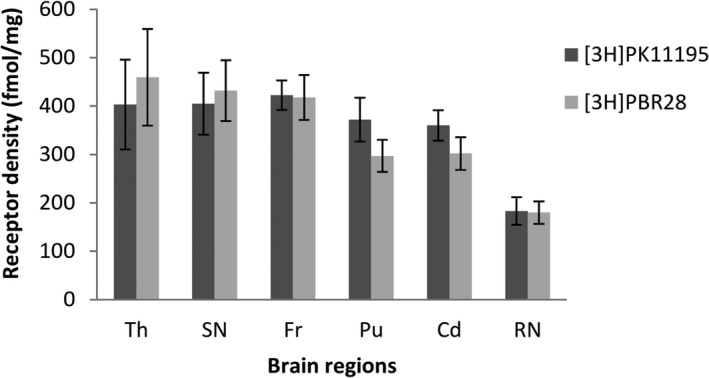
Comparison of translocator protein (TSPO) densities in the normal aged post mortem human brain measured by [3H]PK11195 and [3H]PBR28. Quantitative analysis of 2 nmol/L [3H]PK11195 and 2 nmol/L [3H]PBR28 binding in multiple brain regions of aged human healthy control cases (n = 8, aged 77–92 years). Non‐specific binding was determined by the presence of 1 µmol/L unlabeled PBR28 for [3H]PK11195 and 1 µmol/L PK11195 for [3H]PBR28. Specific receptor binding densities (fmol/mg) were calculated by subtracting nonspecific binding from total binding. Data was calculated from the average of 2–4 adjacent sections of each case and presented as mean ± SEM. Th, Thalamus; SN, Substantia nigra; Fr, Frontal cortex; Cd, Caudate; Pu, Putamen; RN, Red nucleus.
Figure 2.
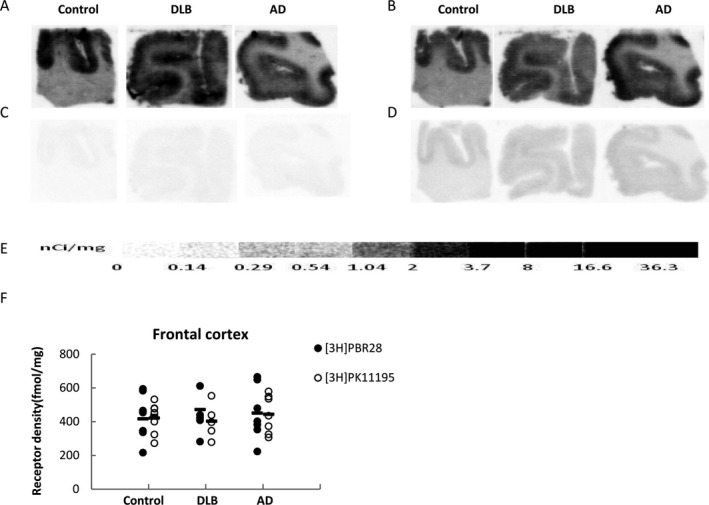
[3H]PBR28 and [3H]PK11195 quantitative autoradiographic analysis of translocator protein (TSPO) density in the frontal cortex of AD, DLB and age‐matched healthy control cases. Representative autoradiograms show total and nonspecific binding of [3H]PBR28 (A, C) and [3H]PK11195 (B, D) in frontal cortex sections. [3H]Microscale standards (ranging from 0 to 36.3 nCi/mg) were also counted (E). Quantitative analysis of TSPO density in frontal cortex (F) using [3H]PBR28 and [3H]PK11195.
Figure 3.
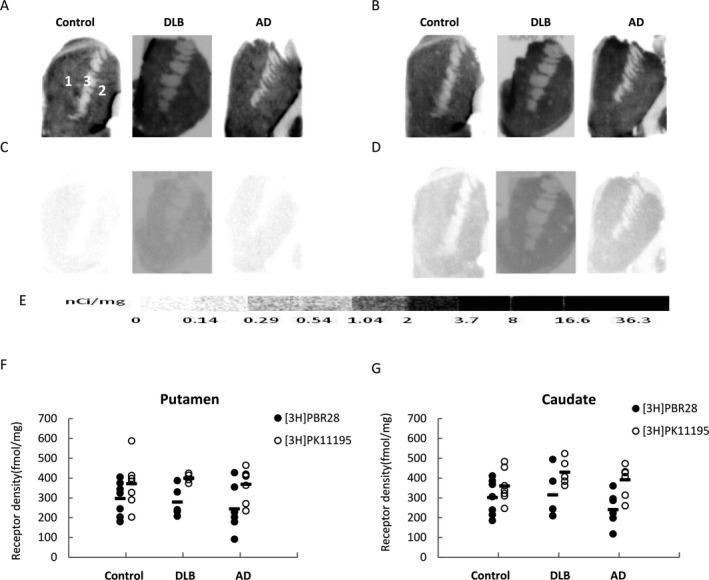
Quantitative autoradiographic analysis of translocator protein (TSPO) density in the striatum of AD, DLB, and age‐matched healthy control cases. Representative autoradiograms show total binding and nonspecific binding of [3H]PBR28 (A, C) and [3H]PK11195 (B, D). CNS anatomical regions are identified on the control striatal section (A): (1) caudate, (2) putamen, (3) internal capsule (IC). [3H]Microscale standards (ranging from 0 to 36.3 nCi/mg) were also counted (E). Quantitative analysis of TSPO density in the putamen (F) and the caudate (G) measured using [3H]PBR28 and [3H]PK11195.
Figure 4.
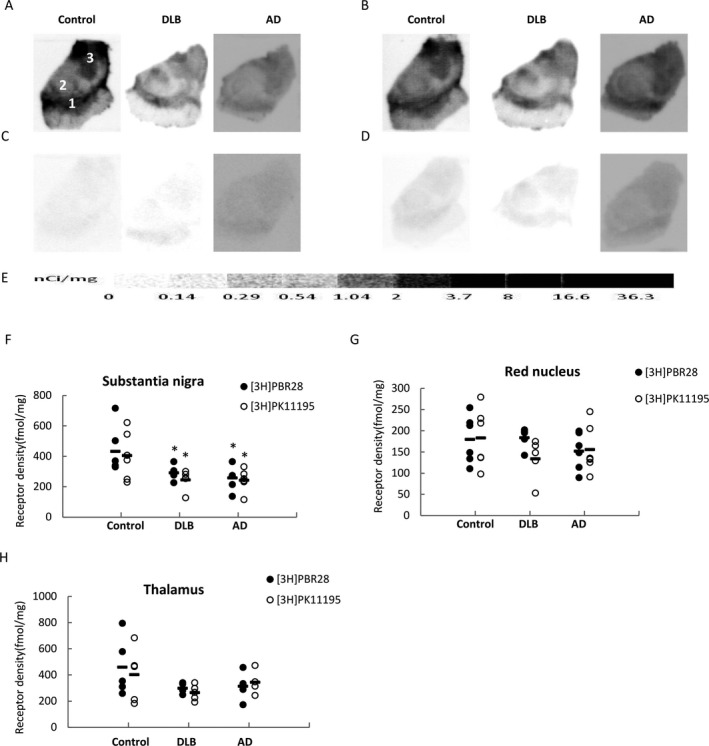
[3H]PBR28 and [3H]PK11195 quantitative autoradiographic analysis of translocator protein (TSPO) density in the substantia nigra, thalamus and red nucleus of AD, DLB, and age‐matched healthy control cases. Representative autoradiograms show total binding and nonspecific binding of [3H]PBR28 (A, D) and [3H]PK11195 (B, C). CNS anatomical regions are identified on the control section (A): (1) substantia nigra, (2) red nucleus, (3) thalamus. [3H]Microscale standards (ranging from 0 to 36.3 nCi/mg) were also counted (E). Quantitative analysis of TSPO density in the substantia nigra (F), red nucleus (G), and thalamus (H). *P < 0.05 compared to control.
Table 2.
[3H]PBR28 and [3H]PK11195 binding densities in the late stage Alzheimer’s Disease (AD) (n = 7) and Dementia with Lewy Bodies (DLB) (n = 5) patients (fmol/mg) compared to age‐matched control (n = 7).
| [3H]PBR28 | [3H]PK11195 | |||||
|---|---|---|---|---|---|---|
| Control | DLB | AD | Control | DLB | AD | |
| Fr | 418 ± 46 | 440 ± 48 | 451 ± 61 | 422 ± 30 | 403 ± 41 | 444 ± 42 |
| Pu | 297 ± 33 | 280 ± 30 | 245 ± 42 | 372 ± 45 | 401 ± 8 | 368 ± 42 |
| Cd | 302 ± 34 | 315 ± 48 | 241 ± 30 | 360 ± 31 | 430 ± 27 | 392 ± 29 |
| SN | 432 ± 63 | 291 ± 20* | 258 ± 26* | 405 ± 64 | 245 ± 27* | 243 ± 29* |
| Th | 459 ± 100 | 298 ± 15 | 314 ± 59 | 403 ± 93 | 265 ± 23 | 345 ± 48 |
| RN | 180 ± 23 | 184 ± 10 | 152 ± 18 | 183 ± 28 | 134 ± 19 | 156 ± 23 |
Fr, Frontal cortex; Pu, Putamen; Cd, Caudate; SN, Substantia nigra; Th, Thalamus; RN, Red nucleus.
Data presented as mean value ± SEM. *P < 0.05 compared to control.
TSPO densities in multiple brain regions of healthy control cases measured by [3H]PK11195 and [3H]PBR28
In order to verify the consistency of [3H]PK11195 and [3H]PBR28 binding, we first compared TSPO densities measured by these two radioligands in different brain regions of control cases. Although the nonspecific binding of PK11195 is higher than [3H]PBR28, the TSPO densities measured by [3H]PK11195 and [3H]PBR28 showed no significant differences across all the brain regions measured (Fig. 1). The regional distribution of TSPO in control brains was similar, whether measured by [3H]PK11195 or [3H]PBR28: the lowest measured TSPO density level was in the RN, a modest level was found in the striatum (putamen and caudate), and the highest levels were found in the frontal cortex, thalamus, and SN (Fig. 1 and Table 2).
TSPO density changes in multiple brain regions of AD, DLB cases compared to age‐matched controls
Frontal cortex
A similar binding pattern of [3H]PK11195 and [3H]PBR28 was observed in the frontal cortex; TSPO binding was mainly located in the grey matter, whereas TSPO binding in the white matter is relatively lower than in the grey matter (Fig. 2A and B). The non‐specific binding of [3H]PBR28 is lower than the binding of [3H]PK11195 (Fig. 2C and D). TSPO binding densities were similar for [3H]PK11195 and [3H]PBR28 in the frontal cortex of the control cases and the AD and DLB cases. Compared to the age‐matched controls, no significant difference in cortical TSPO densities between control and AD or DLB cases was found by either [3H]PK11195 or [3H]PBR28 autoradiography (Fig. 2F).
Striatum
TSPO was found to be homogeneously distributed in the striatum, including in the putamen and the caudate; whereas binding in the internal capsule was very low as measured by both [3H]PK11195 and [3H]PBR28 (Fig. 3A and B). The nonspecific binding was lower with [3H]PBR28 than with [3H]PK11195 (Fig. 3C and D). TSPO density as measured by [3H]PK11195 was higher than that measured with [3H]PBR28 both in the putamen and the caudate, but no significant differences in striatal TSPO densities were found between the control and the AD or DLB cases when measured using either [3H]PK11195 or [3H]PBR28 autoradiography (Fig. 3F and G).
Thalamus
TSPO was abundantly distributed in the thalamus and the density as measured by [3H]PK11195 and [3H]PBR28 gave similar results (Fig. 4A, B and H). However, non‐specific binding of [3H]PK11195 was higher than that of [3H]PBR28 in the thalamus (Fig. 4C and D). The decreased trend of thalamic TSPO binding in AD and DLB compared to the age‐matched controls was found not to be significantly different when measured using [3H]PK11195 and [3H]PBR28 autoradiography (Fig. 4H).
Red nucleus
TSPO density was the lowest in the RN compared to the other regions (Fig. 4G; Table 2). There were no significant changes in the TSPO densities in the red nucleus between the control and the AD or DLB cases when measured using either [3H]PK11195 or [3H]PBR28 autoradiography (Fig. 4G).
TSPO density decreased in the substantia nigra of AD, DLB cases compared to age‐matched control
There was a high level of TSPO in the SN in control cases as measured by [3H]PK11195 and [3H]PBR28 (Figure 1; Fig. 4A and B; Table 2). Compared to the controls, the TSPO density was significantly decreased in AD as measured by both [3H]PBR28 (P = 0.01) and [3H]PK11195 (P = 0.02). A significant reduction in TSPO density was also found for DLB cases compared to that of the age‐matched control measured by either [3H]PBR28 (P = 0.04) or [3H]PK11195 (P = 0.03) (Fig. 4F). TSPO density in the SN measured using either [3H]PK11195 or [3H]PBR28 was similar; a relatively higher nonspecific binding was found with [3H]PK11195 than [3H]PBR28 (Fig. 4C and D).
Regional correlation of TSPO density in the frontal cortex, striatum and substantia nigra measured by [3H]PBR28
TSPO density in the frontal cortex significantly correlated with density in the striatal regions (both putamen and caudate); there was also a significate correlation between TSPO density in the putamen and density in the caudate (Fig. 5A). No such correlations were found between the SN and the frontal cortex, putamen, or caudate (Fig. 5B); this is due to the significant correlation in TSPO density in the SN of AD and DLB cases.
Figure 5.
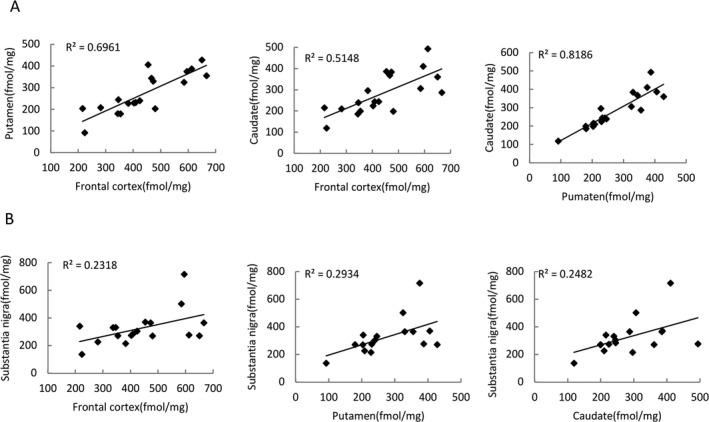
Regional correlation of translocator protein (TSPO) density measured by [3H]PBR28. Regional correlations in TSPO density measured using [3H]PBR28 were performed for all the cases. Significant correlation of TSPO densities was found between the frontal cortex with the putamen and the caudate and between the putamen and the caudate (A). Regional correlation of TSPO densities between substantia nigra with frontal cortex, the putamen, and the caudate are shown in B.
Discussion
In this study, the TSPO density was measured in multiple brain regions of postmortem human brains in a group of late stage AD and DLB cases that had a heavy burden of amyloid beta and NFTs. TSPO density was measured by quantitative autoradiography using two different tritiated ligands, [3H]PK11195 and [3H]PBR28. Brain weight, Braak amyloid, and Braak NFTs stages of AD and DLB cases were significantly different from those of age‐matched healthy controls. However, TSPO density appeared to be at a similar level in multiple brain regions including: frontal cortex, striatum, thalamus and red nucleus. Furthermore, we discovered that TSPO density was significantly decreased in the SN of AD and DLB cases.
TSPO density was measured by [3H]PK11195 and [3H]PBR28 in this study. Although renewed efforts are still ongoing to develop and evaluate more potent and specific TSPO radiotracers,37, 38, 39, 40, 41 [3H]PK11195 is widely used, and is accepted as the “gold standard”, to measure TSPO density both in vitro and in vivo. However, we observed a relatively high nonspecific binding level and lower signal‐noise ratio using [3H]PK11195. [11C]PBR28 is an improved TSPO radioligand with higher binding affinity and selectivity42, 43, 44, 45, 46, 47 which has been successfully used to measure TSPO density in cerebral ischemia48 as well as in multiple sclerosis.49 TSPO densities measured by [3H]PK11195 and [3H]PBR28 in this study appeared to be at similar level, indicating a consistency of measurement for these two radioligands. Human subjects can have variable binding levels in PBR28 PET and autoradiography human study due to polymorphism in the 18 kDa protein; codominance of the trait results in trimodal distribution.50, 51, 52 This is an important issue considering the reduction in TSPO density as measured by [3H]PBR28 in this study, however, it is unlikely that this reduction in TSPO density can be attributed to polymorphism. Two reasons indicate this: (1) In this study the reduction of TSPO density as measured by [3H]PBR28 was only slight (15–20% compared to that of age‐matched control cases), but low affinity binders (a single nucleotide polymorphism (SNP) of PBR, Ala147Thr) have an affinity is 10‐fold lower and while mixed affinity binders show ~ 50% reduction50 compared to the wild type; and (2) TSPO density measured by [3H]PK11195 was at the same level as that of [3H] PBR28 and no low binder phenomenon has been reported for [3H]PK11195. The cases used for our study can be safely assumed to consist of high affinity binders or mixed affinity binders as described previously.45, 51, 52
No significant changes were found in TSPO density of the frontal cortex, striatum, thalamus, or red nucleus in late stage AD and DLB cases compared to that of age‐matched control cases. This is different from most previous PET or autoradiography studies, in which an increase in TSPO density was usually found in the early and middle stage of AD. However, this is in line with a previous PET study using [11C]PK11195 which showed no significant changes of TSPO density in AD compared to that of an age‐matched control.9 It is also in line with another PET study using [11C]vinpocetine, which found a significant increase in TSPO density in the brain of aged healthy controls and aged AD patients compared to that found in young healthy control subjects, but no significant difference between aged AD patients and age‐matched controls.11 Our findings, along with other results of previous studies, indicate that there may be a different pattern of changes in TSPO between early and late stage AD. In early stage AD, TSPO increases as a marker of microglial activation; whereas in late stage AD, this increase in TSPO density disappears. This may reflect the different states of microglia at different AD stages. This is interesting when considering recent findings showing that there were more dysfunctional microglia in the AD brain than in non‐demented, amyloid‐free control subjects29 and that Tau pathology was co‐localized with dystrophic microglial cells.30 It is reasonable to deduce that with the progression of disease, the increase in TSPO density during early stage AD can become slower because of microglial dysfunction, which leads to no significant difference in TSPO density during late stage AD. This different temporal curve of TSPO density changes between the normal aging process and the AD brain may reflect a different microglia reaction pattern. In the AD brain, it seems that microglia are more predisposed to become dystrophic after initial activation than in the normally aging brain, in which a decrease in the up‐regulation of MHCП expression on microglial cell surface, an indicator of microglia activation, was only observed at age> 80 years old in human post‐mortem brain.53
TSPO density was found to be significantly decreased in the SN, indicating a significant microglia dystrophy occurred in this region during the late stages of AD and DLB. This is different from previous reports indicating microglial activation in the SN during early stage DLB and AD cases.54 It is not surprising that decreased TSPO density, which may be a marker of microglia dystrophy, was also found in the SN of the AD brain. This is because a type of SN pathology which includes neuronal loss, Lewy bodies, and NFT has been proven to be present in AD.55, 56, 57, 58, 59, 60, 61, 62 The significant reduction in TSPO density of the SN in the late stages AD and DLB is an interesting phenomenon which may imply an interaction between microglial dystrophy and neuronal loss in the same region. On one hand, dystrophic microglia can accelerate neuronal loss through the dysfunction of phagocytosis leading to more deposition of NFT and amyloid beta as well as through microglia‐mediated neuroinflammation, which is well documented in dystrophic microglial cells. On the other hand, significant neuronal death in the SN may also prompt microglial dystrophy, although this possibility needs to be elucidated further.
It is more likely that the reduction in TSPO density of the SN in AD and DLB cases is mainly attributed to the loss of microglial cells, but it could also be due to downregulation of TSPO on the mitochondrial membrane, or a decreased turnover rate of mitochondria in dystrophic microglia. The complete function of TSPO is not fully understood, but it has been reported that increased expression of TSPO can protect cells from apoptosis through the regulation of the mitochondrial permeability transition pore complex.2, 63, 64, 65 In addition, TSPO can mediate protective effects against ROS damage.66 This is important because oxidative stress and mitochondrial dysfunction are thought to be closely related to neurodegenerative diseases.67, 68, 69, 70 Taken together, the reduction in TSPO density in the late stages of AD and DLB found in this study may not only reflect the loss of microglial cells but also may be a marker of dysfunction of mitochondria in microglial cells, which in turn contributes to microglia apoptosis and eventual neurodegeneration.71
In summary, this study provides quantitative information about TSPO changes in late stage AD and DLB brains, showing different patterns of change compared to that seen in early stage ADs. A presence of change or reduction in TSPO density may reflect dystrophy, senescence and death, or dysfunction of mitochondria in the microglia, in turn contributing to the neuronal loss in the late stage AD. Furthermore, the different pattern of change seen for TSPO density between early and late stage AD may indicate the importance of dynamic monitoring of TSPO through clinical PET imaging; not the initial increase, but rather the decrease in the upregulation of TSPO density may herald the progression of neurodegeneration.
Author Contributions
JX, KRB, TSLB, and DMH involved in study concept and design. JX, JS, and RJP involved in collection of and qualitative analysis of data. JX, JS, RHM, and DMH involved in statistical analysis and interpretation. RJP, JX, and JS involved in preparation of tissue. JX, JCM, TSLB, and DMH obtained funding. JX, JS, and TSLB involved in drafting of the manuscript. JX and JS involved in autoradiography and preparation of figures/tables. JCM involved in critical revision of the manuscript for important intellectual content. JX and TSLB involved in study supervision.
Conflict of Interest
The authors have no conflicts of interest.
Acknowledgments
The authors thank Ms. Erin E. Franklin and Mr. Michael Baxter of the Knight ADRC Neuropathology Core at Washington University School of Medicine, for coordination of the frozen tissue preparation and expert technical assistance. We also thank Ms. Lynne Jones and Mr. William Knight for editorial assistance. Research funded by National Institutes of Health (NIH) R01 NS092865, R01 AG052550, P01 AG026276, P01 AG03991, and P50 AG05681.
Funding Statement
This work was funded by National Institutes of Health grants P01 AG03991 , P50 AG05681, R01 AG052550, and R01 NS092865.
References
- 1. McEnery MW, Snowman AM, Trifiletti RR, Snyder SH. Isolation of the mitochondrial benzodiazepine receptor: association with the voltage‐dependent anion channel and the adenine nucleotide carrier. Proc Natl Acad Sci USA 1992;89:3170–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Casellas P, Galiegue S, Basile AS. Peripheral benzodiazepine receptors and mitochondrial function. Neurochem Int 2002;40:475–486. [DOI] [PubMed] [Google Scholar]
- 3. Gavish M, Bachman I, Shoukrun R, et al. Enigma of the peripheral benzodiazepine receptor. Pharmacol Rev 1999;51:629–650. [PubMed] [Google Scholar]
- 4. Papadopoulos V. In search of the function of the peripheral‐type benzodiazepine receptor. Endocr Res 2004;30:677–684. [DOI] [PubMed] [Google Scholar]
- 5. Wilms H, Claasen J, Röhl C, et al. Involvement of benzodiazepine receptors in neuroinflammatory and neurodegenerative diseases: evidence from activated microglial cells in vitro. Neurobiol Dis 2003;14:417–424. [DOI] [PubMed] [Google Scholar]
- 6. Venneti S, Lopresti BJ, Wiley CA. The peripheral benzodiazepine receptor (Translocator protein 18 kDa) in microglia: from pathology to imaging. Prog Neurogibol 2006;80:308–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cagnin A, Brooks DJ, Kennedy AM, et al. In‐vivo measurement of activated microglia in dementia. Lancet 2001;358:461–467. [DOI] [PubMed] [Google Scholar]
- 8. Edison P, Archer HA, Gerhard A, et al. Microglia, amyloid, and cognition in Alzheimer's disease: an [11C](R)PK11195‐PET and [11C]PIB‐PET study. Neurobiol Dis 2008;32:412–419. [DOI] [PubMed] [Google Scholar]
- 9. Groom GN, Junck L, Foster NL, et al. PET of peripheral benzodiazepine binding sites in the microgliosis of Alzheimer's disease. J Nucl Med 1995;36:2207–2210. [PubMed] [Google Scholar]
- 10. Yasuno F, Ota M, Kosaka J, et al. Increased binding of peripheral benzodiazepine receptor in Alzheimer's disease measured by positron emission tomography with [11C]DAA1106. Biol Psychiat 2008;64:835–841. [DOI] [PubMed] [Google Scholar]
- 11. Gulyás B, Vas Á, Tóth M, et al. Age and disease related changes in the translocator protein (TSPO) system in the human brain: positron emission tomography measurements with [11C]vinpocetine. NeuroImage 2011;56:1111–1121. [DOI] [PubMed] [Google Scholar]
- 12. Kreisl WC, Henter ID, Innis RB. Imaging translocator protein as a biomarker of neuroinflammation in dementia. Adv Pharmacol 2018;82:163–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kreisl WC. Discerning the relationship between microglial activation and Alzheimer's disease. Brain 2017;140:1825–1828. [DOI] [PubMed] [Google Scholar]
- 14. Kreisl WC, Lawrence R, Page E, et al. (11)C‐PBR28 PET detects translocator protein in a patient with astrocytoma and Alzheimer disease. Neurology 2017;88:1001–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kreisl WC, Lyoo CH, Liow JS, et al. Distinct patterns of increased translocator protein in posterior cortical atrophy and amnestic Alzheimer's disease. Neurobiol Aging 2017;51:132–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Venneti S, Wang G, Nguyen J, Wiley CA. The positron emission tomography ligand DAA1106 binds with high affinity to activated microglia in human neurological disorders. J Neuropathol Exp Neurol 2008;67:1001–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Venneti S, Wiley CA, Kofler J. Imaging microglial activation during neuroinflammation and Alzheimer's disease. J Neuroimmune Pharmacol 2009;4:227–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gulyás B, Makkai B, Kása P, et al. A comparative autoradiography study in post mortem whole hemisphere human brain slices taken from Alzheimer patients and age‐matched controls using two radiolabelled DAA1106 analogues with high affinity to the peripheral benzodiazepine receptor (PBR) system. Neurochem Int 2009;54:28–36. [DOI] [PubMed] [Google Scholar]
- 19. Meda L, Cassatella MA, Szendrei GI, et al. Activation of microglial cells by beta‐amyloid protein and interferon‐gamma. Nature 1995;374:647–650. [DOI] [PubMed] [Google Scholar]
- 20. D'Andrea MR, Cole GM, Ard MD. The microglial phagocytic role with specific plaque types in the Alzheimer disease brain. Neurobiol Aging 2004;25:675–683. [DOI] [PubMed] [Google Scholar]
- 21. Masliah E, Hansen L, Adame A, et al. Abeta vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology 2005;64:129–131. [DOI] [PubMed] [Google Scholar]
- 22. Flanary B. The role of microglial cellular senescence in the aging and Alzheimer diseased brain. Rejuvenation Res 2005;8:82–85. [DOI] [PubMed] [Google Scholar]
- 23. Luo XG, Ding JQ, Chen SD. Microglia in the aging brain: relevance to neurodegeneration. Mol Neurodegener 2010;5:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Miller KR, Streit WJ. The effects of aging, injury and disease on microglial function: a case for cellular senescence. Neuron Glia Biol 2007;3:245–253. [DOI] [PubMed] [Google Scholar]
- 25. Streit WJ. Microglia as neuroprotective, immunocompetent cells of the CNS. Glia 2002;40:133–139. [DOI] [PubMed] [Google Scholar]
- 26. Streit WJ. Microglia and neuroprotection: implications for Alzheimer's disease. Brain Res Brain Res Rev 2005;48:234–239. [DOI] [PubMed] [Google Scholar]
- 27. Streit WJ. Microglial senescence: does the brain's immune system have an expiration date? Trends Neurosci 2006;29:506–510. [DOI] [PubMed] [Google Scholar]
- 28. Streit WJ, Xue QS. Life and death of microglia. J Neuroimmune Pharmacol 2009;4:371–379. [DOI] [PubMed] [Google Scholar]
- 29. Flanary BE, Sammons NW, Nguyen C, et al. Evidence that aging and amyloid promote microglial cell senescence. Rejuvenation Res 2007;10:61–74. [DOI] [PubMed] [Google Scholar]
- 30. Streit WJ, Braak H, Xue QS, Bechmann I. Dystrophic (senescent) rather than activated microglial cells are associated with tau pathology and likely precede neurodegeneration in Alzheimer's disease. Acta Neuropathol 2009;118:475–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Flanary BE, Streit WJ. Telomeres shorten with age in rat cerebellum and cortex in vivo. J Anti Aging Med 2003;6:299–308. [DOI] [PubMed] [Google Scholar]
- 32. Flanary BE, Streit WJ. Progressive telomere shortening occurs in cultured rat microglia, but not astrocytes. Glia 2004;45:75–88. [DOI] [PubMed] [Google Scholar]
- 33. Braak H, Alafuzoff I, Arzberger T, et al. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol 2006;112:389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Braak H, Braak E. Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathol 1991;82:239–259. [DOI] [PubMed] [Google Scholar]
- 35. Sun J, Cairns NJ, Perlmutter JS, et al. Regulation of dopamine D(3) receptor in the striatal regions and substantia nigra in diffuse Lewy body disease. Neuroscience 2013;248:112–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Xu J, Hassanzadeh B, Chu W, et al. [3H]4‐(dimethylamino)‐N‐(4‐(4‐(2‐methoxyphenyl)piperazin‐1‐yl) butyl)benzamide: a selective radioligand for dopamine D(3) receptors. II. Quantitative analysis of dopamine D(3) and D(2) receptor density ratio in the caudate‐putamen. Synapse 2010;64:449–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chaney A, Cropper HC, Johnson EM, et al. (11)C‐DPA‐713 versus (18)F‐GE‐180: a preclinical comparison of translocator protein 18 kDa PET tracers to visualize acute and chronic neuroinflammation in a mouse model of ischemic stroke. J Nucl Med 2019;60:122–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chaney A, Williams SR, Boutin H. In vivo molecular imaging of neuroinflammation in Alzheimer's disease. J Neurochem 2019;149:438–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Keller T, López‐Picón FR, Krzyczmonik A, et al. [(18)F]F‐DPA for the detection of activated microglia in a mouse model of Alzheimer's disease. Nucl Med Biol 2018;67:1–9. [DOI] [PubMed] [Google Scholar]
- 40. Narayanaswami V, Dahl K, Bernard‐Gauthier V, et al. Emerging PET radiotracers and targets for imaging of neuroinflammation in neurodegenerative diseases: outlook beyond TSPO. Mol Imaging 2018;17:1536012118792317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zanotti‐Fregonara P, Veronese M, Pascual B, et al. The validity of (18)F‐GE180 as a TSPO imaging agent. Eur J Nucl Med Mol Imaging 2019;46:1205–1207. [DOI] [PubMed] [Google Scholar]
- 42. Imaizumi M, Briard E, Zoghbi SS, et al. Brain and whole‐body imaging in nonhuman primates of [11C]PBR28, a promising PET radioligand for peripheral benzodiazepine receptors. NeuroImage 2008;39:1289–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Frankle WG, Narendran R, Wood AT, et al. Brain translocator protein occupancy by ONO‐2952 in healthy adults: a phase 1 PET study using [(11) C]PBR28. Synapse 2017;71:e21970. [DOI] [PubMed] [Google Scholar]
- 44. Nair A, Veronese M, Xu X, et al. Test‐retest analysis of a non‐invasive method of quantifying [(11)C]‐PBR28 binding in Alzheimer's disease. EJNMMI Res 2016;6:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Owen DR, Guo Q, Kalk NJ, et al. Determination of [(11)C]PBR28 binding potential in vivo: a first human TSPO blocking study. J Cereb Blood Flow Metab 2014;34:989–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Plavén‐Sigray P, Schain M, Zanderigo F, et al. Accuracy and reliability of [(11)C]PBR28 specific binding estimated without the use of a reference region. NeuroImage 2019;188:102–110. [DOI] [PubMed] [Google Scholar]
- 47. Sandiego CM, Gallezot JD, Pittman B, et al. Imaging robust microglial activation after lipopolysaccharide administration in humans with PET. Proc Natl Acad Sci USA 2015;112:12468–12473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Imaizumi M, Kim HJ, Zoghbi SS, et al. PET imaging with [11C]PBR28 can localize and quantify upregulated peripheral benzodiazepine receptors associated with cerebral ischemia in rat. Neurosci Lett 2007;411:200–205. [DOI] [PubMed] [Google Scholar]
- 49. Oh U, Fujita M, Ikonomidou VN, et al. Translocator protein PET imaging for glial activation in multiple sclerosis. J Neuroimmune Pharmacol 2011;6:354–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kreisl WC, Fujita M, Fujimura Y, et al. Comparison of [(11)C]‐(R)‐PK 11195 and [(11)C]PBR28, two radioligands for translocator protein (18 kDa) in human and monkey: implications for positron emission tomographic imaging of this inflammation biomarker. NeuroImage 2010;49:2924–2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Owen DR, Gunn RN, Rabiner EA, et al. Mixed‐affinity binding in humans with 18‐kDa translocator protein ligands. J Nucl Med 2011;52:24–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Owen DR, Howell OW, Tang SP, et al. Two binding sites for [3H]PBR28 in human brain: implications for TSPO PET imaging of neuroinflammation. J Cereb Blood Flow Metab 2010;30:1608–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Overmyer M, Helisalmi S, Soininen H, et al. Reactive microglia in aging and dementia: an immunohistochemical study of postmortem human brain tissue. Acta Neuropathol 1999;97:383–392. [DOI] [PubMed] [Google Scholar]
- 54. McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA‐DR in the substantia nigra of Parkinson's and Alzheimer's disease brains. Neurology 1988;38:1285–1291. [DOI] [PubMed] [Google Scholar]
- 55. Kazee AM, Cox C, Richfield EK. Substantia nigra lesions in Alzheimer disease and normal aging. Alzheimer Dis Assoc Disord 1995;9:61–67. [DOI] [PubMed] [Google Scholar]
- 56. Joachim CL, Morris JH, Selkoe DJ. Clinically diagnosed Alzheimer's disease: autopsy results in 150 cases. Ann Neurol 1988;24:50–56. [DOI] [PubMed] [Google Scholar]
- 57. Mitsuyama Y, Fukunaga H, Yamashita M. Alzheimer's disease with widespread presence of Lewy bodies. Folia Psychiatrica et Neurologica Japonica 1984;38:81–88. [DOI] [PubMed] [Google Scholar]
- 58. Reyes MG, Faraldi F, Chandran R, et al. Histopathology of the substantia nigra in Alzheimer's disease. Panminerva Med 1996;38:8–14. [PubMed] [Google Scholar]
- 59. Rinne JO, Rummukainen J, Paljärvi L, et al. Neuronal loss in the substantia nigra in patients with Alzheimer's disease and Parkinson's disease in relation to extrapyramidal symptoms and dementia. Prog Clin Biol Res 1989;317:325–332. [PubMed] [Google Scholar]
- 60. Rosenblum WI, Ghatak NR. Lewy bodies in the presence of Alzheimer's disease. Arch Neurol 1979;36:170–171. [DOI] [PubMed] [Google Scholar]
- 61. Schneider JA, Bienias JL, Gilley DW, et al. Improved detection of substantia nigra pathology in Alzheimer's disease. J Histochem Cytochem 2002;50:99–106. [DOI] [PubMed] [Google Scholar]
- 62. Tabaton M, Schenone A, Romagnoli P, Mancardi GL. A quantitative and ultrastructural study of substantia nigra and nucleus centralis superior in Alzheimer's disease. Acta Neuropathol 1985;68:218–223. [DOI] [PubMed] [Google Scholar]
- 63. Everett H, Barry M, Sun X, et al. The myxoma poxvirus protein, M11L, prevents apoptosis by direct interaction with the mitochondrial permeability transition pore. J Exp Med 2002;196:1127–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Johnston C, Jiang W, Chu T, Levine B. Identification of genes involved in the host response to neurovirulent alphavirus infection. J Virol 2001;75:10431–10445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Rey C, Mauduit C, Naureils O, Benahmed M. Louisot P, Gasnier F. Up‐regulation of mitochondrial peripheral benzodiazepine receptor expression by tumor necrosis factor alpha in testicular leydig cells. Possible involvement in cell survival. Biochem Pharmacol 2000;60:1639–1646. [DOI] [PubMed] [Google Scholar]
- 66. Carayon P, Portier M, Dussossoy D, et al. Involvement of peripheral benzodiazepine receptors in the protection of hematopoietic cells against oxygen radical damage. Blood 1996;87:3170–3178. [PubMed] [Google Scholar]
- 67. Di Carlo M, Giacomazza D, Picone P, et al. Are oxidative stress and mitochondrial dysfunction the key players in the neurodegenerative diseases? Free Radical Res 2012. [DOI] [PubMed] [Google Scholar]
- 68. Keating DJ. Mitochondrial dysfunction, oxidative stress, regulation of exocytosis and their relevance to neurodegenerative diseases. J Neurochem 2008;104:298–305. [DOI] [PubMed] [Google Scholar]
- 69. Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006;443:787–795. [DOI] [PubMed] [Google Scholar]
- 70. Trushina E, McMurray CT. Oxidative stress and mitochondrial dysfunction in neurodegenerative diseases. Neuroscience 2007;145:1233–1248. [DOI] [PubMed] [Google Scholar]
- 71. Streit WJ, Xue QS. Microglia in dementia with Lewy bodies. Brain Behav Immun 2016;55:191–201. [DOI] [PubMed] [Google Scholar]