

Abstract
Prenatal inflammation is a risk factor for necrotizing enterocolitis (NEC), and it increases intestinal injury in a rat NEC model. We previously showed that maldevelopment of the intestinal microvasculature and lack of vascular endothelial growth factor (VEGF) receptor 2 (VEGFR2) signaling play a role in experimental NEC. However, whether prenatal inflammation affects the intestinal microvasculature remains unknown. In this study, mouse dams were injected intraperitoneally with lipopolysaccharide (LPS) or saline at embryonic day 17. Neonatal intestinal microvasculature density, endothelial cell proliferation, and intestinal VEGF-A and VEGFR2 proteins were assessed in vivo. Maternal and fetal serum TNF concentrations were measured by ELISA. The impact of TNF on the neonatal intestinal microvasculature was examined in vitro and in vivo, and we determined whether prenatal LPS injection exacerbates experimental NEC via TNF. Here we found that prenatal LPS injection significantly decreased intestinal microvascular density, endothelial cell proliferation, and VEGF and VEGFR2 protein expression in neonatal mice. Prenatal LPS injection increased maternal and fetal serum levels of TNF. TNF decreased VEGFR2 protein in vitro in neonatal endothelial cells. Postnatal TNF administration in vivo decreased intestinal microvasculature density, endothelial cell proliferation, and VEGF and VEGFR2 protein expression and increased the incidence of severe NEC. These effects were ameliorated by stabilizing hypoxia-inducible factor-1α, the master regulator of VEGF. Furthermore, prenatal LPS injection significantly increased the incidence of severe NEC in our model, and the effect was dependent on endogenous TNF. Our study suggests that prenatal inflammation increases the susceptibility to NEC, downregulates intestinal VEGFR2 signaling, and affects perinatal intestinal microvascular development via a TNF mechanism.
NEW & NOTEWORTHY This report provides new evidence that maternal inflammation decreases neonatal intestinal VEGF receptor 2 signaling and endothelial cell proliferation, impairs intestinal microvascular development, and predisposes neonatal mouse pups to necrotizing enterocolitis (NEC) through inflammatory cytokines such as TNF. Our data suggest that alteration of intestinal microvascular development may be a key mechanism by which premature infants exposed to prenatal inflammation are at risk for NEC and preserving the VEGF/VEGF receptor 2 signaling pathway may help prevent NEC development.
Keywords: endothelial cells, inflammation, intestinal microvasculature, necrotizing enterocolitis, neonate
INTRODUCTION
Necrotizing enterocolitis (NEC) is the most common gastrointestinal emergency affecting premature infants and, despite significant advances in the field of neonatology over the past 30 years, remains associated with high morbidity and mortality. Besides prematurity, multiple postnatal factors have been identified to contribute to NEC, such as an abnormal bacterial colonization and formula feeding (8, 20). Recently, prenatal events have drawn the attention of clinicians and researchers as predisposing factors for NEC. Conditions that restrict blood oxygen delivery and nutrition to the fetus (e.g., maternal hypertension, preeclampsia, and placental diseases), chorioamnionitis, and maternal infection have been shown to be independently associated with NEC (4, 12, 23, 28). Chorioamnionitis is an acute inflammation of the membranes and chorion of the placenta, typically due to bacterial infection (33). It is a leading cause of prematurity and can be detected in as many as 40–70% of preterm births with premature rupture of membranes or spontaneous preterm labor (33). Interestingly clinical chorioamnionitis is commonly found without amniotic infection (26). Indeed, in 40% of the cases, no bacteria could be identified in the amniotic cavity using either cultivation or molecular microbiology techniques (26). Prenatal administration of Escherichia coli lipopolysaccharide (LPS) to dams has been used to model prenatal inflammation in rats (9, 13) and was found to increase the incidence of experimental NEC in rat pups (13). However, the mechanism by which prenatal inflammation predisposes to NEC remains unclear.
Vascular endothelial growth factor A (VEGF) is a major regulator of angiogenesis. Intestinal VEGF expression is decreased in mouse experimental NEC and in human NEC (27). We also found that inhibiting the kinase activity of VEGF receptor 2 (VEGFR2), the main receptor of VEGF, led to decreased villous endothelial cell proliferation and intestinal microvascular density in neonatal mice (36). Furthermore, VEGFR2 inhibition increased mortality and the incidence of severe NEC (36). In contrast, we showed that pharmacologically preventing the degradation of hypoxia-inducible factor-1α (HIF-1α) increased intestinal VEGF protein expression, attenuated NEC-induced decrease in intestinal villous endothelial cell proliferation, and protected against NEC (6). These studies suggest that lack of VEGFR2 signaling and maldevelopment of the intestinal microvasculature may play an important role in NEC.
Here, we hypothesize that prenatal inflammation impairs the development of the intestinal microvasculature, thus predisposing to NEC. In this study, we first investigated whether prenatal inflammation induced by administering LPS to dams on embryonic day (E) 17 affects intestinal villous microvascular density, permeability, and endothelial cell proliferation in the small intestine of neonatal mice. Second, we determined whether prenatal inflammation altered intestinal VEGF and VEGFR2 protein expression. Third, to assess the mechanism by which prenatal inflammation affected intestinal VEGFR2, we measured maternal and fetal TNF levels after maternal LPS administration. We then examined whether TNF directly affected VEGFR2 protein in isolated small intestinal endothelial cells in vitro and whether administration of TNF to neonatal, dam-fed pups affected the small intestinal microvasculature development, endothelial cell proliferation, and VEGF and VEGFR2 protein expression. Furthermore, we examined whether TNF increased the incidence of NEC in our neonatal mouse NEC model and whether detrimental effects of TNF could be ameliorated by dimethyloxalylglycine (DMOG), a HIF-stabilizing agent that promotes VEGF protein expression. Finally, we determined whether prenatal LPS affected the mortality and incidence of severe intestinal injury in our model and whether this was prevented by anti-TNF antibody administration.
MATERIALS AND METHODS
Materials.
C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, ME). All animal breeding and procedures were approved by the Institutional Animal Care and Use Committee of Northwestern University. Rabbit derived anti-CD31 (cat. no. ab-28364) and rat anti-BrdU (cat. no. ab-6326) antibodies (Ab) were purchased from Abcam (Cambridge, MA) and anti-VEGF Ab that recognizes VEGF-164 and 121 isoforms (cat. no. sc-7269) was obtained from Santa Cruz Biotechnology (Santa Cruz, CA). VEGFR2 (cat. no. 2479) Ab was purchased from Cell Signaling Technology (Danvers, MA). Rat antimouse endomucin (cat. no. 50-5851-80), rabbit anti-Ki-67 (MA5-14520), goat antirabbit Ab (cat. nos. A-11037 and A-11034), goat antirat Ab (cat. no. A-11007), and collagenase IV and attachment factor for endothelial cell culture were obtained from Life Technologies (Grand Island, NY). Endothelial cell culture media VascuLife VEGF Endothelial Medium Complete Kit was obtained from Lifeline Cell Technology (Frederick, MD). Evans blue, albumin, and formamide were purchased from Sigma (St. Louis, MO). DMOG was purchased from Selleck Chemicals (Houston, TX). Recombinant mouse TNF was purchased from PeproTech (Rocky Hill, NJ). Antimouse TNF Ab and rat IgG1 isotype control Ab were obtained from Bio X Cell (West Lebanon, NH).
Animal experiments.
Pregnant dams were injected with 50 μg/kg ip LPS or normal saline (controls) once at E17. Pups were allowed to be delivered naturally, and gestation length and litter size were determined. At 24 h of age, pups were submitted to an experimental NEC protocol that we have established (32). Briefly, naturally delivered C57BL/6 newborn pups were placed in a humidified neonatal incubator at 33°C and were inoculated with 108 colony-forming units of a standardized adult commensal bacteria mixture within 24 h of birth. Pups were then gavaged with 30 μl of Esbilac (PetAg, Hampshire, IL) formula (33 g diluted in 100 ml of water) every 3 h and submitted to brief episodes of asphyxia (100% N2 for 60 s) and cold stress (10 min at 4°C) twice daily. Pups were closely monitored and euthanized by decapitation at 72 h or when showing signs of distress such as severe abdominal distension, respiratory distress, or lethargy. The small intestines were collected, fixed in buffered formalin, and stained by hematoxylin-eosin for histological examination. Microscopic injury was graded as described previously (32) by two independent investigators in a blinded fashion. Severe NEC injury was defined as histological score ≥2. Pups found dead while not under direct observation were excluded from histological analysis. Alternatively, at 2, 4, and 6 h after LPS injection, serum was collected from dams by tail sampling and cesarean section was performed immediately under anesthesia (65 mg/kg ip pentobarbital sodium) before fetal serum was collected by decapitation.
To examine whether TNF promotes NEC development via compromising the VEGF and VEGFR2 pathway, day (D) 0 neonatal mice were injected with 40 mg/kg ip of DMOG (a pharmacological agent that prevents the degradation of HIF-1α) or with vehicle control and injected 16 h later with 200 μg/kg ip of TNF (or vehicle control) on D1. Two hours later, pups were submitted to the NEC model. Intestinal tissues were collected (as explained above) to determine the incidence of severe NEC injury. In some experiments, the intestinal microvasculature was assessed at 48 h after TNF administration (as explained below) or intestinal tissues were collected at 24 h after TNF treatment for endothelial cell proliferation and VEGF/VEGFR2 protein assessment.
To determine whether the negative effect of prenatal LPS on NEC was mediated by TNF, 600 μg of antimouse TNF Ab or isotype control Ab was injected intraperitoneally into pregnant dams 2 h before LPS injection at E17. After their birth, pups were submitted to the NEC model. Animal survival and the incidence of severe NEC (≥2) were determined.
Intestinal microvasculature imaging.
Pups were anesthetized with 65 mg/kg ip pentobarbital sodium, and 500 μl of 40 μg/ml of Alexa Fluor 647-conjugated wheat germ agglutinin (WGA) was injected by intracardiac perfusion. The procedure was performed under microscope and successful perfusion was confirmed by blood vessel blanching and appropriate filling with dye solution. Five minutes later, whole intestinal tissues were collected and fixed in formalin. Small intestinal tissues were cut open, washed in PBS twice, and covered with mounting media including DAPI. Vascular image Z-stacks at 5.81-μm intervals were captured by a Zeiss 880 Confocal Laser Scanning Microscope. Z-stacks were then projected by Zen 2012 software. Vessel area percentage was determined as an index of vascular density using Photoshop software, as previously described (36). Typically, up to three randomly chosen fields (10×) were studied in each sample, and the mean value was calculated.
Intestinal vascular permeability assay.
Pregnant dams were injected intraperitoneally with 50 μg/kg LPS or normal saline (controls) once at E17, and pups were allowed to deliver naturally. At around 12 h of life, pups were anesthetized with pentobarbital sodium (65 mg/kg ip) and injected retro-orbitally with 10 μl of Evans blue albumin (EBA) solution (0.5% Evans blue/40 mg/ml BSA). One hour later, intestinal vascular permeability was measured as previously published (25). Normal saline (500 μl) was infused intracardiacally to purge the dye accumulated into the circulation. Small intestinal tissues were collected immediately after perfusion, followed by careful dissection and removal of the pancreatic and surrounding connective tissues. Small intestinal tissue lysates were prepared. EBA dye that had diffused into the intestinal tissues was extracted with formamide and quantified as an index of intestine vascular permeability by spectrophotometry. Results were compared with serial dilutions of known EBA concentrations and normalized to tissue weight.
In vivo endothelial cell proliferation.
Pups were injected intraperitoneally with 0.3 mg of BrdU 4 h before euthanasia, and small intestinal tissues were collected and fixed in 10% formalin. Intestinal tissue sections were stained with Ab against BrdU and CD31. In each group, proliferating villous endothelial cells, which were positive for CD31 and BrdU, were counted in 3–4 fields/sample from 8 to 10 pups/group (obtained from 3 litters). Alternatively, intestinal tissue slides were stained for Ki-67 and endomucin to identify proliferating endothelial cells.
Western blot analysis.
Small intestines were snap frozen and tissue lysates were prepared by homogenization in lysis buffer (10 mM Tris·Cl, pH 7.6, 150 mM NaCl, 5 mM EDTA, 1 mM PMSF, 1 mM DTT, 1.0% Nonidet P-40, 1% SDS, 0.5% sodium deoxycholate, and Complete Mini Tablet Protease Inhibitor Cocktail from Roche). Protein concentration was determined with the Bradford method. Protein (20–50 μg) was separated by 4–20% gradient SDS-PAGE gel electrophoresis and transferred to nitrocellulose membranes, which were then blocked in 5% milk/Tris-buffered saline-Tween 20. Membranes were incubated at 4°C overnight with primary Ab against VEGF (recognizing isoforms 164 and 120) (1:100) and VEGFR2 (1:1,000), then with horseradish peroxidase-conjugated secondary Ab for 1 h at room temperature. Membrane-bound secondary Ab was detected using the standard Pierce ECL method. β-Actin was detected as endogenous control using the same membrane by reprobing with specific Ab (1:5,000) and detected as above. Detected proteins were quantified by densitometry and normalized to β-actin. The ratio of the obtained values to the control group average is presented.
Immunofluorescence staining.
Formalin fixed, paraffin-embedded tissue sections of small intestinal tissues were prepared and subjected to a pH 8.5 EDTA antigen retrieval procedure for 20 min. After being treated with Triton X-100, sections were blocked with 10% normal goat serum for 1 h at room temperature. Sections were incubated with primary Ab at 4°C overnight, followed by incubation with fluorescent conjugated secondary Ab at room temperature for 1 h. The specific dilution for each Ab was the following: Anti-CD31 (1:50), anti-BrdU (1:50), goat antirabbit secondary antibody (1:500), and goat antirat secondary Ab (1:1,000). Images were captured on a Leica DMR-HC upright microscope.
Small intestinal endothelial cell isolation and culture.
Small intestinal tissues were collected from 1- to 3-day-old pups and cut open into 1–2 cm pieces under a dissection microscope. Epithelial cells were removed by incubation with dissociation buffer (PBS: 2% FBS, 1.0 mM DTT, 5 mM EDTA, 15 mM HEPES) for 30 min at 37°C. Single lamina propria cells were obtained by digesting minced epithelial-free small intestinal tissue pieces with collagenase IV solution (DMEM: 1 mg/ml collagenase, 5 mM CaCl2, 15 mM HEPES) at 37°C for 45–60 min. CD45+ cells were first depleted by incubation with anti-CD45 Ab-coated microbeads. The recovered cells were then incubated with anti-CD31 Ab-coated microbeads, and endothelial cells (CD45-CD31+ cells) were purified by magnetic column selection. Isolated, small intestinal endothelial cells (3 × 105) were cultured in 6-well plates coated with attachment factor in endothelial cell culture media containing 5 ng/ml VEGF, which was prepared according to the manufacturer’s instructions. When reaching 90% confluence (around day 5–6 of culture), neonatal intestinal endothelial cells were incubated with increasing doses of TNF for 12 h. Endothelial cell lysates were prepared in 2× Laemmeli buffer and boiled for 5 min before Western blot analysis.
Enzyme-linked immunosorbent assay.
Serum samples were diluted (1:50) and serum TNF detected by ELISA according to the manufacturer’s instructions (Life Technologies). Samples were allowed to bind to anti-TNF antibody coated on a 96-well plate for 2 h at room temperature. After being washed, the plate was incubated with biotin-conjugated detecting antibody for 1 h and subsequent avidin-conjugated horseradish peroxidase for 30 min. Absorbance was read at 450 nm and corrected at 570 nm after substrate tetramethylbenzidine was added to the plate. Concentration was calculated based on serial dilutions of known standard.
Statistical analysis.
Two-sided Student’s t-test was used for comparison between two groups, and one-way ANOVA was used for comparison of three or more groups. Results are expressed as means ± SE. To evaluate differences in the incidence of severe NEC (Grade ≥2), χ2 analysis was used. Animal survival data were analyzed by log-rank test and median survival determined. Differences were considered statistically significant when P ≤ 0.05.
RESULTS
Prenatal inflammation affects intestinal microvasculature development.
Prenatal inflammation was induced by injecting pregnant dams with 50 μg/kg ip LPS at day 17 of pregnancy. This was the highest dose that did not increase the risk of fetal resorption/abortion/preterm births in our hand and others (11, 34). In a preliminary study, this dose did not significantly affect litter size (6.4 ± 1.1 vs. 7.3 ± 0.5 pups in the LPS and control groups, respectively; not significant), but decreased birth weight [1.34 ± 0.02 vs. 1.46 ± 0.04 g in LPS and control groups, respectively (LPS group: n = 32, control group: n = 18; P < 0.01)]. To determine the effect of prenatal LPS on the neonatal intestinal mucosal microvasculature development, D1 newborn pups born from dams treated at E17 with 50 μg/kg of LPS or saline (controls) were infused intracardiacally with fluorescently-labeled wheat germ agglutinin and the small intestinal microvasculature was examined. In the intestinal submucosa of control pups, we found a complex capillary network with flourishing branches and sprouts between venules and arterioles. However, in pups born from dams treated with LPS, this organized network was markedly reduced (Fig. 1A). Also, the density of the submucosal vascular network was significantly decreased in the prenatal inflammation group compared with controls with a mean vascular area percentage of 34.00 ± 2.98 in pups born from dams treated with LPS compared with 58.30 ± 3.32 in the control group (Fig. 1B).
Fig. 1.

Prenatal lipopolysaccharide (LPS) affects the perinatal development of the intestinal microvasculature. Pregnant dams were injected with lipopolysaccharide (LPS) or normal saline (controls) at embryonic day 17, and pups were allowed to be delivered naturally. Alexa Fluor 647-labeled wheat germ agglutinin was injected by intracardiac perfusion at day 1 of life. The intestinal submucosal (A) and villous (C) microvasculature was imaged at ×10 magnification on formalin-fixed, whole mount tissues. Representative images are presented. A: ×10 images (top); locally magnified images (bottom). Scale bars = 200 μm. B: intestinal submucosal microvascular area density was assessed using Photoshop software and vessel area percentage is presented (***P < 0.001, analyzed using two-sided Student’s t-test). Pups were examined (2–3 per litter) and 3 litters were studied in each group. C: luminal dye leakage noted in pups prenatally treated with LPS (arrowheads).
Prenatal inflammation increases neonatal intestinal microvascular permeability.
Because we noted luminal dye leakage in villi of pups prenatally treated with LPS but not in control pups (Fig. 1C, arrowheads), we tested the hypothesis that prenatal LPS treatment increases intestinal microvascular permeability. To this end, EBA was administered intravascularly by retro-orbital injection, and the small intestinal tissue was assessed 1 h later for dye leakage. We found the mean EBA concentration of the intestinal tissues from pups that were administered LPS prenatally to be significantly higher than in the controls (103.90 ± 9.48 μg/g vs. 69.28 ± 10.65 μg/g) (Fig. 2).
Fig. 2.
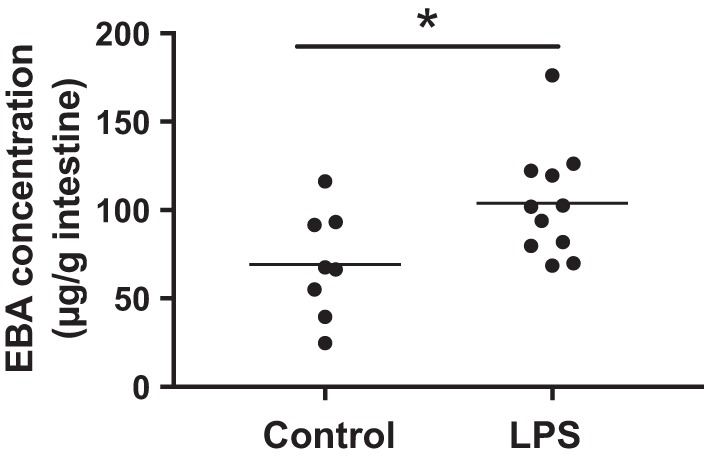
Prenatal lipopolysaccharide (LPS) increases neonatal intestinal microvascular permeability. To measure intestinal microvascular permeability, pups were administered 10 μl of Evans blue albumin (EBA) solution (0.5% Evans blue/40 mg/ml BSA) retro-orbitally, and EBA that had leaked into the small intestinal tissue was quantified 1 h later (n = 11 in pups prenatally treated with LPS; n = 8 in the control group). *P < 0.05, analyzed with two-sided Student’s t-test.
Prenatal inflammation decreases intestinal villous endothelial cell proliferation in neonatal pups.
To determine whether prenatal inflammation affects small intestinal endothelial cell proliferation in neonatal pups, less than 24-h-old pups from dams prenatally injected with LPS or controls at E17 were injected intraperitoneally with BrdU and small intestinal tissues were collected 4 h later. Sections were stained for BrdU and the endothelial cell marker CD31, and intestinal villi were assessed for proliferating endothelial cells (Fig. 3A). We found a marked decrease in the number of proliferating endothelial cells in the intestinal villi of pups from dams that were injected with LPS [5.60 ± 0.76 BrdU+ CD31+ cells per field (×20 magnification)] compared with controls (13.93 ± 0.57) (Fig. 3B).
Fig. 3.

Prenatal lipopolysaccharide (LPS) decreases intestinal villous endothelial cell proliferation. Pregnant dams were intraperitoneally injected with LPS or normal saline at embryonic day 17. Pups were allowed to deliver naturally. Pups that were 24 h old were injected intraperitoneally with BrdU 4 h before euthanasia and tissue collection. Small intestine tissue sections were co-stained with antibodies against BrdU (red-purple) and CD31 (green) to identify proliferating endothelial cells. A: images from pups whose dams were administered prenatal LPS (right) vs. saline (left) (scale bars = 50 μm). B: proliferating endothelial cells of the villi were counted at ×20 magnification in 3–4 fields per sample (n = 15 in each group). ****P < 0.0001. Data represent three independent experiments and were analyzed using two-sided Student’s t-test.
Prenatal inflammation decreases intestinal VEGF and VEGFR2 protein expression in vivo.
When small intestines of 24-h-old pups were assessed for VEGF and VEGFR2 expression by Western blot analysis, we found that pups whose dams received antenatal LPS had significantly lower intestinal VEGF164 isoform (Fig. 4, A and B), no changes of the VEGF120 isoform (data not shown), and markedly decreased VEGFR2 (Fig. 4, A and B) expression compared with controls.
Fig. 4.

Prenatal lipopolysaccharide (LPS) decreases VEGF and VEGF receptor 2 (VEGFR2) protein expression in the small intestine of neonatal mice. Pregnant dams were intraperitoneally injected with LPS or normal saline at embryonic day 17. Pups were allowed to deliver naturally, and small intestines were collected at 24 h. A: intestinal VEGF-164 and VEGFR2 proteins were accessed by Western blot. B: VEGF and VEGFR2 band intensities were quantified by densitometry and normalized to endogenous β-actin. ****P < 0.0001. Data represent three independent experiments and were analyzed using two-sided Student’s t-test.
Maternal LPS increases serum TNF concentration in the fetus.
To examine whether maternal LPS injection triggered a fetal inflammatory response, we measured maternal and fetal TNF 2, 4, and 6 h after maternal LPS administration. We found that prenatal maternal LPS significantly increased serum TNF concentration in both dams and fetuses, which peaked at 2 h after injection and subsided by 6 h (Fig. 5A).
Fig. 5.

Prenatal administration of lipopolysaccharide (LPS) increases maternal and fetal serum TNF. TNF decreases VEGF receptor 2 (VEGFR2) protein expression in neonatal intestinal endothelial cells in vitro. A: cesarean section was performed at indicated time points after LPS injection to the dams at embryonic day 17, and serum was immediately obtained from dams and fetuses. Serum levels of TNF from dams (left) and fetuses (right) were measured by ELISA; ****P < 0.0001. Data were analyzed with one-way ANOVA test and represent three experiments with a total of 6–8 dams per group and 16–30 pups in each group (serum was pooled from 2 to 3 pups). B: endothelial cells isolated from small intestines were cultured until 90% confluence and treated with indicated concentrations of TNF for 12 h. VEGFR2 protein was assessed by Western blot (left). Band densities for each sample were normalized to β-actin and a ratio to the mean control value from endothelial cells treated with culture medium only was calculated (right). *P < 0.05, **P < 0.01. Data represent three independent experiments combined and were analyzed by one-way ANOVA.
TNF decreases expression of VEGFR2 in neonatal intestinal endothelial cells in vitro: TNF affects neonatal intestinal microvasculature development in vivo, which is ameliorated by a HIF-1α stabilizing agent.
To investigate whether TNF directly affects VEGFR2 expression of neonatal small intestinal endothelial cells in vitro, endothelial cells were isolated from day 1 small intestines and incubated with different doses of TNF for 12 h. We found that TNF induced a dose-dependent decrease in endothelial cell VEGFR2 protein (Fig. 5B).
To determine whether TNF affects the neonatal intestinal microvasculature in vivo, D2 pups were injected intraperitoneally with 200 μg/kg of recombinant TNF or PBS (controls) and the intestinal microvasculature density was assessed 24 h later, as explained above. We found that TNF markedly reduced the branching and sprouting of the intestinal submucosal microvessels (Fig. 6A, left) and significantly decreased the vascular density of the network compared with control (mean vascular area percentage = 25.93 ± 2.82 in pups treated with TNF vs. 45.48 ± 2.35 in the control group; Fig. 6A, right). The microvasculature density was preserved when pups were treated with DMOG, a pharmacological agent that prevents the degradation of HIF-1α [38.02 ± 2.64 in pups treated with DMOG+TNF group vs. 25.93 ± 2.82 in pups treated with TNF group (Fig. 6A)].
Fig. 6.

TNF decreases small intestinal microvasculature density, endothelial cell proliferation, and VEGF and VEGF receptor 2 (VEGFR2) protein expression in vivo, which are prevented by dimethyloxalylglycine (DMOG) administration. DMOG attenuates TNF detrimental effects on intestinal injury and mortality in experimental necrotizing enterocolitis (NEC). Pups were administered DMOG or vehicle only (control) intraperitoneally 16 h before intraperitoneal injection of TNF on day 1. A: dye was injected and intestinal tissues were collected 24 h later. Small intestinal submucosal microvasculature images (left) and quantification of microvascular density (right) are shown. Data represent 3 independent experiments from 3 litters (n = 11: control, n = 18: TNF, and n = 12: DMOG+TNF group). Data were analyzed by one-way ANOVA. Scale bar = 200 μm. Intestinal tissues were collected at day 2 (24 h after TNF treatment) and examined by immunofluorescence for endothelial cell proliferation (Ki-67+ endomucin+ cells) (B) or Western blot analysis for VEGF and VEGFR2 expression (C). Data represent 3 separated experiments from 3 litters and were analyzed by one-way ANOVA. D: pups were injected intraperitoneally with TNF or vehicle control and submitted 2 hr later to the NEC model. Animal survival (left) and tissue injury severity score (right) were assessed. Data represent three independent experiments (n = 38: TNF group, n = 31: DMOG+TNF group, n = 32: control group. Log-rank test was used for survival curve and χ2 test for histological score analysis. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. n.s., not significant.
Newborn pups treated with TNF had reduced small intestinal endothelial cell proliferation, and the effect was ameliorated by pretreatment with DMOG (Fig. 6B). Pups treated with TNF had decreased intestinal VEGF-164 and VEGFR2 protein expression, which was prevented by DMOG administration (Fig. 6C).
TNF increases the incidence of severe intestinal injury and the mortality in experimental NEC, which is prevented by DMOG administration.
TNF injection significantly lowered survival in pups submitted to the NEC protocol compared with injection of vehicle only (P < 0.01), and the effect was blocked when DMOG was administered before TNF (P < 0.05 compared with TNF group), with median survival of 27.5, 48, and 42 h in TNF, NEC only, and DMOG+TNF groups, respectively (Fig. 6D). The incidence of severe intestinal tissue injury was higher in the TNF group (22/37) compared with the NEC only group (7/31) (χ2 = 9.38, P < 0.01). We observed a trend toward a decrease in the incidence of severe intestinal injury in the DMOG group, although this was not statistically significant (10/28; χ2 = 3.60, P = 0.058) (Fig. 6D).
Prenatal inflammation increases the incidence of severe intestinal injury and the mortality in experimental NEC, which are improved by neutralizing TNF in dams.
Dams injected with LPS (50 μg/kg) or saline at E17 (14) were allowed to deliver naturally, and their pups were submitted to a NEC model, as previously described (32). We found a significant increase in mortality (median survival of 28 h in the prenatal LPS group compared with 58.5 h in the control group, Fig. 7A). Furthermore, the incidence of severe NEC (score ≥ 2) was increased in the prenatal LPS group (24/37) compared with the control group (9/24; χ2 = 4.39; Fig. 7, A and B). To examine the role of TNF in this process, TNF was neutralized by injecting dams intraperitoneally with 600 μg of anti-TNF 2 h before LPS injection. When compared with treatment with control antibody, TNF neutralization was found to improve animal survival and to significantly decrease the incidence of severe NEC [12/36 in the anti-TNF-treated group vs. 23/34 in control group (Fig. 7C)].
Fig. 7.

Prenatal administration of lipopolysaccharide (LPS) to dams decreases survival and increases the incidence of severe intestinal injury in neonatal mice submitted to a necrotizing enterocolitis (NEC) model, which are improved by neutralizing TNF. Dams injected with 50 μg/kg LPS or saline at embryonic day 17 were allowed to deliver naturally. Pups were submitted to a NEC protocol and euthanized when showing signs of distress. Mortality was recorded and intestinal tissues were collected for histology. A: animal survival curves (left) and histological injury scores (right) (n = 39 in the prenatal LPS group and n = 24 in the control group). Data were obtained from three separate experiments. (B: representative histological images from each group are presented (Scale bar = 100 μm). C: pregnant dams were injected with anti-TNF neutralizing antibody or control antibody at E17 2 h before LPS injection. Pups were submitted to the experimental NEC protocol, and survival (left) and histological tissue injury (right) were assessed (n = 36: anti-TNF antibodies and n = 34: control antibodies group). Data were obtained from three separate experiments. *P < 0.05, **P < 0.01, ****P < 0.0001. Log-rank test was used for survival curve and χ2 test for histological score analysis.
DISCUSSION
There is increasing clinical evidence that prenatal inflammation contributes to NEC (4, 12, 23, 28). In a large cohort of ~10,000 very-low-birth-weight infants, NEC was found to be associated with chorioamnionitis (12). Also, although rarer in this age group, a retrospective case-control study involving full-term or near-term infants found a higher incidence of chorioamnionitis in NEC patients compared with non-NEC controls (21). Placentas from surgically treated NEC patients were more frequently found to have vasculopathy and villitis associated with fetal inflammatory response compared with non-NEC, high-risk cases (23). Clinical chorioamnionitis is commonly present without evidence of amniotic infection (26), suggesting that inflammatory factors rather than bacteria itself may be sufficient to negatively impact the fetus. Intraperitoneal administration of LPS to rodent dams has been used to reproduce a maternal-to-fetal transmission of inflammation that occurs during chorioamnionitis (9). It has been shown to induce a fetal inflammatory response that peaks at 2 h and decreases by 6 h (9). Although prenatal LPS administration to rats has been shown to increase the severity of intestinal injury in a neonatal rat NEC model (13), the mechanism remains poorly understood.
In this study, we found that prenatal LPS given at E17 significantly increased the mortality and incidence of severe NEC in neonatal mice. We investigated the mechanism and provide new evidence that prenatal inflammation affects the intestinal microvasculature, which we recently showed to play a pivotal role in NEC (36). Indeed, we report the following: 1) prenatal LPS administration to the dams inhibited intestinal villous endothelial cell proliferation, increased intestinal vascular permeability, and decreased intestinal mucosal microvascular density; 2) prenatal LPS injection decreased VEGF and VEGFR2 expression in neonatal pup intestines; 3) in vitro TNF, which we found to be increased in the fetal serum after maternal LPS injection, decreased VEGFR2 expression of isolated neonatal small intestinal endothelial cells; 4) in vivo TNF decreases VEGF and VEGFR2 expression in the intestine and decreases intestinal endothelial cell proliferation and microvascular density; and 5) pups from dams prenatally injected with LPS had an increased incidence of severe NEC compared with controls, and the effect was attenuated by neutralizing TNF. Together, our findings suggest that prenatal inflammation increases the susceptibility to NEC, at least in part, by inducing the production of cytokines such as TNF, which, in addition to its other proinflammatory effects, affects intestinal VEGFR2 signaling and perinatal intestinal microvascular development.
Blood flow impairment is thought to play a role in NEC pathogenesis (24). Indeed, in a review of 84 histological specimens of NEC, the most consistent and severe finding present in all reviewed cases, together with inflammation and bacterial overgrowth, was coagulation necrosis (3). Decreased microvascular blood flow and increased microvascular dysfunction were found in rats submitted to a NEC model compared with controls (10, 38). However, besides functional vascular disturbances, recent evidence suggests that defective microvascular development causing inappropriate blood supply to intestinal tissues may be a key mechanism leading to NEC, and impaired microvasculature may also interfere with and compromise blood flow. Recently, our laboratory has shown that VEGFR2 signaling in the perinatal intestine may play an important role in NEC. Indeed, VEGF, a regulator of angiogenesis, and its main receptor VEGFR2 are strongly expressed in the intestine at the end of gestation in neonatal mice (36), but their expression decreases dramatically after birth when the neonate transitions from a low-oxygen blood concentration in utero to a higher oxygen concentration (36). VEGF, VEGFR2, and phospho-VEGFR2 expression are significantly reduced in experimental NEC (27, 36). In intestinal tissues of infants with NEC, the number of VEGF+ cells was found to be decreased compared with controls (27). Inhibition of VEGFR2 kinase activity decreased intestinal villous endothelial cell proliferation and increased mortality and the incidence of severe intestinal injury in a neonatal mouse NEC model (36). Interestingly, we have evidence here that prenatal inflammation not only predisposes to NEC, but also affects intestinal microvasculature development by a TNF-dependent mechanism.
Here, we show that prenatal LPS injection to dams decreased VEGFR2 and VEGF-164 expression in the intestine of neonatal pups. However, as in experimental NEC (36), VEGF-120 was unaffected (data not shown). In our previous study, VEGF-164 was found to be the major isoform of VEGF, with a very low level of VEGF-120 and undetectable VEGF-188 isoform expression (36). Interestingly, relative expression of these isoforms differs among different tissues (29). Conflicting results have been reported regarding the impact of prenatal inflammation on the neonatal microvasculature. Indeed, intra-amniotic injection of LPS has been shown to decrease lung tissue VEGF and VEGFR2 protein and pulmonary vascular density at birth (31). Also, intra-amniotic administration of LPS decreased VEGF and VEGFR2 protein in the lungs of neonatal lambs (17). However, prenatal LPS E15 increased lung neovascularization in mice in another study (22). Notably, the placenta of human neonates with chorioamnionitis and funisitis was found to have decreased VEGF and VEGFR2 expression (19). It was suggested that the resulting impaired placental angiogenesis could contribute to several neonatal morbidities affecting premature infants, such as bronchopulmonary dysplasia and retinopathy of prematurity (19). LPS was found to impair neonatal retinal vascular development by decreasing vessel extension, reducing capillary density, and inducing localized overgrowth of abnormal retinal vessels in one study (15), whereas another study reported an early increased retinal vessel development (35). In adults during acute lung injury, LPS has been shown to induce endothelial cell apoptosis and to decrease VEGF and VEGFR2 (16).
Mechanisms by which prenatal LPS affects the fetus and increases its susceptibility to NEC are unknown. Whether prenatal LPS crosses the placenta has been controversial. In a study conducted in pregnant mice and using intravenous injection of 125I-labeled LPS, radioactivity was found to be increased in the fetuses 1 h after maternal administration at D14–16, suggesting that it may be crossing the placenta (18). However, in two other studies done in pregnant rats, LPS was found not to cross the placenta (2, 14). A recent report in mice also showed no placental crossing of LPS (11). We show here that serum TNF is increased in the fetus during maternal prenatal inflammation within 2 h and that TNF decreases small intestinal endothelial cell VEGFR2 expression in vitro and in vivo. Other inflammatory factors are also likely to be involved in this process. Interestingly, TNF does not cross the placenta in pregnant rats (7), and prenatal cytokines (e.g., TNF, IL-1β, and IL-6) do not appear to cross the placenta at term in humans (1). This suggests that LPS-induced, maternal inflammation results in production of inflammatory mediators (e.g., TNF) by the fetal tissues, which affects intestinal endothelial cell VEGFR2 protein expression and microvasculature development. Interestingly, although we show here in neonatal mice that blocking TNF prevented the negative impact of prenatal LPS on the incidence of severe NEC, blocking TNF postnatally in rat pups submitted to the NEC protocol did not improve the incidence of severe NEC in our model (unpublished data).
Our data show that prenatal inflammation causes impaired development of the neonatal small intestinal microvasculature and that neonatal pups exposed to prenatal inflammation are at increased risk for NEC. A causal relationship link between the two events needs further studies.
Other mechanisms may play a role in the susceptibility to NEC of pups exposed prenatally to LPS. For instance, we found here that prenatal LPS not only affected intestinal microvascular development, but also increased intestinal vascular permeability, which may promote the inflammatory response by increasing inflammatory cell recruitment. Also, nonvascular effects of prenatal LPS, such as an effect on intestinal epithelial cells, could contribute to the increased incidence of NEC, as LPS has been shown to increase intestinal epithelial paracellular permeability in vitro (5).
In summary, we found that alteration of the intestinal microvascular development may be a key mechanism by which premature infants exposed to prenatal inflammation are at increased risk for NEC. Therapeutic approaches to preserve intestinal VEGF and VEGFR2 expression and intestinal microvasculature development during prenatal inflammation (such as using HIF-1α regulators) may help prevent NEC in high-risk premature infants.
GRANTS
This study was supported by funding from the Stanley Manne Children’s Research Institute, the Ann & Robert H. Lurie Children’s Hospital of Chicago, and National Institutes of Health Grants RO1 DK116568 (to I. G. De Plaen) and RO1GM122406 and RO1GM117628 (to X.-D. Tan).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
I.G.D.P. conceived and designed research; X.Y. and E.M. performed experiments; X.Y. and E.M. analyzed data; X.Y. and I.G.D.P. interpreted results of experiments; X.Y. prepared figures; X.Y. drafted manuscript; X.-D.T. and I.G.D.P. edited and revised manuscript; I.G.D.P. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Marissa Docter for technical support.
This manuscript is original, has not been previously published, and has not been submitted for publication elsewhere while under consideration.
REFERENCES
- 1.Aaltonen R, Heikkinen T, Hakala K, Laine K, Alanen A. Transfer of proinflammatory cytokines across term placenta. Obstet Gynecol 106: 802–807, 2005. doi: 10.1097/01.AOG.0000178750.84837.ed. [DOI] [PubMed] [Google Scholar]
- 2.Ashdown H, Dumont Y, Ng M, Poole S, Boksa P, Luheshi GN. The role of cytokines in mediating effects of prenatal infection on the fetus: implications for schizophrenia. Mol Psychiatry 11: 47–55, 2006. doi: 10.1038/sj.mp.4001748. [DOI] [PubMed] [Google Scholar]
- 3.Ballance WA, Dahms BB, Shenker N, Kliegman RM. Pathology of neonatal necrotizing enterocolitis: a ten-year experience. J Pediatr 117: S6–S13, 1990. doi: 10.1016/S0022-3476(05)81124-2. [DOI] [PubMed] [Google Scholar]
- 4.Been JV, Lievense S, Zimmermann LJ, Kramer BW, Wolfs TG. Chorioamnionitis as a risk factor for necrotizing enterocolitis: a systematic review and meta-analysis. J Pediatr 162: 236–42.e2, 2013. doi: 10.1016/j.jpeds.2012.07.012. [DOI] [PubMed] [Google Scholar]
- 5.Bein A, Zilbershtein A, Golosovsky M, Davidov D, Schwartz B. LPS induces hyper-permeability of intestinal epithelial cells. J Cell Physiol 232: 381–390, 2017. doi: 10.1002/jcp.25435. [DOI] [PubMed] [Google Scholar]
- 6.Bowker RM, Yan X, Managlia E, Liu SXL, Marek C, Tan XD, De Plaen IG. Dimethyloxalylglycine preserves the intestinal microvasculature and protects against intestinal injury in a neonatal mouse NEC model: role of VEGF signaling. Pediatr Res 83: 545–553, 2018. doi: 10.1038/pr.2017.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carbó N, López-Soriano FJ, Argilés JM. Tumour necrosis factor-alpha does not cross the rat placenta. Cancer Lett 128: 101–104, 1998. doi: 10.1016/S0304-3835(98)00057-3. [DOI] [PubMed] [Google Scholar]
- 8.De Plaen IG. Inflammatory signaling in necrotizing enterocolitis. Clin Perinatol 40: 109–124, 2013. doi: 10.1016/j.clp.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dell’Ovo V, Rosenzweig J, Burd I, Merabova N, Darbinian N, Goetzl L. An animal model for chorioamnionitis at term. Am J Obstet Gynecol 213: 387.e1–387.e10, 2015. doi: 10.1016/j.ajog.2015.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Downard CD, Grant SN, Matheson PJ, Guillaume AW, Debski R, Fallat ME, Garrison RN. Altered intestinal microcirculation is the critical event in the development of necrotizing enterocolitis. J Pediatr Surg 46: 1023–1028, 2011. doi: 10.1016/j.jpedsurg.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 11.Fricke EM, Elgin TG, Gong H, Reese J, Gibson-Corley KN, Weiss RM, Zimmerman K, Bowdler NC, Kalantera KM, Mills DA, Underwood MA, McElroy SJ. Lipopolysaccharide-induced maternal inflammation induces direct placental injury without alteration in placental blood flow and induces a secondary fetal intestinal injury that persists into adulthood. Am J Reprod Immunol 79: e12816, 2018. doi: 10.1111/aji.12816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.García-Muñoz Rodrigo F, Galán Henríquez G, Figueras Aloy J, García-Alix Pérez A. Outcomes of very-low-birth-weight infants exposed to maternal clinical chorioamnionitis: a multicentre study. Neonatology 106: 229–234, 2014. doi: 10.1159/000363127. [DOI] [PubMed] [Google Scholar]
- 13.Giannone PJ, Nankervis CA, Richter JM, Schanbacher BL, Reber KM. Prenatal lipopolysaccharide increases postnatal intestinal injury in a rat model of necrotizing enterocolitis. J Pediatr Gastroenterol Nutr 48: 276–282, 2009. doi: 10.1097/MPG.0b013e31818936b8. [DOI] [PubMed] [Google Scholar]
- 14.Goto M, Yoshioka T, Ravindranath T, Battelino T, Young RI, Zeller WP. LPS injected into the pregnant rat late in gestation does not induce fetal endotoxemia. Res Commun Mol Pathol Pharmacol 85: 109–112, 1994. [PubMed] [Google Scholar]
- 15.Hong HK, Lee HJ, Ko JH, Park JH, Park JY, Choi CW, Yoon CH, Ahn SJ, Park KH, Woo SJ, Oh JY. Neonatal systemic inflammation in rats alters retinal vessel development and simulates pathologic features of retinopathy of prematurity. J Neuroinflammation 11: 87, 2014. doi: 10.1186/1742-2094-11-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jesmin S, Zaedi S, Islam AM, Sultana SN, Iwashima Y, Wada T, Yamaguchi N, Hiroe M, Gando S. Time-dependent alterations of VEGF and its signaling molecules in acute lung injury in a rat model of sepsis. Inflammation 35: 484–500, 2012. doi: 10.1007/s10753-011-9337-1. [DOI] [PubMed] [Google Scholar]
- 17.Kallapur SG, Bachurski CJ, Le Cras TD, Joshi SN, Ikegami M, Jobe AH. Vascular changes after intra-amniotic endotoxin in preterm lamb lungs. Am J Physiol Lung Cell Mol Physiol 287: L1178–L1185, 2004. doi: 10.1152/ajplung.00049.2004. [DOI] [PubMed] [Google Scholar]
- 18.Kohmura Y, Kirikae T, Kirikae F, Nakano M, Sato I. Lipopolysaccharide (LPS)-induced intra-uterine fetal death (IUFD) in mice is principally due to maternal cause but not fetal sensitivity to LPS. Microbiol Immunol 44: 897–904, 2000. doi: 10.1111/j.1348-0421.2000.tb02581.x. [DOI] [PubMed] [Google Scholar]
- 19.Kramer BW, Kaemmerer U, Kapp M, Herbst D, Marx A, Berg D, Groneck PA, Speer CP. Decreased expression of angiogenic factors in placentas with chorioamnionitis after preterm birth. Pediatr Res 58: 607–612, 2005. doi: 10.1203/01.PDR.0000175641.39056.7A. [DOI] [PubMed] [Google Scholar]
- 20.Lin PW, Nasr TR, Stoll BJ. Necrotizing enterocolitis: recent scientific advances in pathophysiology and prevention. Semin Perinatol 32: 70–82, 2008. doi: 10.1053/j.semperi.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 21.Martinez-Tallo E, Claure N, Bancalari E. Necrotizing enterocolitis in full-term or near-term infants: risk factors. Biol Neonate 71: 292–298, 1997. doi: 10.1159/000244428. [DOI] [PubMed] [Google Scholar]
- 22.Miller JD, Benjamin JT, Kelly DR, Frank DB, Prince LS. Chorioamnionitis stimulates angiogenesis in saccular stage fetal lungs via CC chemokines. Am J Physiol Lung Cell Mol Physiol 298: L637–L645, 2010. doi: 10.1152/ajplung.00414.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moore SW, Arnold M, Wright C. Necrotizing enterocolitis and the placenta - a key etiological link. J Pediatr Surg 48: 359–362, 2013. doi: 10.1016/j.jpedsurg.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 24.Nankervis CA, Giannone PJ, Reber KM. The neonatal intestinal vasculature: contributing factors to necrotizing enterocolitis. Semin Perinatol 32: 83–91, 2008. doi: 10.1053/j.semperi.2008.01.003. [DOI] [PubMed] [Google Scholar]
- 25.Radu M, Chernoff J. An in vivo assay to test blood vessel permeability. J Vis Exp 73: e50062, 2013. doi: 10.3791/50062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Romero R, Miranda J, Chaemsaithong P, Chaiworapongsa T, Kusanovic JP, Dong Z, Ahmed AI, Shaman M, Lannaman K, Yoon BH, Hassan SS, Kim CJ, Korzeniewski SJ, Yeo L, Kim YM. Sterile and microbial-associated intra-amniotic inflammation in preterm prelabor rupture of membranes. J Matern Fetal Neonatal Med 28: 1394–1409, 2015. doi: 10.3109/14767058.2014.958463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabnis A, Carrasco R, Liu SX, Yan X, Managlia E, Chou PM, Tan XD, De Plaen IG. Intestinal vascular endothelial growth factor is decreased in necrotizing enterocolitis. Neonatology 107: 191–198, 2015. doi: 10.1159/000368879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Seliga-Siwecka JP, Kornacka MK. Neonatal outcome of preterm infants born to mothers with abnormal genital tract colonisation and chorioamnionitis: a cohort study. Early Hum Dev 89: 271–275, 2013. doi: 10.1016/j.earlhumdev.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 29.Stalmans I, Ng YS, Rohan R, Fruttiger M, Bouché A, Yuce A, Fujisawa H, Hermans B, Shani M, Jansen S, Hicklin D, Anderson DJ, Gardiner T, Hammes HP, Moons L, Dewerchin M, Collen D, Carmeliet P, D’Amore PA. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J Clin Invest 109: 327–336, 2002. doi: 10.1172/JCI0214362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tang JR, Seedorf GJ, Muehlethaler V, Walker DL, Markham NE, Balasubramaniam V, Abman SH. Moderate postnatal hyperoxia accelerates lung growth and attenuates pulmonary hypertension in infant rats after exposure to intra-amniotic endotoxin. Am J Physiol Lung Cell Mol Physiol 299: L735–L748, 2010. doi: 10.1152/ajplung.00153.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tian R, Liu SX, Williams C, Soltau TD, Dimmitt R, Zheng X, De Plaen IG. Characterization of a necrotizing enterocolitis model in newborn mice. Int J Clin Exp Med 3: 293–302, 2010. [PMC free article] [PubMed] [Google Scholar]
- 33.Tita AT, Andrews WW. Diagnosis and management of clinical chorioamnionitis. Clin Perinatol 37: 339–354, 2010. doi: 10.1016/j.clp.2010.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toyama RP, Xikota JC, Schwarzbold ML, Frode TS, Buss ZS, Nunes JC, Funchal GD, Nunes FC, Walz R, Pires MM. Dose-dependent sickness behavior, abortion and inflammation induced by systemic LPS injection in pregnant mice. J Matern Fetal Neonatal Med 28: 426–430, 2015. doi: 10.3109/14767058.2014.918600. [DOI] [PubMed] [Google Scholar]
- 35.Tremblay S, Miloudi K, Chaychi S, Favret S, Binet F, Polosa A, Lachapelle P, Chemtob S, Sapieha P. Systemic inflammation perturbs developmental retinal angiogenesis and neuroretinal function. Invest Ophthalmol Vis Sci 54: 8125–8139, 2013. doi: 10.1167/iovs.13-12496. [DOI] [PubMed] [Google Scholar]
- 36.Yan X, Managlia E, Liu SX, Tan XD, Wang X, Marek C, De Plaen IG. Lack of VEGFR2 signaling causes maldevelopment of the intestinal microvasculature and facilitates necrotizing enterocolitis in neonatal mice. Am J Physiol Gastrointest Liver Physiol 310: G716–G725, 2016. doi: 10.1152/ajpgi.00273.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yu X, Radulescu A, Zorko N, Besner GE. Heparin-binding EGF-like growth factor increases intestinal microvascular blood flow in necrotizing enterocolitis. Gastroenterology 137: 221–230, 2009. doi: 10.1053/j.gastro.2009.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]