

Abstract
Using tetradecyltrimethylammonium bromide (TTAB) as a surfactant denaturant, and augmentation of different spectroscopic data, helped to detect the intermediates of hemoglobin (Hb) during unfolding process. UV–vis, fluorescence, and circular dichroism spectroscopy were used simultaneously to monitor different aspects of hemoglobin species from the tertiary or secondary structure points of view. Application of the multivariate curve resolution–alternating least square (MCR–ALS), using the initial estimates of spectral profiles and appropriate constraints on different parts of augmented spectroscopic data, showed good efficiency for characterization of intermediates during Hb unfolding. These results indicated the existence of five protein species, including three intermediate-like compounds in this process. The unfolding pathway in the presence of TTAB included conversion of oxyhemoglobin into deoxyhemoglobin, and then ferrylhemoglobin, ferrihemoglobin or aquamethemoglobin, which finally transformed into hemichrome. This is the first application of chemometric analysis on the merged spectroscopic data related to chemical denaturation of a protein. These types of analysis in multisubunit proteins not only increase the domain of information, but also can reduce the ambiguities of the obtained results.
Keywords: Folding intermediate, Chemical unfolding, Chemometrics, Hemoglobin, Spectroscopy, Curve resolution
GRAPHICAL ABSTRACT
Introduction
Hemoglobin (Hb) is a heterogeneous protein, existing as a complex mixture of several quaternary structures, conformations, and heme-binding states [1]. The structure of Hb comprises two pairs of heme containing α and β subunits in a tetrahedral arrangement. The non-covalent bonds stabilizing this complex include nonpolar and van der Waals interactions, hydrogen bonds, and salt bridges [2]. Hemoglobins are among the most thoroughly studied multi-subunit proteins in recent years [3,4], but there are few previous data on the occurrence of transient species during the unfolding of Hb quaternary structure. Most conventional and current spectroscopic techniques generate only structural information averaged across the entire protein population present in the cell [1–2,5]. As proteins increase in size (and length) or have more than one subunit, the folding process becomes more complex, and often involves numerous intermediates.
The story of protein folding paradox started by Levinthal [6] who challenged the idea of Anfinsen et al. [7] indicating that proteins fold in a direct process, just dictated by energetic constraints. Later these ideas resulted in the theoretical representation of the folding process. The folding process can be mathematically simulated using a funnel-like energetic landscape, where the protein is allowed to fold to the state or states of lowest energy through a large number of pathways and intermediates [8].
Hb is a model system for studying multi-subunit proteins, and although it has been extensively investigated, its exact folding and assembly pathways remained unresolved [1]. The heterotetra-meric structure of Hb is in equilibrium with αβ dimers as well as α and β monomers [9]. Electrostatic interactions play an important role in guiding these assembly processes [10]. The exact heme-binding state of the dimeric αβ intermediate(s) (αhβh,αhβa,αaβh or αaβa) is still under investigation [2], but it appears that more than one possible and accessible assembly pathway leads from the isolated subunits to the native Hb [11].
In vitro, conversion of folded to unfolded states of proteins can be induced upon addition of chemical denaturants or other denaturants like heat, and monitored experimentally by several methods. These methods are common approaches to gather information about folding mechanisms and pathways. Surfactants are probably the most effective chemical denaturants, on the millimolar scale, which have been extensively utilized [12–14]. The interactions between surfactants and proteins have been studied, and the focus has been on the formation of surfactant–protein complex structures and changes in the protein structure [15–17].
Venkatesh et al. studied the effect of cetyl trimethylammonium bromide (CTAB) and sodium n-dodecyl sulfate (SDS) on the heme release from Hb. They concluded that CTAB was more effective than SDS in releasing heme from Hb, due to the stronger hydrophobicity of CTAB [17]. Liu et al. investigated the impact of interactions between Hb and surfactants, with the same hydrophobic tail and different charged head groups, on Hb structure. They concluded that the charge of a surfactant and pH value of the buffer can increase the formation of hemichrome below the cmc of surfactant, and the exposure of heme above the cmc. When the Hb surface charge is the opposite of that of the surfactant the interaction is both electrostatic and hydrophobic. However, when Hb surface charge is the same as that of the surfactant the interaction is mainly hydrophobic [18].
Mojtahedi et al. studied the interaction of n-dodecyltrimethyl-ammonium bromide (DTAB) with oxyhemoglobin A and oxyhemoglobin S using UV–visible absorption spectra and chemometric resolution techniques [5]. Oxyhemoglobins (A and S) could transform to partial oxidized state (ferrihemoglobin) by DTAB, and finally transform to fully oxidized hemichrome. Hemichrome mole fractions of HbS were more than HbA because of more hydrophobic interaction of DTAB–HbS in the second set of binding site relative to DTAB–HbA. The effect of SDS as an anionic surfactant on hemoglobin was also studied, and it was shown that the reaction products are aquamethemoglobin and hemichrome [19]. Hemichromes are believed to be on the degradation pathway of HbA once oxidation has occurred [5,19].
One of the essential points in realizing the mechanism of protein folding is characterization of the folding intermediates. However, the highly cooperative process of protein folding makes the lifetime of the transient intermediates too short. Thus, obtaining information about the intermediate structures is very challenging using common experimental techniques. Multivariate resolution methods as an effective chemometric tool are sets of mathematical approaches, which could be used for analyzing and interpreting spectroscopic data during monitoring of a chemical process. The aim of resolution methods is the extraction of chemical and/or physical information from the experimental data. Such information may include the presence of intermediate populations, the equilibrium and rate constants, and the spectra for each of the intermediates [20].
Multivariate curve resolution–alternating least squares (MCR–ALS), which is one of the most commonly used resolution technique, has been used to study “thermal unfolding” of proteins [21–23]. In the current work, we applied MCR–ALS on merged spectroscopic data of UV–visible, CD and fluorescence from the ‘‘chemical unfolding’’ study of Hb in the presence of tetradecyltrimethylammonium bromide (TTAB), as an effective denaturant in two different initial concentrations of Hb. The benefit of the applied approach in such a complex system is analyzing a data set containing information about both the secondary and tertiary structure of protein species, which diminish or generate during unfolding process. The resolved spectra provided information regarding the nature of these intermediates.
Materials and methods
Reagents and solutions
Tetradecyltrimethylammonium bromide (TTAB 99%, Sigma), and all other salts required for purification of Hb and preparation of buffers were obtained from Merck and were of analytical grade. All solutions were prepared with double distilled water unless stated otherwise. A phosphate buffer solution (50 mM, pH 7.4) was prepared using monobasic and dibasic phosphate salts in double distilled water. Phosphate buffer was used as a diluent for all test solutions.
Hemoglobin purification
Blood was collected from a healthy and non-smoker donor of genotype HbA [24], and anions were removed as reported by Riggs [25]. Plasma components were removed by centrifugation. The packed red cells were washed off by adding ten volumes of an isotonic saline solution (0.9% NaCl) and centrifuged at 4 °C for 15 min at 10,000 rpm. After removing the supernatant, five volumes of phosphate buffer (200 mM, pH 7.4) was added to the remaining and centrifuged at 5000 rpm for 15 min. The washed packed cells were lysed with five volume of deionized water and centrifuged at 4 °C for 10 min at 18,000 rpm. In this step, stroma was removed. The Hb solution was then brought to about 20% saturation with ammonium sulfate salt, left standing for 15 min, and centrifuged at 2 °C for 1 h at 14,000 rpm. The obtained supernatant was then dialyzed by phosphate buffer (50 mM, pH 7.4) at 4°C for 48 h, which was changed every eight hours. Concentration of Hb sample was determined from its absorbance at 414 nm (Soret band) using heme absorption coefficients (ε) of 125 mM−1 – cm−1. SDS–PAGE (15%) was used to examine the purity of Hb and was compared with Hb obtained from Sigma, and a single band was detected (data not shown).
Circular dichroism spectroscopy
The pure spectra of TTAB in CD and all other spectroscopic methods have been distracted from Hb + TTAB spectra or the equal amount of TTAB was added to the blank solution depending on the spectroscopic method.
The spectra in the far-UV range from 190 to 250 nm and in the near-UV range from 251 to 340 were obtained using a CD spectrophotometer (Jasco J 715, Japan) with an increment equal to 1.0 nm at 25 °C.
In the far-UV CD, 500 μl of 12 μM and 20 μM Hb solutions were placed, respectively, in CD cuvette and titrated with 20 μl solution of 10 mM TTAB as 1 μl fractions. Because of an instrumental problem, the CD spectrum before 200 nm was very noisy and thus were eliminated, and only the range of 200–250 nm were used. The CD data were collected in data matrices (D1 and D5) of the size of 21 × 51 where 21 was the number of spectrum recorded after each addition of TTAB to Hb (Hb in the presence of TTAB in the range of 0–380 μM) and 51 was the number of channels in the wavelength range. In the near-UV CD, 3 ml of 12 μM and 20 μM Hb solutions were placed, respectively, in CD cuvette and titrated with 120 μl solution of 10 mM TTAB as 6 μl fractions. The near- UV CD data was collected in the range of 251–340 nm. These CD data were collected in data matrices (D2 and D6) of the size of 21 × 90 where 21 was the number of spectrum recorded after each addition of TTAB to Hb (Hb in the presence of TTAB in the range of 0–380 μM) and 90 was the number of channels in the wavelength range. The first spectrum in both series belonged to Hb, before the addition of TTAB.
Fluorescence spectroscopy
Fluorescence measurements were made using a fluorescence spectrophotometer (Cary Eclipse, Varian Co, Australia). Fluorescent emission spectra were scanned from 295 to 445 nm with 1.0 nm increment at excitation wavelength (λEx) of 280 nm. Instrument settings, i.e. the slit widths (excitation at 10 nm; emission at 10 nm), scan speed (600 nm min−1) and excitation voltage (medium), were kept constant for all experiments. Titration process was exactly as far-UV CD at 25 °C. Here, there were also 21 samples (Hb in the presence of TTAB in the range of 0–380 μM) recorded in 151 channels of emission. Thus, the fluorescence data were collected in data matrices (D3 and D7) of the size of 21 × 151.
UV–vis spectrophotometry
The UV–vis spectra of Hb with TTAB in 50 mM phosphate buffer solution were recorded using a UV–vis spectrophotometer (Cary 100, Varian Co, Australia) at 25 °C in the range of 250–650 nm with an increment equal to 1.0 nm in a 1-cm quartz cell. Titration process was exactly as far-UV CD and fluorescence spectroscopy. Finally, the UV–vis data were collected in data matrices (D4 and D8) of the size of 21 × 401, where 21 is the number of spectrum recorded after each addition of TTAB to Hb collected in rows (Hb in the presence of TTAB in the range of 0–380 μM), and 401 is the number of wavelengths.
Data analysis
The spectra recorded during the chemical unfolding process of Hb protein by TTAB were arranged in a data matrix D, whose rows are the spectra of protein after addition of each amount of TTAB (in the range of 0–380 μM). The columns describe the variation of instrumental signal (absorbance, elipticity and fluorescence versus TTAB concentration) for each wavelength. Thus, the data matrices of far and near-UV CD, fluorescence, and UV–vis spectroscopy of Hb (20 μM) titrated with TTAB are named D1–D4, respectively. D5–D8 were equivalence of D1–D4 obtained with different initial concentration of Hb (12 μM). It is important to emphasize that the range of surfactant concentration was similar in all 8 types of data matrices. The experimental conditions for each data set are summarized in Table 1.
Table 1.
Characteristics of different data matrices included in augmentation (range of TTAB in all eight data matrices was 0–380 μM).
| Data Matrix | Hb concentration (μM) |
Spectroscopic method |
Wavelength range (nm) |
|---|---|---|---|
| D1 | 20 | Far CD | 200–250 |
| D2 | 20 | Near CD | 251–340 |
| D3 | 20 | Fluorescence | 295–445 |
| D4 | 20 | UV-vis | 250–650 |
| D5 | 12 | Far CD | 200–250 |
| D6 | 12 | Near CD | 251–340 |
| D7 | 12 | Fluorescence | 295–445 |
| D8 | 12 | UV-vis | 250–650 |
The data obtained by augmentation of these matrices was analyzed by MCR–ALS strategy [26]. The details of this well-known resolution method could be found in the literature [20]. In this mixture analysis method, the raw spectroscopic data (D) is decomposed to two matrices, one of those contained profiles related to the evolution along the rows, or concentration profile (C), and the other along the columns of each pure component in D, or spectral profile (S). The general expression could be shown in this simple equation:
| (1) |
where E is the residual error matrix. However, C and S could be obtained with a mathematical decomposition (singular value decomposition), but some chemical/physical constraints are used in an iterative process to give meaningful results with minimum lack-of-fit error. In MCR–ALS, selecting the number of component (chemical rank), using suitable initial estimate for iteration and applying appropriate constraints are three critical steps. In the current work, singular value decomposition (SVD) with different statistical parameters was used to determine the number of components. Both evolving factor analysis (EFA) and simple-to-use interactive self-modeling mixture analysis (SIMPLISMA) [27] were used to generate concentration and spectral initial estimates. However, SIMPLISMA gave better results and convergence criteria. Thus, the results of MCR-ALS using initial spectral estimate are presented here.
In this protein unfolding study, the concentration profiles related to the different protein species were forced to be non-negative and to obey the form of a closed system. Because of monotonic variation of TTAB concentration along the titration experiments, unimodality constraint was also introduced to the concentration profiles. Selectivity was also applied at the beginning of the titration (zero concentration of surfactant), where only the native conformation of Hb supposed to be present. The constraints on the spectral profiles were applied according to different properties of spectroscopic techniques. Thus, fluorescence, near CD and UV–vis spectra were forced to be non-negative, whereas far CD spectra remained unconstrained because of the natural occurrence of negative ellipticities in this technique. Selectivity constraint was not used for spectral profiles because there was no evidence of the presence of selective wavelengths for any of these four spectroscopic methods. All four spectroscopic data were smoothed before analysis by Savitzky–Golay algorithm (not derivative 2nd polynomial; window width = 7 and 9). As it will be discussed later, an augmentation strategy was applied to the data, and because of different scale for signals in fluorescence, UV–vis and CD, these spectroscopic data were scaled before augmentation. Near-UV CD, fluorescence and UV–vis spectra were scaled between 0 and +1.0, but far-UV CD spectra were scaled between –1.0 and +1.0. All data manipulations and analysis steps were done in MATLAB (version 7, Math work, Inc., http://www.mathworks.com, USA). MCR-ALS analysis was performed using the codes written by Tauler research group [28].
Results and discussion
Spectroscopic data
UV–vis spectrum of oxyhemoglobin is characterized by two peaks, α (575 nm) and β (540 nm). The α peak is usually slightly greater [5], and undergoes changes by addition of TTAB (Fig. 1a). The spectra at different concentrations of TTAB showed isosbestic points of about 585, 525, 435 and 395 nm, which suggested different species being involved during chemical unfolding by TTAB, including native state, final unfolded state, and intermediates (Fig. 1a). Evolving peaks around 565 and 365 are very obvious from the figure. No remarkable shift was observed in the Soret band (414 nm) by addition of TTAB.
Fig. 1.
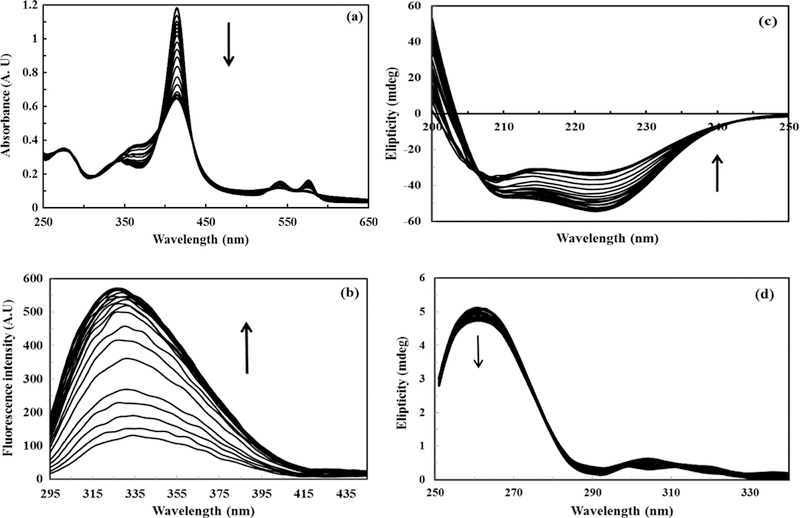
Spectra of hemoglobin (12 μM in phosphate buffer pH, 7.4) interacting with different concentrations of TTAB (0–380 μM) at 25 °C: (a) UV–vis spectra, (b) fluorescence spectra, (c) far CD spectra, and (d) near CD spectra.
With the addition of TTAB, the fluorescence intensity of Hb was remarkably increased (Fig. 1b). The fluorescence of Hb is mainly due to tryptophan (Trp), and the efficient energy transfer from Trp to heme significantly quenched the protein fluorescence emission [29]. Thus, the fluorescence intensity of Hb in water is very low [18]. However, in the presence of TTAB, the hydrophobic chains of the surfactant molecule may penetrate inside the hydrophobic moiety of Hb, exposing the heme. Thus, the fluorescence quenching of Trp with heme was depressed and the fluorescence intensity of Hb was increased. Fig. 1c shows the far-UV CD spectra of Hb, indicating that the secondary structure content of Hb was lost. Fig. 1d indicates the near-UV CD which represents third structure of Hb, reflects the environments of the aromatic amino acid side chains and thus gives information about the tertiary structure of the protein. Signals in the region from 250–270 nm are assigned to phenylalanine residues, signals from 270–290 nm are assigned to tyrosine, and those from 280–300 nm are assigned to tryptophan. The native folding of Hb is being lost as can be seen by adding more TTAB, the signal changes in all three regions which mean side chains are losing their chiral environments.
As a sample only, the recorded spectra of Hb in one initial concentration (12 μM) is shown in Fig. 1, and other spectra are shown in supplementary material.
Chemometric analysis
The UV–visible, fluorescence, and CD spectra were used as experimental data for the chemometric analysis. The number of total components and their mole fractions in the interaction mixture during the interaction with TTAB was determined and used to resolve the concentration and spectral profiles of these components. UV–vis, near-UV CD and fluorescence spectroscopy were the techniques proposed to monitor changes in tertiary structure, and Far-UV CD served to monitor modifications of the secondary structure, which was sensitive to rotations and changes of peptide bonds. These have specific angle values for each of the secondary structural motifs. Thus, augmentation of these data could obtain rich and informative results.
Here the augmentation strategy was performed on the data before analysis, because augmentation could remove rank deficiency in highly overlapped components, reduce ambiguity in the resultant profiles, and increase information extraction from the complex systems [30,31]. Generally data matrices could be augmented row-wise, column-wise or row- and column-wise. In the case of either row- or column-wise augmentation, only one direction of the array (rows and columns, respectively) needs to be common in all the merged data matrices. In the titration of Hb with TTAB as a chemical denaturant, two series of experiences were performed using two concentrations of Hb, and each titration was followed with three different spectroscopic data. In each series of experiences (on Hb with initial concentration of 20 μM and 12 μM, respectively), the spectral profiles in the data matrices of fluorescence, CD and UV–vis were different, but concentration profiles in these matrices were common. Thus, we had two series of data, which should be augmented row-wise. Finally, these two row-wise augmented data were column wise augmented and a row-and-column wise augmented dataset were resulted, which were used as the input of MCR-ALS. A scheme about the data augmentation could be found in Fig. 2. Please also see Table 1 for information about different parts of the data.
Fig. 2.
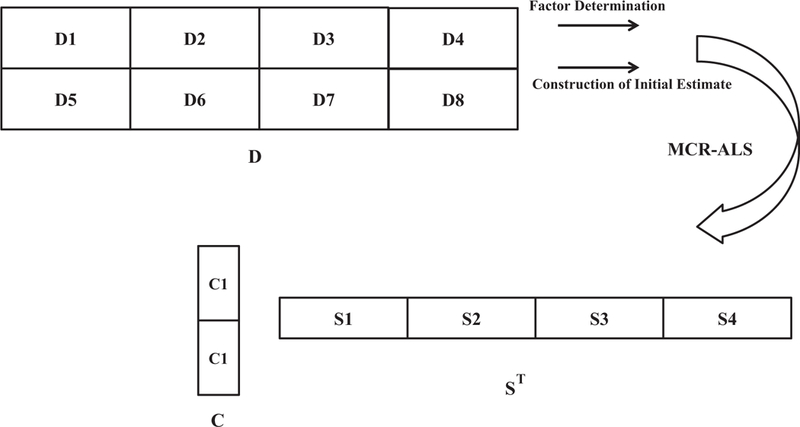
Row-and-column wise augmentation and resolution scheme. Information about different parts of D (D1–D8) is shown.
A critical step of curve resolution is the determination of the number of significant components. There are several methods for the determination of the number of components (chemical rank) in two-way data. In the current contribution, principal component analysis, based on singular value decomposition (SVD) algorithm, was used. Different statistical parameters were calculated to estimate the number of significant components in the augmented multi-spectroscopic data [32]. Some of these like eigenvalues (EV), reduced eigenvalues (REVs), residual standard deviation (RSD), root mean square error (RMSE), Chi-square (χ2) and residual percentage variance (RPV) are shown in Table 2. According to these parameters, and also limit of noise (which was obtained using the analysis of blank augmented data of all three spectroscopic methods after smoothing similar to original data) it was concluded that the number of significant factors in the titration of Hb with our chemical denaturant agent was five.
Table 2.
Different statistics used to determine number of factors (protein components) based on SVD.
| No. factors | log10(eig) | log10(REV) | RSD | RMS | chiR | RPV |
|---|---|---|---|---|---|---|
| 1 | 3.490 | −0.980 | 0.376 | 0.091 | 3189.795 | 7.305 |
| 2 | 2.369 | −2.090 | 0.074 | 0.018 | 124.265 | 0.285 |
| 3 | 0.575 | −3.873 | 0.058 | 0.014 | 75.005 | 0.172 |
| 4 | 0.446 | −3.990 | 0.041 | 0.010 | 38.414 | 0.088 |
| 5a | −0.008 | −4.434 | 0.034 | 0.008 | 25.602 | 0.059 |
| 6 | −0.173 | −4.585 | 0.027 | 0.007 | 16.802 | 0.038 |
| 7 | −0.562 | −4.962 | 0.024 | 0.006 | 13.212 | 0.030 |
| 8 | −0.681 | −5.067 | 0.022 | 0.005 | 10.478 | 0.024 |
| 9 | −0.833 | −5.207 | 0.019 | 0.005 | 8.555 | 0.020 |
| 10 | 3.490 | −0.980 | 0.376 | 0.091 | 3189.795 | 7.305 |
Optimum number of factors.
As it was noted previously SIMPLISMA was used to generate initial spectral estimate, and MCR–ALS was run with appropriate logical constraints. Finally, as it is schematically shown in Fig. 2, a spectral profile (with four parts, each related to one of the spectroscopic technique), and a concentration profile (with two parts, related to experiences on two different concentrations of protein). In order to make sure that five components exist in the data, we compared the lack of fit error of analysis of compounds from 1 to 8. These results indicated that the existence of five components is the best answer (not shown).
The spectral profiles of UV–vis, fluorescence, far CD and near CD related to five protein components (P1–P5) are shown in Fig. 3a–d, respectively. It is apparent that two of these five components are native and final-denatured form of Hb and three others are some intermediate-like structures. Additionally, Fig. 4a and b shows concentration profiles of each of these species in the reaction mixture. It is important to note that spectral and concentration profiles of the system were resolved simultaneously as it was noted graphically in Fig. 2, but it is represented separately in four and two parts in Figs. 3 and 4, respectively. Concentration profiles in Fig. 4a, b proved that the unfolding process of Hb is related to initial concentration of Hb. This is supported by the fact that when the molar ratio of TTAB/Hb differs, the unfolding pathways, and the manner of variations in concentration profiles of involved protein species (intermediate-like components) also changed.
Fig. 3.
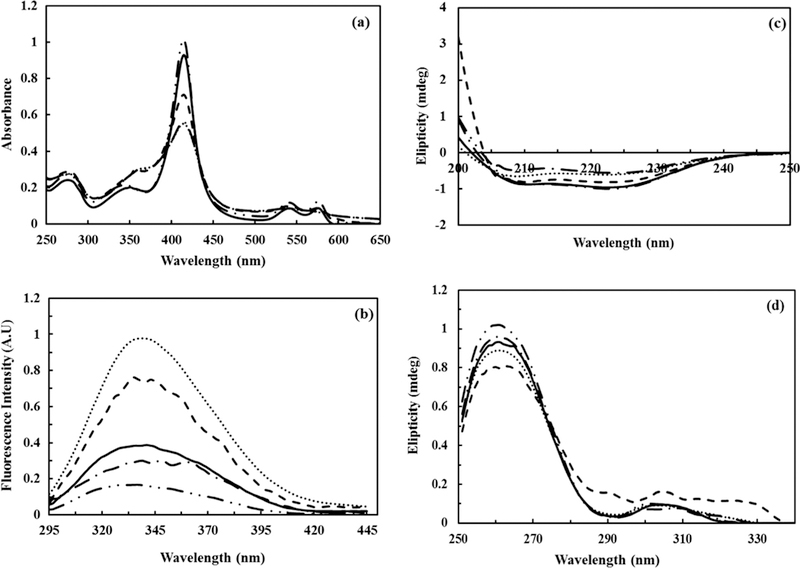
Spectral profiles of components resolved by MCR–ALS, in the denaturation path of hemoglobin (12 μM) upon interaction with TTAB (0–380 μM): (a) UV–vis spectra, (b) fluorescence spectra, (c) far CD spectra, and (d) near CD spectra. P1 (long dashed dot dot — •• —): oxyhemoglobin, P2 (dash dot – • – •): deoxyhemoglobin, P3 (dash – – –): ferrylhemoglobin, P4 (solid—): methemoglobin, and P5 (dot • • • •): hemichrom.
Fig. 4.
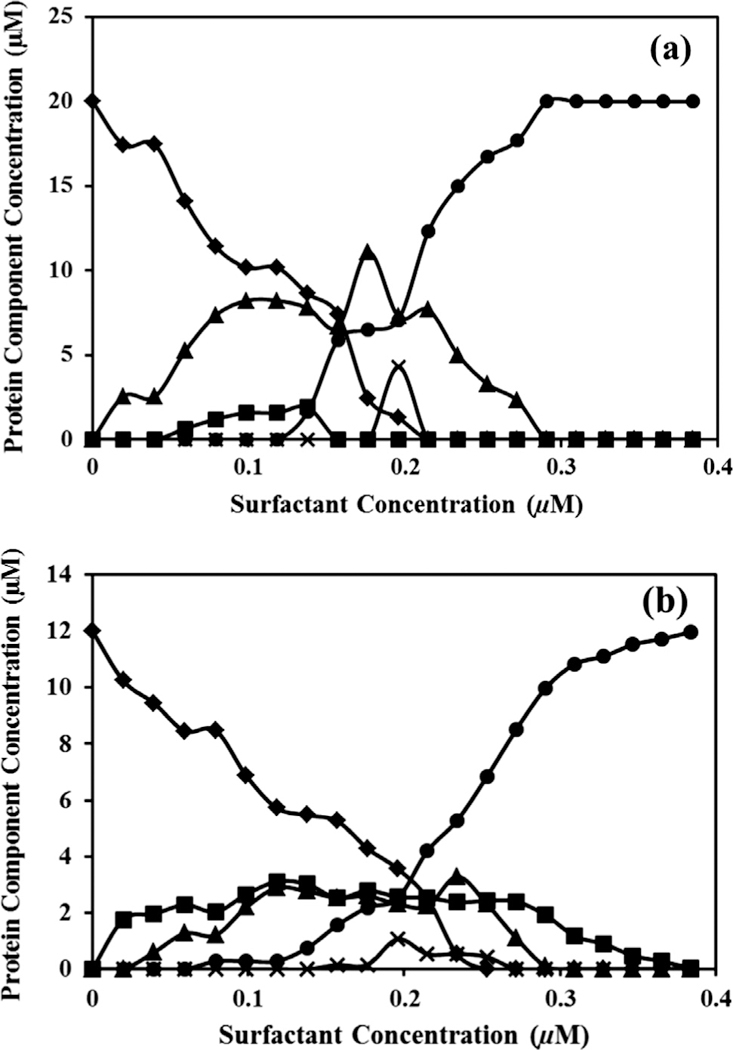
Separated concentration profiles resolved by MCR–ALS in Hb–DTAB solution: (a) initial Hb concentration 20 μM and (b) initial Hb concentration 12 μM. (♦): P1, (▲): P2, (×): P3, (■): P4, and (•): P5.
According to Fig. 4a, in the experience with initial concentration of Hb equal to 20 μM three intermediate-like protein species (P2–P4) were emerged before 0.2 μM of TTAB. With increased concentration of TTAB, these intermediate-like structures decreased and the final species (P5) were generated, which appeared to be the unfolded structure. However, it should be noted that by increasing the concentration of P5, the concentration of all three intermediates (P2–P4) reached zero. These compounds completely disappear at the end of titration. Intermediates P2 and P4 are in equilibrium until the concentration of TTAB is not more than 0.15 mM after this concentration P4 disappears and P3 emerges.
Fig. 4b shows that in the concentration profile related to the experience with lower initial concentration of Hb (12 μM), P2–P4 appeared before 0.2 μM of TTAB by decreasing the amounts of native structure (P1). Contrary to Fig. 4a, all intermediates are in equilibrium before 0.2 mM and native structure (P1) reaches to zero later than 0.25 mM of TTAB. In both series of experiments P5 terminated the unfolding process as the final unfolded protein specie. The interesting fact about these two series is intermediate P3 which actually has the lowest concentration in titration. It appears just for a short period suggesting being least stable specie in the reaction pot. One of the major problems in curve resolution analysis is the rotational ambiguity, which could compromise the accuracy of output analysis. Different approaches are reported to check this ambiguity and also the visualization of feasible solutions. One approach commonly used is MCRBANDS [33], and details could be found in the original reference [33]. However, it should be noted that in MCRBANDS a maximum and a minimum value of the relative contribution function (fmax and fmin, respectively) is calculated for each component of the resolved data. The difference of fmax and fmin should be zero in the case of the results with no serious rotational ambiguity (unique solutions). It is important to use the similar constraint of the MCR–ALS analysis during the check of ambiguity by MCRBANDS. Unfortunately, one of the limitations of MCR-BANDS is the calculation of feasible solution of augmented data arrays that need different kind of constraints in different parts of the data. For example, in the current work the non-negativity constraint was required and applied in fluorescence and UV–vis spectra, but was not applied on CD spectra because of its negative nature. Thus, using a perfect test of ambiguity in the resolved profiles was not possible. To have a brief estimate from the rotational ambiguity of the Hb unfolding system, MCR-BANDS was used with similar constraints (even non-negativity) on all parts of the data obtained from merging different spectroscopic data. It was observed that there were no differences between the fmax and fmin values for all five resolved components (Table 3). Thus, the resolved concentration and spectral profiles from the augmented spectroscopic data are relatively reliable.
Table 3.
Relative contribution function maximum and minimum values (fmin and fmax) and their differences for the five resolved protein species’ profiles and brief check of their rotational ambiguity.
| P1 | P2 | P3 | P4 | P5 | |
|---|---|---|---|---|---|
| fmax | 0.4234 | 1.092 | 0.1918 | 0.1877 | 1.309 |
| fmin | 0.4234 | 1.092 | 0.1918 | 0.1877 | 1.309 |
| fmax – fmin | 0 | 0 | 0 | 0 | 0 |
In Fig. 3a, all partially unfold and unfold species (P2–P4) showed changes in Q-band (500–600), Soret band (410–414), and aromatic region (250–300), which are formed before producing final unfolded Hb (P5). These results were in agreement with the literature that the transition from a fully reduced hemoglobin (α2+β2+)2 to a fully oxidized α3+β3+)2 one involves intermediates, called valence hybrids [5]. Here we should emphasize that the emerging of intermediates may not be in the same sequence in different pathways, in agreement with previous findings that multi-domain proteins generally follow multiple pathways in biological systems [34]. Here we studied two concentrations of Hb reacted with TTAB, and it was observed that each of these experiences went through different pathways. In both Fig. 4a and b the sequence of protein species are alike. However, the amount of intermediates in each scanned point differed. The only cause for these differences is the rate of changes in the ratio of TTAB and Hb. This subtle change induced different pathways for unfolding, so more deviation may be expected in different denaturing environment.
According to the original UV–vis spectra (Fig. 1a) the emerging peak at 365 nm suggested dimerization of free hemes liberated during protein denaturation by oxidation. These expulsed hemes further interacted with the protein to form hemichrome [35]. Again based on Fig. 1a, α and β peaks slightly moved to lower wavelengths as the concentration of TTAB increased, which is suggestive of the formation of hemichrom in which the distal histidine or an external ligand occupies the sixth coordination position of the ferric heme [35].
Chemometric analysis also resolved three intermediate-like species with their pure spectra, the first one (P2; Fig. 3a) had a peak around 560 nm, indicating a charge transfer between ligand and metal. The change in the spectrum shape implies that a five coordinated high-spin specie, deoxyhemoglobin, was formed [35]. The deoxyhemoglobin species are the preferred form for many kinds of oxidants, due to its susceptibility to the accelerated anion induced autoxidation [36]. The CD and fluorescence data also showed that P2 has a very similar tertiary structure as OxyHb (P1), and the secondary structures were changed.
After addition of more TTAB, the absorption peak at 560 nm disappeared and another intermediate-like species (P3) emerged. A new peak at 580 nm was recognized again, which is indicative of a transient high spin ferryl heme species accompanied by heme dimerization, and was detected because of a sharp peak at 365 nm [35]. As mentioned earlier, the peaks at about 560 and 580 nm are indicative of the formation of deoxyhemoglobin (P2) and ferryl hemoglobin (P3) species, respectively. The met-species were formed from this anion-induced autoxidation (P4). The maximum wavelength red shift in P4 confirmed the more folded state of intermediate relative to the native state, and indicated that a folded structure was formed. When TTAB concentration increased, the shielding of intramolecular electrostatic forces in the native state by the positive polar head of TTAB-binding reflected the intrinsic force that favored the formation of a more compact state (T conformation).
Methemoglobin in aqueous solution has a six coordinated Fe(III) center in which a water molecule occupies the O2 binding site, called aquamet. Methemoglobin is quickly converted into the hemichrome [37], which is dominant, as can be seen in final results of chemometric analysis (P5 species). The formation of new species, characterized by a Soret and Q bands at 537 and 555 nm, was observed and was named the final unfolded specie. These maxima are indicative of a six coordinated low-spin heme with ligand coordinated to the sixth position of the heme iron called hemichrome [5]. As the formation of hemichrome means the movement of the distal histidine imidazole towards the metal center, Moreira et al. [5,38] concluded that the aquamet-hemichrome transition must be due to the reduced ability of the nitrogen of the distal histidine imidazole to stabilize the coordinated water molecule. This makes the water molecule easily dissociate from the first coordination space of the iron.
It was previously reported that two binding sets exist in Hb-surfactant complexes. The first is electrostatic interaction, and the second has hydrophobic nature [18]. Once all the ionic sites are saturated the hydrophobic contribution predominates, and both of which can affect the microenvironment around heme and break the hydrogen bond between the imidazole and water molecule, indicating the formation of hemichrome [18]. The dimerization of hemin is also apparent with the appearance of a 370 nm band. Hemichrom is the endproduct and was confirmed by the CD and Fluorescence (Fig. 3b and c). As the hydrophobic tail enters the heme moiety, the heme prosthetic group comes out and fluorescence quenching is increased [18]. Since during Hb autoxidation the oxyhemoglobin is oxidized to valency hybrids (partial oxidized), the oxygenated chains of each tetramer are then easily oxidized to the final hemichrom form [5]. P3 is a rare intermediate that is produced and consumed during oxidation. As can be seen in UV–visible 500–600 nm region, the α- and β-peaks heights were decreased. The high reduction in absorption intensity at 540 nm relative to oxyhemoglobin confirmed that hemichrome was going to be formed [39]. Thus, TTAB can induce oxyhemoglobin to first convert to deoxyhemoglobin and ferrylhemoglobin, and then ferri-hemoglobin and hemichrome. The nature of ferrihemoglobin and hemichrome of Hb are electrostatic and hydrophobic, respectively [5]. Furthermore, up to the formation of ferrihemoglobin the process was enhanced by electrostatic forces induced by the initial ionic binding. The hemichrome was subsequently increased by the hydrophobic effects induced by a second set of TTAB-binding sites [38]. Mojtahedi et al. showed that the interaction between ionic head of DTAB as a cationic surfactant and polar part of Hb induced ferrihemoglobin species [5]. They could only detect one intermediate during denaturation of Hb, and showed that Hb passes through a three states during unfolding process.
Contrary to Mojtahedi et al., using the contribution of merged spectroscopic techniques, we found that the pathway for Hb unfolding includes five states and the concentration profiles depended on Hb/TTAB concentrations. These results showed that the analysis of merging spectroscopic data could obtain richer domains of information because the data used contained different secondary and tertiary structural characteristics of protein species involved. By increasing initial Hb concentration, the TTAB/Hb decreases in each point during addition of TTAB, and so does the rate of unfolding. Thus, more intermediates are trapped and extracted by the help of chemometrics. Beside experimental techniques like single molecule spectroscopy and other computational methods, this method appears to be valuable in revealing the hidden information. Undoubtedly, folding intermediates provide valuable information into the fluctuations from the native structure that may be important in regulating biological functions. Intermediates are often the species in misfolding processes that lead to aggregation and diseases [40].
Conclusion
The use of multivariate resolution methods is an appropriate combination to study and interpret protein unfolding pathways and intermediates. These types of studies uncover rare events that are masked by averaging in bulk studies. The mechanism of the process and the nature of all species involved can be estimated. Here we showed that oxyhemoglobin converts to other forms in the presence of TTAB. MCR–ALS results proved that the denaturation of Hb process included some intermediate-like species that their variation could not be the same at different Hb to TTAB ratios. Consequently, through the denaturation path in the presence of TTAB, oxyhemoglobin was first converted into a five coordinated high-spin species called deoxyhemoglobin, and then ferrylhemoglobin. This specie changes to partial ferrihemoglobin or aquamethemoglobin, which transforms into a fully oxidized low-spin species called hemichrome. However, at lower concentration of Hb, one of the last intermediate-like species (P4; partial ferrihemoglobin or aquamethemoglobin) was stabilized enough to survive as one of the final denatured component of Hb. This component showed an almost similar secondary and tertiary structure as fully oxidized hemichrome (P5) based on resolved CD and fluorescence spectral profiles. It appears that augmentation strategy reduced the ambiguity and increased the information domain of a complex denaturation system. Although the exact folding/unfolding pathway for heterotetrameric Hb has remained unresolved, possible mechanisms were discussed here. This combined analysis of chemical denaturation of a protein provides strong evidence for the utility of this methodology for quantitatively determining the number and qualitatively the structure of the intermediates upon any chemical unfolding.
Supplementary Material
HIGHLIGHTS.
Merging spectroscopic data gave rich information about chemical unfolding of hemoglobin.
MCR-ALS was applied to uncover hemoglobin species involved in an unfolding pathway.
Hb unfolding pathway included oxyHb, deoxyHb, ferrylHb, aquametHb and hemichrome.
Concentration profiles and spectral profiles of protein species were extracted.
Acknowledgments
The support of Research Council of University of Tehran, Center of Excellence in Biothermodynamics (CEBiotherm), Iran National Science Foundation (INSF), Iran National Elites Foundation (INEF) and UNESCO Chair in Interdisciplinary Research in Diabetes, Iran Society of Biophysical Chemistry are gratefully acknowledged.
Footnotes
Appendix A. Supplementary material
Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.saa.2014.10.120.
References
- [1].Simmons DA, Wilson DJ, Lajoie GA, Doherty-Kirby A, Konermann L, Biochemistry 43 (2004) 14792–14801. [DOI] [PubMed] [Google Scholar]
- [2].Boys BL, Konermann L, J. Am. Soc. Mass Spectrom. 18 (2007) 8–16. [DOI] [PubMed] [Google Scholar]
- [3].Chen T, Zhu S, Shang Y, Ge C, Jiang G, Spectrochim. Acta A 93 (2012) 125–130. [DOI] [PubMed] [Google Scholar]
- [4].He W, Dou H, Li Z, Wang X, Wang L, Wang R, Chang J, Spectrochim. Acta A 123 (2014) 176–186. [DOI] [PubMed] [Google Scholar]
- [5].Mojtahedi M, Parastar H, Jalali-Heravi M, Chamani J, Chilaka FC, Moosavi-Movahedi AA, Colloids Surf., B 6 (2008) 183–191. [DOI] [PubMed] [Google Scholar]
- [6].Levinthal C, J. Chim. Phys. 65 (1968) 44–45. [Google Scholar]
- [7].Anfinsen CB, Haber E, Sela M, White FH, Proc. Natl. Acad. Sci. USA47 (1961) 1309–1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dill KA, Chan HS, Nat. Struct. Biol. 4 (1997) 10–19. [DOI] [PubMed] [Google Scholar]
- [9].Kellett GL, Gutfreund H, Nature 227 (1970) 921–926. [DOI] [PubMed] [Google Scholar]
- [10].Yamaguchi T, Yang Y, McDonald MJ, Adachi K, Biochem. Biophys. Res. Commun. 270 (2000) 683–687. [DOI] [PubMed] [Google Scholar]
- [11].Yip YK, Waks M, Beychok S, Proc. Natl. Acad. Sci. USA 74 (1977) 64–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Moosavi-Movahedi AA, Housaindokht MR, Moghadasi J, Thermochim. Acta 219 (1993) 143–150. [Google Scholar]
- [13].Moosavi-Movahedi AA, J. Iran. Chem. Soc. 2 (2005) 189–196. [Google Scholar]
- [14].Nazari K, Saboury AA, Moosavi-Movahedi AA, Thermochim. Acta 302 (1997) 131–135. [Google Scholar]
- [15].Chakraborty A, Seth D, Setua P, Sarkar N,J. Phys. Chem. B 110 (2006) 16607–16617. [DOI] [PubMed] [Google Scholar]
- [16].Orioni B, Roversi M, La Mesa C, Asaro C, Asaro F, Pellizer G, D’Errico G, J. Phys. Chem. B 110 (2006) 12129–12140. [DOI] [PubMed] [Google Scholar]
- [17].Venkatesh B, Venkatesh S, Jayadevan S, Rifkind JM, Manoharan PT, Pept. Sci. 80 (2005) 18–25. [DOI] [PubMed] [Google Scholar]
- [18].Liu W, Guo X, Guo R, Int. J. Biol. Macromol. 41 (2007) 548–557. [DOI] [PubMed] [Google Scholar]
- [19].Dayer MR, Moosavi-Movahedi AA, Norouzi P, Shamsipour M, Chaichi MJ, Ghourchian H, Colloids Surf., B 30 (2003) 139–146. [Google Scholar]
- [20].Garrido M, Rius FX, Larrechi MS, Anal. Bioanal. Chem. 390 (2008) 2059–2066. [DOI] [PubMed] [Google Scholar]
- [21].Nevea S, Juan A, Tauler R, Anal. Chim. Acta 446 (2001) 185–195. [Google Scholar]
- [22].Navea S, De Juan A, Tauler R, Anal. Chem. 74 (2002) 6031–6039. [DOI] [PubMed] [Google Scholar]
- [23].Navea S, De Juan A, Tauler R, Anal. Chem. 75 (2003) 5592–5601. [DOI] [PubMed] [Google Scholar]
- [24].William RC, Tsay KY, Anal. Biochem. 54 (1973) 137–145. [DOI] [PubMed] [Google Scholar]
- [25].Riggs A, Meth. Enzym. 76 (1981) 5–29. [DOI] [PubMed] [Google Scholar]
- [26].De Juan A, Tauler R, Anal. Chim. Acta 500 (2003) 195–210. [Google Scholar]
- [27].Windig W, Chemom. Intell. Lab. Syst. 36 (1997) 3–16. [Google Scholar]
- [28].Jaumot J, Gargallo R, De Juan A, Tauler R, Chemom. Intell. Lab. Syst. 76 (2005) 101–110. [Google Scholar]
- [29].Santiago PS, Moreira LM, Almeida EV, Tabak M, Biochim. Biophys. Acta 1770 (2007) 506–517. [DOI] [PubMed] [Google Scholar]
- [30].Peré-Trepat E, Petrovic M, Barceló D, Tauler R, Anal. Bioanal. Chem. 378 (2004) 642–654. [DOI] [PubMed] [Google Scholar]
- [31].Jaumot J, Escaja N, Gargallo R, González C, Pedroso E, Tauler R, Nucl. Acids Res. 30 (2002) e92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Elbergali A, Nygren J, Kubista M, Anal. Chim. Acta 379 (1999) 143–158. [Google Scholar]
- [33].Jaumot J, Tauler R, Chemom. Intell. Lab. Syst. 103 (2010) 96–107. [Google Scholar]
- [34].Aggarwal V, Kulothungan SR, Balamurali MM, Saranya SR, Varadarajan R, Ainavarapu SRK, J. Biol. Chem. 286 (2011) 28056–28065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Chilaka FC, Nwamba CO, Moosavi-Movahedi AA, Cell Biochem. Biophys. 60 (2011)187–197. [DOI] [PubMed] [Google Scholar]
- [36].Macdonald V, Meth. Enzym. 231 (1994) 480–490. [DOI] [PubMed] [Google Scholar]
- [37].Tsuruga M, Matsuoka A, Hachimori A, Sugawara Y, Shikama K, J. Biol. Chem. 273 (1998) 8607–8615. [DOI] [PubMed] [Google Scholar]
- [38].Moreira LM, Poli AL, Costa-Filho AJ, Imasato H, Biophys. Chem. 124 (2006) 62–72. [DOI] [PubMed] [Google Scholar]
- [39].Harrington JP, Newton P, Crumpton T, Keaton L, Int. J. Biochem. 25 (1993) 665–670. [DOI] [PubMed] [Google Scholar]
- [40].Tsytlonok M, Itzhaki LS, Arch. Biochem. Biophys. 531 (2013) 14–23a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.