

SUMMARY
Horizontal transmission of the microbiota between different individuals is widely used to normalize the microbiota in laboratory mice. However, little is known about the dynamics of microbial communities and the level of microbiota transmission after cohousing. We extensively analyzed the fecal microbiota in Jackson and Taconic C57BL/6 mice to study horizontal transmission after weaning. Changes in the microbiota were clearly detected on day 3, almost plateaued on day 7, and resulted in near-comparable composition by day 28 after cohousing. Notably, the transmission of bacterial species was asymmetric in kinetics and abundance, resulting in a microbiota that is more similar to that of Jackson mice than Taconic. Several operational taxonomic units (OTUs) increased their abundance rapidly after cohousing in Taconic mice whereas several OTUs including two mucus-associated bacteria increased their abundance with delayed kinetics in Jackson mice. These studies provide insight into the dynamics and normalization of the microbiota during horizontal transmission.
Graphical Abstract
In Brief
Caruso et al. present a comprehensive analysis of horizontal transmission of the fecal microbiota between mouse populations from two common vendors after cohousing. These studies validate the requirement of cohousing for microbiota normalization and will guide researchers who perform experiments with the gut microbiota.
INTRODUCTION
The gastrointestinal tract of vertebrates is colonized by large numbers of microorganisms, including bacteria, fungi, archaea, and protozoa, a community that is collectively referred to as microbiota. The composition of the gut microbiota exhibits wide inter- and intra-individual variability (Eckburg et al., 2005; Human Microbiome Project Consortium, 2012). The mutualistic symbiosis between the host and the microbial community provides a nutritious and hospitable environment to the microbes in the gut, and the microbiota, in turn, contributes to several host physiological processes. Indeed, the gut microbiota performs both digestive and metabolic functions, promotes the intestinal epithelial barrier and the development of immune system, and confers protection against pathogen colonization (Bäckhed et al., 2005; Pickard et al., 2017; Round and Mazmanian, 2009). Alteration in the composition of the gut microbiota, known as dysbiosis, has been associated with several chronic metabolic and intestinal diseases (Bouter et al., 2017; Greenblum et al., 2012; Peterson et al., 2008; Qin et al., 2010; Sartor and Wu, 2017). Furthermore, intestinal microbes can influence cellular processes at sites distant from the intestine (Schroeder and Bäckhed, 2016). For example, gut dysbiosis can regulate cardiovascular and neurological processes and disease (Gregory et al., 2015; Hsiao et al., 2013; Kim et al., 2017; Zhu et al., 2016). Hence, the critical role the gut microbiota plays in host physiology needs to be considered for the experimental design and interpretation of results in animal models.
Mouse models are fundamental tools for studying the contribution of microbes to human health and disease. To determine the role of a specific gene in vivo, many studies compare mutant mice deficient in a particular gene and wild-type animals. However, several studies have identified confounding variables that, if uncontrolled, can complicate the interpretation of results (McCafferty et al., 2013). For example, mice of the same strain, housed in different cages at the same facility, harbor a diverse microbiota (Hoy et al., 2015; Rogers et al., 2014) that can account for up to 30% of variance in the composition of the gut microbiota (Hildebrand et al., 2013). Furthermore, the same line of mice from different facilities can harbor very different microbiotas (Hufeldt et al., 2010; Ivanov et al., 2008). Same strains of mice purchased from different vendors also have a distinct microbiota (Ericsson et al., 2015; Ivanov et al., 2009). Although initial evidence suggested that differences in microbial community were driven by host genotype (Vijay-Kumar et al., 2010), recent studies indicate that the major contributor in shaping the composition of gut microbiota is maternal transmission (Ubeda et al., 2012). Indeed, the microbiota of littermate mice is more similar than the microbiota of genetically identical mice that differ in maternal origin (Ley et al., 2005). Thus, differences in phenotypes seen in mutant mice when compared to wild-type animals could be explained by differences in microbiota rather than genotype. Hence, standardization of microbiota in mouse models is essential to minimize the occurrence of confounding factors, and several approaches aimed at controlling for microbial diversity have been developed (Franklin and Ericsson, 2017; Mooser et al., 2018).
Controlling for colonization of maternal microbiota by heterozygous crosses allows having proper controls when comparing different genotypes (Stappenbeck and Virgin, 2016). Breeding heterozygous parents assures that the whole litter experiences the same microbial and environmental exposures from birth regardless of genotype. However, this technique has multiple drawbacks, including necessity for genotyping, overproduction of heterozygotes, and difficulty in having proper controls in the case of doubly mutant mice. An alternative approach to control for maternal vertical transmission involves cross-fostering of rodents to a foster mother, which allows pups to be exposed to the microbiota of the nursing dam right after birth (Daft et al., 2015). However, this technique has some practical issues as well, such as the necessity for large breeding colonies and coordinated breedings. Another approach involves rederivation via surgical embryo transfer into a foster dam (Franklin and Ericsson, 2017). Pups generated via this technique are exposed to the maternal microbiota immediately at birth and acquire microbial communities by natural means. However, this approach requires considerable expertise and involves potentially cost-limiting surgery. Finally, it is now well established that microbiota can be horizontally transferred from one mouse to another by simply co-housing the animals. During cohousing, animals may feed on feces (as a result of coprophagy) or ingest feces by self-grooming. Cohousing is now a widely accepted method for normalizing microbial communities; it is a simple and convenient approach, which allows for separately breeding distinct mouse lines and then cohousing upon weaning. However, the temporal dynamics of microbial community transfer and the level of microbiota normalization between different host populations after cohousing are largely unknown. Moreover, there is no standardized method of cohousing in terms of duration and timing post-weaning required for microbiota normalization. Here, we provide detailed analyses of bacterial horizontal transmission during cohousing of laboratory mice from two common vendors Taconic Biosciences (Tac) and Jackson Laboratories (Jax) as a comprehensive resource for microbiota normalization in laboratory mice.
RESULTS
General Features of the Fecal Microbiota in Jax and Tac Mice
We used C57BL/6 (B6) mice from Jax and Tac, two common mouse vendors, to study the transmission of the microbiota after cohousing. To obtain basic knowledge about the composition of the fecal microbiota in Jax and Tac mice before the cohousing, we first assessed the microbiota in 3-week-old mice from four different Jax and Tac mothers at weaning. The composition of the fecal microbiota was determined by analysis of ~30,000 paired reads per mouse of the ~250-bp v4 region of the 16S rRNA gene by Illumina MiSeq sequencing (Figure 1A). Using the ≥ 97% nucleotide sequence identity level, we detected 5,348 operational taxonomic units (OTUs) with 289 ± 54 OTUs per mouse in 297 fecal samples of 58 mice used in this study (Table S1). The bacterial populations in feces from both Jax and Tac mice were rich in the taxonomic families Porphyromona-daceae, Prevotellaceae, Rikenellaceae, and Bacteroidaceae belonging to the phylum Bacteroidetes and Lachnospiraceae, as well as Ruminococcaceae and Lactobacillaceae of the phylum Firmicutes (Figure 1A). The α diversity, including Shannon diversity and evenness indexes, as well as OTU richness in the fecal microbiota of 3-week-old Jax and Tac mice were similar, whereas the phylogenetic diversity (a biodiversity index used to characterize the structure and diversity of ecological communities) was different between these two mouse populations (p = 0.0176) (Figure 1B). Notably, the β diversity by the Bray-Curtis dissimilarity index (BC) that measures the diversity in the microbial community (difference in taxonomic abundance profiles) within 3-week-old Jax mice and Tac mice was different (Figure 1C). Importantly, non-metric multidimensional scaling (NMDS) plot obtained from BC indexes of individual mice revealed the Jax and Tac populations had significantly distinct fecal microbiotas (Figures 1C and 1D). We also detected some differences in the β diversity among individual mice and litters (cages) of both Jax and Tac populations (Figure 1D). No clear differences in the microbiota composition were detected in mice of different genders at weaning (Figure 1D). The abundance of 52 OTUs in the fecal microbiota were significantly different between Jax and Tac mice as determined by linear discriminant analysis effect size (LEfSe) analysis p < 0.05 with q < 0.05 of the false discovery rate (Figure 1E), which reflects differences in higher-level taxons between Jax and Tac mice (Figure S1). No additional OTUs contributed to the differences in higher-level taxons between Jax and Tac mice (data not shown).
Figure 1. Distinct Structure of the Microbial Community in Jax and Tac Mice.

(A) Relative abundance of dominant bacterial families in feces collected from 3-week-old Jax (n = 23) and Tac (n = 20) mice; av, average. Numbers indicate family identities.
(B and C) Average α diversity (B, Shannon diversity, OTU richness, and phylogenetic diversity indexes) and box plot for α diversity (B, Shannon evenness) of fecal microbiota composition from Jax and Tac mice. *p < 0.05; n.s., not significant (two-tailed unpaired t test for Shannon index, OTU richness, and phylogenetic diversity; two-tailed Mann-Whitney U test for Shannon evenness). β diversity (C, Bray-Curtis dissimilarity) of the fecal microbiota within Jax and Tac mice, and that between the two groups. ****p < 0.0001 (Kruskal-Wallis test followed by Dunn’s multiple comparisons test). Data are expressed as mean ± SEM for Shannon diversity, OTU richness, and phylogenetic diversity indexes in (B). Boxes represent the interquartile range between the first and third quartiles (25th and 75th percentiles, respectively), the horizontal line inside the box defines the median, and whiskers represent the lowest and highest values for Shannon evenness in (B) and for Bray-Curtis dissimilarity in (C). Data are pooled from four independent experiments in both (B) and (C).
(D) NMDS plot of β-diversity values (Bray-Curtis dissimilarity indexes) of microbiota in fecal pellets from 3-week-old pups (circles; n = 23 and n = 20 for Jax and Tac, respectively) and mothers (squares; n = 4 each) at weaning is shown with the minimal stress (stress = 0.276383) after 1,000 iterations. Overall, the difference in β diversity between microbiota of Jax (bluish fill) and Tac (reddish fill) pups was statistically significant with p < 0.0001 (permutational multivariate analysis of variance [PERMANOVA]).
(E) The OTUs that were differentially abundant in fecal microbiota of 3-week-old Jax and Tac mice are shown with linear discriminant analysis (LDA) values of LEfSe with p < 0.05, positive false discovery rate q < 0.05, and maximal abundance cut < 1%. Their abundances are depicted by heatmaps. Each colored box represents the abundance of the indicated OTU in individual mice shown with cage identity number at the bottom.
Common and Distinct Microbiota Changes after Weaning in Jax and Tac Mice
We next examined the impact of cohousing in the composition of the fecal microbiota in Jax and Tac mice. In these experiments, we cohoused 3-week-old Jax and Tac mice at weaning or kept non-cohoused littermates in different cages as sentinel controls and monitored changes in the microbiota for up to 4 weeks (Figure 2A). Sequential analysis of the β diversity by the BC index on days 0, 1, 3, 7, 21, or 28 after weaning revealed that the composition of the microbiota changed overtime in both cohoused and sentinel (non-cohoused) mice from both the Jax and Tac populations, although the changes were more dramatic in cohoused Jax and Tac mice (Figure 2B). The β diversity of the microbiota of sentinel Jax mice was reduced between days 0 and 28 (Figure 2C), which may be explained by the fact that sentinel mice of the same litter remained in the same cage (Figure 2A). To test the cage effect, we compared the BC indexes within littermates and non-littermates of sentinel Jax populations that were housed in separate cages. Indeed, we found a reduction of the β diversity within littermates, which were kept in the same cages, suggesting cage-dependent and -independent effects on the reduction of the β diversity in sentinel Jax mice. In contrast, no reduction of the β diversity was observed in both littermates and non-littermates Tac mice (Figure 2C). These results suggest the existence of both cage-dependent and -independent effects on β-diversity changes. Furthermore, cage-independent reduction in β diversity during the first 2 months of life is not a universal event but rather is dependent on the mouse population.
Figure 2. Microbiota Composition Changes during Cohousing of Jax and Tax Mice.
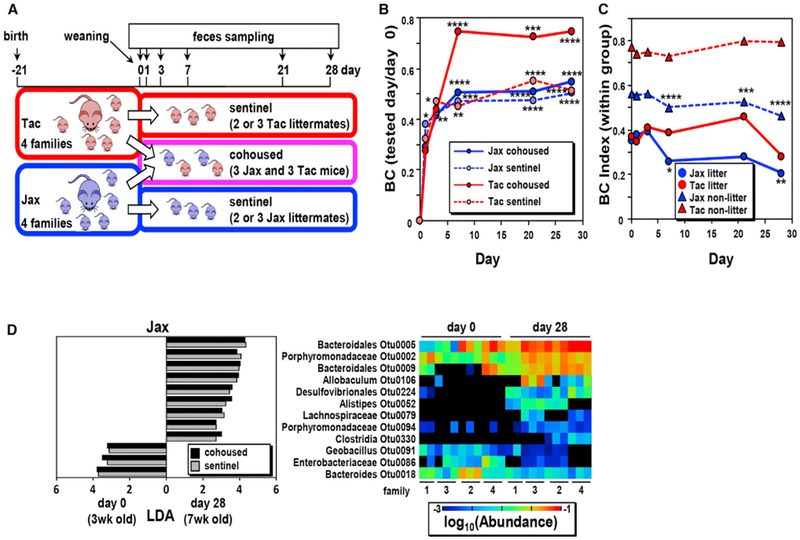
(A) Schematic representation of the experimental design used to characterize the microbiota composition of cohoused (n = 12 Jax and n = 10 Tac in four families)and sentinel (non-cohoused) mice (n = 11 Jax and n = 10 Tac in four families) over time. Pups were weaned at day 21 of age and kept as sentinels or cohoused for 28 days. The composition of the fecal microbiota of the pups and mothers was analyzed on days 0, 1, 3, 7, 21, and 28 after cohousing.
(B) Changes in microbiota composition from day 0 (weaning). β diversities (BC index) of fecal microbiota between day 0 and the indicated day in the same mice.
(C) β diversity between day 0 and the indicated days within the same group of Jax and Tac littermates and non-littermates. Data are expressed as the median from four independent experiments in both (B) and (C). *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 (Friedman test followed by Dunn’s multiple comparisons test).
(D) Age-dependent and cohousing-independent microbiota changes after weaning in Jax mice. OTUs whose abundance was significantly increased or decreased in both cohoused and sentinel Jax are shown (left). Heatmap of the abundance of indicated OTUs in sentinel Jax mice at days 0 and 28 (right).
See also Figure S2.
To determine the dominant bacteria that are responsible for the cage-independent reduction of microbial β diversity in sentinel Jax mice, we analyzed individual OTUs that were differentially abundant (with p < 0.05 of LEfSe during the β-diversity reduction) in the microbiota of Jax cohoused and sentinel mice on day 0 versus day 28. Twelve OTUs were differentially abundant at days 0 and 28 in cohoused and sentinel Jax mice (Figure 2D). These OTUs include six Bacteroidetes, two Proteo-bacteria, and four Firmicutes (Figure 2D). These OTU differences were not associated with any particular cage, thus ruling out a cage-dependent effect. The abundance changes of these OTUs during 28 days of cohousing significantly impacted the β-diversity index because the combined contribution of these OTUs was as high as 11.5% ± 0.3% of Σ|xA − xB| (xA and xB are the abundances of individual OTUs in microbiotas A and B, respectively) at days 0 and 28. Importantly, the bacterial OTUs that changed from the age of 3 weeks to day 28 after weaning represented roughly 16%–32% of the total bacteria in the Jax mice, but only 7.4%–12.4% in Tac mice in both cohoused and sentinel mice (Figure S2). These results indicate that Jax mice undergo more microbiota changes than Tac mice after weaning.
Normalization ofthe Microbiota by Cohousing in Jax and Tac Mice
As expected, the β diversity of the fecal microbiota between Jax and Tac mice was reduced after cohousing (Figure 3A). We found differences in the BC index as early as 3 days after cohousing and the BC indexes of the fecal microbiotas between Jax and Tac mice reached almost a plateau within 7 days after cohousing (Figure 3A). At day 28, both cohoused and sentinel mice showed a reduction of the BC index, which can be explained by age- and cage-dependent microbiota changes after weaning (Figure 2). In contrast, no differences were observed in the α diversity as determined by the Shannon and phylogenetic indexes of the fecal microbiota between cohoused and sentinel Jax or Tac mice, except for a slight difference in α diversity between days 0 and 21 or 28 in sentinel Jax mice (Figures 3B and S3A). All the bacterial OTUs that were present in the fecal microbiota at different abundances in both Jax and Tac mice at day 0 reached a comparable level of abundance within 21 days, except for Lachnospiraceae OTU0114 (Figure 3C). NMDS plot of the BC indexes from individual mice also showed that extended cohousing (up to 28 days) normalized the microbiota composition in Jax and Tac mice (Figure 3D). Additional analysis showed that the difference in β diversity between the microbiota of cohoused Tac mice and sentinel Jax mice is reduced compared to that between cohoused Tac mice and sentinel Tac animals (Figure 3E). These results indicate that the microbiota of cohoused mice resembled more that of Jax than that of Tac mice.
Figure 3. Microbiota Normalization during Cohousing.
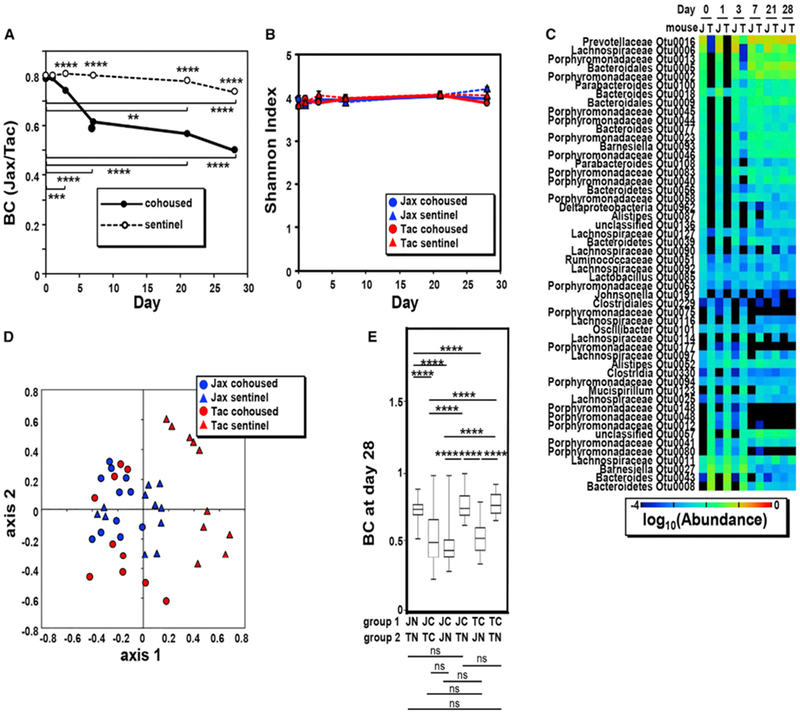
(A) β-diversity BC index between fecal microbiotas of cohoused and sentinel mice during 4 weeks (the numbers of cohoused and sentinel Jax and Tac mice are the same as shown in Figure 2). Data are expressed as the median from four independent experiments. The asterisks above symbols indicate differences between cohoused and sentinel populations. ****p < 0.0001 (Kruskal-Wallis test followed by Dunn’s multiple comparisons test), whereas asterisks with bars indicate the differences between day 0 and the indicated days. **p < 0.01, ***p < 0.001, and ****p < 0.0001 (Friedman test followed by Dunn’s multiple comparisons test).
(B) α diversity (Shannon diversity index) of fecal microbiotas of cohoused and sentinel mice during 4 weeks. Data are expressed as mean ± SEM from four independent experiments. All α diversities of fecal microbiotas, except sentinel Jax mice on day 0 versus day 21 and on day 0 versus day 28 (p < 0.05) (one-way ANOVA followed by Dunnett’s multiple comparisons test), show no difference over time.
(C) Heatmap of the average abundance of indicated OTUs that were significantly different in Jax and Tac mice before cohousing.
(D) NMDS plot of BC indexes between cohoused (circle) and sentinel (triangle) Jax (blue) and Tac (red) mice at day 28 after cohousing is shown with the minimal stress (stress = 0.252948) after 1,000 iterations. See also Table S1.
(E) Boxplots showing BC indexes between cohoused (C) and non-cohoused (N) Jax (J) and Tac (T) mice at day 28 after cohousing. ****p < 0.0001; n.s., not significant (Kruskal-Wallis test followed by Dunn’s multiple comparisons test). See also Figure S3. Boxes represent the interquartile range between the first and third quartiles (25th and 75th percentiles, respectively), the horizontal line inside the box defines the median, and whiskers represent the lowest and highest values.
We next compared microbial communities associated with vertical and horizontal microbiota transmission. To this end, we performed cross-fostering experiments between Jax and Tac mice within 2 days after birth and compared the β diversity between the microbiotas of foster mothers and fostered pups at weaning on day 21 and 3 weeks after weaning. Consistent with a previous study (Daft et al., 2015), Jax or Tac pups fostered to Tac or Jax mothers, respectively, acquired a microbiota that resembled that of the foster mothers, but not that of their biological mothers at weaning (Figure S3B). By comparing age-matched fostered or cohoused pups (both 6 weeks old), we observed that the microbiota β diversity between mothers and cross-fostered pups was lower than that between mothers and pups that were cohoused for 3 weeks after weaning (Figure S3B). These results suggest that cross-fostering is more efficient than cohousing of mice in equilibrating the microbiota.
Different Kinetics and Asymmetric Microbiota Transmission after Cohousing
We further determined the changes in bacterial OTUs after cohousing by pairwise comparison between cohoused and sentinel mice. To discover OTUs that exhibit differential abundance, we used LEfSe, and the findings were verified by false discovery rate analysis (Pedamallu et al., 2016; Segata et al., 2011). Because LEfSe analysis does not perform multiple hypotheses correction in identifying features enriched in a group, we performed comparative marker selection using GENE-E to identify significantly enriched features in each group. LEfSe analysis revealed multiple OTUs that were increased or decreased in cohoused versus sentinel mice 28 days after cohousing (see Figure 4A for Jax and Figure 4B for Tac). Using a similar comparison between cohoused and sentinel mice at different time points, we found that many of these bacteria in Tac mice altered their abundance within 7 days whereas only four OTUs increased their abundance by day 7 in Jax mice after cohousing (Figures 4A and 4B; Table S1). In contrast, most of the OTUs increased their abundance with delayed kinetics (by day 28) in cohoused Jax mice compared to cohoused Tac mice (Figures 4A and 4B). Overall, these results indicate that individual bacterial OTUs change their abundance with different kinetics during horizontal transmission of the microbiota in cohoused mice. Therefore, we further characterized the changes of bacterial populations that were either shared (common core) or unique to Jax and Tac mice during cohousing. Eleven OTUs (common core) were consistently detected at all time points in both cohoused and sentinel Jax and Tac mice at a total abundance of 8.2% ± 3.7%, whereas 34 and 13 OTUs were selectively detected in only sentinel Jax or Tac animals (Jax or Tac core) at 41% ± 14% and 10.6% ± 5.1% total abundance, respectively (Figure 5A). The dominant OTUs in the common as well as Jax and Tac cores mostly belonged to the Porphyromonadaceae and Lachnospiraceae families. Several bacterial families were detected only in the Jax microbiota core whereas Deferribacteraceae that include a single OTU Mucispirillum was only found in the Tac microbiota core (Figure 5B). Totals of 1,114 and 1,115 OTUs were uniquely found in Jax and Tac mice, respectively, although these OTUs were not present in all individual mice of each population (Figure 5A). The remarkable OTU variation unique to Jax and Tac mice did not lead to the presence of additional families (Figure 5B). We found that Jax- and Tac-specific core OTUs represented roughly 41% and 11% of the microbiota, respectively (Figure 5C), whereas the combined abundance of Jax- or Tac-unique OTUs represented < 2.2% of the total microbiota (Figure 5D). Seven days after cohousing, the combined abundance of the Jax core OTUs was dramatically increased in Tac mice, whereas a clear increase in abundance of the Tac core in Jax mice was not observed (Figure 5C). In contrast to core members, unique OTUs in both Jax and Tac mice increased 21 days after cohousing (Figure 5D). Jax unique OTUs reached an abundance in cohoused Tac mice that was comparable to that of sentinel Jax mice (Figure 5D). In contrast, the level of transmission of Tac-unique OTUs to Jax mice was limited when compared with sentinel Tac mice (Figure 5D). These results indicate differential kinetics and asymmetric transmission of the core and unique members of the microbiota in Jax and Tac mice after cohousing. Importantly, analysis of the dominant OTUs, belonging to families specifically found in the core and unique bacterial populations showed that the kinetics of individual OTU abundance was often different from that of the overall abundance in individual groups during cohousing (Figure S4). Collectively, these observations suggest the existence of complex bacterial interactions within individual bacterial groups (core and unique populations).
Figure 4. Asymmetric Kinetics of OTU Changes in Jax and Tac Mice after Cohousing.
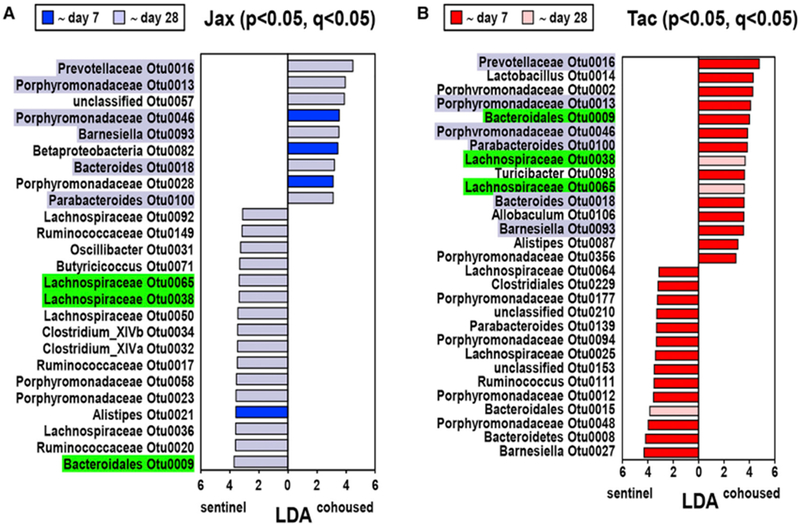
LEfSe values of OTUs that were differentially abundant between the microbiota of sentinel and cohoused (for 28 days) mice with p<0.05, and maximal abundance cut < 1%. OTUs with a false positive rate q < 0.05 are shown (the numbers of cohoused and sentinel Jax and Tac mice are the same as shown in Figure 2). Data are pooled from four independent experiments. OTUs differences between cohoused and sentinel Jax (A) and Tac (B) mice that increase their abundance rapidly within 7 days (in dark blue and red) or with delayed kinetics (in light blue and pink) after cohousing. LDA and p values of OTUs at all time points analyzed in this study are available in Table S1. OTUs whose abundance is significantly different between cohoused and sentinel mice in both Jax and Tac are highlighted in blue and green. OTUs whose abundance is higher in cohoused compared to sentinel Jax mice are highlighted in blue, whereas OTUs whose abundance is lower in cohoused Jax mice are highlighted in green. No OTUs whose abundance is lower in cohoused Tac mice compared with sentinel Tac animals were part of the group of OTUs that were differentially abundant in cohoused versus sentinel Jax mice. See also Table S1.
Figure 5. Kinetics of Core and Population-Specific Microbial Groups during Cohousing.
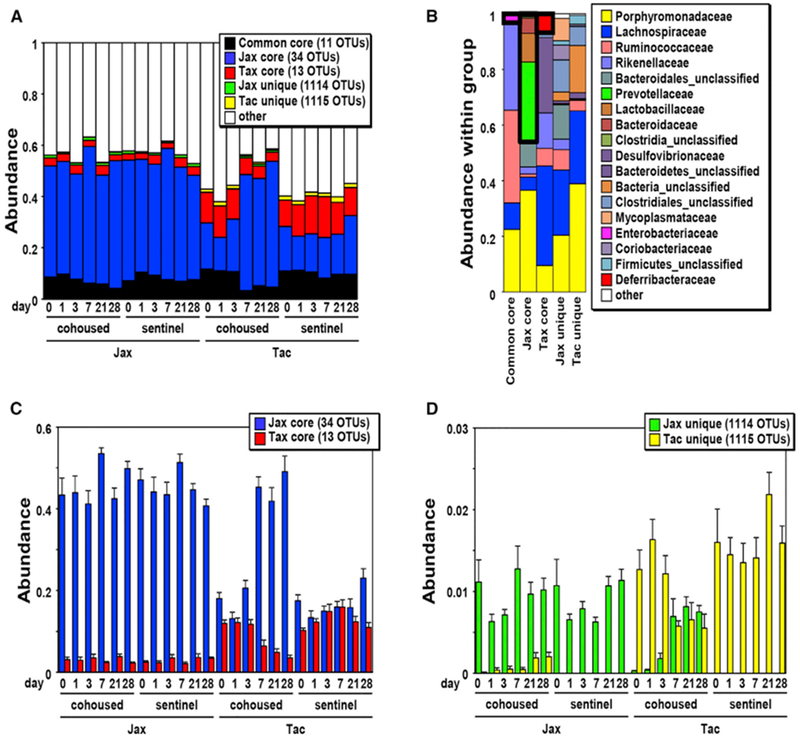
(A and B) OTUs were classified into six groups: OTUs that were commonly found in all Jax and Tac pups (common core), OTUs that were found in all Jax pups (Jax core), OTUs that were found in all Tac pups (Tac core), and OTUs that were selectively found in at least one of Jax or Tac mice (Jax or Tac unique, respectively). The numbers of cohoused and sentinel Jax and Tac mice are the same as shown in Figure 2. (A) Overall coverage of all groups in average abundance in cohoused and sentinel Jax and Tac mice at the indicated day. (B) Relative abundance (average) of bacterial families within each group is shown. The families that were specifically found with the cut value of average abundance < 10−5 in each group are shown in boxes with thick black lines.
(C and D) The abundance of population-specific core (C) and unique (D) OTUs, shown in (A), are magnified to better reveal the differences in abundance. Data are expressed as mean ± SEM from four independent experiments in both (C) and (D).
See also Figure S4.
Clustering Analysis Reveals Potential Positive and Negative Interactions among Community Members
The asymmetric nature of OTU transfer between Jax and Tac mice during cohousing suggests that specific bacteria may regulate the colonization of other community members during horizontal transmission. We, therefore, performed comprehensive clustering analysis using SparCC correlation coefficients between all mice that were used in this study to reveal potential positive or negative interactions within the microbial community (Table S2). We identified four highly correlated co-abundance groups (CAGs) with |SparCC| > 0. 62 (top 1% in correlation) and all CAGs contained negatively correlated OTU pairs with SparCC| < −0. 62 (Figure 6A). The largest cluster contained 27 OTUs and represented 26.9% ± 0.8% of the total abundance in cohoused mice (group 1 in Figure 6A). Notably, 12 of the 27 OTUs were found in the bacterial populations that changed during cohousing (Figures 4, 6A, and 6B upper panel). The remaining three clusters contained a total of 24 OTUs and represented from 13.7% to 5.3% of the total bacteria (Figure 6A). These clusters, in particular group 4, were different from cluster 1 in that they were characterized by an almost mutually exclusive distribution; the presence of a particular OTU correlated with the absence or near absence of another OTU (Figures 6A and 6B lower panel). Notably, all clusters contained OTUs that belong to the same family, suggesting negative interactions between taxonomically similar bacteria in the major clusters (Figure 6A).
Figure 6. Negative Correlations in OTU Abundance in Jax and Tac Mice.
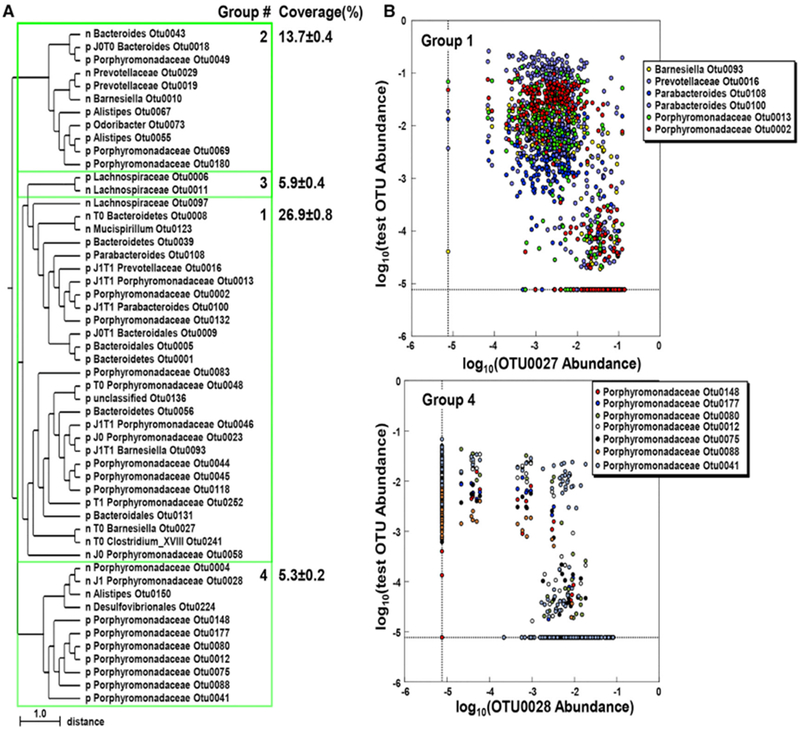
(A) Clustering analysis of OTU abundance. OTUs with the highest SparCC correlation coefficient absolute values (> 0.623, top 1%) and maximal abundance >1% in the microbiota of Jax and Tac mice were hierarchically clustered. Co-abundance groups 1-4 were defined with the same SparCC correlation coefficients absolute values (|SparCC| > 0.623) for positively (p) and negatively (n) correlated bacteria, respectively. OTUs that changed during cohousing are labeled as J0 for increased in Jax mice, J1 for decreased in Jax mice, T0 for increased in Tac mice, and T1 for decreased in Tac mice at day 28 after cohousing, according to Figure 4. The closest pairs of negatively correlated OTUs are shown with their distances on the tree. All correlations shown are significant with p < 0.001.
(B) Distribution of negatively correlated OTUs in individual mice (circle). The upper and lower panels show six representative pairs of negatively correlated OTUs ingroup 1 (each OTU shown inside the box was tested versus OTU0027) and group 4 (each OTU shown inside the box was tested versus OTU0028), respectively.
See also Table S2.
DISCUSSION
In this study, we utilized two widely used mice populations from two vendors, Jax and Tac, to investigate the dynamics of the transmission of microbial communities by cohousing, an approach that is increasingly performed by laboratories around the world. Jax and Tac mice harbor different microbiota that can differentially impact on immune homeostasis and host defense (Ivanov et al., 2008, 2009). However, the extent of normalization and kinetics of OTU transfer have not been studied in detail. Using Jax and Tac mice, we provide detailed analyses of microbiota normalization by horizontal and vertical transmission as a resource to laboratories whose animal research can be affected by resident microbes. Our results show that cohousing results in near-complete normalization of the fecal microbiotas of the mice within 28 days, with the majority of bacterial OTUs normalized after 7 days of horizontal transmission. However, our studies do not rule out that the possibility that differences in the microbiota composition may exist at other specific locations or niches within the intestine even after cohousing. One limitation of the study is that our 16S rRNA gene sequencing-based analysis lacks the resolution to identify changes at species and strain level. Therefore, a more comprehensive approach is needed to understand changes at lower taxonomic rank. Our study has revealed that the microbiota of 3-week-old mice undergo significant changes after weaning in the absence of cohousing, so it was important to take into account these changes to accurately assess the changes in the abundance of individual OTUs resulting from cohousing. These age-dependent changes impacted the overall community structures and intra-host diversity in cohoused mice. Because the level of bacterial OTUs changes and the capacity of bacterial transmission may differ in young and adult mice, similar cohousing studies need to be performed in older mice to be sure that the features of bacterial horizontal transmission in cohoused young mice can be extended to older mouse populations. Nevertheless, many research studies use 5-8-week-old mice so our analyses can be valuable to many investigators conducting studies in a laboratory setting. However, long-term microbiota changes following separation of cohoused mice remain to be determined. Although our findings provide detailed analyses of bacterial horizontal transmission during cohousing of laboratory mice, extrapolations for non-murine microbiome studies (i.e., humans and humanized mice) require additional work.
Previous studies have characterized the dynamics of a self-contained microbial community in inbred Jax mice during their first year of life (Schloss et al., 2012). These early studies showed that the α diversity of the fecal microbiota did not change early (0–9 days) after weaning when compared to a late sampling period (141–150 days post weaning). Consistently, our data indicate that the α diversity of microbial communities of cohoused and sentinel Jax and Tac mice did not show obvious differences over time. Contrary to these results, Hoy et al. (2015) showed wide variation in α diversity within individual Jax mice overtime (sampled for over 200 days). It is, however, noteworthy that Hoy and colleagues studied a population of unrelated adult mice without controlling for maternal effects, which could explain, at least in part, the differences in results. It is also possible that the discrepancy reflects the fact that the same line of mice can harbor distinct microbiotas when housed at different facilities (Hufeldt et al., 2010).
Consistent with previous studies (Hoy et al., 2015; Schloss et al., 2012), we found a shift in the community structure over time in individual mice after weaning. Our sequential analysis of the β diversity over time (up to 28 days after weaning) revealed that the composition of fecal microbiota changed as a function of time in both cohoused and non-cohoused (sentinel) mice. Thus, our findings suggest the presence of both cage-dependent and -independent effects on β-diversity changes. Overall, these results may be explained by ecological mechanisms (e.g., competition among species) that occur after changes in nutrient availability at weaning or immune-mediated effects, which may have an impact on the microbial composition. Furthermore, it is conceivable that stochastic drifts of community structure may also occur over time, even in well-controlled mouse populations.
An important finding was the observation that bacterial species do not transfer and accumulate uniformly in mice after cohousing. For example, several Jax core OTUs increased their abundance rapidly in Tac mice whereas Tac core OTUs, such as Mucispirillum OTU00123, accumulated with delayed kinetics in Jax mice after cohousing. The transmission of Tac-unique OTUs was suppressed in cohoused Jax mice, whereas the abundance of Jax-unique OTUs reached a comparable level in cohoused Jax and Tac mice. Importantly, β-diversity analyses suggest that the microbiota of Tac mice become similar to that of Jax mice after cohousing. The reason that accounts for the differential kinetics and asymmetry of horizontal transmission in Jax and Tac mice is unclear, but it could be explained partly by established interactions among resident bacteria in the gut of the host prior to cohousing. Although unlikely, we cannot rule out the possibility that minor differences in the genetic background between C57BL/6 Jax and Tac mice may play a role in the different kinetics and asymmetric horizontal transfer of the microbiota after cohousing (Mekada et al., 2009). Importantly, reduction in the abundance of some community members was strongly associated with an increase in the abundance of other community members, suggesting potential competition or another type of direct or indirect negative interaction between bacterial symbionts. These potentially competing bacteria include Bacteroidetes species, which can utilize similar nutrients and antimicrobial molecules including type VI secretion system effectors for ecological niche competition (Chatzidaki-Livanis et al., 2014, 2016; Martens et al., 2009; Roelofs et al., 2016; Russell et al., 2014; Verster et al., 2017). Consistently, we found that many of the negatively correlated OTUs belonged to the same family and utilized similar nutrients and antimicrobial molecules that often target highly related species (Gillor et al., 2009; Kommineni et al., 2015; Sweeney et al., 1996). Kinetic and clustering analyses of individual OTUs suggest that differential and complex interactions of symbiotic bacteria play a role in the regulation of their abundance in the gut. Further work is needed to understand the interactions between symbiotic bacteria.
In summary, we provide evidence that cohousing for ~28 days can result in a near-comparable composition of the fecal microbiota of the two mouse populations. Consistent with previous reports (Daft et al., 2015), we found that cross-fostering within 2 days after birth is more effective than 28 days of cohousing in normalizing the microbiota of mouse populations. Finally, crossing of heterozygous mice to generate genetically distinct littermates is another method of ensuring the normalization of the microbiota when the observed phenotype or the function of specific genes could be affected by the microbial community (Stappenbeck and Virgin, 2016; Ubeda et al., 2012). These approaches to normalize the microbial community are important when performing mouse studies that may be impacted by differences in the microbiota.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCING SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Naohiro Inohara (ino@umich.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
C57BL/6J mice were originally purchased from Jackson Laboratory (Jax) and C57BL/6NTac animals were obtained from Taconic Biosciences (Tac). Both lines of mice were left to acclimate for at least 4 weeks after purchasing and then bred in-house at the University of Michigan (Jax and Tac parents > 1 generation at the time of the experiments).
All mice were weaned on day 21 after birth and feces, for day 0 post weaning, were collected as the animals were transferred to new cages. Mice were maintained in specific pathogen free conditions and were given a single type of autoclaved chow from a single source (LabDiet 5L0D; autoclavable laboratory rodent diet), as well as water ad libitum. All mice were housed in the same room in filter top, corncob bedding, cages. Cage changing occurred every 14 days and was performed in a laminar flow hood by a single individual. All animal protocols used in this study were approved by the University Committee on Use and Care of Animals at the University of Michigan. The sample size (3 mice/group) was determined to be the optimal size for statistical analysis while allowing for independent repeats. Animals were randomly assigned to experimental groups (i.e., cohoused or sentinel mice) during cohousing experiments. The investigators were not blinded to allocation during experiments and analyses unless otherwise indicated.
METHOD DETAILS
Study Design
Jax and Tac mothers were used both as a reference to assess the microbial community at baseline and to determine the similarity of community structures between mothers and offspring. For cohousing experiments, age-matched Jax and Tac female mice were cohoused (up to 6 mice/cage) at weaning (21-day old) until 7 weeks of age. Non-cohoused male and female siblings were used as sentinel controls (up to 3 mice/cage for both Jax and Tac) to ensure that both cohoused and non-cohoused mice belong to the same mother. No single-housed mice were included in this study. A total of 26 females (14 Jax and 12 Tac) and 17 males (9 Jax and 8 Tac and) were used for these experiments. Fecal samples were collected at weaning (day 0) and on day 1, 3, 7, 21 and 28 after co-housing. Additional fecal pellets were collected from sentinel mice and from each mother. Fresh fecal pellets, collected soon after defecation, were placed in a sterile DNA-free 2 mL screw cap tube and stored at −80°C. These experiments were performed four times (4 unrelated mouse families) to verify reproducibility. Each experiment required 5 cages, of which 2 cages were used to house either Jax or Tac parents, 1 cage held cohoused pups (Jax and Tac mice) and the last 2 cages held non-cohoused sentinel mice either Jax or Tac mice. A total of 40 cages were used in cohousing experiments.
Cross-Fostering Experiments
For cross-fostering experiments, Jax-born and Tac-born pups were fostered to Tac or Jax mothers, respectively. The day on which pups were born was considered day 0 and pups were fostered between day 0 and day 2. Foster mothers were selected based on the presence of a healthy and well-fed litter. Foster mothers were removed from the cages and placed in a holding pen. The fostered pups were gently covered with the nesting material and bedding of the foster mother’s cage. Mothers were then introduced in their original cage with the fostered pups. Fostered pups were weaned when they were 21 days old and then separately housed based on the gender. Fecal samples were collected from pups and mothers at weaning and 21 days after weaning. These experiments were performed twice (2 unrelated mouse families) to verify reproducibility.
Fecal Pellet DNA Extraction and 16S rRNA Gene Sequencing and Sequence Curation
Fecal DNA was extracted using a commercially available kit (E.Z.N.A stool DNA kit, Omega Biotek). We generated amplicons of the V4 region within the 16S rRNA gene and sequenced both ends of the fragments using an Illumina MiSeq instrument (Kozich et al., 2013). PCR and library preparation were performed by the University of Michigan Microbial Systems Molecular Biology Lab as previously described by Kozich et al. (2013). The V4 region of the 16S rRNA gene was amplified using barcoded dual-index primers (Kozich et al., 2013). Each primer consists an Illumina adaptor, an 8-nt index sequence, a 10-nt pad sequence, a 2-nt linker, and the V4 specific primers F515 and R806 (Kozich et al., 2013). Mixture for PCR assays contains: 5 μL of each of the 4 μM equimolar primers, 0.15 μL AccuPrime High Fidelity Taq polymerase, 2 μL of 10× AccuPrime PCR II buffer (both from Thermo Fisher Scientific, USA), 11.85 μL of sterile PCR-grade water and 1 μL of the DNA template. The PCR conditions used consisted of 2 min of denaturation at 95°C, followed by 30 cycles each consisting of 95°C for 20 s, 55°C for 15 s and 72°C for 5 min, followed by 72°C for 10 min. Each PCR reaction was normalized to the lowest concentration of the pooled plates using a SequalPrep normalization plate kit (Thermo Fisher Scientific, USA). The normalized PCR reactions were quantified using a KAPA Library Quantification kit for Illumina platforms (Kapa Biosystems, USA). The Agilent Bioanalyzer high-sensitivity DNA analysis kit (Agilent, USA) was used to determine the amplicon size. The pooled amplicon library was sequenced using an Illumina MiSeq apparatus with a MiSeq Reagent 222 kit V2 (Illumina, USA). The libraries were prepared according to the Illumina protocol for 2nM libraries: ‘Preparing Libraries for Sequencing on the MiSeq’ (part 15039740, Rev. D). The paired end sequences were curated using Mothur (v.1.40.5) (Kozich et al., 2013; Schloss et al., 2009) as previously described (Hasegawa et al., 2014). In detail, paired end sequences were assembled into contigs using ≈250 bp contigs and the resulting Fasta sequences were screened by screen.seqs with maxambig = 0 and maxlength = 275. After getting unique sequences by unique.seqs, their numbers were counted by count.seqs and aligned to the 16S rRNA V4 database, which was extracted from the SILVA 16S rRNA reference file release 132 (Pruesse et al., 2007) using pcr.seqs with the options start = 11894, end = 25319, keepdots = F. The aligned sequences were extracted by screen.seq with the option start = 1968, end = 11550, maxhomop = 8, followed by filter.seqs with the options “vertical=T” and “trump=.,” and unique.seqs. The sequences were clustered using pre.cluster with the options diffs = 2. Possible chimeric sequences were removed by the UCHIME algorithm chimera.uchime followed by remove.seqs (Edgar et al., 2011). The sequences were binned into OTUs at > 97% identity level and taxonomically assigned using classify.seqs with 16S rRNA gene training set version 16 of Ribosomal Database Project (Wang et al., 2007). Non-bacterial sequences were removed using remove.lineage with the option taxon = Chloroplast-Mitochondria-unknown-Archaea-Eukaryota, followed by get.groups. The distances were calculated by dist.seqs using the resulting sequences and the option cutoff = 0.20. Sequences were assigned to OTUs by cluster.split using the options splitmethod = classify and taxlevel = 4, cutoff = 0.15. The matrix table of OTUs was generated by make.shared, followed by taxonomical classification using classify.otu. The results were assembled into one file and the relative abundances of individual OTUs and higher taxons were calculated by Microsoft Excel. The α-diversity (Shannon diversity, evenness indexesand phylogenetic diversity index generated by the command summary.single) β-diversity (Bray-Curtis dissimilarity index generated by the command dist.shared), NMDS plot of β-diversity values generated by the command nmds with the options mindim = 2, maxdim = 3 and iters = 1000, and LEfSe Linear discriminant analysis (LDA) values of OTUs were all determined using Mothur (Costello et al., 2009; Schloss et al., 2009). Positive false discovery rate between two groups was calculated with the Metastats command of Mothur. Comparative marker selection was used to complement LEfSe analysis via GENE-E (https://www.broadinstitute.org/cancer/software/GENE-E/) using default settings and 10,000 permutations. Heatmaps of OTUs were visualized by MeV (Saeed et al., 2006). To screen positively and negatively correlated bacterial populations in Jax and Tac mice, we used SparCC (Friedman and Alm, 2012). SparCC correlation coefficients of OTUs detected at maximal abundance > 0.5% level in all Jax and Tac mice were included in this study. OTUs that showed negative and positive correlation with another OTU were selected when they exhibited maximal | SparCC | > top 1% and “pseudo p” = 0. The OTUs were hierarchically clustered with Euclidean distance by average linkage with MeV and co-abundance groups were defined by maximal | SparCC | cut > top 1% in Excel. After hierarchical clustering of co-abundance groups by MeV, the newick file of the resulting phylogenetic tree was exported from MeV and visualized by TreeGraph 2 (Stöver and Müller, 2010). To label individual OTUs, we chose the lowest taxons that were not identified as “unclassified.” For example, we labeled “Porphyromonadaceae OTU0001” for OTU0001 annotated to Phylum Bacteroidetes, Class Bacteroidia, Order Bacteroidales, Porphyromonadaceae, Genus Porphyromonadaceae-unclassified. A detailed list of commands used to analyze the data, including the commands used in Mothur, are included in Table S1 (worksheet#C). Raw sequences are available via Short-Read Archive (SRA) with BioProject number PRJNA531699.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analyses were performed using GraphPad Prism software version 8 (GraphPad Software Inc.). Past 3.22 was used for PERMANOVA calculation based on 9999 permutations. Shapiro-Wilk normality test was used to test the assumption of normal distribution (GraphPad Software Inc.). Differences between two groups were evaluated using Two-tailed Unpaired t test or Mann-Whitney U test for normally or non-normally distributed datasets, respectively. Comparison of more than two groups was performed with one-way ANOVA followed by Dunnett’s multiple comparisons test for normally distributed datasets or by either Kruskal-Wallis or Friedman test both followed by Dunn’s multiple comparisons test for data with non-normal distributions. For highly repetitive comparison between two groups, we further performed false positive rate analysis using metastats of Mothur after removal of the OTUs that were not included in tested dataset. PERMANOVA was used for analysis of Bray-Curtis indexes for Figure 1D. Differences at p < 0.05 and q < 0.05 were considered significant.
DATA AND SOFTWARE AVAILABILITY
Raw sequences are available via NCBI SRA with BioProject: PRJNA531699.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Critical Commercial Assays | ||
| E.Z.N.A stool DNA kit | Omega Biotek | Cat#: D4015-02 |
| AccuPrimeTaq DNA Polymerase, high fidelity kit | Thermo Fisher Scientific | Cat#12346086 |
| High-sensitivity DNA analysis kit | Agilent | Cat#5067-4626 |
| KAPA Library Quantification Kit for Illumina platforms | KAPA Biosystems | Cat# KK4873 |
| SequalPrep Normalization Plate Kit, 96-well | Thermo Fisher Scientific | Cat#A1051001 |
| 500 cycle MiSeq V2 Reagent kit | Illumina | Cat#MS-102-2003 |
| Deposited Data | ||
| Raw and analyzed data | This paper; Table S1 | PRJNA531699 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | Stock No: JAX: 000664 |
| Mouse: C57BL/6NTac | Taconic Biosciences | Model: Black 6 (B6NTac), B6 |
| Oligonucleotides | ||
| 16S rRNA gene Illumina sequencing primers | Kozich et al., 2013 | PubMed 23793624 |
| Software and Algorithms | ||
| GraphPad Prism version 7 | GraphPad software | https://www.graphpad.com/ |
| Microsoft Excel | Microsoft | https://products.office.com/en-us/excel |
| Mothur v.1.40.5 | Schloss et al., 2009 | https://www.mothur.org/ |
| SILVA 16S rRNA database | Pruesse et al., 2007 | https://mothur.org/wiki/Silva_reference_files |
| UCHIME | Edgar et al., 2011 | http://drive5.com/usearch/manual/uchime_algo.html |
| 16S rRNA gene training set version 9 of Ribosomal Database Project | Wang et al., 2007 | https://mothur.org/wiki/RDP_reference_files |
| MeV | Saeed et al., 2006 | http://mev.tm4.org |
| Past 3 | Hammer et al., 2001 | https://folk.uio.no/ohammer/past/ |
| TreeGraph 2 | Stöver and Müller, 2010 | http://treegraph.bioinfweb.info/ |
| GENE-E | N/A | https://www.broadinstitute.org/cancer/software/GENE-E/ |
| Other | ||
| PicoLab Laboratory Rodent Diet Irradiated 5L0D | LabDiet | Cat#: 0067138 |
| Illumina MiSeq platform | Illumina | Illumina |
Highlights.
The microbiota changes in a cage-dependent and -independent manner after weaning
Extended cohousing of two mouse populations can nearly normalize the fecal microbiota
Transfer kinetics of individual microbes is different during horizontal transmission
Microbial transmission between two mouse populations is asymmetrical
ACKNOWLEDGMENTS
The authors thank the Sequencing Core of the University of Michigan Host Microbiome Initiative for support, Lisa Haynes for animal husbandry, Melody Zeng for review of the manuscript, and Tailor Mathes for generating the slider image. R.C. was supported by a career development award from the Crohn’s and Colitis Foundation of America, an EMBO long-term fellowship, and a scholarship for training abroad from the Italian Group for the Study of Inflammatory Bowel Diseases. This work was supported by a senior research award from the Crohn’s and Colitis Foundation of America to N.I. and NIH grants DK091191 and DK095782 to G.N
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.05.042.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, and Gordon JI (2005). Host-bacterial mutualism in the human intestine. Science 307, 1915–1920. [DOI] [PubMed] [Google Scholar]
- Bouter KE, van Raalte DH, Groen AK, and Nieuwdorp M (2017). Role of the Gut Microbiome in the Pathogenesis of Obesity and Obesity-Related Metabolic Dysfunction. Gastroenterology 152, 1671–1678. [DOI] [PubMed] [Google Scholar]
- Chatzidaki-Livanis M, Coyne MJ, and Comstock LE (2014). An antimicrobial protein of the gut symbiont Bacteroides fragilis with a MACPF domain of host immune proteins. Mol. Microbiol 94, 1361–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatzidaki-Livanis M, Geva-Zatorsky N, and Comstock LE (2016). Bacteroides fragilis type VI secretion systems use novel effector and immunity proteins to antagonize human gut Bacteroidales species. Proc. Natl. Acad. Sci. USA 113, 3627–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello EK, Halloy SR, Reed SC, Sowell P, and Schmidt SK (2009). Fumarole-supported islands of biodiversity within a hyperarid, high-elevation landscape on Socompa Volcano, Puna de Atacama, Andes. Appl. Environ. Microbiol 75, 735–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daft JG, Ptacek T, Kumar R, Morrow C, and Lorenz RG (2015). Cross-fostering immediately after birth induces a permanent microbiota shift that is shaped by the nursing mother. Microbiome 3, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M, Gill SR, Nelson KE, and Relman DA (2005). Diversity ofthe human intestinal microbial flora. Science 308, 1635–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, and Knight R (2011). UCHIME improves sensitivity and speed ofchimera detection. Bioinformatics 27, 2194–2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ericsson AC, Davis JW, Spollen W, Bivens N, Givan S, Hagan CE, McIntosh M, and Franklin CL (2015). Effects of vendor and genetic background on the composition of the fecal microbiota of inbred mice. PLoS ONE 10, e0116704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franklin CL, and Ericsson AC (2017). Microbiota and reproducibility of rodent models. Lab Anim. (NY) 46, 114–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman J, and Alm EJ (2012). Inferring correlation networksfrom genomic survey data. PLoS Comput. Biol 8, e1002687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillor O, Giladi I, and Riley MA (2009). Persistence ofcolicinogenic Escherichia coli in the mouse gastrointestinal tract. BMC Microbiol. 9, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenblum S, Turnbaugh PJ, and Borenstein E (2012). Metagenomicsystems biology ofthe human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 109, 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory JC, Buffa JA, Org E, Wang Z, Levison BS, Zhu W, Wagner MA, Bennett BJ, Li L, DiDonato JA, et al. (2015). Transmission of atherosclerosis susceptibility with gut microbial transplantation. J. Biol. Chem 290, 5647–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammer Q, Harper DAT, and Ryan PD (2001). PAST: Paleontological statistics software package for education and data analysis. Palaeontologia Electronica 4, 1–9. [Google Scholar]
- Hasegawa M, Yada S, Liu MZ, Kamada N, Munoz-Planillo R, Do N, Nunez G, and Inohara N (2014). Interleukin-22 regulates the complement system to promote resistance against pathobionts after pathogen-induced intestinal damage. Immunity 41, 620–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildebrand F, Nguyen TL, Brinkman B, Yunta RG, Cauwe B, Vandenabeele P, Liston A, and Raes J (2013). Inflammation-associated enterotypes, host genotype, cage and inter-individual effects drive gut microbiota variation in common laboratory mice. Genome Biol. 14, R4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoy YE, Bik EM, Lawley TD, Holmes SP, Monack DM, Theriot JA, and Relman DA (2015). Variation in TaxonomicComposition ofthe Fecal Microbiota in an Inbred Mouse Strain across Individualsand Time. PLoSONE 10, e0142825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao EY, McBride SW, Hsien S, Sharon G, Hyde ER, McCue T, Codelli JA, Chow J, Reisman SE, Petrosino JF, et al. (2013). Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 155, 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hufeldt MR, Nielsen DS, Vogensen FK, Midtvedt T, and Hansen AK (2010). Variation in the gut microbiota of laboratory mice is related to both genetic and environmental factors. Comp. Med 60, 336–347. [PMC free article] [PubMed] [Google Scholar]
- Human Microbiome Project Consortium (2012). Structure, function and diversity ofthe healthy human microbiome. Nature 486, 207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Frutos Rde.L., Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, and Littman DR (2008). Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4, 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. (2009). Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim H, Yim YS, Ha S, Atarashi K, Tan TG, Longman RS, Honda K, Littman DR, Choi GB, and Huh JR (2017). Maternal gut bacteria promote neurodevelopmental abnormalities in mouse offspring. Nature 549, 528–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kommineni S, Bretl DJ, Lam V, Chakraborty R, Hayward M, Simpson P, Cao Y, Bousounis P, Kristich CJ, and Salzman NH (2015). Bacteriocin production augments niche competition by enterococci in the mammalian gastrointestinal tract. Nature 526, 719–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozich JJ, Westcott SL, Baxter NT, Highlander SK, and Schloss PD (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol 79, 5112–5120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE, Backhed F, Turnbaugh P, Lozupone CA, Knight RD, and Gordon JI (2005). Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 102, 11070–11075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC, Koropatkin NM, Smith TJ, and Gordon JI (2009). Complex glycan catabolism by the human gut microbiota: the Bacteroidetes Sus-like paradigm. J. Biol. Chem 284, 24673–24677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCafferty J, MGhlbauer M, Gharaibeh RZ, Arthur JC, Perez-Chanona E, Sha W, Jobin C, and Fodor AA (2013). Stochastic changes over time and not founder effects drive cage effects in microbial community assembly in a mouse model. ISME J. 7, 2116–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mekada K, Abe K, Murakami A, Nakamura S, Nakata H, Moriwaki K, Obata Y, and Yoshiki A (2009). Genetic differences among C57BL/6 substrains. Exp. Anim 58, 141–149. [DOI] [PubMed] [Google Scholar]
- Mooser C, Gomez de Agiiero M, and Ganal-Vonarburg SC (2018). Standardization in host-microbiota interaction studies: challenges, gnotobiology as a tool, and perspective. Curr. Opin. Microbiol 44, 50–60. [DOI] [PubMed] [Google Scholar]
- Pedamallu CS, Bhatt AS, Bullman S, Fowler S, Freeman SS, Durand J, Jung J, Duke F, Manzo V, Cai D, et al. (2016). Metagenomic Characterization of Microbial Communities In Situ Within the Deeper Layers of the Ileum in Crohn’s Disease. Cell Mol. Gastroenterol. Hepatol 2, 563–566.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson DA, Frank DN, Pace NR, and Gordon JI (2008). Metagenomic approaches for defining the pathogenesis of inflammatory bowel diseases. Cell Host Microbe 3, 417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickard JM, Zeng MY, Caruso R, and Nunez G (2017). Gut microbiota: Role in pathogen colonization, immune responses, and inflammatorydisease. Immunol. Rev 279, 70–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, and Glockner FO (2007). SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNAsequence data compatiblewith ARB. Nucleic Acids Res. 35, 7188–7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. ; MetaHIT Consortium (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roelofs KG, Coyne MJ, Gentyala RR, Chatzidaki-Livanis M, and Comstock LE (2016). Bacteroidales Secreted Antimicrobial Proteins Target Surface Molecules Necessary for Gut Colonization and Mediate Competition In Vivo. MBio 7, e01055–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers GB, Kozlowska J, Keeble J, Metcalfe K, Fao M, Dowd SE, Mason AJ, McGuckin MA, and Bruce KD (2014). Functional divergence in gastrointestinal microbiota in physically-separated genetically identical mice. Sci. Rep 4, 5437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Round JL, and Mazmanian SK (2009). The gut microbiota shapes intestinal immune responses during health and disease. Nat. Rev. Immunol 9, 313–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell AB, Wexler AG, Harding BN, Whitney JC, Bohn AJ, Goo YA, Tran BQ, Barry NA, Zheng H, Peterson SB, et al. (2014). A type VI secretion-related pathway in Bacteroidetes mediates interbacterial antagonism. Cell Host Microbe 16, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed AI, Bhagabati NK, Braisted JC, Liang W, Sharov V, Howe EA, Li J, Thiagarajan M, White JA, and Quackenbush J (2006). TM4 microarray software suite. Methods Enzymol. 411, 134–193. [DOI] [PubMed] [Google Scholar]
- Sartor RB, and Wu GD (2017). Roles for Intestinal Bacteria, Viruses, and Fungi in Pathogenesis of Inflammatory Bowel Diseases and Therapeutic Approaches. Gastroenterology 152, 327–339.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol 75, 7537–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schloss PD, Schubert AM, Zackular JP, Iverson KD, Young VB, and Petrosino JF (2012). Stabilization of the murine gut microbiome following weaning. Gut Microbes 3, 383–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder BO, and Bäckhed F (2016). Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med 22, 1079–1089. [DOI] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stappenbeck TS, andVirgin HW (2016). Accountingfor reciprocal host-microbiome interactions in experimental science. Nature 534, 191–199. [DOI] [PubMed] [Google Scholar]
- Stöver BC, and Müller KF (2010). TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinformatics 11, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweeney NJ, Klemm P, McCormick BA, Moller-Nielsen E, Utley M, Schembri MA, Laux DC, and Cohen PS (1996). The Escherichia coli K-12 gntP gene allows E. coli F-18 to occupy a distinct nutritional niche in the streptomycin-treated mouse large intestine. Infect. Immun 64, 3497–3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda C, Lipuma L, Gobourne A, Viale A, Leiner I, Equinda M, Khanin R, and Pamer EG (2012). Familial transmission rather than defective innate immunity shapes the distinct intestinal microbiota of TLR-deficient mice. J. Exp. Med 209, 1445–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verster AJ, Ross BD, Radey MC, Bao Y, Goodman AL, Mougous JD, and Borenstein E (2017). The Landscape of Type VI Secretion across Human Gut Microbiomes Reveals Its Role in Community Composition. Cell Host Microbe 22, 411–419.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijay-Kumar M, Aitken JD, Carvalho FA, Cullender TC, Mwangi S, Srinivasan S, Sitaraman SV, Knight R, Ley RE, and Gewirtz AT (2010). Metabolic syndrome and altered gut microbiota in mice lacking Toll-like receptor 5. Science 328, 228–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, and Cole JR (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol 73, 5261–5267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu W, Gregory JC, Org E, Buffa JA, Gupta N, Wang Z, Li L, Fu X, Wu Y, Mehrabian M, et al. (2016). Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 165, 111–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequences are available via NCBI SRA with BioProject: PRJNA531699.