

Supplemental Digital Content is available in the text.
Keywords: gene expression; hypertrophy, left ventricular; metabolism; protein biosynthesis; protein folding; proteostasis; ribosomes
Abstract
Rationale:
Gene expression profiles have been mainly determined by analysis of transcript abundance. However, these analyses cannot capture posttranscriptional gene expression control at the level of translation, which is a key step in the regulation of gene expression, as evidenced by the fact that transcript levels often poorly correlate with protein levels. Furthermore, genome-wide transcript profiling of distinct cell types is challenging due to the fact that lysates from tissues always represent a mixture of cells.
Objectives:
This study aimed to develop a new experimental method that overcomes both limitations and to apply this method to perform a genome-wide analysis of gene expression on the translational level in response to pressure overload.
Methods and Results:
By combining ribosome profiling (Ribo-seq) with a ribosome-tagging approach (Ribo-tag), it was possible to determine the translated transcriptome in specific cell types from the heart. After pressure overload, we monitored the cardiac myocyte translatome by purifying tagged cardiac myocyte ribosomes from cardiac lysates and subjecting the ribosome-protected mRNA fragments to deep sequencing. We identified subsets of mRNAs that are regulated at the translational level and found that translational control determines early changes in gene expression in response to cardiac stress in cardiac myocytes. Translationally controlled transcripts are associated with specific biological processes related to translation, protein quality control, and metabolism. Mechanistically, Ribo-seq allowed for the identification of upstream open reading frames in transcripts, which we predict to be important regulators of translation.
Conclusions:
This method has the potential to (1) provide a new tool for studying cell-specific gene expression at the level of translation in tissues, (2) reveal new therapeutic targets to prevent cellular remodeling, and (3) trigger follow-up studies that address both, the molecular mechanisms involved in the posttranscriptional control of gene expression in cardiac cells, and the protective functions of proteins expressed in response to cellular stress.
Meet the First Author, see p 368
Changes in levels of specific key proteins play a fundamental role during the pathogenesis of heart failure, independent from the cause.1 Genome-wide expression profiling has thus emerged as a key tool for examining cardiac pathophysiology and for studying the molecular basis of the regulation of gene expression during disease development or progression. Most expression profiling is performed nowadays by mRNA sequencing (RNA-seq), which report on the transcripts that are present in tissues. Analyses of transcript abundance have revealed important transcriptional regulatory programs in various areas of cardiac biology and pathophysiology.2–4
However, such studies suffered from 2 method-based limitations. First, due to lack of appropriate tools, most studies were incapable of determining cell-type–specific gene expression changes in intact hearts. Importantly, the heart consists of ≈70% nonmyocytes (ie, fibroblasts and endothelial cells) and only 30% cardiac myocytes.5 Second, studies analyzing transcript levels ignore the significant contribution of control mechanisms affecting protein synthesis at the level of translation and therefore provide an incomplete picture of gene expression control. The correlation between transcript and protein levels is frequently poor, which is partly due to the regulation of gene expression at the level of translation.6 Direct analyses of translation would provide a more accurate and complete measure of gene expression in tissues compared with analyzing transcript levels alone. These measurements would reveal posttranscriptional programs and permit the dissection of underlying regulatory mechanisms.
Ribosome profiling (Ribo-seq; ribosome footprinting) overcomes the limitation of classical expression analysis as it directly quantifies the number of translating ribosomes, thereby integrating control at the level of transcription and translation. Ribo-seq allows for deep sequencing of mRNA fragments that are protected by the ribosome (ribosome-protected fragments, RPFs).7 The RPFs, aligned to the genome, reveal the position of ribosomes at the nucleotide level, adding considerable detail to the analysis of mRNA translation. Identification of the translatome with nucleotide resolution has previously been inaccessible with polyribosomes sequencing approaches (such as TRAP-seq [translating ribosome affinity purification sequencing]).8,9 Accordingly, detection of translation start-sites, and additionally the identification of novel open reading frames, are only possible with Ribo-seq. Actively translating ribosomes move along the transcript in steps of 3-nucleotide (nt; codon size), which is used as a basis to infer translation.10 Translation events found by Ribo-seq are not limited to the prediction of annotated protein-coding genes. This means that identification of RPFs from long noncoding RNAs, for example, may indicate RNA translation. This cannot be said for such species of RNAs identified associated with polyribosomes in polyribosome sequencing, which has already been established in the cardiac context.9 In contrast, polyribosome sequencing monitors the translational status of an entire transcript and enables the measurement of differences in translation of alternate transcript isoforms. Therefore, the approaches clearly differ but complement each other to provide insight into molecular mechanisms of translational control. Moreover, measuring expression dynamics with Ribo-seq is a closer proxy for changes in protein levels, compared with RNA data.11
Furthermore, by combining Ribo-seq with a ribosome-tagging approach, it is possible to determine translatomes of specific cell types from complex tissues. In this study, cell-type–specific analysis of translated mRNAs is achieved by using the Ribo-tag mouse to affinity purify tagged ribosomes from a complex lysate of homogenized heart tissue. The Ribo-tag mouse, developed by Sanz et al,8 uses Cre-loxP-dependent recombination and was previously used to demonstrate the usefulness of the Ribo-tag approach through isolation of cell-specific polyribosomes from specific neurons starting with whole-brain homogenates and Sertoli cell mRNAs from whole testis. Until now, only a few studies have combined the Ribo-tag approach with Ribo-seq in vivo,12,13 which, to the best of our knowledge, has never been done in the cardiac context.
In this study, we successfully established cell-type–specific Ribo-seq in the heart, which will allow investigations of gene expression control at the level of translation in different cardiac cells. We used the Ribo-tag system to purify and identify actively translating transcripts from cardiac myocytes and endothelial cells. We employed this technique to profile gene expression in cardiac myocytes in response to pressure overload. We found that translational control determines early gene expression changes in response to cardiac stress in cardiac myocytes and predict important roles for translational control by regulatory upstream open reading frames (uORFs).
Therefore, the method and the findings in this report represent an important and valuable advance in the development of novel techniques to facilitate understanding of gene expression control in complex tissues. Moreover, the analysis of ribosome profiles on distinct transcripts can be used as a starting point for further studies of mechanisms of translational regulation during pathological remodeling and heart failure.
Methods
A detailed description of the methods is available in the Online Data Supplement. The authors declare that all supporting data are available within the article (and in the Online Data Supplement). Raw sequencing data have been made publicly available at the SRA and can be accessed under: PRJNA484227.
Animal Tissues
All experiments were performed in 9-week-old male mice. The Ribo-tag mice were purchased from Jackson Laboratory (JAX ID 011029). The Ribo-tag mouse was bred to the αMHC-Cre mice or to Cdh5-CreERT2 mouse lines to obtain cardiac myocyte or endothelial Rpl22HA homozygous mice, respectively. Transverse aortic constriction (TAC) surgery was performed as previously described.14 Animals were randomly assigned to the experimental groups. Sham-operated mice were euthanized at time points matching to TAC surgery time points. Institutional Animal Care and Use Committee approval was obtained for all animal studies.
Parallel Generation of Ribo-seq and RNA-seq Libraries
Ribo-seq and RNA-seq libraries were prepared for each biological replicate. Ribosome footprints were generated after immunoprecipitation of cardiac myocyte-specific monosomes with anti-HA (hemagglutinin) magnetic beads after treating the lysate with RNAse I (ribonuclease I). Libraries were generated according to the mammalian Ribo-seq kit (Illumina). Barcodes were used to perform multiplex sequencing and create sequencing pools containing at least 8 different samples and always an equal amount of both RNA and RPF libraries. Sample pools were sequenced on the HiSeq 2500 platform using 50-bp sequencing chemistry.
Statistics
In vivo experiments were performed on 3 to 7 biological replicates (mice) for each treatment. Throughout the studies, the investigators were blinded to the sample group allocation during the experiment and analysis of the experimental outcome. Statistical analysis was performed using GraphPad Prism 7.0 (Graphpad Software Inc; www.graphpad.com) or R. All data sets were tested for normality of distribution using the Shapiro-Wilks test (threshold P<0.05). For normal distributed data, values shown are mean±SEM. Statistical analysis of data involving 2 groups was performed using unpaired 2 tailed t test, for more than 2 groups, 1-way ANOVA with the Bonferroni test applied to correct for multiple comparisons. For not normally disturbed data, a nonparametric test was used to test for significance between different groups. A Mann-Whitney U test was performed when comparing 2 groups. A Kruskal-Wallis test was used when comparing multiple groups (more than 2), followed by a Dunn multiple test comparison. Read count data generated by RNA-seq and Ribo-seq methods were modeled with the negative binomial distribution, which is the standard for modeling transcriptome data from next-generation sequencing methods.15,16 Experiments involving treatment conditions and batch effect were analyzed while taking full account of biological variability.17
Results
Ribosome Profiling in the Mouse Heart
Ribo-seq has emerged as a global, quantitative technique to study gene expression at the level of translation. Ribo-seq extends ribosome footprinting18 to next-generation sequencing platforms and identifies the position of ≈30-nt footprints in the transcripts, which are protected against nuclease digest. Conversion of these mRNA fragments to DNA allows massively parallel sequencing, which gives information about transcripts that are actively translated (Figure 1A). The Ribo-tag mouse can be used to define the translatome (translated transcriptome) of a specific cell type in the heart by combining affinity tagging of ribosomes with Ribo-seq or TRAP-seq (Figure 1B). The Ribo-tag mouse carries a floxed Rpl22 (a component of the large ribosomal subunit) allele with a wild-type C-terminal exon followed by an identical C-terminal exon with 3 copies of the HAepitope inserted before the stop codon. After crossing with a cell-type–specific Cre recombinase-expressing mouse line, Cre will activate recombination of the floxed exon, leading to the synthesis of HA-Rpl22-tagged ribosomes in the desired cell type. This makes possible the precipitation of HA-tagged ribosomes for polysome profiling (targeted purification of polysomal mRNA; TRAP-seq) or the combination of the ribosome-tagging approach with Ribo-seq, following RNAse digestion of precipitated polysomes.
Figure 1.
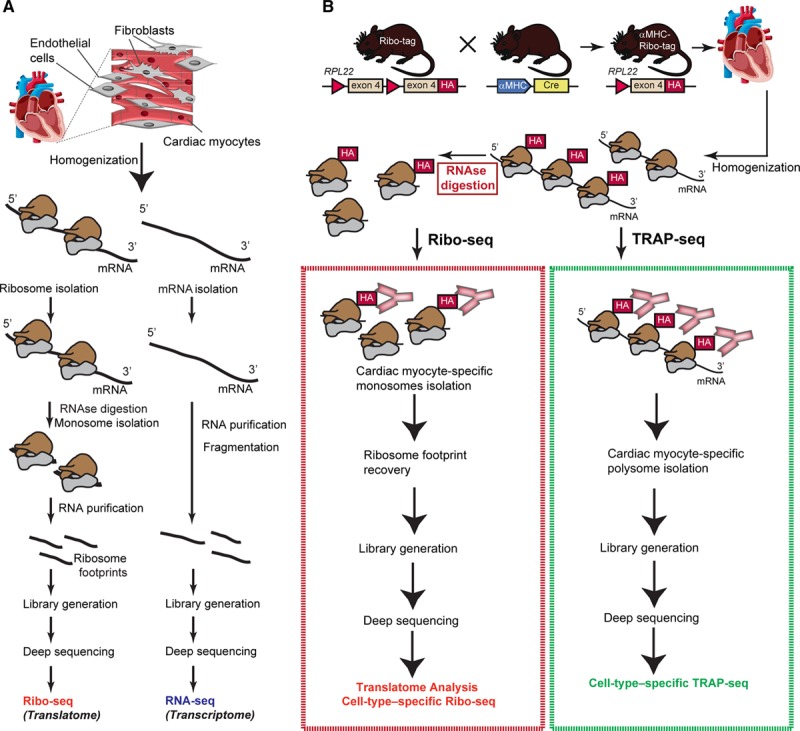
Ribosome profiling in the mouse heart. A, Overview of Ribo-seq and RNA-seq. In Ribo-seq, a small fragment inside the ribosome is protected against RNAse digestion and used for deep-sequencing. B, Overview of the Ribo-tag mouse and schematic drawing of Ribo-seq strategy for cell-type–specific ribosome profiling. A cell-specific promotor (αMHC [alpha-myosin heavy chain] for cardiac myocytes) drives the expression of Cre which induces cell-type–specific HA-tagged RPL22. HA (hemagglutinin)-tagged ribosomes from left ventricle lysates are isolated by affinity purification, subjected to ribosome footprint isolation (Ribo-seq) or polysome profiling (translating ribosome affinity purification [TRAP]-seq) for subsequent deep sequencing and read mapping.
We first established Ribo-seq on mouse whole heart tissue and predicted active translation of ORFs with a previously published computational approach (Rp-bp10; Figure 2A). Digestion of precipitated mRNA-ribosome complexes with RNase produces ≈30-nt regions of transcripts that are covered by ribosomes and protected against enzymatic digestion. These RPFs are size-selected, isolated, converted into cDNA, deep-sequenced, and finally mapped back to the genome to provide quantitative information on mRNA translation. After purification of the RPFs from left ventricular lysates (Online Figure I), libraries were constructed for high-throughput sequencing using the Illumina Ribo-seq kit, per manufacturer’s instructions.
Figure 2.
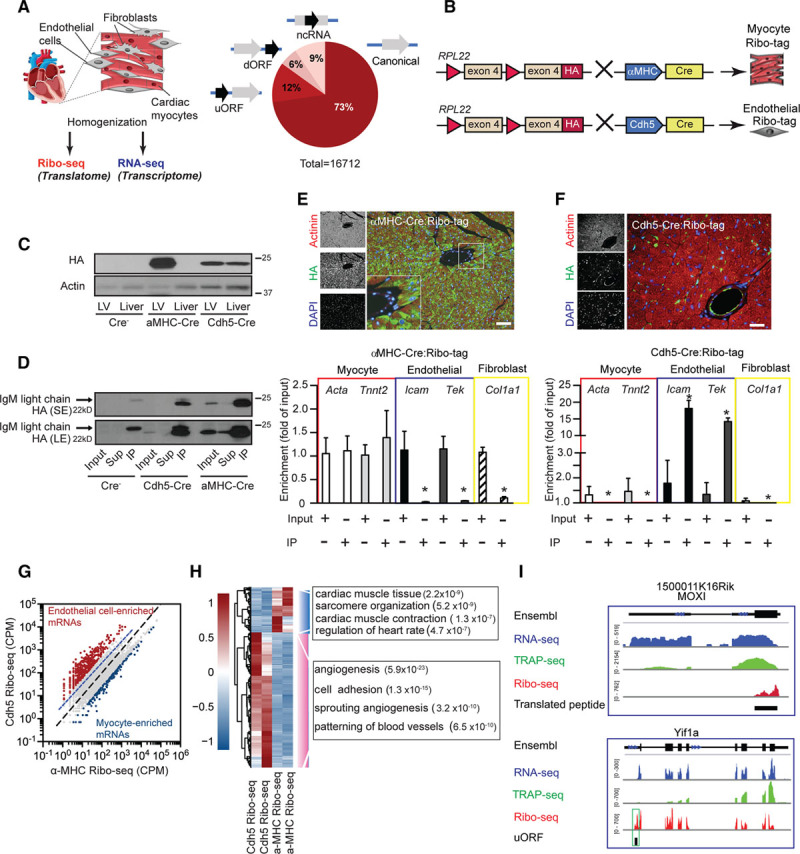
Cell-type–specific ribosome profiling in the mouse heart. A, Overview of Ribo-seq and RNA-seq approach combined with Ribo-tag. Predicted actively translated open reading frames (ORFs) from Ribo-seq data of mouse ventricular tissue and predicted active translation of using the Rp-bp approach.10 B, Schematic drawing of Ribo-seq strategy for cell-type–specific ribosome profiling. C, Immunoblotting of αMHC-Cre:Ribo-tag and Cdh5-CreERT2:Ribo-tag left ventricle and liver lysates. D, Immunoblot of αMHC-Cre:Ribo-tag and Cdh5-CreERT2:Ribo-tag left ventricle lysates after anti-HA (hemagglutinin) immunoprecipitation. E (upper), Immunofluorescence microscopy of heart sections from αMHC-Cre:Ribo-tag hearts. HA antibody (green), actinin (red), and nuclei (blue). Scale bar =100 μm. E (lower), Enrichment of transcripts after anti-HA immunoprecipitation from αMHC-Cre:Ribo-tag lysates. N=3; *P<0.01. F (upper), Immunofluorescence microscopy of heart sections from Cdh5-CreERT2:Ribo-tag hearts. Scale bar =100 μm. F (lower), Enrichment of transcripts after anti-HA immunoprecipitation from Cdh5-CreERT2:Ribo-tag left ventricle lysates. n=3; *P<0.01. Two-tailed Student unpaired t test for E and F. G, Comparison of αMHC-Cre and Cdh5-Cre Ribo-seq libraries. Endothelial-enriched transcripts (red dots) and myocyte-enriched transcripts (blue dots; log2-fold change >2). N=2 individual libraries for each condition. H, Clustering analysis of endothelial and myocyte-enriched transcripts. Hierarchical clustering analysis was performed with the R package ‘pheatmap’ using ‘ward.D2’ and ‘euclidean’ distance algorithm. Scale: scaled gene expression. Enrichment of significant gene ontology terms in the group of regulated genes (Fisher exact test, −log10 P value). I, Ribo-seq (red), RNA-seq (blue), and TRAP (translating ribosome affinity purification)-seq (green) coverage plots for the M. musculus genome loci containing MOXI (ncRNA with a translating ORF) and Yif1a (reads mapped to the 5′ UTR). dORF indicates downstream open reading frame; LE, long exposure; MOXI, micropeptide regulator of β-oxidation; ncRNA, noncoding RNA; SE, short exposure; uORF, upstream open reading frame; and UTR, untranslated regions.
High-quality Ribo-seq libraries have specific properties that distinguish them from conventional RNA-seq data. First, reads map mostly to the coding sequence. Second, Ribo-seq libraries have distinct read-length distribution, which usually peaks at 29 nt. Third, Ribo-seq reads resulting from translating ribosomes exhibit a 3-nt periodicity along the transcript (Online Figure II). The vast majority of ORFs in protein-coding transcripts overlapped with known canonical coding regions (coding), while about 20% of the translated ORFs come from annotated 5′ untranslated regions (UTRs) or noncoding regions (uORF and ncRNA), and few translated ORFs are located in the 3′ UTR of annotated coding regions (dORF [downstream open reading frames]). These results are consistent with ribosome profiling studies in other mammalian cells,12,19 demonstrating feasibility of cell-type–specific Ribo-seq on heart tissue. Similarly, Ribo-seq reads are found in the 5′ UTR of 2787 transcripts (Online Data Set).
Cell-Type–Specific Purification of Ribosomes in the Heart Using the Ribo-Tag Mouse
We crossed the Ribo-tag mouse with mice expressing Cre recombinase under the control of the αMHC (α-myosin heavy chain) promoter that is specifically induced in cardiac muscle cells (αMHC-Cre:Ribo-tag; Figure 2B). For endothelial cell-specific tagging of ribosomes, we crossed the Ribo-tag mouse with Cdh5-CreERT2 mice20 expressing the Cre recombinase under the control of the Cdh5 (cadherin 5) promoter (Cdh5-CreERT2:Ribo-tag). Incorporation of the HA-tagged Rpl22 in actively translating ribosomes was confirmed by immunoblots of polysome fractions and by immunostaining in isolated cardiac myocytes (Online Figure III). Immunoblots confirmed expression of HA-tagged ribosomes in heart lysates from αMHC-Cre:Ribo-tag mice, whereas expression of HA-tagged ribosomes could be found in both heart and liver lysates from Cdh5-CreERT2:Ribo-tag animals, in line with the presence of endothelial cells in both liver and heart tissue (Figure 2C). Cell-type–specific analysis of translation was achieved by affinity purification of HA-tagged ribosomes from lysates of homogenized left ventricular tissue. An optimized immunoprecipitation protocol (Material in the Online Data Supplement) resulted in efficient Rpl22-HA pulldown (Figure 2D). Expression of HA-tagged ribosomes in cardiac myocytes or endothelial cells was also confirmed by immunocytofluorescence microscopy of cardiac sections (Figure 2E and 2F). HA immunoprecipitations were used to isolate HA-RPL22-bound transcripts from αMHC-Cre:Ribo-tag hearts and compared with total mRNA. Quantitative RT-polymerase chain reaction for transcripts of endothelial cells (Tie2 and Icam) or fibroblasts (Col1a1) showed that these transcripts were depleted (P<0.01) in HA-tagged ribosomes isolated from αMHC-Cre:Ribo-tag hearts. Instead, cardiac myocyte-specific transcripts (Acta, Tnnt2) were preserved after anti-HA immunoprecipitation (Figure 2E). Conversely, canonical endothelial transcripts were enriched (P<0.01) in Cdh5-CreERT2:Ribo-tag hearts, where cardiac myocyte markers were depleted (P<0.01; Figure 2F). Having established cell-type–specific ribosome affinity purification, we next established cell-type–specific Ribo-seq of cardiac myocytes and endothelial cells, in vivo. Lysates from Ribo-tag hearts were first digested with RNase I, and then HA-tagged ribosome-mRNA complexes were isolated. After purification of the RPFs, libraries were constructed for high-throughput sequencing using the Illumina Ribo-seq kit, per manufacturer’s instructions.
Quality control of cell-type–specific libraries confirmed characteristic features of Ribo-seq libraries. Only periodic fragment lengths that showed a distinctive triplet periodicity were kept for downstream analysis,10 and a typical library yielded around 2 to 3 million usable (periodic) reads (Online Figure IV). Next, after mapping the reads, we counted the reads mapping to the coding region and analyzed first how well those correlated among 4 different libraries from αMHC-Cre:Ribo-tag purified ribosomes. Pearson correlation values were consistently high, ranging from r = 0.97 to r = 0.99, highlighting the quality and reproducibility of our data. Moreover, we performed mass spectrometry-based quantification of total protein abundance from isolated adult mouse myocytes, and the Ribo-seq data had a higher predictive value of final protein levels in isolated myocytes than RNA-seq (Pearson correlation coefficient r = 0.63 versus 0.55 (Online Figure IV).
We also generated Ribo-seq libraries from isolated neonatal rat ventricular myocytes to compare our cell-type–specific Ribo-seq libraries to data from isolated cardiac myocytes, in addition to whole heart Ribo-seq libraries. Comparison of these data sets confirmed a high overlap of cardiac myocytes transcripts in our cell-type–specific libraries (Online Figure V). Importantly, transcripts that could only be detected (n=672) in whole heart Ribo-seq libraries were enriched (P<0.001) in functional categories including inflammatory response and chemotaxis, suggesting that those are indeed coming from the nonmyocyte fraction in the heart (Online Figure V). Next, we compared Ribo-seq libraries from αMHC-Cre:Ribo-tag to endothelial-specific Ribo-seq libraries from Cdh5-CreERT2:Ribo-tag mice. We found hundreds of transcripts specifically enriched (log2-fold change >2) in Cdh5-CreERT2:Ribo-tag libraries (endothelial transcripts) or αMHC-Cre:Ribo-tag libraries (cardiac myocytes transcripts; Figure 2G). To discover the classes of transcripts preferentially translated in endothelial cells, we performed a gene ontology analysis for genes whose translation is higher (log2-fold change >2) in endothelial cells compared with cardiac myocytes (Figure 2H). Analysis with the biological process component category showed that endothelial-enriched transcripts generally encode proteins that function in processes of angiogenesis or cell adhesion. In contrast, transcripts encoding cardiac myocyte function (cardiac muscle contraction and sarcomere) were depleted from the endothelial cell translatome (log2-fold change <2). Transcripts with the highest ribosome binding (highest 500) were strongly enriched in metabolic control (P<0.001) and regulation of muscle contraction (P<0.001), further confirming cardiac myocyte-enriched transcripts in libraries from αMHC-Cre:Ribo-tag hearts (Online Table I). Overall, these data indicate that Cre-activated HA-tagging followed by Ribo-seq are unbiased means to determine cell-type–specific translatomes in the heart. Moreover, we also generated RNA-seq libraries from transcripts associated with polyribosomes (TRAP-seq). A direct comparison between RNA-seq (blue), TRAP-seq (green), and Ribo-seq (red) read coverage along a previously identified ncRNA with a translating ORF (MOXI [micropeptide regulator of β-oxidation])21 shows specific lack of reads in the untranslated region and read coverage in the coding region only in Ribo-seq libraries (Figure 2I). Moreover, only Ribo-seq allows the identification of uORFs in the 5′ UTR of transcripts. Figure 2I shows read coverage in the 5′ UTR of one of these genes, Yif1a, predicting the potential presence of an uORF.
Collectively, these data illustrate the value of the Ribo-tag approach for cell-type–specific analysis of translated ORFs in the heart and suggest that Ribo-seq is more informative than RNA-seq in terms of overall protein abundance.
Translational Control in Response to Pressure Overload
We next used the cell-type–specific Ribo-seq approach to study translational regulation in cardiac myocytes in response to acute and chronic pressure overload by TAC, a commonly used experimental model for pressure overload-induced cardiac hypertrophy (Figure 3A). We decided to monitor cell-type–specific translation at 3 different time points: an early time point (3 hours after TAC) to monitor the acute response to pressure overload, an intermediate time point (2 days after TAC) based on previous published findings that found increases in protein synthesis and remodeling as early as 2 days after pressure overload,22,23 and a chronic time point (2 weeks after TAC), when severe cellular and molecular remodeling has occurred, but cardiac function is still preserved.
Figure 3.
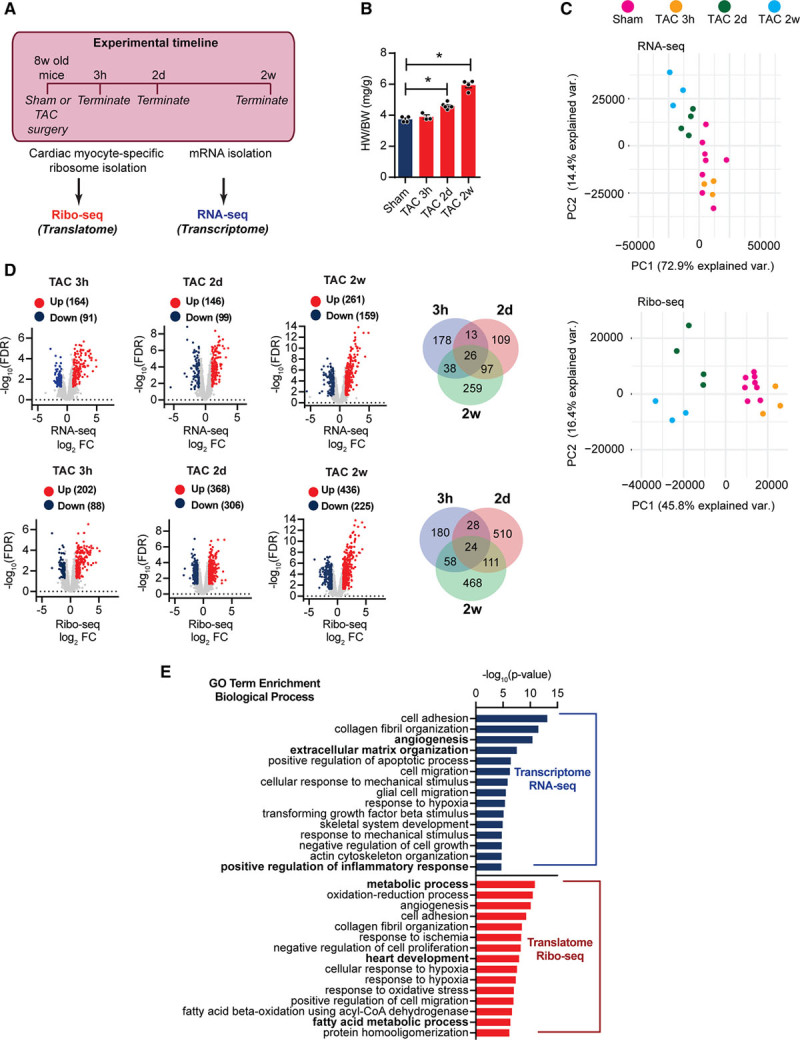
Cardiac myocyte-specific Ribo-seq identifies myocyte translational regulation during cardiac stress by pressure overload. A, Experimental strategy for identification of the translatome and transcriptome during cardiac growth. B, Heart-weight-to-body-weight ratio (HW/BW) 3h, 2d, and 2w after transverse aortic constriction (TAC) surgery. N≥3 for each time point; *P<0.01. One-way ANOVA. C, Principal component analysis of RNA-seq and Ribo-seq libraries after TAC surgery. N≥3 for each time point. D, Translational or transcriptional control in response to TAC surgery. Transcripts were considered significant when false discovery rate <0.05 and Ribo-seq log2-fold change of count per million >1 (upregulated) or <−1 (down-regulated). Venn diagram shows relative relationship between TAC 3h, TAC 2d, and TAC 2w regulation. N≥3 for each time point. E, Enrichment of gene ontology terms (biological process) in differentially expressed transcripts after TAC surgery. −log10 P values, Fisher exact test. FC indicates fold change.
First, we confirmed molecular and cellular phenotypic alterations in our mouse model. Increased heart-weight-to-body-weight ratios were observed 2 days after TAC surgery (Figure 3B). Ribo-seq and RNA-seq in vivo were used to unveil dynamic translational and transcriptional changes under pressure overload stress for 3 hour, 2 day, and 2 week TAC, as well as matching sham surgeries. Information about library quality and samples are provided in Online Figure VI. Global features of transcriptional and translational response to TAC surgery were analyzed by principal component analysis. Interestingly, prominent distinctions could be observed between transcriptional and translational changes after TAC, as translational profiles were clearly different from those in transcription (Figure 3C).
Significantly differentially expressed genes (DEGs; false discovery rate [FDR] <0.05; count per million cutoff >1.0 in all samples, log2-fold change >1) were detected from RNA-seq and Ribo-seq data sets at different time points after TAC or sham surgery (Figure 3D, Material in the Online Data Supplement). Hundreds of DEGs were identified at each time point, with only a small overlap between the different time points (Venn diagrams in Figure 3D). To determine distinct molecular signatures that are associated with responses to pressure overload, we performed pathway analysis of the transcriptional (RNA-seq) and translational (Ribo-seq) DEGs (720 DEGs on RNA-seq, 1379 on Ribo-seq) in TAC-operated animals. Analysis with biological process component category revealed clear differences between RNA-seq and Ribo-seq data and showed that DEGs from Ribo-seq encode proteins that function in processes including metabolism, fatty acid metabolic processes, and muscle-specific processes, whereas DEGs from RNA-seq data regulated extracellular matrix organization or angiogenesis (Figure 3E).
In line, comparison of differences in transcript levels (log2-fold change RNA-seq) to changes in the translatome (log2-fold change Ribo-seq) 3 hours after TAC surgery (Figure 4A) indicated that most transcripts were not significantly (FDR >0.05) regulated at this time point (gray dots), but subsets of transcripts were differentially expressed (FDR <0.05, log2-fold change >1) at the level of transcription only (blue dots), or translation only (red dots), or both (green dots; Figure 4A). Similarly, translational regulation was assessed in αMHC-Cre:Ribo-tag mice 2 days after TAC or sham surgery. DEGs were identified between TAC and sham-operated mice in RNA-seq and Ribo-seq samples. Compared with sham-operated hearts, we identified 306 genes that were significantly less translated (Figure 3D, blue dots) and 368 genes that were more translated (red dots) in TAC-operated hearts (Online Data Set). Comparing differences in transcript levels (log2-fold change RNA-seq) to changes in translation (log2-fold change Ribo-seq) showed that around 400 significantly up- or down-regulated transcripts by Ribo-seq are regulated on the translational level (Figure 4B). Examination of changes in the DEGs showed that specifically at TAC 2D compared with sham about 30% of DEGs were regulated on the transcriptome level, and about 70% were regulated on the translatome level (pie chart in Figure 4B). Importantly, we followed DEGs at different time points at different levels of regulation. These data could not show a substantial delay between alterations of transcript abundance and ribosomal footprint density (Online Figure VII). We identified 225 genes that were significantly less translated (FDR <0.05) and 436 genes that were more translated (red dots) in chronic (2 weeks) TAC-operated hearts (Figure 3D; Online Data Set). In contrast to earlier time points, 30% of DEGs were regulated on the translational level, and 70% were regulated on the transcriptional level (pie chart in Figure 4C). Gene set enrichment analyses identified Kyoto Encyclopedia of Genes and Genomes terms for DEGs that are translationally regulated when compared with transcriptionally regulated DEGs, suggested that specific networks of translational regulated transcripts altered expression of key regulatory genes involved in metabolism, protein synthesis, and signaling cascades at both early and late time points after TAC surgery (Figure 4D).
Figure 4.
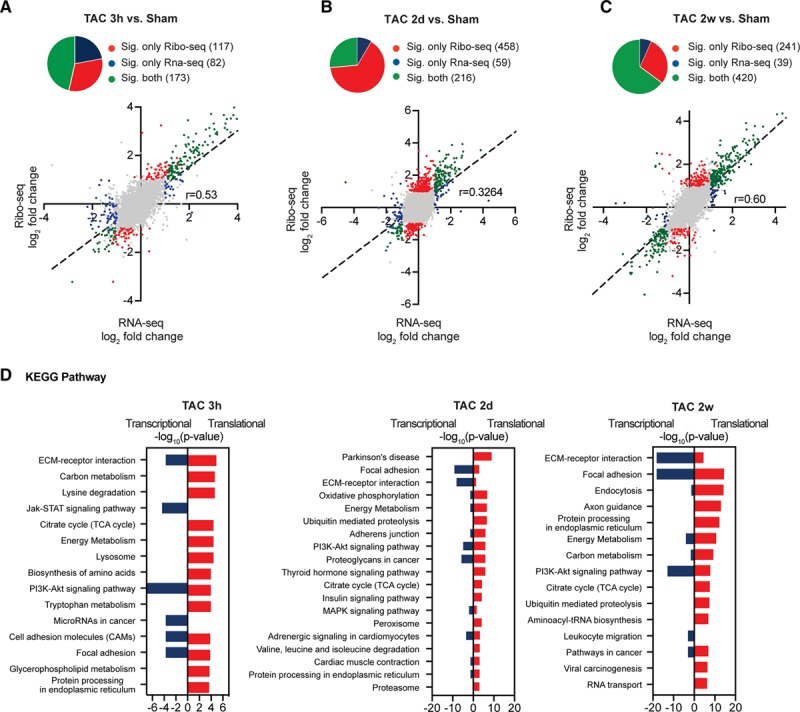
Regulated transcript networks after acute and chronic cardiac stress. A–C, Scatter plots of Ribo-seq vs RNA-seq in sham- and transverse aortic constriction (TAC)-operated mice. Transcripts were considered significant when false discovery rate <0.05. Gray dots indicate no significant change. Significant change at translational level is shown in red, at transcriptional level in blue, and regulation at both translational and transcriptional levels in green. N≥3 for each time point. D, Enrichment of Kyoto Encyclopedia of Genes and Genomes (KEGG) terms for differentially expressed genes. −log10 P values, Fisher exact test. ECM indicates extracellular matrix; FDR, false discovery rate; STAT, signal transducer and activator of transcription; and TCA, tricarboxylic acid cycle.
Widespread Translational Control During Cardiac Hypertrophy
To identify categories of translationally regulated transcripts, we clustered transcriptionally or translationally regulated DEGs into groups that are jointly regulated in a time point-specific stage (Figure 5). Translatome and transcriptome reprogramming during growth response were visualized by k-means clustering in which the expression trends were categorized into 10 clusters. Each cluster is distinctively composed of genes with specific biological functions as highlighted by the cluster-specific enrichment of functional genes sets (gene ontology term biological processes). Genes that encode proteins involved in fibroblast regulation and response to mechanical stimulus were enriched (P<0.001) in cluster 10, representing genes that are differentially regulated very early after TAC (3 hours). Translationally regulated transcripts specific for an early response to TAC in cluster 6 and 8 in which genes showed peak expression 2 days after TAC control protein folding, translation, response to endoplasmic reticulum (ER) stress, transcriptional regulation, and metabolic control. Sustained transcriptional regulation at 2 days and 2 weeks after TAC involved transcripts regulating heart contraction and cellular response to mechanical stimulus (cluster 7). In contrast, translationally down-regulated genes specific after TAC (cluster 3) regulate proteolysis, proteasomal function, and chromatin modifications. Genes regulating mRNA transport, autophagy, and intracellular transport are shown in cluster 4, which represents genes that are less translated after TAC, suggesting a major contribution of translationally regulated clusters for early and long-term adaption to cardiac stress. Transcriptionally regulated genes (cluster 1, 2, and 5) are highly enriched for angiogenesis (P<0.001), cell migration (P<0.001), and tricarboxylic acid cycle metabolism (cluster 9). These data indicate that a specific acute response to cardiac stress is regulated by translation, while chronic stress is regulated at the translational and transcriptional levels, in agreement with the finding that both mechanisms act together on the same transcripts in the later TAC time points.
Figure 5.
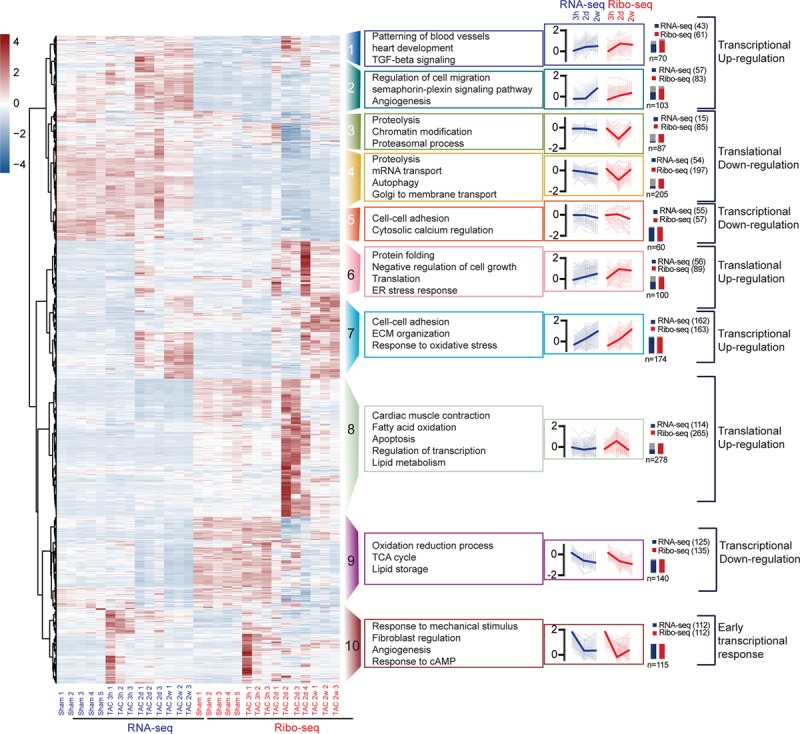
Regulated transcript networks after acute and chronic hemodynamic stress. Unbiased clustering analysis of Ribo-seq and RNA-seq of DEGs 3h, 2d, and 2w after transverse aortic constriction (TAC) surgery. Different colors indicate different clusters. Lines of each gene are transparent, and the lines of average values are in bold in each cluster. Numbers indicate significant genes by RNA-seq or Ribo-seq in the cluster and the total number of genes within the cluster (n). Scale: scaled gene expression. cAMP indicates cyclic adenosine monophosphate; ECM, extracellular matrix; ER, endoplasmic reticulum; TCA, tricarboxylic acid cycle; and TGF, transforming growth factor.
We next aimed to validate our findings using quantitative mass spectrometry-based proteomics from isolated myocytes 2D post-TAC or sham surgery. Again, Ribo-seq data had a higher predictive value of final protein levels in isolated myocytes after TAC surgery than RNA-seq (Pearson correlation coefficient r = 0.60 versus 0.51 (Figure 6A). From 10 microgram sample amounts, we quantified 1783 proteins. Comparing the fold change in protein abundance of translationally regulated transcripts 2D postsurgery (121 out of 458 detected on protein level, highlighted in red) correlated better with the Ribo-seq than RNA-seq (Pearson correlation coefficient r = 0.45 versus 0.19; Figure 6B). In contrast, transcriptionally regulated transcript (180 out of 276, highlighted in green) from Ribo-seq and RNA-seq data correlated similarly with changes in protein abundance (Figure 6B). Increases of individual total protein abundance from translationally regulated clusters, such as translation or protein folding, as well as transcriptionally regulated transcripts, could be confirmed in the proteomics data set (Figure 6C). Coverage plots show representative transcripts of translationally regulated transcripts (EeF2, Rps5, and Trdn) or transcriptional upregulation (Manf; Figure 6D). In addition, selected differentially translated genes were validated in an independent cohort of mice 2D after TAC by immunoblotting (Online Figure VIII).
Figure 6.
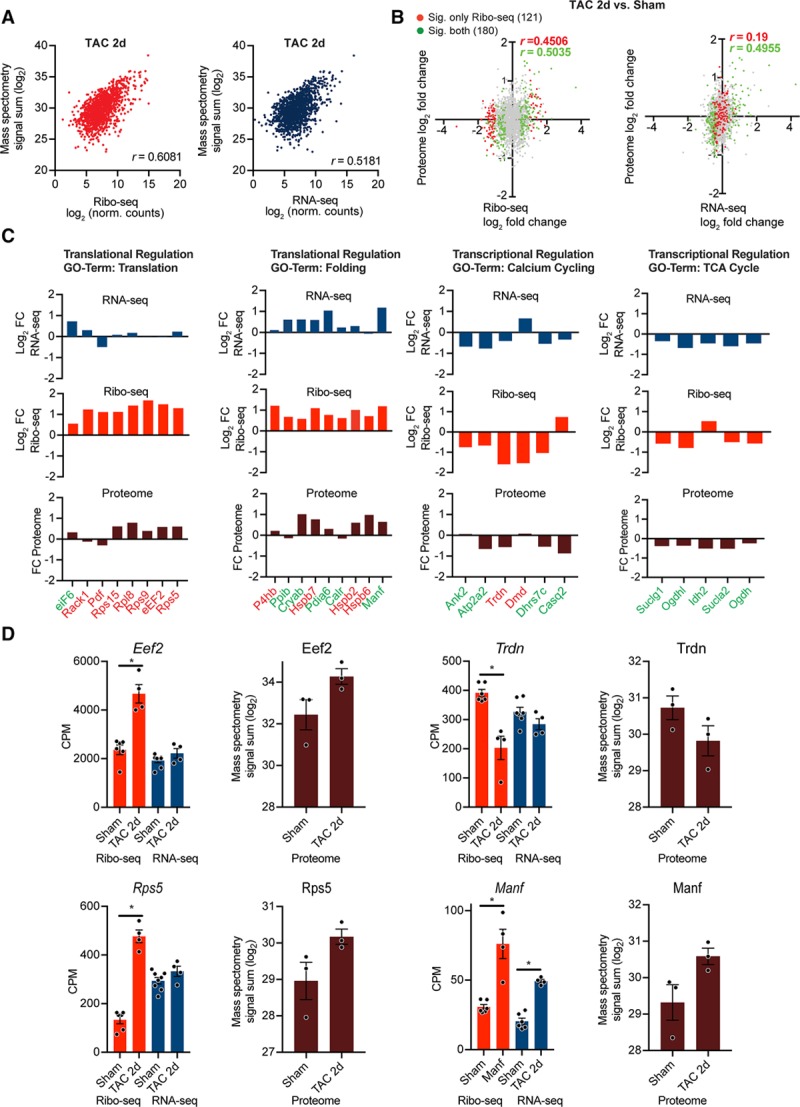
Mass spectrometry-based validation of translationally regulated transcripts. A, Gene-based scatterplot showing the correlation between Ribo-seq (red) or RNA-seq expression levels (blue) and protein abundance derived from isolated myocytes 2 days post surgery. Correlation coefficients are Pearson r values. B, Scatter plot of Ribo-seq or RNA-seq vs changes in protein abundances after transverse aortic constriction (TAC) surgery. Gray dots indicate no significant change, differentially expressed genes (DEGs) at translational level (Ribo-seq) are shown in red, DEGs at transcriptional level (RNA-seq) in green. Correlation coefficients are Pearson’s r values. C, Transcriptional and translational regulation of DEGs and changes in overall protein abundance of genes related to different biological processes from identified clusters in Figure 5. Translationally regulated transcripts (Ribo-seq, false discovery rate [FDR] <0.05) are highlighted in red, transcriptionally regulated transcripts (RNA-seq, FDR <0.05) in green. D, Examples for translational regulation (eEF2, Rps5, and Trdn) and transcriptional regulation (Manf). N=7 Sham, n=4 TAC. Mass spectrometry was performed on myocytes from n≥3 individual mice after TAC or Sham surgery. *FDR <0.05. CPM indicates counts per million.
Overall, these analyses suggest that assessment of cell-type–specific translation with Ribo-seq may provide a more accurate and complete measure of gene expression in the heart than the picture that is presented by analyzing only transcript levels.
Regulatory uORFs as Translational Repressors During Cardiac Hypertrophy
Translational regulation is often attributable to structural characteristics of the transcript, especially of the 5′ UTR, also known as a leader sequence (Figure 7A). uORFs are known regulatory genetic elements.24 By taking advantage of the nucleotide resolution of the Ribo-seq data, uORFs were detected in cardiac myocytes in the Ribo-tag mouse hearts. The proportions of the different ORF types are shown in Figure 7B. Around 85% of the predicted ORFs are canonical annotated coding regions or variants, while about 15% of the ORFs come from annotated 5′ leader regions. One example is shown in Figure 7C. Fbxl3 is less translated 2 weeks after TAC (FDR <0.05) and shows increased read coverage in the uORF in the 5′ UTR, whereas transcription is unchanged. Next, we analyzed the proportion of transcripts with uORFs in the 10 identified clusters (Figure 5). Importantly, transcripts in cluster 6 and 8 (translational upregulation) have less uORFs than transcripts in cluster 3 and 4 (translational downregulation; Figure 7D). Indeed, comparing the differences in transcript levels by RNA-seq and translated transcripts by Ribo-seq highlighted strong translational regulation of transcripts containing an uORF (Figure 7E, P<0.001). In line, transcripts containing an uORF were less translated in response to TAC surgery, whereas transcript levels were unchanged (Figure 7E and 7F).
Figure 7.
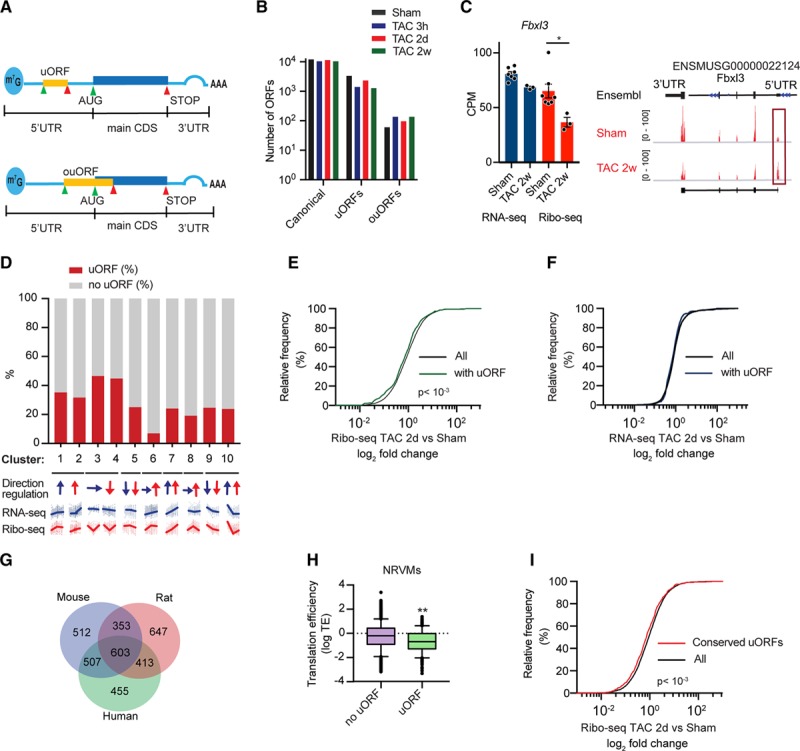
Upstream open reading frame (ORF) expression after transverse aortic constriction (TAC) surgery. A, Scheme of RNA and regulatory elements. B, Number of different ORF types between the different conditions. N≥3 for each time point. C, Ribo-seq coverage plots for the M. musculus genome locus containing Fblx3. N≥3 for each time point; *false discovery rate <0.05. D, Percentage of transcripts with uORF in different clusters (Figure 5). Arrows indicate direction and transcriptional (blue) or translational regulation (red). E, Cumulative fraction of transcripts relative to their fold change of Ribo-seq. F, Cumulative fraction of transcripts relative to their fold change of RNA-seq. P<0.001, Mann-Whitney U test. G, Venn diagram showing conserved uORFs across 3 different species. H, Transcripts with uORFs show decreased translational efficiency in myocytes. N=2; P<0.001, Mann-Whitney U test. Whiskers represent 5% to 95% CI. I, Cumulative fraction of transcripts relative to their fold change of Ribo-seq for conserved uORFs in response to TAC. P<0.001, Mann-Whitney U test. AUG indicates start codon; CDS, coding sequence; CPM, counts per million; NRVMs, neonatal rat ventricular myocytes; ouORF, overlapping upstream ORF; STOP, stop codon; uORF, upstream ORF; and UTR, untranslated region.
To identify human cardiac uORFs, we applied RNA-seq and Ribo-seq to human left ventricular tissue of 2 end-stage patients with dilated cardiomyopathy (Online Figure IX and Material in the Online Data Supplement) and identified more than 20 000 ORFs including uORFs in dilated cardiomyopathy heart tissue (Online Data Set). Sequenced RPFs showed expected size distributions and displayed the 3-nt codon periodicity characteristic of actively translating ribosomes (Online Figure IX). In total, we identified 1372 uORFs in human cardiac tissue. We also identified uORFs in Ribo-seq libraries from neonatal rat ventricular myocytes to identify conserved uORFs across 3 different species. We identified 603 transcripts with uORFs conserved across all 3 species (Figure 7G and Online Table II). To examine the sequence conservation of uORFs, we used the phastCons scores calculated from the vertebrate genome alignment. By this measure, uORFs were less well conserved than coding regions, but more than flanking 5′ UTR regions (Online Figure X). This intermediate conservation is supported by our finding that hundreds of predicted uORFs are only present in one species (Figure 7G). Moreover, Ribo-seq libraries from neonatal rat ventricular myocytes showed that transcripts with the presence of a uORF in the leader sequence have a significantly decreased translational efficiency, further confirming the overall repressive function of uORFs in cardiac myocytes (Figure 7H, P<0.01). In line, transcripts with conserved uORFs across different species were also less translated in response to TAC surgery (Figure 7I, P<0.001). These data indicate novel insight into the regulatory potential within the mRNA leader sequence in cardiac myocytes and their capacity to modulate translation. In addition, microRNAs (miRNAs) are small noncoding RNAs that extensively regulate gene expression partly by suppressing translation. In line, miRNA target motif analysis identified several enriched (P<0.05) miRNA motifs in translationally regulated DEGs in clusters 3 and 4 (Online Table III).
uORFs Regulate Translation of Key Proteins Involved in Energy Homeostasis
Although the exact mechanisms underlying translation of uORF and their effects on canonical downstream ORF expression are still unknown, these elements may be under selective control during stress conditions and may confer stress-regulated modulation of translation of the canonical ORF. To identify potential translationally controlled transcripts containing an uORF involved in early cardiac remodeling, we focused our next analysis on transcripts in cluster 3 and 4, since around 40% of the transcripts in this cluster contain an uORF (Figure 7D). We found a decrease in translation of transcripts with an uORF within cluster 3 and 4 after TAC (Figure 8A, P<0.001), whereas transcription was unchanged (Figure 8B). Fifty-one genes were significantly (FDR <0.05) less translated at 2 days and 2 weeks after TAC (Figure 8C). These genes were enriched in processes, including cellular growth control (P<0.001) and metabolic control (P<0.01). Among those, 2 transcripts are interesting key regulators of cell growth, autophagy, and metabolism (Flcn and Fnip1). Fnip1 (folliculin-interacting protein) mutants and Fnip1 deficient mice develop cardiomyopathies with severe metabolic defects.25 Similarly, loss of folliculin (Flcn) causes severe cardiac hypertrophy with deregulated energy homeostasis with cardiac failure.26 Flcn and Fnip1 form a complex and bind to AMPK (AMP-activated kinase), a critical energy-sensing molecule that broadly regulates metabolic pathways. Both Fnip1 and Flcn have been shown to be endogenous regulators of AMPK,27 and the Fnip/Flcn pathway regulates cardiac myocyte cell growth through modulation of the AMPK–mechanistic target of rapamycin (mTOR) axis.26 Importantly, the predicted uORF in the leader sequence of the Fnip gene was conserved between human and mouse tissue (Figure 8D). Fnip1 and Flcn were significantly less translated at 2 days and 2 weeks after TAC (FDR <0.05), whereas Flcn transcription was unchanged and Fnip1 transcription is only significantly decreased 2 weeks after TAC (FDR <0.05; Figure 8E). Finally, we confirmed decreased expression of FNIP1 and FLCN in human cardiomyopathies, whereas transcripts levels were unchanged (Figure 8G and 8H; P<0.05).
Figure 8.
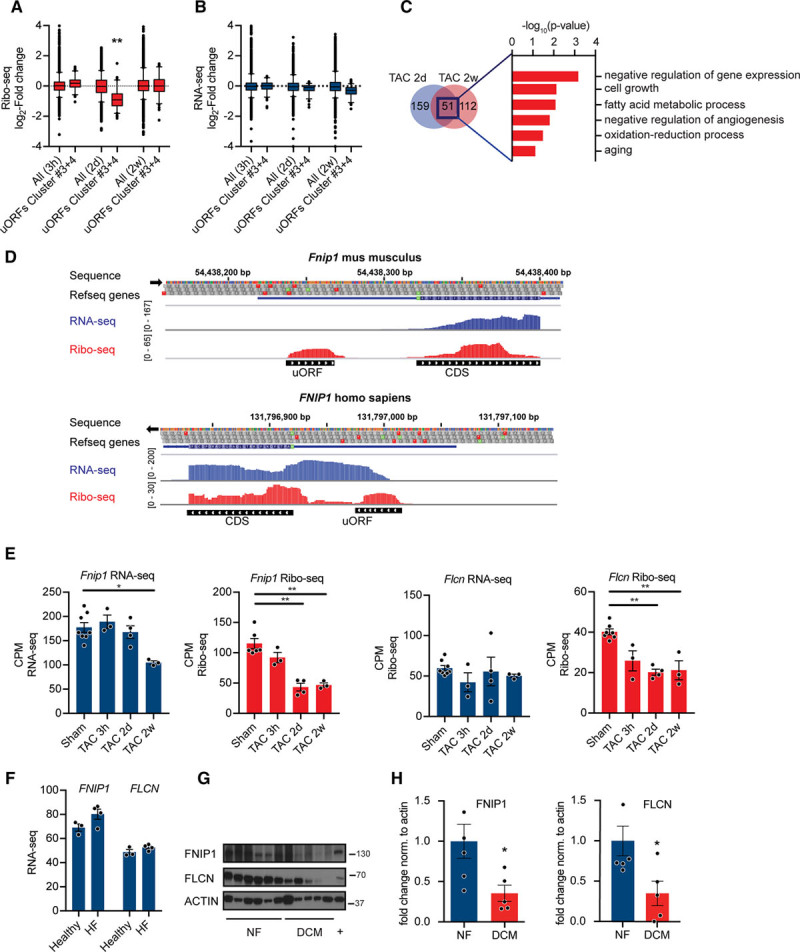
Regulation of gene expression by upstream open reading frame (uORFs). A, Ribo-seq counts per million (CPM) for transcripts in cluster 4 at different time points after transverse aortic constriction (TAC) surgery. N≥3 for each time point; **P<0.01 Kruskal-Wallis test. Whiskers represent 5% to 95% CI. B, RNA-seq CPM for transcripts in cluster 4 at different time points after TAC surgery. N≥3 for each time point. C, Venn diagram showing overlap between significantly less translated transcripts containing a regulatory uORF at 2d and 2w after TAC. Enrichment of gene ontology terms (biological process) in differentially expressed transcripts after TAC surgery with uORFs. −log10 P values, Fisher exact test. D, Ribo-seq (red), RNA-seq (blue), and coverage plots for the H. sapiens and M. musculus genome loci containing FNIP1 (folliculin-interacting protein 1) with reads mapped to the 5′ UTR. E, Ribo-seq CPM and RNA-seq CPM for Fnip1 and Flcn (folliculin) at different time points after TAC surgery. *False discovery rate (FDR) <0.05, **FDR <0.01. F, RNA-seq counts for FNIP1 and FLCN in human heart samples. N=4. G, Immunoblots and (H) quantification for FNIP1 and FLCN in human heart samples confirming decreased expression in dilated cardiomyopathies (DCM). P<0.05; n≥5. Two-tailed Student unpaired t test. CDS indicates coding sequence.
Collectively, these data indicate that translational control contributes to early changes in gene expression of key regulators of cardiac metabolism. Mechanistically, regulatory uORFs could affect translation of canonical downstream ORFs.
Discussion
Compared with our substantial understanding of transcriptional networks and their regulation, knowledge of translational regulation during cardiac stress is still in its infancy. Previous studies have shown an increase in global protein synthesis in models of acute pressure overload,22,23 but the relative role of the translatome between steady-state and adaptation in response to stress in the heart, and a genome-wide overview for translational regulation, such as exists for transcriptional regulation, were missing. It is becoming clear that absolute abundances of transcripts often poorly correlate with those of proteins, and recent reports confirmed that cardiac disease gene expression profiles had only a limited commonality at the transcriptome and proteome levels.28 In line with this notion, an integrated approach combining measurements of transcript abundance, protein abundance, and protein turnover in the hypertrophied myocardium supported the notion that both transcriptional and posttranscriptional mechanisms affect complex disease development.3
We present here, to the best of our knowledge, for the first time a cell-type–specific Ribo-seq technique to study translational control, in vivo, in cardiac myocytes. Previous studies have used the Ribo-tag technology combined with polysome sequencing in the heart,9,29 but the combination of Ribo-tag with Ribo-seq has only been done only in few studies.12,13,30 Polysome profiling suffers from a number of drawbacks. Connecting co-sedimentation of a transcript with polysomes to its translation is rather a crude assumption, as several other large ribonucleoprotein complexes can co-sediment with polysomes without active translation and many mRNAs associated with ribosomes are not actively translated.31,32 Moreover, polysome profiling cannot identify additional ORFs along the transcript, such as upstream ORFs in the 5′ UTR, which can independently undergo translation and act as translational regulators of the main canonical protein-coding ORF. Ribo-seq enables identification of all translating ORFs, which can reveal translation occurring outside of canonical ORFs, translation from alternative initiation sites, usage of non-AUG (alternative) start codons, or small ORFs which reside in previously annotated noncoding RNAs.
Ribo-seq has a few drawbacks. First, since only a fraction of total cellular mRNA undergoes active translation at any given time point, library generation requires sufficient input material, and sample preparation, and bioinformatic analyses make Ribo-seq experiments costly and time-consuming. Moreover, RNA-binding proteins have been shown to regulate translation elongation rates by stalling of ribosomes along the ORF.33 Stalling of ribosome could be misinterpreted as increased ribosome footprint density in Ribo-seq experiments.
Overall, both methods, TRAP-seq and Ribo-seq, will enable researchers to map and measure mRNA translation, in vitro, and, in vivo. As these methods become more readily available to different researchers, it is likely that multiple approaches will be used in combination for a more comprehensive assessment of translational regulation. Cell-type–specific Ribo-seq can also be used for cell-type–specific gene expression analysis of genetically modified mouse models such as knock-out or transgenic animals. Another fascinating development will be profiling translation of transcripts from specific subcellular compartments such as the ER or mitochondria.34,35
Applying Ribo-seq in vivo after TAC, we identified subsets of mRNA that are regulated at the translational level. The relative contribution of translational control was especially high at the earlier time points after TAC surgery, whereas chronic stress is regulated at the translational and transcriptional levels, in agreement with the finding that both mechanisms act together on the same transcripts in the later TAC time points. This suggests that translation of mRNAs is important for stressed cells by triggering a rapid and specific response to stress before transcriptional changes influence cellular fate.36
Our study supports previous studies of pathological hypertrophy showing aberrant transcriptional activation resulting in a transcriptional remodeling in response to stress.2,4 The present findings extend these findings towards an additional cytosolic gene expression remodeling process controlled at the level of mRNA translation. On pressure overload, myocytes activated specific translationally controlled modules, which involved protein quality control, translational regulation, stress response, metabolic remodeling, as well as transcriptional activation. This could be partly explained by structural characteristics of transcripts, especially of the 5′ UTR. Specifically, our data show early translational upregulation of ribosomal proteins as well as initiation and elongation factors such as Eef2 (elongation factor 2), Eif6 (eukaryotic translation initiation factor 6), components of the Eif3 (eukaryotic translation initiation factor 3) complex, as well as regulatory proteins such as 4-EBP1 (eIF4E-binding protein 1) early after TAC surgery. Increased protein synthesis is one of the cardinal features of cardiac myocyte hypertrophy22 and regulated at the level of initiation and elongation. Increased expression of Eef2 has been shown to promote cellular growth by increased protein synthesis,37,38 possibly linking translational elongation to pathological hypertrophy in response to TAC. In contrast, 4-EBP1 is a well-known translational inhibitor,39 suggesting a negative feedback mechanism to prevent further protein synthesis controlled at the level of translation. Increase in protein synthesis is coordinate with increase in protein folding demand, resulting in subsequent transcriptional and translational upregulation of proteins involved in protein folding and response to ER stress. While the molecular mechanisms underlying the maintenance of ER proteostasis during cardiac myocyte growth are not well understood, it was recently shown that the ER stress transcription factor ATF6 (activating transcription factor 6α) upregulated Ras homolog enriched in brain, an activator of mTORC1, linking ER stress and mTOR activation, which could explain how proteostasis is achieved during hypertrophic growth.40
The most notable change in the metabolic profile of hypertrophied hearts is an increased reliance on glucose with an overall reduced oxidative metabolism. Coordinate with a large number of previous studies; we observed downregulation of genes involved in mitochondrial oxidative metabolism during the development of pathological hypertrophy. Intriguingly, components of the translational machinery, as well as regulators of protein synthesis, such as mTOR, have been linked to metabolic regulation. Eif6 and Eif3, both upregulated early after TAC, have been shown to regulate translation of regulators of lipogenesis and glycolysis, as well as mitochondrial proteins,41,42 whereas mTOR activates a complete metabolic regulatory network.43 Ribo-seq now enables us to investigate the specific translational network downstream of initiation and elongation factors or other components of the translational machinery, in vivo, in disease models. This will allow further characterization of the cross-talk between protein synthesis and myocyte energetics. Both processes are heavily dysregulated in cardiac disease states independent from the cause. Improved understanding of translational control could result in novel therapeutic avenues based on specific translational inhibition. Our data also support the idea that metabolic remodeling precedes, triggers, and maintains structural and functional remodeling of the heart.44 Moreover, it also suggests that early translational control regulates gene expression of metabolic genes in response to acute pathological stress.
Mechanistically, we identified regulatory uORFs in myocytes as cis-acting elements for translational regulation. uORFs are sequences defined by an initiation codon with a termination codon located upstream or downstream of the main start codon. We identified hundreds of potentially regulatory uORFs, including uORFs in significantly differentially regulated transcripts. While the individual effect of a specific uORF on translation of the coding sequence is unclear, our data support the overall repressive effect of a uORFs on the coding sequence, since translation of transcripts containing uORFs was repressed. Specifically, we identified regulatory uORFs in key proteins regulating autophagy and metabolism—Flcn and Fnip1. Previous work has highlighted the critical role of both Fnip1 and Flcn in regulating the integrity of the AMPK-mTOR signaling pathway in myocytes; however, it was unknown that expression of both proteins is decreased at the translational level in response to pressure overload. Decreased expression of Flcn and Fnip1 complex could contribute to dysregulation of the AMPK-mTOR signaling pathway early after pressure overload. Importantly, several studies have suggested AMPK as a promising target for inhibition of cardiac hypertrophy.45,46
Further characterization of the mechanism by which altered uORF-mediated translational control of Flcn and Fnip1 affects cell fate might uncover new therapeutic targets. In line, recent studies showed that antisense oligonucleotide technology against uORFs increased translation of the canonical downstream ORF both in vitro and in vivo.47 This could be especially very relevant for cardiac diseases, which often result from decreased protein expression that is, for Flcn and Fnip1, which are decreased in expression during disease progression. Hence, one novel strategy for enhancing expression could be by blocking uORFs.
In summary, we describe a new method to analyze cell-type–specific gene expression at the translational level in the mouse heart. Applying this technology, our study provides a comprehensive characterization of translational regulation in cardiac myocytes in response to pressure overload, in vivo. We identified dynamic translational control of fundamental cellular processes, such as translational and metabolic regulators, as well as ER stress response. The magnitude of translational control early after the induction of pathological stress, involving hundreds of transcripts only regulated on the translational level, suggests that regulatory mechanisms and transcript-specific translational regulators bestow a cell-type–specific gene expression program that is crucial for adaptation to stress conditions.
Knowing which genes are expressed provides a foundation to discover novel mechanisms across all areas of cardiovascular pathophysiology. Understanding how changes in gene expression at multiple levels of regulation allow the heart to adapt the translatome and thus cellular function to stress conditions will hopefully also result in new therapeutic concepts.
Sources of Funding
H.A. Katus, J. Burghaus, M. Völkers, S. Doroudgar, T. Jakobi, and C. Dieterich acknowledge the DZHK (German Center for Cardiovascular Research) Partner Site Heidelberg/Mannheim. C. Dieterich and T. Jakobi acknowledge funding from the Klaus-Tschira Stiftung GmbH. S. Doroudgar acknowledges the European Society of Cardiology Basic Research Fellowship and the DZHK Excellence Program. M. Völkers acknowledges the DFG (German Research Foundation, DFG VO 1659 2/1 and DFG VO 1659 4/1). C. Hofmann acknowledges the German Cardiac Society (DGK).
Disclosures
None.
Supplementary Material
{kind=link}
Nonstandard Abbreviations and Acronyms
- αMHC
- α-myosin heavy chain
- AMPK
- AMP-activated kinase
- Cdh5
- cadherin 5
- DEGs
- differential expressed genes
- 4-EBP1
- eIF4E-binding protein 1
- ER
- endoplasmic reticulum
- Flcn
- folliculin
- Fnip1
- folliculin-interacting protein
- HA
- hemagglutinin
- mTOR
- mechanistic target of rapamycin
- ncRNA
- noncoding RNA
- nt
- nucleotide
- RNAse I
- ribonuclease I
- RPF
- ribosome protected mRNA fragments
- RPL22
- 60S ribosomal protein L22
- TAC
- transverse aortic constriction
- TRAP
- translating ribosome affinity purification
- uORF
- upstream open reading frame
Current address for B. Malone: NEC Laboratories Europe, Kurfürsten-Anlage 36, 69115 Heidelberg, Germany.
C.D. and M.V. contributed equally to this article.
For Sources of Funding and Disclosures, see page 447.
The online-only Data Supplement is available with this article at https://www.ahajournals.org/doi/suppl/10.1161/CIRCRESAHA.119.314817.
Novelty and Significance
What Is Known?
Aberrant changes in myocardial gene expression represent a fundamental feature of both diseased human hearts as well as many animal models of cardiac remodeling.
Transcript levels often poorly correlate with synthesis levels of the encoded proteins.
Ribo-seq overcomes limitations of classical expression analysis as it directly quantifies the number of translating ribosomes, thereby integrating gene expression control at the level of transcription and translation.
What New Information Does This Article Contribute?
Cell-type–specific translation in cardiac myocytes was monitored by purifying tagged cardiac myocyte ribosomes and subjecting the ribosome-protected mRNA fragments to deep sequencing (Ribo-seq).
By combining Ribo-seq with a ribosome-tagging approach, it was possible to determine changes in the translatome in response to hemodynamic stress in cardiac myocytes, in vivo.
Subsets of transcripts were regulated at the translational level in myocytes in response to pressure overload induced by transverse aortic constriction.
Regulatory upstream open reading frames (ORF) in cardiac myocytes are cis-acting elements for translational regulation in response to transverse aortic constriction.
Numerous gene expression studies based on transcript profiling in human heart failure or experimental heart failure models have been performed, providing large data sets describing the networks of transcriptional regulation after pathological stress. In contrast, the contribution of translational control to gene expression changes and phenotypic changes, in vivo, in the heart are poorly understood. In this study, we have mapped the dynamic translational response of cardiac myocytes to pathological stress by using cell-type–specific Ribo-seq. We could show that translation of subsets of transcripts is critical for cardiac myocyte remodeling by triggering a rapid and specific response to transverse aortic constriction independent of transcript changes. We identified dynamic translational control of fundamental cellular processes, such as translational regulation, endoplasmic reticulum stress response, and metabolism. Mechanistically, regulatory upstream open reading frames have an overall repressive effect on the translation of the main ORF. Cell-type–specific Ribo-seq can be used as starting point for studies of mechanisms of translational regulation. Potential mechanisms are the stress-induced relief of ribosomes paused on mRNAs, altered translation initiation kinetics, control of translation initiation by upstream open reading frames, use of alternative reading frames or start codons, as well as discovery of translation outside of the annotated coding regions.
References
- 1.Mudd JO, Kass DA. Tackling heart failure in the twenty-first century. Nature. 2008;451:919–928. doi: 10.1038/nature06798. doi: 10.1038/nature06798. [DOI] [PubMed] [Google Scholar]
- 2.Preissl S, Schwaderer M, Raulf A, Hesse M, Grüning BA, Köbele C, Backofen R, Fleischmann BK, Hein L, Gilsbach R. Deciphering the epigenetic code of cardiac myocyte transcription. Circ Res. 2015;117:413–423. doi: 10.1161/CIRCRESAHA.115.306337. doi: 10.1161/CIRCRESAHA.115.306337. [DOI] [PubMed] [Google Scholar]
- 3.Lau E, Cao Q, Lam MPY, Wang J, Ng DCM, Bleakley BJ, Lee JM, Liem DA, Wang D, Hermjakob H, et al. Integrated omics dissection of proteome dynamics during cardiac remodeling. Nat Commun. 2018:9, 120. doi: 10.1038/s41467-017-02467-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thum T, Condorelli G. Long noncoding RNAs and microRNAs in cardiovascular pathophysiology. Circ Res. 2015;116:751–762. doi: 10.1161/CIRCRESAHA.116.303549. doi: 10.1161/CIRCRESAHA.116.303549. [DOI] [PubMed] [Google Scholar]
- 5.Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D’Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, et al. Revisiting cardiac cellular composition. Circ Res. 2016;118:400–409. doi: 10.1161/CIRCRESAHA.115.307778. doi: 10.1161/CIRCRESAHA.115.307778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 7.Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sanz E, Yang L, Su T, Morris DR, McKnight GS, Amieux PS. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc Natl Acad Sci USA. 2009;106:13939–13944. doi: 10.1073/pnas.0907143106. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou P, Zhang Y, Ma Q, Gu F, Day DS, He A, Zhou B, Li J, Stevens SM, Romo D, et al. Interrogating translational efficiency and lineage-specific transcriptomes using ribosome affinity purification. Proc Natl Acad Sci USA. 2013;110:15395–15400. doi: 10.1073/pnas.1304124110. doi: 10.1073/pnas.1304124110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malone B, Atanassov I, Aeschimann F, Li X, Großhans H, Dieterich C. Bayesian prediction of RNA translation from ribosome profiling. Nucleic Acids Res. 2017;45:2960–2972. doi: 10.1093/nar/gkw1350. doi: 10.1093/nar/gkw1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schafer S, Adami E, Heinig M, Rodrigues KEC, Kreuchwig F, Silhavy J, van Heesch S, Simaite D, Rajewsky N, Cuppen E, et al. Translational regulation shapes the molecular landscape of complex disease phenotypes. Nat Commun. 2015;6:7200. doi: 10.1038/ncomms8200. doi: 10.1038/ncomms8200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao X, Wan J, Liu B, Ma M, Shen B, Qian SB. Quantitative profiling of initiating ribosomes in vivo. Nat Methods. 2015;12:147–153. doi: 10.1038/nmeth.3208. doi: 10.1038/nmeth.3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hornstein N, Torres D, Das Sharma S, Tang G, Canoll P, Sims PA. Ligation-free ribosome profiling of cell type-specific translation in the brain. Genome Biol. 2016;17:149. doi: 10.1186/s13059-016-1005-1. doi: 10.1186/s13059-016-1005-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Doroudgar S, Völkers M, Thuerauf DJ, Khan M, Mohsin S, Respress JL, Wang W, Gude N, Müller OJ, Wehrens XH, et al. Hrd1 and ER-Associated Protein Degradation, ERAD, are critical elements of the adaptive ER Stress Response in cardiac myocytes. Circ Res. 2015;117:536–546. doi: 10.1161/CIRCRESAHA.115.306993. doi: 10.1161/CIRCRESAHA.115.306993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biol. 2010;11:R106. doi: 10.1186/gb-2010-11-10-r106. doi: 10.1186/gb-2010-11-10-r106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li W, Wang W, Uren PJ, Penalva LOF, Smith AD. Riborex: fast and flexible identification of differential translation from ribo-seq data. Bioinformatics. 2017;33:1735–1737. doi: 10.1093/bioinformatics/btx047. doi: 10.1093/bioinformatics/btx047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 2012;40:4288–4297. doi: 10.1093/nar/gks042. doi: 10.1093/nar/gks042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Steitz JA. Polypeptide chain initiation: nucleotide sequences of the three ribosomal binding sites in bacteriophage R17 RNA. Nature. 1969;224:957–964. doi: 10.1038/224957a0. doi: 10.1038/224957a0. [DOI] [PubMed] [Google Scholar]
- 19.Ingolia NT, Lareau LF, Weissman JS. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. doi: 10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y, Nakayama M, Pitulescu ME, Schmidt TS, Bochenek ML, Sakakibara A, Adams S, Davy A, Deutsch U, Lüthi U, et al. Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature. 2010;465:483–486. doi: 10.1038/nature09002. doi: 10.1038/nature09002. [DOI] [PubMed] [Google Scholar]
- 21.Makarewich CA, Baskin KK, Munir AZ, Bezprozvannaya S, Sharma G, Khemtong C, Shah AM, McAnally JR, Malloy CR, Szweda LI, et al. MOXI Is a mitochondrial micropeptide that enhances fatty acid β-oxidation. Cell Rep. 2018;23:3701–3709. doi: 10.1016/j.celrep.2018.05.058. doi: 10.1016/j.celrep.2018.05.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zimmer HG, Steinkopff G, Gerlach E. Changes of protein synthesis in the hypertrophying rat heart. Pflugers Arch. 1972;336:311–325. doi: 10.1007/BF00586956. [DOI] [PubMed] [Google Scholar]
- 23.Souders CA, Borg TK, Banerjee I, Baudino TA. Pressure overload induces early morphological changes in the heart. Am J Pathol. 2012;181:1226–1235. doi: 10.1016/j.ajpath.2012.06.015. doi: 10.1016/j.ajpath.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morris DR, Geballe AP. Upstream open reading frames as regulators of mRNA translation. Mol Cell Biol. 2000;20:8635–8642. doi: 10.1128/mcb.20.23.8635-8642.2000. doi: 10.1128/mcb.20.23.8635-8642.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Siggs OM, Stockenhuber A, Deobagkar-Lele M, et al. Mutation of Fnip1 is associated with B-cell deficiency, cardiomyopathy, and elevated AMPK activity. Proc Natl Acad Sci USA. 2016;113:E3706–E3715. doi: 10.1073/pnas.1607592113. doi: 10.1073/pnas.1607592113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hasumi Y, Baba M, Hasumi H, Huang Y, Lang M, Reindorf R, Oh HB, Sciarretta S, Nagashima K, Haines DC, et al. Folliculin (Flcn) inactivation leads to murine cardiac hypertrophy through mTORC1 deregulation. Hum Mol Genet. 2014;23:5706–5719. doi: 10.1093/hmg/ddu286. doi: 10.1093/hmg/ddu286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wada S, Neinast M, Jang C, Ibrahim YH, Lee G, Babu A, Li J, Hoshino A, Rowe GC, Rhee J, et al. The tumor suppressor FLCN mediates an alternate mTOR pathway to regulate browning of adipose tissue. Genes Dev. 2016;30:2551–2564. doi: 10.1101/gad.287953.116. doi: 10.1101/gad.287953.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foster DB, Liu T, Kammers K, O’Meally R, Yang N, Papanicolaou KN, Talbot CC, Jr, Cole RN, O’Rourke B. Integrated omic analysis of a guinea pig model of heart failure and sudden cardiac death. J Proteome Res. 2016;15:3009–3028. doi: 10.1021/acs.jproteome.6b00149. doi: 10.1021/acs.jproteome.6b00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen Q, Zhang H, Liu Y, Adams S, Eilken H, Stehling M, Corada M, Dejana E, Zhou B, Adams RH. Endothelial cells are progenitors of cardiac pericytes and vascular smooth muscle cells. Nat Commun. 2016;7:12422. doi: 10.1038/ncomms12422. doi: 10.1038/ncomms12422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gonzalez C, Sims JS, Hornstein N, Mela A, Garcia F, Lei L, Gass DA, Amendolara B, Bruce JN, Canoll P, et al. Ribosome profiling reveals a cell-type-specific translational landscape in brain tumors. J Neurosci. 2014;34:10924–10936. doi: 10.1523/JNEUROSCI.0084-14.2014. doi: 10.1523/JNEUROSCI.0084-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meijer HA, Thomas AAM. Control of eukaryotic protein synthesis by upstream open reading frames in the 5′-untranslated region of an mRNA. Biochem J. 2002;367:1–11. doi: 10.1042/BJ20011706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Juntawong P, Girke T, Bazin J, Bailey-Serres J. Translational dynamics revealed by genome-wide profiling of ribosome footprints in arabidopsis. Proc Natl Acad Sci USA. 2014;111:E203–E212. doi: 10.1073/pnas.1317811111. doi: 10.1073/pnas.1317811111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Darnell JC, Van Driesche SJ, Zhang C, Hung KY, Mele A, Fraser CE, Stone EF, Chen C, Fak JJ, Chi SW, et al. FMRP stalls ribosomal translocation on mRNAs linked to synaptic function and autism. Cell. 2011;146:247–261. doi: 10.1016/j.cell.2011.06.013. doi: 10.1016/j.cell.2011.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Williams CC, Jan CH, Weissman JS. Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science. 2014;346:748–751. doi: 10.1126/science.1257522. doi: 10.1126/science.1257522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reid DW, Nicchitta CV. LOCAL TRANSLATION. Comment on “ principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling”. Science. 2015;348:1217. doi: 10.1126/science.aaa7257. doi: 10.1126/science.aaa7257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature. 2012;485:109–113. doi: 10.1038/nature11083. doi: 10.1038/nature11083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakamura J, Aoyagi S, Nanchi I, Nakatsuka S, Hirata E, Shibata S, Fukuda M, Yamamoto Y, Fukuda I, Tatsumi N, et al. Overexpression of eukaryotic elongation factor eEF2 in gastrointestinal cancers and its involvement in G2/M progression in the cell cycle. Int J Oncol. 2009;34:1181–1189. [PubMed] [Google Scholar]
- 38.Pott LL, Hagemann S, Reis H, Lorenz K, Bracht T, Herold T, Skryabin BV, Megger DA, Kälsch J, Weber F, et al. Eukaryotic elongation factor 2 is a prognostic marker and its kinase a potential therapeutic target in HCC. Oncotarget. 2017;8:11950–11962. doi: 10.18632/oncotarget.14447. doi: 10.18632/oncotarget.14447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hay N, Sonenberg N. Upstream and downstream of mTOR. Genes Dev. 2004;18:1926–1945. doi: 10.1101/gad.1212704. doi: 10.1101/gad.1212704. [DOI] [PubMed] [Google Scholar]
- 40.Blackwood EA, Hofmann C, Santo Domingo M, Bilal AS, Sarakki A, Stauffer W, Arrieta A, Thuerauf DJ, Kolkhorst FW, Müller OJ, et al. ATF6 regulates cardiac hypertrophy by transcriptional induction of the mTORC1 activator, rheb. Circ Res. 2019;124:79–93. doi: 10.1161/CIRCRESAHA.118.313854. doi: 10.1161/CIRCRESAHA.118.313854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shah M, Su D, Scheliga JS, Pluskal T, Boronat S, Motamedchaboki K, Campos AR, Qi F, Hidalgo E, Yanagida M, et al. A transcript-specific eIF3 complex mediates global translational control of energy metabolism. Cell Rep. 2016;16:1891–1902. doi: 10.1016/j.celrep.2016.07.006. doi: 10.1016/j.celrep.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Brina D, Miluzio A, Ricciardi S, Clarke K, Davidsen PK, Viero G, Tebaldi T, Offenhäuser N, Rozman J, Rathkolb B, et al. eIF6 coordinates insulin sensitivity and lipid metabolism by coupling translation to transcription. Nat Commun. 2015;6:8261. doi: 10.1038/ncomms9261. doi: 10.1038/ncomms9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Düvel K, Yecies JL, Menon S, Raman P, Lipovsky AI, Souza AL, Triantafellow E, Ma Q, Gorski R, Cleaver S, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taegtmeyer H. Genetics of energetics: transcriptional responses in cardiac metabolism. Ann Biomed Eng. 2000;28:871–876. doi: 10.1114/1.1312187. [DOI] [PubMed] [Google Scholar]
- 45.Gélinas R, Mailleux F, Dontaine J, Bultot L, Demeulder B, Ginion A, Daskalopoulos EP, Esfahani H, Dubois-Deruy E, Lauzier B, et al. AMPK activation counteracts cardiac hypertrophy by reducing O-GlcNAcylation. Nat Commun. 2018;9:374. doi: 10.1038/s41467-017-02795-4. doi: 10.1038/s41467-017-02795-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circ Res. 2007;100:474–488. doi: 10.1161/01.RES.0000258446.23525.37. doi: 10.1161/01.RES.0000258446.23525.37. [DOI] [PubMed] [Google Scholar]
- 47.Liang XH, Shen W, Sun H, Migawa MT, Vickers TA, Crooke ST. Translation efficiency of mRNAs is increased by antisense oligonucleotides targeting upstream open reading frames. Nat Biotechnol. 2016;34:875–880. doi: 10.1038/nbt.3589. doi: 10.1038/nbt.3589. [DOI] [PubMed] [Google Scholar]