

Abstract
Background
The purpose of this study was to investigate the role and mechanism of steroid receptor coactivator-interacting protein (SIP) in an astrocyte model of 1-methyl-4-phenylpyridinium (MPP+)-induced Parkinson’s disease.
Material/Methods
To perform our study, a Parkinson’s disease cell model was established by treating the rat glioblastoma cell line C6 with MPP+. SIP was overexpressed in C6 cells using SIP-plasmid. Cell viability and apoptosis were analyzed using MTT assay and flow cytometer respectively. Tumor necrosis factor (TNF)-α and interleukin (IL)-1β levels were detected using enzyme linked immunosorbent assay and quantitative reverse transcription PCR. Besides, lactate dehydrogenase (LDH) release, reactive oxygen species (ROS) production, and superoxide dismutase (SOD) enzyme activity were determined in the present study. For protein and mRNA detection, western blot assay, and qRT-PCR were performed respectively.
Results
SIP was decreased in MPP+-induced C6 cells. SIP overexpression relieved MPP+-induced cytotoxicity of C6 cells, displayed as increased cell viability and reduced cell apoptosis and reduced LDH release. Besides, SIP inhibited MPP+-induced inflammatory response and oxidative stress, evidenced by decreased levels of inflammatory factors (TNF-α and IL-1β), reduced ROS generation and enhanced SOD activity. Moreover, MPP+-induced nuclear factor-κB activation was inhibited by SIP overexpression.
Conclusions
SIP was downregulated in Parkinson’s disease and it played a protective role in the development Parkinson’s disease, thus may be a promising target for Parkinson’s disease treatment.
MeSH Keywords: Apoptosis, Astrocytes, Neurogenic Inflammation, Oxidative Stress, Parkinson Disease
Background
Neurodegenerative diseases are seriously jeopardizing the health of the elderly. The World Health Organization has announced that as the phenomenon of aging population becomes more prominent, by 2040 neurodegenerative diseases will become the second most dangerous group of diseases to human health following cardiovascular diseases [1]. Parkinson’s disease (PD) is the second most common neurodegenerative disease named by James Parkinson in 1817. The prevalence of PD in people over 65 years of age is 1–2%, while the risk of PD in people over 85 years of age is increased by nearly 4%. Therefore, it is imperative to further understand the pathological mechanism of PD and develop effective treatment methods.
The main pathological features of PD are the deposition of Lewy bodies and loss of dopaminergic neurons in substantia nigra pars compacta (SNc) [2–4]. Enhanced oxidative stress and inflammatory responses [5,6], protein misfolding and aggregation [7], and dysfunction of the ubiquitin-proteasome system [8] are involved in the pathogenesis of PD, but the exact mechanism leading to degeneration of dopaminergic neurons remains unclear. Current drugs for the treatment of PD are mainly based on improving dopamine concentration in the brain or activating dopamine receptors to improve the motor symptoms of PD [9]. These drugs include levodopa (L-Dopa), dopamine agonist, and monoamine oxidase B inhibition agent, and amantadine [10,11]. However, there are still no effective drugs to slow down or terminate the pathological process of PD. Therefore, it is urgent to find effective molecular targets for PD or to further elucidate the pathological mechanism of PD, so as to provide more experimental and clinical basis for the development of new PD treatment methods.
The role of nuclear factor-κB (NF-κB) signaling in immunity and inflammation is evolutionarily conserved [12]. In addition, the contribution of NF-κB to neurodegeneration in PD has been well documented [13,14]. Steroid receptor co-activator-interacting protein (SIP) is a new inhibitor of the NF-κB family that interacts with the p65 protein of the NF-κB family through the PEST domain [15]. Previous study has reported that SIP could relieve acute pancreatitis induced myocardial damage by inhibiting inflammatory response [16]. Another study indicated that SIP inhibited caspase-independent apoptosis by inhibiting apoptosis-inducing factor (AIF) [17]. To date, as we known, the role of SIP in the development of PD has not been clarified.
The current study aimed to investigate the role of SIP in astrocyte model of 1-methyl-4-phenylpyridinium (MPP+)-induced PD, and to explore the underlying molecular mechanism.
Material and Methods
Cell culture and establishment of astrocyte model of Parkinson’s disease (PD)
The rat glioblastoma cell line C6 was purchased from ATCC (Manassas, VA, USA). C6 cells were grown in DMEM (GIBCO, Grand Island, NY, USA) supplemented with 10% fetal bovine serum (FBS, GIBCO, Grand Island, NY, USA), 1% streptomycin-penicillin solution, and 2 mM L-glutamine, and incubated at 37°C in a humidified atmosphere containing 5% CO2.
C6 cells were treatment with various concentrations of MPP+ (0, 100, 200, 300, 400, 500, 750, and 1000 μM) at 37°C for indicated times (0–48 hours).
The astrocyte model of MPP+-induced PD was conducted by treating C6 cells with 500 μM MPP+ for 24 hours [18].
Cell transfection
C6 cells were seeded in 6-well plates (1×106 cells per well) and cultured at 37°C for 24 hours. Then the C6 cells were transfected with control-plasmid, SIP-plasmid using Lipofectamine 2000 reagent following the manufacturer’s instructions. Cells without any treatment were considered the control group. Cell transfection efficiency was detected 24 hours later. To determine the role of SIP on PD in vitro, C6 cells were divided into 4 groups: control group (C6 cells without any treatment); MMP group (C6 cells treated with 500 μM MPP+ for 24 hours); MMP+control-plasmid group (C6 cells were transfected with control-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours); MMP+SIP-plasmid group (C6 cells were transfected with SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours).
3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay
C6 cells were treated with various concentrations of MPP+ (0, 100, 200, 300, 400, 500, 750, and 1000 μM) at 37°C for 24 hours, or the cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours. Subsequently, C6 cell viability was determined using the 3-(4,5-dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Cell viability was detected by measuring the absorbance at 570 nm using FLUOstar® Omega Microplate Reader (BMG LABTECH, Ortenberg, Germany).
Cell apoptosis assay
C6 cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours. To analyze C6 cell apoptosis, we performed the Annexin V-fluorescein isothiocyanate (FITC)/propidium iodide (PI) apoptosis detection kit (Cat no. 70-AP101-100; MultiSciences, Hangzhou, China) according to the manufacturer’s instructions. After specific treatment, C6 cells were collected by 0.25% Trypsin, and then incubated with 5 μL Annexin V- FITC and 5 μL PI for 30 minutes in dark at room temperature. At last, flow cytometer (BD Biosciences) was performed to analyze cell apoptosis.
Enzyme linked immunosorbent assay (ELISA)
C6 cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours. The levels of TNF-α and IL-1β in supernatant of C6 cells were detected by using enzyme-linked immunosorbent (ELISA) kits (Cell Signaling Technology Inc., Danvers, MA, USA) according to the relevant manuals.
Lactate dehydrogenase (LDH) assay
C6 cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours. Then, we used the LDH in vitro toxicology assay kit (TOX-7, Sigma-Aldrich, St. Louis, MO, USA) to measure the amount of cytoplasmic LDH released into the culture medium. A TRIAD Series Multimode Detector (Dynex Technologies Inc., Chantilly, VA, USA) was used to detect the absorbance at 490 nm. The calculated value was the relative intensity of absorbance compared to the control cells.
Measurement of reactive oxygen species (ROS) and superoxide dismutase (SOD)
C6 cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours. Then, ROS level was detected by using ROS assay kit (Beyotime, Jiangsu, China) according to the manufacturer’s instructions. And we evaluated the SOD enzyme activities by performing commercial detection kits (Cayman Chemical, MI, USA) following with the manufacture’s introduction.
Western blot assay
C6 cells were treated with various concentrations of MPP+ (0, 100, 200, 300, 400, 500, 750, and 1000 μM) at 37°C for 24 hours, or treated with 500 μM MPP+ at 37°C for 6, 12, 24, and 48 hours respectively, then the protein level of SIP was measured by western blotting. Besides, C6 cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours, then the protein levels of SIP, AIF, caspase-3, and p-p65 in C6 cells were detected. For western blot assay, C6 cells were washed twice with phosphate-buffered saline (PBS), collected and lysed in lysis buffer (Beijing Solarbio Science & Technology Co., Ltd., Beijing, China). Supernatants, which were used as the total cell lysates, were collected by centrifuging lysates at 12 000 rpm for 10 minutes at 4°C. We used the bicinchoninic acid assay kit (BCA; Pierce Biotechnology, Rockford, IL, USA) to detect the concentrations of protein samples. Equal amount of protein samples (30 μg per lane) were separated on 12% sodium dodecyl sulfate (SDS)-polyacrylamide gels and electrotransferred onto a PVDF membrane (Millipore, Bellerica, MA, USA). After blocking with 5% skim milk at room temperature for 1.5 hours, the membrane was incubated with primary antibodies (anti-SIP, anti-AIF, anti-caspase-3, anti-p65, anti-p-p65, and anti-β-actin) overnight at 4°C. Then, the membrane was incubated with a second antibody at room temperature for 3 hours. Finally, we used the enhanced chemiluminescence reagent (ECL, Thermo Scientific, Logan, UT, USA) to determine the immunoreactive bands.
Quantitative reverse transcription polymerase chain reaction (qRT-PCR)
Total RNAs were collected from cultured cells by using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) following with the manufacturer’s instructions. The cDNAs were synthesized by using a PrimeScript RT Reagent Kit (Takara Bio, Inc.) according to protocols of the manufacturer. Then, SYBR® Premix Ex Taq™ II (Takara Bio, Inc.) was used to analyze the synthesized cDNAs. GAPDH was served as an internal control. The relative mRNA levels were calculated using the 2−ΔΔCT method [19].
Statistical analysis
All the experiments in the current study were performed 3 times. Data were displayed as mean ± standard deviation (SD). SPSS 17.0 statistical software (SPSS, Inc., Chicago, IL, USA) was used for all statistical analyses. Differences between groups were evaluated by Student’s t-tests and one-way ANOVA followed by NSK tests. A value of P<0.05 was considered statistically significant.
Results
SIP was downregulated in MPP+-induced C6 cell PD model
To detect SIP expression level in our PD cell model, various concentrations of MPP+ (0, 100, 250, 500, 750, and 1000 μM) were added to C6 cells for 24 hours or 500 μM MPP+ for indicated times followed by western blot and qRT-PCR analysis. As shown in Figure 1, compared with the control group, both protein and mRNA expression of SIP were reduced in MPP+ treated C6 cells in a dose- and time-dependent manner. The data revealed that SIP may be involved in PD pathogenesis.
Figure 1.

SIP expression in MMP+ treated C6 cells. C6 cells were treated with various concentrations of MPP+ (0, 100, 250, 500, 750, and 1000 μM) for 24 hours, then SIP mRNA (A) and protein (B) expression level in C6 cells was detected using qRT-PCR and western blotting, respectively. C6 cells were treated with 500 μM MPP+ for indicated times (0, 6, 12, 24, and 48 hours), then SIP mRNA (C) and protein (D) expression level in C6 cells was detected using qRT-PCR and western blotting, respectively. Data were displayed as mean ±SD. *, ** P<0.05, 0.01 versus control. SIP – steroid receptor coactivator-interacting protein; qRT-PCR – quantitative reverse transcription polymerase chain reaction; MPP+ – 1-methyl-4-phenylpyridinium; SD – standard deviation.
SIP inhibited MPP+-induced C6 cell cytotoxicity
The functional effect of SIP on MPP+ induced C6 cells was explored. C6 cells were treated with various concentrations of MPP+ (0, 100, 250, 500, 750, and 1000 μM) at 37°C for 24 hours, or the cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours. Cell transfection efficiency was detected by western blotting and qRT-PCR, and the results indicated that compared with the control group, SIP-plasmid significantly increased SIP protein and mRNA level (Figure 2A, 2B). First, cell viability was determined by MTT assay. As shown in Figure 2C, MPP+ caused cytotoxicity in C6 cells. Compared with the control group, SIP-over-expression significantly increased the reduced cell viability of C6 cells induced by MPP+ (Figure 2D).
Figure 2.

Effect of SIP on MMP+ treated C6 cell viability. (A) C6 cells were transfected with control-plasmid or SIP-plasmid for 24 hours, then mRNA level of SIP was measure by qRT-PCR. (B) C6 cells were transfected with control-plasmid or SIP-plasmid for 24 hours, then protein levels of SIP were measure by western blotting. (C) C6 cells were treated with various concentrations of MPP+ (0, 100, 250, 500, 750, and 1000 μM) for 24 hours, then cell viability was detected using MTT assay. (D) C6 cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours, then cell viability was detected using MTT assay. Data were displayed as mean ±SD. *, ** P<0.05, 0.01 versus control. # P<0.05 versus MMP+ treatment alone group. SIP – steroid receptor coactivator-interacting protein; qRT-PCR – quantitative reverse transcription polymerase chain reaction; MPP+ – 1-methyl-4-phenylpyridinium; SD – standard deviation.
C6 cell apoptosis was also detected to explore the effect SIP on MPP+ induced C6 cell cytotoxicity. Results indicated that compared with the control group, MPP+ significantly induced C6 cell apoptosis, while SIP-over-expression significantly decreased cell apoptosis (Figure 3A, 3B). Meanwhile, pro-apoptotic protein caspase-3 and apoptosis inducing factor (AIF) were determined in current study. As expect, the increased protein expression of caspase-3 and AIF caused by MPP+ treatment was inhibited by SIP upregulation (Figure 3C).
Figure 3.

Effect of SIP on MMP+ treated C6 cell apoptosis. C6 cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours, then cell apoptosis was analyzed by FCM (A); and the cell apoptosis rate was calculated and presented (B). The protein level of caspase-3 and AIF was detected using western blotting (C). Data were displayed as mean ±SD. ** P<0.01 versus control. ## P<0.01 versus MMP+ treatment alone group. SIP – steroid receptor coactivator-interacting protein; MPP+ – 1-methyl-4-phenylpyridinium; FCM – flow cytometer; AIF – apoptosis-inducing factor; SD – standard deviation.
In the current study, we analyzed cell permeability by detecting LDH release in cell culture medium to monitor cell membrane integrity. As shown in Figure 4, after exposed to 500 μM MPP+ for 24 hours, the LDH released from cells significantly increased, and SIP over-expression notably decreased LDH release.
Figure 4.
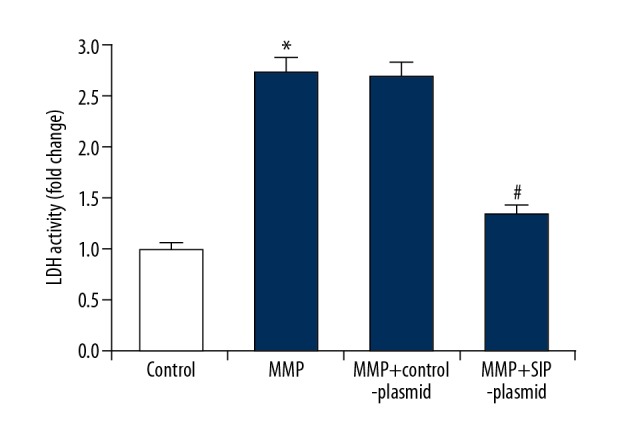
Effect of SIP on LDH activity in MMP+ treated C6 cells. C6 cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours, then LDH was measured and the data were presented as fold of the control group. Data were displayed as mean ±SD. * P<0.05 versus control. # P<0.05 versus MMP+ treatment alone group. SIP – steroid receptor coactivator-interacting protein; MPP+ – 1-methyl-4-phenylpyridinium; LDH – lactate dehydrogenase; SD – standard deviation.
SIP prevented MPP+-induced C6 cell inflammation
To explore whether SIP had an effect on inflammation, the expression level of inflammatory factors TNF-α and IL-1β were detected using ELISA. As shown in Figure 5, compared with the control group, 500 μM MPP+ treatment significantly enhanced the expression levels of TNF-α and IL-1β in C6 cells, while SIP upregulation notably eliminated these enhancements.
Figure 5.

(A, B) Effect of SIP on TNF-α and IL-1β production in MMP+ treated C6 cells. C6 cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours, then the level of TNF-α and IL-1β in the supernatants of C6 cells was detected using ELISA. Data were displayed as mean ±SD. ** P<0.01 versus control. ## P<0.01 versus MMP+ treatment alone group. SIP – steroid receptor coactivator-interacting protein; TNF – tumor necrosis factor; IL – interleukin; MPP+ – 1-methyl-4-phenylpyridinium; ELISA – enzyme linked immunosorbent assay; SD – standard deviation.
SIP prevented MPP+-induced C6 cell oxidative stress
Furthermore, ROS generation and antioxidant enzymes SOD activity were determined in C6 cells. The results of our study indicated that SIP upregulation attenuated MPP+ medicated ROS generation and SOD activity inhibition in C6 cells (Figure 6).
Figure 6.

(A, B) Effect of SIP on oxidative stress in MMP+ treated C6 cells. C6 cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours, then ROS generation and antioxidant enzymes SOD activity were determined respectively. Data were presented as fold of the control group. Data were displayed as mean ±SD. ** P<0.01 versus control. ## P<0.01 versus MMP+ treatment alone group. SIP – steroid receptor coactivator-interacting protein; MPP+ – 1-methyl-4-phenylpyridinium; ROS – reactive oxygen species; SOD – superoxide dismutase; SD – standard deviation.
SIP prevented MPP+-induced NF-κB pathway activation in C6 cells
Previous studies indicated that NF-κB was involved in the development of PD and SIP was an inhibitor of the NF-κB pathway [13–15]. Therefore, in our study we finally investigated the effect of SIP on the NF-κB pathway in MPP+ induced C6 cells. As expected, we found that compared with the control group, MPP+ treatment significantly promoted the activation of the NF-κB pathway in C6 cells, evidenced by enhanced protein level of p-p65. SIP over-expression notably decreased the protein level of p-p65 in C6 cells in comparison with the MPP+ treatment alone cells (Figure 7).
Figure 7.

Effect of SIP on NF-κB pathway in MMP+ treated C6 cells. C6 cells were transfected with control-plasmid and SIP-plasmid for 2 hours prior to treatment with 500 μM MPP+ for 24 hours, then SIP protein (A) and mRNA (B) level was detected using qRT-PCR and western blotting, and p-p65 protein (A, C) level was measured using western blot assay. Data were displayed as mean ±SD. ** P<0.01 versus control. ## P<0.01 versus MMP+ treatment alone group. SIP – steroid receptor coactivator-interacting protein; NF-κB – nuclear factor-κB; MPP+ – 1-methyl-4-phenylpyridinium; qRT-PCR – quantitative reverse transcription polymerase chain reaction; SD – standard deviation.
Discussion
In the present study, we demonstrated that the protein and mRNA levels of SIP were downregulated in MPP+ treated C6 cells compared with the control cells, indicating the downregulation of SIP in PD. SIP over-expression significantly eliminated MPP+ induced cell viability inhibition, cell apoptosis, LDH activity enhancement, inflammatory factors TNF-a and IL-1β levels up-regulation, SOD activity elevation and ROS generation reduction in C6 cells. Moreover, MPP+-induced NF-κB activation in C6 cells was repressed by SIP over-expression. These findings suggested that SIP may play a protective role in PD development by inhibiting C6 cell inflammatory response and oxidative stress via repressing the NF-κB signaling pathway. Evidence collected in this study support that SIP may be a novel and promising therapeutic target for PD treatment.
PD is a common neurodegenerative disease that severely affects the quality of life of patients. The most important pathological changes in the pathological process of PD are the progressive loss of dopaminergic neurons, which is associated with activation of glial cells, increased levels of cytokines, and accumulation of a-synuclein within neurons [2]. At present, how to effectively delay the loss of neurons and restore their function has become a bottleneck restricting PD therapeutic breakthrough. A large number of astrocytes proliferate and activate during PD pathology [20,21]. As the largest number of immune cells in the brain, astrocytes are closely connected with neurons, microglia, and blood vessels in the brain and play an important central role in the regulation of neuroimmune inflammation. Astrocytes regulate neuronal function via the modulation of synaptic transmission and plasticity, secretion of growth factors, uptake of neurotransmitters, and regulation of extracellular ion concentrations and metabolic support of neurons [22,23]. Over-proliferation of activated astrocytes reduces the secretion of neurotrophic factors and increases the secretion of pro-inflammatory cytokines, thereby inducing neuronal damage [24–26]. Therefore, elucidating the mechanism of neuroinflammation in pathological processes of PD and balancing the release of astrocytes against neurotrophic factors and inflammatory factors is expected to provide new intervention strategies and drug targets for the prevention and treatment of PD.
The involvement of NF-κB in the development of PD is well studied [13,14]. However, SIP, a novel NF-κB family inhibitor [15], has not been studied in PD. This study was conducted to explore the role of SIP during PD progress in vitro. To perform our investigation, a PD cell model was established by treating the rat glioblastoma cell line C6 with MPP+. MPP+ has been reported can induce cytotoxicity of astrocytes and it was widely used for PD cell model induction [18,27,28]. Then, we firstly studied the expression of SIP in MPP+ treated C6 cells, and explored the effect of SIP on MPP+ induced cytotoxicity in C6 cells. The findings suggested that SIP was downregulated in MPP+ treated C6 cells and over-expression of SIP significantly reduced MPP+ induced C6 cell cytotoxicity, evidenced by increased cell viability, decreased cell apoptosis and reduced LDH release.
Enhanced oxidative stress and neuroinflammation [5,6] are involved in the pathogenesis of PD. We then explored whether SIP can relieve PD during repressing oxidative stress and inflammatory response. We found that oxidative stress and inflammation induced by MPP+ were inhibited by SIP over-expression in C6 cells. In addition, another important discovery of the current study was that MPP+ significantly induced NF-κB activation in C6 cells and this activation was inhibited by SIP upregulation.
Taken together, this study for the first time indicted that SIP was downregulated in MPP+ induced PD cell model, and that it could inhibit MPP+ induced cytotoxicity, oxidative stress, and inflammatory response in C6 cells.
Conclusions
SIP played a neuroprotective effect on the astrocyte model of MPP+-induced PD in vitro through inhibiting astrocyte inflammatory response and oxidative stress via repressing the NF-κB signaling pathway. Therefore, SIP may be a novel and promising therapeutic target for PD treatment.
Acknowledgment
Thanks to Professor Lihua Zhang, the chief physician of the Chuzhou Central Hospital for his guidance and help.
Footnotes
Conflict of interests
None.
Source of support: Departmental sources
References
- 1.Valera E, Masliah E. Therapeutic approaches in Parkinson’s disease and related disorders. J Neurochem. 2016;139:346–52. doi: 10.1111/jnc.13529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang Q, Liu Y, Zhou J. Neuroinflammation in Parkinson’s disease and its potential as therapeutic target. Transl Neurodegener. 2015;4:19. doi: 10.1186/s40035-015-0042-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.He Y. Imaging brain networks in neurodegenerative diseases. CNS Neurosci Ther. 2015;21:751–53. doi: 10.1111/cns.12466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baggio HC, Segura B, Junque C. Resting-state functional brain networks in Parkinson’s disease. CNS Neurosci Ther. 2015;21:793–801. doi: 10.1111/cns.12417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katunina EA, Titova NV, Malykhina EA, et al. [Oxidative stress and Parkinson’s disease: Mechanisms and perspectives of treatment]. Zh Nevrol Psikhiatr Im S S Korsakova. 2015;115:141–45. doi: 10.17116/jnevro201511571141-145. [in Russian] [DOI] [PubMed] [Google Scholar]
- 6.Dursun E, Gezen-Ak D, Hanagasi H, et al. The interleukin 1 alpha, interleukin 1 beta, interleukin 6 and alpha-2-macroglobulin serum levels in patients with early or late onset Alzheimer’s disease, mild cognitive impairment or Parkinson’s disease. J Neuroimmunol. 2015;283:50–57. doi: 10.1016/j.jneuroim.2015.04.014. [DOI] [PubMed] [Google Scholar]
- 7.Paleologou KE, El-Agnaf OM. alpha-Synuclein aggregation and modulating factors. Subcell Biochem. 2012;65:109–64. doi: 10.1007/978-94-007-5416-4_6. [DOI] [PubMed] [Google Scholar]
- 8.Opattova A, Cente M, Novak M, Filipcik P. The ubiquitin proteasome system as a potential therapeutic target for treatment of neurodegenerative diseases. Gen Physiol Biophys. 2015;34:337–52. doi: 10.4149/gpb_2015024. [DOI] [PubMed] [Google Scholar]
- 9.Sybertz E, Krainc D. Development of targeted therapies for Parkinson’s disease and related synucleinopathies. J Lipid Res. 2014;55:1996–2003. doi: 10.1194/jlr.R047381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Connolly BS, Lang AE. Pharmacological treatment of Parkinson disease: A review. JAMA. 2014;311:1670–83. doi: 10.1001/jama.2014.3654. [DOI] [PubMed] [Google Scholar]
- 11.Seppi K, Weintraub D, Coelho M, et al. The movement disorder society evidence-based medicine review update: Treatments for the non-motor symptoms of Parkinson’s disease. Mov Disord. 2011;26:S42–80. doi: 10.1002/mds.23884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ruland J. Return to homeostasis: downregulation of NF-kappaB responses. Nat Immunol. 2011;12:709–14. doi: 10.1038/ni.2055. [DOI] [PubMed] [Google Scholar]
- 13.Ghosh A, Roy A, Liu X, et al. Selective inhibition of NF-kappaB activation prevents dopaminergic neuronal loss in a mouse model of Parkinson’s disease. Proc Natl Acad Sci USA. 2007;104:18754–59. doi: 10.1073/pnas.0704908104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glass CK, Saijo K, Winner B, et al. Mechanisms underlying inflammation in neurodegeneration. Cell. 2010;140:918–34. doi: 10.1016/j.cell.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wu G, Wang DD, Sun LY. [Mechanism involved in cytoplasmic sequestration of p65 by SIP]. Chinese Journal of Biochemistry and Molecular Biology. 2011;27:728–36. [in Chinese] [Google Scholar]
- 16.Chen X, Zhu B. Steroid receptor coactivator-interacting protein (sip) suppresses myocardial injury caused by acute pancreatitis. Med Sci Monit. 2018;24:3204–11. doi: 10.12659/MSM.906968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang D, Liang J, Zhang Y, et al. Steroid receptor coactivator-interacting protein (SIP) inhibits caspase-independent apoptosis by preventing apoptosis-inducing factor (AIF) from being released from mitochondria. J Biol Chem. 2012;287:12612–21. doi: 10.1074/jbc.M111.334151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ramalingam M, Kim SJ. Protective effects of activated signaling pathways by insulin on C6 glial cell model of MPP+-induced Parkinson’s disease. J Recept Signal Transduct Res. 2017;37:100–7. doi: 10.3109/10799893.2016.1171342. [DOI] [PubMed] [Google Scholar]
- 19.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔt method. Methods. 2011;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 20.Yamada T, Kawamata T, Walker DC, McGeer PL. Vimentin immunoreactivity in normal and pathological human brain tissue. Acta Neuropathol. 1992;84:157–62. doi: 10.1007/BF00311389. [DOI] [PubMed] [Google Scholar]
- 21.Lu M, Sun XL, Qiao C, et al. Uncoupling protein 2 deficiency aggravates astrocytic endoplasmic reticulum stress and nod-like receptor protein inflammasome activation. Neurobiol Aging. 2014;35:421–30. doi: 10.1016/j.neurobiolaging.2013.08.015. [DOI] [PubMed] [Google Scholar]
- 22.Forno LS, DeLanney LE, Irwin I, et al. Astrocytes and Parkinson’s disease. Prog Brain Res. 1992;94:429–36. doi: 10.1016/s0079-6123(08)61770-7. [DOI] [PubMed] [Google Scholar]
- 23.Ramalingam M, Kim SJ. The protective effects of insulin on hydrogen peroxide-induced oxidative stress in C6 glial cells. Biomol Ther. 2009;17:395–402. [Google Scholar]
- 24.Osborn LM, Kamphuis W, Wadman WJ, Hol EM. Astrogliosis: A integral player in the pathogenesis of Alzheimer’s disease. Prog Neurobiol. 2016;144:121–41. doi: 10.1016/j.pneurobio.2016.01.001. [DOI] [PubMed] [Google Scholar]
- 25.Sofroniew MV. Astrocyte barriers to neurotoxic inflammation. Nat Rev Neurosci. 2015;16:249–63. doi: 10.1038/nrn3898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tanaka T, Kai S, Matsuyama T, et al. General anesthetics inhibit LPS-induced IL-lbeta expression in glial cells. PLoS One. 2013;8:e82930. doi: 10.1371/journal.pone.0082930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu EY, Langston JW, Di Monte DA. Toxicity of the 1-methyl-4-phenyl-2,3-dihydropyridinium and 1-methyl-4-phenylpyridinium species in primary cultures of mouse astrocytes. J Pharmacol Exp Ther. 1992;262:225–30. [PubMed] [Google Scholar]
- 28.Soh Y, Kim JA, Sohn NW, et al. Protective effects of quinic acid derivatives on tetrahydropapaveroline-induced cell death in C6 glioma cells. Biol Pharm Bull. 2003;26:803–7. doi: 10.1248/bpb.26.803. [DOI] [PubMed] [Google Scholar]