

Abstract
The local environments surrounding the luminophores can significantly influence their photophysical properties. Herein, we report the self-assembly of a highly emissive platinum (II)-based metallacage. In order to accommodate the connectivity of the platinum (II) building block used in the self-assembly process, the luminophore-containing building block adopts a highly twisted geometry relative to its free form, leading to the emergence of an emissive transition with a radiative rate constant an order of magnitude higher than that of the free luminophore. This increased rate constant is the primary driver for the 10-fold increase in quantum yield from 4.2% to 40%. Model complexes with platinum or methyl groups bound to the nitrogen were synthesized. These complexes had lower quantum yields (10% and non-emissive, respectively) due mainly to decreases in radiative rate constants. Computational studies were conducted and indicated that the excited state of the ensembles, as well as the model complexes, is a charge transfer to the pyridyl groups, in contrast to the free luminophore which involves the diphenyl sulphone moiety. The differences in quantum yield can be explained by a twist in the chromophore upon coordination of platinum or methylation on the pyridyl group, leading to intersystem crossing to a triplet state. This state then becomes more emissive with the addition of platinum that increases the radiative rate constant via the heavy atom effect. The formation of a metallacage also decreases the nonradiative rate constant by inhibiting the intramolecular motions of the incorporated luminophore.
Graphical Abstract
INTRODUCTION
The design and synthesis of luminescent molecules have drawn significant interest due to their promising applications as optoelectronic devices, imaging agents, and biosensors.1 Photophysical properties of many luminescence molecules are sensitive to local environments, which includes the distance and orientation of the luminophores, the conformation of individual luminophores and their mobility.2 Various photophysical phenomenon, such as charge/electron transfer, excimer/exciplex formation, and aggregate formation are influenced by these parameters.2 Understanding and controlling these parameters is crucial for the development of materials with photophysical properties favorable for applications.
Coordination-driven self-assembly is a simple yet efficient approach for the synthesis of discrete supramolecular coordination complexes (SCCs) with well-defined sizes and shapes via the spontaneous formation of metal-ligand bonds between Lewis-acidic metal acceptors and Lewis-basic organic donors.3 The versatility of coordination-driven self-assembly allows the incorporation of precursor building blocks with diverse topologies and functionalities into SCCs, which give rise to extraction, separation, biomedical, and catalytic functions.3-4 SCCs that display emergent, light-emitting characteristics have also been synthesized. The use of platinum as a building block in coordination-driven self-assembly promotes intersystem crossing in the luminophore via the heavy atom effect.5 When luminophores that display aggregation-induced emission are used as the building block, supramolecular systems with an efficient solution and solid dual-state emission can be obtained. The formation of rigid supramolecular ensembles efficiently restricts the intramolecular motions of the luminophores incorporated.6
A molecule used as a building block for coordination-driven self-assembly may adopt a different geometry than it otherwise would as a free molecule in solution. The new geometry of the molecule, in turn, can change the nature of the photophysics of the ligand, specifically changing the orbitals involved in transitions between the ground and excited states.7 These changes may be complementary to other photophysical behavior induced by SCC formation as mentioned above, leading to favorable synergetic effects.
Herein, luminophore 1 that comprised an electron-deficient diphenyl sulfone moiety, electron-sufficient phenothiazine moieties, and pyridyl groups that can be utilized as the structural linker for metal-ligand coordination interactions was prepared. 1 exhibits aggregation-induced enhanced emission (AIEE) due to the free intramolecular motions of the phenothiazine moieties.8 Taking advantage of the pyridyl groups, the coordination-driven self-assembly of 1 with a 60° di-platinum motif 2 and a mono-platinum motif 3 in an appropriate ratio leads to the formation of [2+4] metallacage 4 and [1+4] acyclic ensemble 5 (Scheme 1). To compare the effect of connecting motifs on the photophysical properties, methylation at the pyridyl N-atoms of 1 was conducted to afford methylpyridinium salt 6. The steady-state solution photophysical profiles of 1, 4, 5, and 6 were examined, followed by computational studies. They revealed that the formation of supramolecular ensembles via coordination-driven self-assembly results in a significant enhancement in the emission efficiency of the luminophore in dilute solutions by the activation of emissive transition that exhibit higher radiative decay rate (kr) relative to the free luminophore 1 or its non-self-assembled analog 6.
Scheme 1.

Self-Assembly of SCCs and structure of 6
RESULTS AND DISCUSSION
The organic donor 1 was readily synthesized by a Pd-catalyzed Suzuki coupling. Stirring a mixture of 1 with a 60° di-Pt(II) acceptor 2 and a mono-Pt(II) acceptor 3 in appropriate stoichiometry ratios in CH2Cl2/CH3COCH3 (v:v = 1:1) led to the formation of the self-assembled [2+4] metallacage 4 and [1+4] acyclic ensemble 5, respectively. Multinuclear NMR (1H and 31P) analyses were employed to examine the formation of 4 and 5. In the 1H NMR spectra of both SCCs 4 and 5, protons of the pyridine rings exhibit a downfield shift relative to the donor 1 (Figures 1a-c), suggesting the loss of electron density upon the formation of the metal-ligand bond.3 The 31P{1H} spectra of 4 and 5 exhibited sharp singlets (14.93 ppm for 4 and 14.91 ppm for 5) with concomitant 195Pt satellites (Figures 1d-e), indicating a single phosphorus environment and the formation of discrete, highly symmetric supramolecular ensembles. Diffusion-ordered NMR spectroscopy (DOSY) confirmed the formation of a single supramolecular ensemble upon self-assembly. Metallacage 4 exhibited a single band with a diffusion coefficient of D = 4.1 × 10−11 m2 s−1 (Figure S16), indicating the formation of a product with an estimated diameter of 4.8 nm. In contrast, the acyclic ensemble showed 5 showed a diffusion coefficient of D = 9.6 × 10−11 m2 s−1 (Figure S17), suggesting an estimated diameter of 2.1 nm. Electrospray ionization time-of-flight mass spectrometry (ESI-TOF-MS) studies were then employed to provide further evidence for the molecularity of the SCCs. For metallacage 4, four peaks corresponding to intact [2+4] SCC with positive charge owing to the loss of trifluoromethanesulfonate group (-OTf) are observed in the ESI-TOF-MS spectrum, including m/z = 1649.08 for [M−4OTf]4+, m/z = 1289.39 for [M−5OTf]5+, m/z = 1049.40 for [M−6OTf]6+, and m/z = 878.18 for [M−7OTf]7+ (Figures 1f, S9). Similarly, two peaks are assigned consistent with the formation of a [1+4] assembly (m/z = 1626.56 for [M−2OTf]2+ and 1034.65 for [M−3OTf]3+) in the ESI-TOF-MS spectrum of 5 (Figure 1g , S12). All identified peaks are isotopically resolved and are in good agreement with their simulated theoretical distributions. Methylpyridinium salt 6 was readily synthesized by methylation of 1 using methyl iodide, followed by counter-ion exchange using NaOTf. The formation of 6 is confirmed using multinuclear NMR (1H and 13C) experiments and ESI-TOF-MS (Figure S13-S15).
Figure 1.

Characterization of SCCs. (a-c) Partial 1H NMR spectra (500 MHz, DMSO-d6, 298 K) of 4 (a) organic ligand 1 (b) and 5 (c). (d-e) 31P{1H} NMR spectra (121 MHz, DMSO-d6, 298 K) of 4 (d) and 5 (e). (f-g) Experimental (red) and calculated (blue) ESI-TOF-MS spectra of 4 [M – 5OTf]5+ (f) and 5 [M – 3OTf]3+ (g).
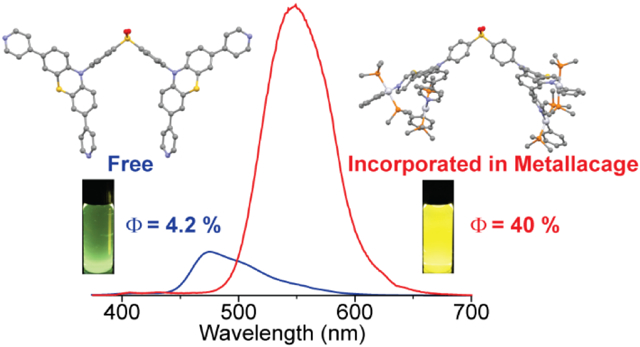
Figure 2 depicts the UV-vis absorption and emission profiles of 1, 4, 5, and 6 in a dilute THF solution at room temperature. The emission maximum (λemmax), luminescence quantum yield (Φem), luminescence lifetime, and radiative and nonradiative decay rates are summarized in Table 1. 1 display the main absorption peaks by the phenothiazine substituted diphenyl sulfone backbone structure (286 nm and 340 nm) and a long wavelength absorption band centered at 375 nm. The emission maximum of 1 in THF is observed at 475 nm, with a Φem of 4.2% in deoxygenated THF and a lifetime of 5.17 ns. The kr and knr were calculated to be 8.1×106 s−1 and 1.9×108 s−1, respectively. In addition, 1 displays a typical AIEE behavior because the gradual increase in the ratio of hexane—a poor solvent for 1—in the mixed solution of THF and hexane result in a continuous increase in the luminescence intensity of the mixture (Figure S18). The solid-state Φem is 9.0% for 1, which is more than double Φem in a dilute solution, confirming the AIEE character of 1.
Figure 2.

Photophysical properties of 1, 4, 5 and 6 in deoxygenated THF. (a) Molar absorption coefficients. (b) Emission spectra (c = 1 × 10−6 M).
Table 1.
Photophysical properties of compounds 1, 4, 5 and 6 in deoxygenated THF.
| λabsmax (nm) |
λemmax (nm) |
Φem (%)a |
τ (ns)b | kr (s−1) | knr (s−1) | |
|---|---|---|---|---|---|---|
| 1 | 286, 340, 375 | 475 | 4.2 | 5.17 | 8.1×106 | 1.9×108 |
| 4 | 313, 361, 420 | 551 | 40 | 5.26 | 7.6×107 | 1.1×108 |
| 5 | 305, 363, 420 | 555 | 9.7 | 5.41 | 1.8×107 | 1.7×108 |
| 6 | 328, 502 | - | - | - | - | - |
Emission quantum yield measured via an integrating light sphere (see the Supporting Information for details)
Excited-state lifetime measured by time-correlated single photon counting (see the Supporting Information for details)
SCCs 4 and 5 show stronger absorption bands at short wavelengths because of the introduction of platinum motifs with large conjugated systems. Notably stronger and redshifted low-energy absorption bands centered at 420 nm are observed for the SCCs. The emission spectra of 4 and 5 exhibit a significant redshift to 551 nm and 555 nm, respectively. The Φem of 4 and 5 are 40% and 9.7% in THF, respectively, which are significantly higher than the Φem of 1 in THF. The luminescence lifetimes of 4 and 5 are 5.26 ns and 5.41 ns, respectively, slightly longer than that of 1. The emission intensities of 4 and 5 were found to be sensitive to oxygen (Figure S21), suggesting the involvement of triplet excited state in their emission. These phenomena are in sharp contrast to common metal-containing fluorophores in which the heavy-atom effect manifests itself in decreased excited-state lifetimes and lowered quantum yields owing to increased amounts of intersystem crossing processes to dark triplet excited states.9 4 exhibits a higher kr (7.6×107 s−1) and a lower knr (1.1×108 s−1) relative to that of 1, whereas 5 displays less significant changes in both kr (1.7×107 s−1) and knr (1.7×108 s−1). Increasing the ratio of hexane in the mixed solution of THF and hexane of SCCs 4 and 5 lead to enhancements in their luminescence intensity (Figures S19 and S20), suggesting the AIEE behavior of the SCCs. The absorption spectrum of 6 shows the appearance of a new band centered at 522 nm, and no emission is observed upon photoexcitation of 6.
To elucidate the distinctive emission behavior between the compounds in dilute solutions, we performed geometry optimizations, single point calculations, and time-dependent density functional theory (TD-DFT) calculations on 1, 5, and 6 to determine how the frontier orbitals of the ligand change when the geometry is adjusted to accommodate the self-assembly.
The optimized geometry of 1 (1-Opt) has the phenothiazine arms of the ligand in-plane with one another and pointed above and below the bridging diphenyl sulfone (Figure 3). This is in stark contrast to the geometry the ligand must take to accommodate the designed self-assembly. The lowest energy transition from the TD-DFT with appreciable oscillator strength is a π → π* transition on the phenothiazine arms which also to an extent involve the phenyl groups of the bridging diphenyl sulfone. The LUMO, which is almost entirely diphenyl sulfone-based, is not involved in the transition at all. (Figure 4, Tables S1-S2) When 1 is held in the geometry necessary for self-assembly (1-SAGeo) (Figure 3), without the addition of any other atoms, the LUMO, while still being of mostly sulfone character, now extends out to the phenothiazine arms as well (Figure 4). The result of this is that the lowest energy transition now has significant charge transfer character from the phenothiazine to the diphenyl sulfone (Tables S3-S4). Methylation of 1 to form 6 twists one the phenothiazine arms 90° out of plane with the other arm (Figure 3). The effect of methylation is to render a smaller HOMO-LUMO energy gap as compared to 1 (Figure 4). The HOMO to LUMO transition has significant charge transfer characteristics from the central phenothiazine to the now methylated pyridyl rings, while the diphenyl sulfone is not involved at all (Tables S9-S10).
Figure 3.

Geometries of 1-Opt, 1-SAGeo, 1-SAGeo+Pt, 5-Opt, and 6-Opt. Hydrogen atoms are omitted for clarity.
Figure 4.

Calculated HOMO-LUMO energy gaps of 1-Opt, 1-SAGeo, 1-SAGeo+Pt, 5-Opt, and 6-Opt.
The addition of platinum to the self-assembly geometry of 1 (1-SAGeo+Pt) (Figure 3) results in LUMO and LUMO + 1 orbitals that are split between the phenothiazine and the pyridyl ligands while the HOMO and HOMO − 1 orbitals are concentrated just on the phenothiazine (Figure 4). This sets up a charge transfer excited state similar to that of 6. The lowest energy triplet excited state, which can now be accessed due to intersystem crossing made possible by the spin-orbit coupling induced by the platinum, has the same character (Tables S7-S8). The transition and resulting triplet excited state is much the same for 5 (Figure 4, Tables S5-S6).
During the assembly of 4, the ligand 1 is forced into a geometry that it would not otherwise take in either its free form or when the pyridyls are methylated (6) or coordinated to a mono-platinum building block (5) (Figure 3). Any manipulation to the pyridyl nitrogen drives the photophysical properties of the ligand from a π → π* state to a charge transfer state between the phenothiazine arms and the pyridyl group. Two aromatic systems twisted in respect to one another has been known to induce intersystem crossing.10 A plausible explanation for the lack of emission from 6 would be that the twist induced by methylation allows intersystem crossing to the triplet excited state, however, does not make the triplet to singlet transition back to the ground state sufficiently allowed enough to beat out the nonradiative pathways. By the addition of platinum (5), both intersystem crossing and the oscillator strength of the emissive transition is increased due to the heavy atom effect. Finally, the self-assembly 4 has a higher quantum yield due to an increase in the kr which most likely stems from the ligand being held in a different orientation relative to 5, which has the same ratio of platinum centers to linkers, as well as the inhibition of the intramolecular motions by the formation of a rigid cage structure that decreases the knr.
CONCLUSIONS
In summary, coordination-driven self-assembly is utilized for the preparation of a light-emitting metallacage with a Φem in dilute solution higher than that of the parent luminophore. The addition of either platinum or methyl to the pyridyl groups of 1 changes the nature of the excited state from a π → π* state to a triplet charge transfer state. This triplet charge transfer state is non-emissive in 6, but the transition becomes sufficiently allowed in 5 by the introduction of platinum. Self-assembly into 4 increases the quantum yield in solution 4-fold primarily due to the further twisting of the luminophore, which increases the radiative rate constant. Restriction of intramolecular motions by the formation of rigid cage structure of 4 also contributed to the enhancement in quantum yield by decreasing non-radiative decay rate. This study shows the potential of coordination-driven self-assembly for the preparation of supramolecular systems with emergent photophysical properties due to its synthetic efficiency and high structural versatility, which enables the facile modulation of the local environments surrounding the luminophores.
Supplementary Material
ACKNOWLEDGMENT
X.L. thanks the NSF (CHE-1506722) and NIH (1R01GM128037–01) for their support. P.J.S. thanks NIH (Grant R01 CA215157) for financial support. T.R.C. thanks the University at Buffalo and the State University of New York (SUNY) Research Foundation for financial support.
Footnotes
Supporting Information. The supporting information is available free of charge via the Internet at http://pubs.acs.org.
Synthetic methods for the compounds, NMR, ESI-TOF-MS..
The authors declare no competing financial interest.
REFERENCES
- 1. (a).Basabe-Desmonts L; Reinhoudt DN; Crego-Calama M Design of Fluorescent Materials for Chemical Sensing. Chem. Soc. Rev. 2007, 36, 993. [DOI] [PubMed] [Google Scholar]; (b) Schaferling M The Art of Fluorescence Imaging with Chemical Sensors. Angew. Chem. Int. Ed. Engl. 2012, 51, 3532. [DOI] [PubMed] [Google Scholar]; (c) Ostroverkhova O Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116, 13279. [DOI] [PubMed] [Google Scholar]
- 2. (a).Mei J; Leung NLC; Kwok RTK; Lam JWY; Tang BZ Aggregation-Induced Emission: Together We Shine, United We Soar! Chem. Rev. 2015, 115, 11718. [DOI] [PubMed] [Google Scholar]; (b) Sasaki S; Drummen GPC; Konishi G.-i. Recent Advances in Twisted Intramolecular Charge Transfer (Tict) Fluorescence and Related Phenomena in Materials Chemistry. Journal of Materials Chemistry C 2016, 4, 2731. [Google Scholar]; (c) Wu J; Liu W; Ge J; Zhang H; Wang P New Sensing Mechanisms for Design of Fluorescent Chemosensors Emerging in Recent Years. Chem. Soc. Rev. 2011, 40, 3483. [DOI] [PubMed] [Google Scholar]; (d) Dias FB; Santos J; Graves DR; Data P; Nobuyasu RS; Fox MA; Batsanov AS; Palmeira T; Berberan-Santos MN; Bryce MR; Monkman AP The Role of Local Triplet Excited States and D-a Relative Orientation in Thermally Activated Delayed Fluorescence: Photophysics and Devices. Advanced Science 2016, 3, 1600080. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Mako TL; Racicot JM; Levine M Supramolecular Luminescent Sensors. Chem. Rev. 2018. [DOI] [PubMed] [Google Scholar]; (f) Pan M; Liao W-M; Yin S-Y; Sun S-S; Su C-Y Single-Phase White-Light-Emitting and Photoluminescent Color-Tuning Coordination Assemblies. Chem. Rev. 2018, 118, 8889. [DOI] [PubMed] [Google Scholar]
- 3.Cook TR; Stang PJ Recent Developments in the Preparation and Chemistry of Metallacycles and Metallacages Via Coordination. Chem. Rev. 2015, 115, 7001. [DOI] [PubMed] [Google Scholar]
- 4. (a).Yoshizawa M; Klosterman JK; Fujita M Functional Molecular Flasks: New Properties and Reactions within Discrete, Self-Assembled Hosts. Angew. Chem. Int. Ed. Engl. 2009, 48, 3418. [DOI] [PubMed] [Google Scholar]; (b) Croue V; Goeb S; Salle M Metal-Driven Self-Assembly: The Case of Redox-Active Discrete Architectures. Chem Commun (Camb) 2015, 51, 7275. [DOI] [PubMed] [Google Scholar]; (c) Jongkind LJ; Caumes X; Hartendorp APT; Reek JNH Ligand Template Strategies for Catalyst Encapsulation. Acc. Chem. Res. 2018, 51, 2115. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ward MD; Hunter CA; Williams NH Coordination Cages Based on Bis(Pyrazolylpyridine) Ligands: Structures, Dynamic Behavior, Guest Binding, and Catalysis. Acc. Chem. Res. 2018, 51, 2073. [DOI] [PubMed] [Google Scholar]; (e) Jansze SM; Severin K Clathrochelate Metalloligands in Supramolecular Chemistry and Materials Science. Acc. Chem. Res. 2018, 51, 2139. [DOI] [PubMed] [Google Scholar]; (f) Zhang D; Ronson TK; Nitschke JR Functional Capsules Via Subcomponent Self-Assembly. Acc. Chem. Res. 2018, 51, 2423. [DOI] [PubMed] [Google Scholar]; (g) Wu GY; Chen LJ; Xu L; Zhao XL; Yang HB Construction of Supramolecular Hexagonal Metallacycles Via Coordination-Driven Self-Assembly: Structure, Properties and Application. Coord. Chem. Rev. 2018, 369, 39. [Google Scholar]; (h) Hong CM; Bergman RG; Raymond KN; Toste FD Self-Assembled Tetrahedral Hosts as Supramolecular Catalysts. Acc. Chem. Res. 2018, 51, 2447. [DOI] [PubMed] [Google Scholar]; (i) Pan M; Wu K; Zhang J-H; Su C-Y Chiral Metal–Organic Cages/Containers (Mocs): From Structural and Stereochemical Design to Applications. Coord. Chem. Rev. 2019, 378, 333. [Google Scholar]
- 5.Zhang YZ; Fulong CRP; Hauke CE; Crawley MR; Friedman AE; Cook TR Photophysical Enhancement of Triplet Emitters by Coordination-Driven Self-Assembly. Chem. Eur. J. 2017, 23, 4532. [DOI] [PubMed] [Google Scholar]
- 6.Yan X; Cook TR; Wang P; Huang F; Stang PJ Highly Emissive Platinum(Ii) Metallacages. Nat. Chem 2015, 7, 342. [DOI] [PubMed] [Google Scholar]
- 7.Harris DC; Bertolucci MD Symmetry and Spectroscopy: An Introduction to Vibrational and Electronic Spectroscopy. Dover Publications, New York, 1989. [Google Scholar]
- 8. (a).Xu S; Liu T; Mu Y; Wang YF; Chi Z; Lo CC; Liu S; Zhang Y; Lien A; Xu J An Organic Molecule with Asymmetric Structure Exhibiting Aggregation-Induced Emission, Delayed Fluorescence, and Mechanoluminescence. Angew. Chem. Int. Ed. Engl. 2015, 54, 874. [DOI] [PubMed] [Google Scholar]; (b) Gan S; Luo W; He B; Chen L; Nie H; Hu R; Qin A; Zhao Z; Tang BZ Integration of Aggregation-Induced Emission and Delayed Fluorescence into Electronic Donor–Acceptor Conjugates. J. Mater. Chem. C 2016, 4, 3705. [Google Scholar]
- 9.Kaloudi-Chantzea A; Karakostas N; Raptopoulou CP; Psycharis V; Saridakis E; Griebel J; Hermann R; Pistolis G Coordination-Driven Self Assembly of a Brilliantly Fluorescent Rhomboid Cavitand Composed of Bodipy-Dye Subunits. J. Am. Chem. Soc. 2010, 132, 16327. [DOI] [PubMed] [Google Scholar]
- 10. (a).Schmidt K; Brovelli S; Coropceanu V; Beljonne D; Cornil J; Bazzini C; Caronna T; Tubino R; Meinardi F; Shuai Z; Brédas J-L Intersystem Crossing Processes in Nonplanar Aromatic Heterocyclic Molecules. J. Phys. Chem. A 2007, 111, 10490. [DOI] [PubMed] [Google Scholar]; (b) Nagarajan K; Mallia AR; Muraleedharan K; Hariharan M Enhanced Intersystem Crossing in Core-Twisted Aromatics. Chem. Sci. 2017, 8, 1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.