

In Brief
A highly multiplexed CRISPRi screen uncovers gene-enhancer relationships at scale.
SUMMARY
Over one million candidate regulatory elements have been identified across the human genome, but nearly all are unvalidated and their target genes uncertain. Approaches based on human genetics are limited in scope to common variants and in resolution by linkage disequilibrium. We present a multiplex, expression quantitative trait locus (eQTL)-inspired framework for mapping enhancer-gene pairs by introducing random combinations of CRISPR/Cas9-mediated perturbations to each of many cells, followed by single-cell RNA sequencing (RNA-seq). Across two experiments, we used dCas9-KRAB to perturb 5,920 candidate enhancers with no strong a priori hypothesis as to their target gene(s), measuring effects by profiling 254,974 single-cell transcriptomes. We identified 664 (470 high-confidence) cis enhancer-gene pairs, which were enriched for specific transcription factors, non-housekeeping status, and genomic and 3D conformational proximity to their target genes. This framework will facilitate the large-scale mapping of enhancer-gene regulatory interactions, a critical yet largely uncharted component of the cis-regulatory landscape of the human genome.
Graphical Abstract
INTRODUCTION
Consequent to an era of biochemical surveys of the human genome (e.g., Encyclopedia of DNA Elements [ENCODE]) and “common variant” human genetics (i.e., genome-wide association study [GWAS] and expression quantitative trait locus [eQTL] studies), we are awash in candidate regulatory elements and phenotype-linked haplotypes, respectively (ENCODE Project Consortium, 2012; MacArthur et al., 2017). Determining whether and how each candidate regulatory element is truly functional, as well as pinpointing which variant(s) are causal for each genetic association, will require functional characterization of vast numbers of sequences.
We and others have recently adapted cell-based CRISPR/Cas9 genetic screens to evaluate candidate regulatory sequences in their native genomic context (Canver et al., 2015; Diao et al., 2016, 2017; Fulco et al., 2016; Gasperini et al., 2017; Klann et al., 2017; Korkmaz et al., 2016; Rajagopal et al., 2016; Sanjana et al., 2016). However, two aspects of these studies limit their scalability. First, they focus on the regulation of a single gene per experiment, typically entailing the development of a gene-specific assay. Second, each cell is a vehicle for one CRISPR-mediated perturbation, with the specificity-conferring guide-RNAs (gRNAs) usually introduced via lentivirus at a low multiplicity of infection (MOI). With millions of candidate regulatory elements and ~20,000 regulated genes in the human genome, these limitations preclude the comprehensive dissection of the cis-regulatory architecture of even a single cell line.
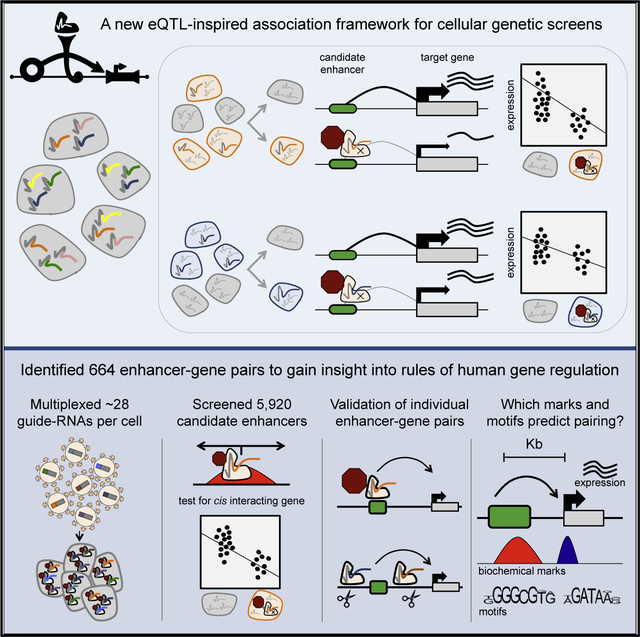
Here, we introduce a framework (Figure 1A) designed to overcome both limitations. First, by using single-cell RNA sequencing (scRNA-seq) instead of gene-specific assays, one experiment can globally capture perturbations to gene expression (Adamson et al., 2016; Datlinger et al., 2017; Dixit et al., 2016; Hill et al., 2018; Jaitin et al., 2016; Xie et al., 2017), with no strong a priori hypothesis as to the target gene of each regulatory element tested. Second, by introducing gRNAs at a high MOI, each individual cell acquires a unique combination of perturbations against the isogenic background of a cell line. Introducing multiple perturbations per cell markedly increases power (Figure 1B). An association framework inspired by eQTL studies (Morley et al., 2004; Stranger et al., 2012) is used to map cis and trans effects by comparing gene expression in the subset of cells that contain a given gRNA to those that lack that guide. This strategy is analogous to conventional eQTL studies, but with individuals replaced by cells, variants replaced by unique combinations of gRNAs per cell to induce multiplex CRISPR-interference (CRISPRi), and tissue-level RNA-seq replaced by scRNA-seq. However, unlike eQTL studies, the resolution of our screen is not constrained by linkage disequilibrium, nor is it limited to studying sites in which common genetic variants happen to exist. Although we recognize the imperfection of the analogy given that a reverse genetic screen using CRISPRi is far from equivalent to mapping the natural genetic variation that underlies QTLs, the fact that we were directly inspired by the eQTL framework led us to originally term this method “crisprQTL mapping.”
Figure 1. Multiplex Enhancer-Gene Pair Screening.

(A) Enhancer-gene pairs are screened by introducing random combinations of CRISPR/Cas9 candidate enhancer perturbations to each of many cells, followed by scRNA-seq to capture expression levels of all transcripts. Then, all candidate enhancers are tested against any gene by correlating presence of any perturbation with reduction of any transcript.
(B) Multiplex perturbations increase power to detect changes in expression in single-cell genetic screens while greatly reducing the number of cells that need to be profiled. Power calculations on simulated data show that increasing the number of perturbations per cell increases power to detect changes in expression, including for genes with low (0.10 mean UMIs per cell), medium (0.32), or high (1.00) mean expression. x axis corresponds to the simulated % repression of target transcript.
RESULTS
A Proof-of-Concept Multiplex Enhancer-Gene Pair Screen Targeting 1,119 Candidate Enhancers
To establish the feasibility of the assay formerly known as crisprQTL mapping, we targeted 1,119 candidate enhancers in the chronic myelogenous leukemia cell line K562, with CRISPRi as our mode of perturbation. For CRISPRi, we used a nuclease-inactive Cas9 tethered to the KRAB repressor domain to induce heterochromatin across an ~1–2 kb window around a gRNA’s target site (Thakore et al., 2015). The 1,119 candidate enhancers were all intergenic DNase I hypersensitive sites (DHSs) representing various combinations of H3K27 acetylation, p300, GATA1, and RNA Pol II binding (Figure 2A). Candidate enhancers were required to fall within the same toplogically associated domain (TAD) as at least one gene from the top decile of K562 expression and were collectively distributed across 510 TADs on every chromosome (Rao et al., 2014). 5,611 of the 12,984 genes expressed in K562 cells fell within 1 Mb of at least one candidate enhancer (K562-expressed genes defined as those observed in at least 0.525% of cells profiled in this pilot experiment).
Figure 2. Pilot Multiplex Enhancer-Gene Pair Screen Testing 1,119 Candidate Enhancers in K562 Cells.

(A) 1,119 candidate enhancers were chosen based on intersection of enhancer-associated features and each targeted by two gRNAs.
(B) Schematic of this multiplex enhancer-gene pair screening method. (i) gRNAs were cloned into a lentiviral vector, and delivered to K562 cells at a high MOI. (ii) scRNA-seq was performed on these cells, with concurrent capture of the multiple gRNAs present in each cell. (iii) For each candidate enhancer, cells were partitioned based on whether or not they contained a gRNA targeting it. (iv) For each such partition, we tested for differential expression between the two populations for any gene within 1 Mb of the candidate enhancer.
(C) gRNAs were delivered to K562 cells at a high MOI, with median of 15 ± 11.3 gRNAs identified per cell.
(D) A total of 47,650 single cell transcriptional profiles were generated. Each perturbation was identified in a median of 516 ± 177 cells.
(E) Quantile-quantile plot of the differential expression tests. Distributions of observed versus expected p values for candidate enhancer-targeting gRNAs (orange) and NTC gRNAs (gray; downsampled) are shown.
(F) Expression of selected TSS (top row) and β-globin LCR positive controls (bottom row). Nearly all targeted TSSs, and all positive controls, showed significant differential expression of the expected target genes between cells with (+) versus without (−) targeting gRNAs, in contrast with NTCs. Percent changes and p values show the effect size and significance of differential expression of the denoted target gene between these cell groups. See also Figure S1 and Table S1.
Two gRNAs were designed to target each candidate enhancer. Additional pairs of gRNAs served as positive controls (targeting the transcription start sites [TSSs] of genes sampled from the top decile of K562 expression, or alternatively hyper-sensitivity sites of the α-globin locus control region [LCR]) and negative controls (50 non-targeting controls or “NTC” that target nowhere or in a gene desert) (Table S1A).
This gRNA library was cloned into the lentiviral CROP-seq vector modified to include a CRISPRi-optimized backbone (Chen et al., 2013; Datlinger et al., 2017; Hill et al., 2018), and K562 cells were transduced at a high MOI (Figure 2B). After 10 days to allow for effective CRISPRi, the transcriptomes of 47,650 single cells were profiled. With a targeted amplification protocol (Adamson et al., 2016; Dixit et al., 2016; Hill et al., 2018), we identified a median of 15 ± 11.3 gRNAs per cell (Figure 2C). Each candidate enhancer or control was targeted in a median of 516 ± 177 cells (Figure 2D). For each targeted element, we partitioned the 47,650 cells based on whether they did or did not contain gRNA(s) targeting it. We then tested for a reduction in the expression of each K562-expressed gene within 1 Mb of that element (Figure 2B) (Stranger et al., 2012). We also tested the 50 NTCs against all K562-expressed genes within 1 Mb of any targeted candidate enhancer. For perspective, with a “one gRNA per cell” framework, achieving equivalent power would require profiling the transcriptomes of ~715,000 single cells.
A quantile-quantile plot showed an excess of significant associations involving the targeting of candidate enhancers relative to NTC controls (Figure 2E). We defined a 3.5% empirical false discovery rate (FDR) threshold based on the NTC tests as they are subject to the same sources of error as the element-targeting gRNAs. At this threshold, 94% (357 of 381) of TSS-targeting positive controls repressed their associated genes, as did all β-globin LCR controls (examples shown in Figure 2F). Additionally, we re-identified a known enhancer 3.6 kb upstream of GATA1 (Fulco et al., 2016).
At this same threshold, targeting of 11% of candidate enhancers (128 of the 1,119) repressed 1+ gene(s) within 1 Mb. As there were 13 candidate enhancers whose targeting impacted more than one gene (Figure S1A), this analysis yielded a total of 145 enhancer-gene pairs (Table S1B). Of the 105 downregulated target genes (Figure S1B), 26 were impacted by targeting of more than one of the 128 candidate enhancers (Figure S1A).
We examined the characteristics of paired enhancers whose targeting significantly impacted expression of 1+ genes in cis. We found paired candidate enhancers to be enriched for high chromatin immunoprecipitation sequencing (ChIP-seq) peak strength (based on average enrichment in ChIP-seq peak region) for enhancer-associated histone modifications (H3K27ac, logistic regression p value = 4e 5, candidate enhancers in the top quintile were 1.4-fold more likely to be paired than those in the bottom quintile), certain co-activators (p300, p value = 4e–16, 1.1-fold) and lineage-specific transcription factors (TFs) (GATA1 p value = 2e–7, 1.4-fold; GATA2 p value = 3e–10, 1.5-fold; SMAD1 p value = 1e–6, 1.4-fold; TAL1 p value = 6e–6, 1.1-fold; CCNT2 p value = 3e–7, 1.4-fold), whereas RNA Pol II and H3K4me1 were not associated (Figure S1C). Using these features, as well as average enrichment within the DHS and whether each had been previously validated in vivo (Visel et al., 2007), we trained a multivariate logistic regression classifier to distinguish the 128 paired candidate enhancers from the 991 candidate enhancers for which we did not identify a target gene, achieving an AUPR of 0.31 (area under precision-recall curve; median from 5-fold cross validation; Figure S1D).
A Scaled Multiplex Enhancer-Gene Pair Screen Targeting 5,779 Candidate Enhancers
To demonstrate this approach at a substantially greater scale, we performed a second experiment targeting five times as many candidate enhancers (n = 5,779). First, two-thirds of these (n = 3,853) were new DHSs chosen by the classifier trained on the first experiment (Figures 3A and S1D). Second, as this set may be biased toward annotations used to select the initially targeted candidate enhancers (Figure 2A), we also targeted 948 exploratory DHSs chosen independent of the model (see STAR Methods). Third, we re-targeted 978 of the 1,119 initially targeted pilot candidate enhancers, including the aforementioned candidate enhancers paired with target genes in the pilot. Altogether, candidate enhancers targeted in this scaled experiment were within 1 Mb of 10,560 of 13,135 K562-expressed genes. As previously, we designed two gRNAs per candidate enhancer. However, to evaluate whether poorly efficacious gRNAs might contribute to false negatives, we designed an additional two gRNAs for 377 of the 978 re-targeted candidate enhancers (Figure 3B). Finally, in addition to gRNA pairs targeting 5,779 candidate enhancers, we included the same positive and negative control gRNA pairs targeting 381 TSSs, the globin LCR, and 50 NTC pairs (Table S2A).
Figure 3. Multiplex Enhancer-Gene Pair Screening at Scale in K562 Cells.

(A) For a scaled experiment, gRNAs were designed to target a total of 5,779 candidate enhancers. Characteristics are shown for 3,853 sites chosen by a model informed by the hits identified in the pilot experiment.
(B) 948 exploratory candidate enhancers were sampled from K562 DHSs. 978 candidate enhancers from the pilot were re-targeted with the same gRNA pair, and 377 of these were also targeted with a second, alternative gRNA pair.
(C) gRNAs were again delivered to K562 cells, but at a higher MOI than the pilot experiment (median 28 ± 15.3 gRNAs identified per cell).
(D) A total of 207,324 single cell transcriptional profiles were generated. Each perturbation was identified in a median of 915 ± 280 single cells.
(E) Q-Q plot of the differential expression tests. Distributions of observed versus expected p values for candidate enhancer-targeting gRNAs that were correlated with decrease in target gene expression (orange) and NTC gRNAs (gray; downsampled) are shown.
(F) Histogram of the number of target genes impacted by each candidate enhancer identified as part of a pair (10% empirical FDR).
(G) Histogram of the number of paired candidate enhancers detected as regulating each target gene (10% empirical FDR).
(H) Effect sizes for the 664 enhancer-gene pairs that pass a <0.1 empirical FDR, the 470 high-confidence enhancer-gene pairs, and the 97% of TSS controls that are detected as repressing their target genes.
See also Figure S2E and Table S2.
K562 cells were transduced at an even higher MOI than in the proof-of-concept experiment. We profiled the transcriptomes of 207,324 single cells and identified a median of 28 ± 15.3 gRNAs per cell (Figure 3C). Each candidate enhancer was targeted in a median of 915 ± 280 single cells (Figure 3D). Testing for associations as previously, a quantile-quantile plot again showed an inflation of significant associations involving the targeting of candidate enhancers (Figure 3E). Using the NTCs to set a more inclusive empirical FDR of 10%, 97% (369 of 381) of TSS-targeting positive controls repressed their associated genes, as did all β-globin LCR controls. At this same threshold, of the 5,779 candidate enhancers, we identified 600 as repressing 1+ gene(s) within 1 Mb. These included 397/3,853 model-selected candidate enhancers (10%), 35/948 systematically sampled exploratory DHS (4%), and 168/978 previously targeted candidate enhancers (17%). As targeting of 53/600 candidate enhancers downregulated more than one gene (Figure 3F), we collectively identified a total of 664 enhancer-gene pairs (Table S2B). As 113 genes were downregulated by targeting of more than one candidate enhancer, these pairs involved 479 target genes (Figure 3G). These ranged in effect size from −1.4% to −97.5% target gene repression (Figure 3H).
To evaluate reproducibility, we compared our results for the 978 candidate enhancers targeted in both experiments. Applying the same empirical FDR threshold of 10% to each dataset, 187/978 were identified as paired candidate enhancers in the pilot experiment, and 168/978 as paired candidate enhancers in the scaled experiment. Of these, 105 were identified in both experiments (hypergeometric test of overrepresentation p value 7e–45; 3.3-fold enriched over expectation). The pairs identified in both experiments had stronger effect sizes (median 25% versus 13% repression), better correlated effect sizes (Spearman’s rho for % repression: 0.82 versus 0.13; Figure S2A), and involved more highly expressed genes (median0.90 versus 0.63 UMIs per cell), than pairs identified in only one experiment.
As noted above, an additional pair of gRNAs for 377/978 re-targeted candidate enhancers were included in this experiment, to facilitate evaluation of the extent to which poorly efficacious gRNAs might contribute to false negatives. In the scaled experiment at a 10% empirical FDR, 109/377 of the original gRNA pairs and 119/377 of the new gRNA pairs mediated enhancer-gene pairs. Of these, 84 were directed at the same candidate enhancers, a highly significant overlap (hypergeometric test of overrepresentation p value 4e–33; 2.4-fold enriched over expectation). Furthermore, the effect sizes on the most highly repressed genes for gRNA pairs targeting the same candidate enhancer were well-correlated (Spearman’s rho for % repression: 0.73; Figure S2B). Overall, this analysis suggests that targeting candidate enhancers with more than two gRNAs could modestly increase our sensitivity.
Due to the noise from variability in expression levels, effect sizes, and gRNA quality, we defined a high-confidence subset of reproducible enhancer-gene pairs as those identified in both experiments at the 10% empirical FDR (112 pairs; 359/381 [94%] of targeted TSSs also met this criteria), as well as those internally reproducible between the 2 independently targeting gRNAs for candidate enhancers only tested in the scaled experiment (358 pairs; 337/381 [88%] of targeted TSSs also met this criteria). Putting these sets together, we annotated 470 enhancer-gene pairs as high-confidence (Table S2B), involving 441 candidate enhancers (Figure S2C) and impacting expression of 353 target genes. These ranged in effect size from −7.9% to −97.5% (Figure 3H). We use this high-confidence subgroup for all summary analyses described below, unless otherwise noted. Of note, 24 candidate enhancers are paired with multiple target genes (Figure S2D); it is possible that some of these pairings represent indirect effects (e.g., if a gene that is the primary target of the enhancer is involved in the regulation of the other gene).
Replication or Validation of 22 Selected Enhancer-Gene Pairs in Singleton Experiments
We next sought to individually replicate 15 enhancer-gene pairs with a range of effect sizes (−10% to −81%) and 6 “null” candidate enhancers not paired with any target gene (Table S3A). We transduced K562 cells separately with small pools of gRNAs targeting individual candidate enhancers, and investigated the impact on gene expression via bulk RNA-seq (Table S3A). For 12/15 replication experiments targeting candidate enhancers associated with downregulation of a target gene, the effect sizes were similar in magnitude and direction of effect (Figures 4A–4D and S5). For all 9 experiments predicted to cause >30% repression, replication effects were also significant in a test of differential expression (cis adjusted p value <0.1). Of the 6 lines targeting a “null” candidate enhancer, none significantly decreased expression of a gene located within 1 Mb of the target (cis adjusted p values >0.1).
Figure 4. Replication and Validation of Selected Enhancer-Gene Pairs in Singleton Experiments.

(A–D) For each singleton replication experiments of enhancer-gene pairs, bulk RNA-seq was performed on CRISPRi+ K562 cells transduced with gRNAs targeting (purple) e-PRKCB (A), e-PTGER3 (B), e-GYPC (C), e-NMU (D), or the TSSs (dark red) of their respective target genes. Target gene expression in the singleton-target cell lines (red/purple) as compared to replication experiments in which the other 4 candidate enhancers or TSSs were targeted (gray). Eleven other singleton CRISPRi experiments are summarized in Figure S5.
(E–G) To validate three enhancer-gene pairs by sequence deletion, monoclonal lines were generated with full deletion of the locus’s genomic sequence in three to six independent clones (e-NMU, E; e-CITED2, F; and e-GLUL, G), followed by bulk RNA-seq. See also Figure S4A.
(H) NMU-targeting cells were phenotyped by fluorophore-labeling of intracellular NMU transcripts by RNA flowFISH. (ii–iii) Singleton CRISPRi targeted cells as in (D). (iv–v) A heterogeneous pool of cells engineered such that a portion (based on deletion efficiency) harbor full or scanning deletions of e-NMU (see also Figures S4B and S4C).
Although the field often refers to singleton independent re-testing via CRISPRi as “validation,” it is a recapitulation of the modality of perturbation of the screen and perhaps better classified as another form of replication. Therefore, we also performed a more stringent validation by generating 3+ monoclonal homozygous deletion lines for each of 3 enhancers (effect size in scRNA-seq screen: e-NMU = −81%, e-CITED2 = −35%, e-GLUL = −21%; Figure S4; Table S3B). All three selected enhancers are quite distal from the gene whose expression they regulate (>50 kb). These homozygously deleted lines all had the expected and magnitude of direction of effect (Figure 4E–4G), indeed with stronger effect sizes than seen by CRISPRi perturbation in the scRNA-seq screen (effect size with deletion: e-NMU = −100%, e-CITED2 = −57%, e-GLUL = 67%; Table S3B).
In our validations of the NMU candidate enhancer (“e-NMU”), we also applied RNA flowFISH (Choi et al., 2018) and again observed decreased NMU expression in singleton CRISPRi populations targeting NMU’s TSS (−79% less NMU than untreated cells) and e-NMU (−73% less NMU, Figure 4H, ii–iii). We also used flowFISH to phenotype a heterogeneous pool of cells that harbored a mix of full, partial, or no deletions of e-NMU, generated by transient transfection of flanking pairs of gRNAs. 12% of the cells showed reduced NMU expression in comparison to untreated cells (Figure 4H, iv), which is in-line with expected full deletion efficiency (Gasperini et al., 2017). Cells were sorted into bins of low, medium, or high NMU expression. PCR of the e-NMU locus revealed enrichment of the full deletion in the low and medium NMU bins, whereas full deletion was rarer in the high NMU bin (Figure S4B). To further dissect e-NMU, we additionally transfected with 19 gRNAs interspersed every ~100 bp across e-NMU to generate deletions of diverse lengths and locations, inducing reduction of NMU expression in 35% of cells compared to untreated (Figure 4H, v). PCR of e-NMU again showed a similar enrichment of longer deletions in the cells with lower NMU expression (Figure S4C).
In summary, of the high confidence pairs that we re-tested by singleton CRISPRi and/or singleton CRISPR-mediated deletion, 13/16 matched with respect to both their direction and magnitude of effect size, whereas 3/16 failed to validate. This false-positive rate is consistent with the 10% FDR that we used to assign a threshold for calling pairs (p value on whether 3/16 disagrees with 10% FDR = 0.21).
Selected Examples of Enhancer-Gene Pairs
We highlight four of the enhancer-gene loci in Figure 5. An “e-” prefix is used to denote candidate enhancers that we targeted in singleton replication experiments. In the scaled experiment, we targeted four candidate enhancers across the region upstream of PRKCB. The furthest of these (Figure 5A, i; 50 kb upstream) did not have an effect, but candidate enhancers 32, 14, and 9 kb upstream of the TSS were associated with repression of PRKCB (Figure 5A, ii–iv). The strongest of these, located 14 kb upstream, was also targeted and replicated in both the pilot and singleton experiments (“e-PRKCB”, Figure 4A and Figure 5A, iii).
Figure 5. Highlighted Examples of Enhancer-Gene Pairs.

(A) Three candidate enhancers (labeled ii–iv) that reside 32, 14, and 9 kb upstream of PRKCB were paired with PRKCB, but a fourth (i) that lies 50 kb upstream was not (shown: hg19 chr16:23791225–23851797; iii is e-PRKCB in Figure 4A).
(B) A single candidate enhancer (e-PTGER3 in Figure 4B) located 371 kb downstream of PTGER3 was paired with PTGER3 (shown: chr1:71104684–71582921).
(C) Two candidate enhancers paired with GYPC (ii–iii) lie in the 11 kb region upstream of GYPC. However, a third candidate enhancer (i) immediately adjacent to (ii) was not paired with GYPC (shown: chr1:71104684–71582921; ii is e-GYPC in Figure 4C).
(D) Targeting five candidate enhancers (i–v) located 30.5, 87, 93.4, 94.1, and 97.6 kb upstream of NMU, significantly reduced expression of NMU (shown: chr1:71104684–71582921; iii-iv is e-NMU in Figure 4D).
Target genes’ normalized expression presented on log scale.
Asterisks denote the candidate enhancers that were targeted as part of a singleton replication experiment (Figure 4). + and - denote thecells from the at-scale screen with or without gRNAs targeting that locus. Percent changes and p values denote the size and significance of a differential expression between these cell groups.
In the pilot, scaled, and singleton replication experiments, we targeted only one candidate enhancer within 1 Mb of PTGER3 (“e-PTGER3,” Figure 4B and Figure 5B), located 371 kb downstream of the PTGER3 TSS. In each of the three experiments, targeting of e-PTGER3 consistently repressed expression of PTGER3.
We targeted three candidate enhancers in the region upstream of GYPC, a human erythrocyte membrane protein. Targeting of candidate enhancers 4.5 kb upstream (Figure 5C, iii) and 10 kb (“e-GYPC”, Figure 4C and Figure 5C, ii) upstream of GYPC’s TSS resulted in its repression in the scaled experiment. Interestingly, a candidate enhancer so close to e-GYPC as to likely be unresolvable from it by CRISPRi (Figure 5C, i) did not result in repression of GYPC in the scaled experiment, potentially attributable to poor gRNA quality or another source of false negatives.
Targeting of multiple candidate enhancers decreased expression of the same gene, NMU, which encodes neuromedin U, a neuropeptide that plays roles in inflammation as well as erythropoiesis (Gambone et al., 2011). One candidate enhancer was associated with light repression of NMU (Figure 5D, i; located 30.5 kb upstream of the NMU TSS). An additional four candidate enhancers were located in close proximity to one another, but nearly 100 kb upstream of the NMU TSS (“e-NMU”, Figure 4D and Figure 5D, ii–v; located 87, 93.4, 94.1, and 97.6 kb upstream). Because of their proximity, these closely located candidate enhancers internally replicate e-NMU within the scaled experiment, in contrast to the neighboring candidate enhancers of e-GYPC.
Insights into the Properties of Human Enhancers and Their Target Genes
Distance between Paired Enhancers and Promoters
We find that of the class of enhancers surveyed here (nonintronic, unbuffered by other enhancers), paired enhancers are separated from the TSS of their target genes by a median distance of 24.1 kb (Figure 6A, top row). Note that this analysis is restricted only to high-confidence pairs that fall upstream of their target genes (n = 354), to avoid bias from the length of the gene body consequent to the fact that we avoided targeting intronic candidate enhancers for which CRISPRi might directly inhibit transcription. Upstream and downstream enhancers do not exhibit large differences in their effect size distributions (Figure S2E). Given that we tested for associations against all genes within 1 Mb of each candidate enhancer (Figure 6A, fourth row; median distance of 440.2 kb, similarly restricted to upstream tests), this supports a very strong role for proximity in governing enhancer-promoter choice. Nonetheless, 153/770 (33%) of enhancer-gene pairs involved skipping of at least one closely located TSS of another K562-expressed gene (Figure 6B). Interestingly, low-confidence enhancer-gene pairs (i.e., the subset of the 600 that were not high-confidence and also fall upstream; n = 127) were also enriched for proximity to their target genes, suggesting that a substantial proportion of these are bona fide enhancers (Figure 6A, second row; median distance of 45.0 kb).
Figure 6. Characteristics of K562 Enhancer-Gene Pairs.

(A) Paired candidate enhancers fall close to target genes. Distribution of distances between the paired candidate enhancers and their target gene’s TSS (top row, high confidence pairs; second row, lower confidence pairs), the TSS of whatever K562-expressed gene is closest (third row), or the TSS of every K562-expressed gene within 1 Mb (fourth row). Plotted with respect to gene orientation. Of the 470 high confidence pairs, this plot displays only the 354 that fall upstream of the target genes (as the gRNA library does not include candidate enhancers within 1 kb of any gene body, downstream enhancers are biased to fall further from the target TSS). A TSS-focused zoom of this plot is included as Figure S5E.
(B) 317 of 470 high-confidence pairs target the most proximal K562-expressed gene. Target genes are ranked by their absolute distance to the paired candidate enhancer (1 = closest, 2 = second closest, etc.).
(C) This framework captures regulatory effects on genes from a broad range of expression levels (expression = mean transcript UMIs/cell in the entire 207,324 cell dataset, for 13,135 K562-expressed genes, 10,560 of these within 1 Mb of a targeted candidate enhancer in the scaled experiment, and 470 high-confidence enhancer-gene pairs). See also Figure S5D.
(D) Paired candidate enhancers tend to fall in enhancer-associated ChIP-seq peaks that show stronger signals. All ChIP-seq peaks that overlap the scaled experiment’s 5,779 candidate enhancers were divided into quintiles defined as the average enrichment in ChIP-seq peak region (0 = no such peak overlaps the candidate enhancer, 1 = lowest, 5 = highest). Histograms of the proportion of which candidate enhancers in each quintile that were paired with a target gene are shown for the eight most-enriched ChIP-seq datasets.
(E) Enhancer-gene pairs interact more frequently in K562 Hi-C data (left, fractional ranking of enhancer-gene pairs’ Hi-C interaction-frequency against all other possible interactions at similar distances within the same TAD, K-S test against a uniform distribution p value <2e - 16), as compared to two control distributions: paired target gene TSSs paired with a shuffled genomic locus (middle: K-S test versus actual enhancer-gene pairs distribution = p value 2e - 7) or paired candidate enhancers paired with a shuffled genomic locus (right, K-S test versus actual enhancer-gene pairs distribution = p value 1e - 9). See also Figures S5B and S5C.
See also Table S4.
Of our 359 “positive control” TSSs whose targeting successfully repressed the expected gene in both experiments, 35 reduced expression of 1+ additional genes (45 apparent promoter-promoter relationships in total). 15 of these 45 involved overlapping promoters (TSSs within 1 kb), such that the observed effect of CRISPRi is likely direct. As for the remaining 30, one possibility is that these represent examples of promoters acting as enhancers, as recently reported (Diao et al., 2017; Fulco et al., 2016). Additionally, as repressive epigenetic effects may spread a few kilobases from the target site, it is possible that CRISPRi of promoters may be silencing proximal enhancers as well. However, these 30 are largely not enriched for proximity to affected genes (Figure S5A; median distance of 405.3 kb, similarly restricted to upstream tests), in contrast with enhancer-gene pairs (median distance = 24.1 kb). We therefore hypothesize that these are more likely consequent to trans effects of repressing the primary target of these TSS-targeting gRNAs. In other words, rather than these gRNA-targeted promoters acting as noncoding regulatory elements of other genes, the reduction in protein levels of the targeted gene may secondarily affects the expression of other genes.
Characteristics of Target Genes
The 353 genes included in 1+ 470 high-confidence enhancer-gene pairs had several notable characteristics. First, their expression levels are distributed similarly to the full set of 10,560 genes against which we tested (Figure 6C), suggesting we are reasonably well-powered to detect regulatory effects on even modestly expressed genes. Second, housekeeping genes were underrepresented, relative to all tested genes (hypergeometric test p value = 3e–5 and 2.1-fold depleted using the housekeeping gene list of Eisenberg and Levanon [2013]; hypergeometric test p value = 2e–6 and 3.9-fold depleted using the housekeeping gene list of Lin et al. [2017]). Similar depletions of housekeeping genes are observed when we instead compare paired target genes to the K562-expressed genes most proximal to tested candidate enhancers. Although these analyses support the view that a prevailing characteristic of housekeeping genes may be a dearth of distal regulatory elements (Ganapathi et al., 2005; Gasperini et al., 2017), we cannot fully rule out that the possibility that this result is influenced by our choice of candidate enhancers to target. Finally, paired target genes were enriched for genes with roles in leukocyte migration and differentiation, consistent with distal enhancers shaping the expression of K562-specific genes (Table S4A).
Characteristics of Paired Enhancers
We also examined the characteristics of the candidate enhancers for which targeting significantly impacted expression of 1+ genes in cis. First, as compared with the full set of 5,779 candidate enhancers targeted in either or both experiments, we tested if the 441 high-confidence candidate enhancers were enriched for strong peaks in 169 K562 ChIP-seq datasets (ENCODE Project Consortium, 2012). We identified 87 that were significantly enriched (threshold of an adjusted p value <0.005), but the eight most significant were co-activators (p300 logistic regression p value = 1e–46, candidate enhancers in the top quintile were 1.8-fold more likely to be paired than those in the bottom quintile; BRD4 p value = 2e–33, 1.6-fold), an enhancer-associated histone modification H3K27ac (p value = 8e–37, 1.6-fold), the MYC activator TBL1XR1 (p value = 2e–34, 1.5-fold), and line-age-specific TFs (TAL1 p value = 2e–33, 1.6-fold; GATA2 p value = 1e–31, 1.5-fold; DPF2 p value = 5e–31, 1.5-fold; RNF2 p value = 2e–33, 1.5-fold; Figure 6D). Other expected enhancer-associated marks also exhibited significant enrichment (CCNT2 p value = 4e–21, 1.3-fold; H3K4me1 p value =1e–19, 1.8-fold; MYC p value = 2e–12, 1.3-fold; Table S4B). However, many of these features are correlated, and BRD4, H3K4me1, TRIM24, p300, H3K27ac, ETS1, and ZNF274 were the only significant predictors in a multivariate logistic regression (p value <0.01, Table S4B). Of note, high conservation as measured by median phyloP scores (Pollard et al., 2010) was not enriched in these candidate enhancers as compared to all tested candidate enhancers (independent logistic regression p values >0.5).
Second, we examined whether paired enhancers were more likely to intersect with K562 super-enhancers. Overall, 474 of the 5,779 candidate enhancers that we tested fell within 65 K562 super-enhancers (Cao et al., 2017); however, a much higher proportion of high-confidence paired enhancers belonged to this set (102/441). Several super-enhancers contained multiple targeted enhancers that were paired with the same gene. More specifically, 20 genes were linked with two candidate enhancers, and 6 genes were linked with three or four candidate enhancers, that were located within the same super-enhancer (Table S4C).
Third, we evaluated enrichment of TF motifs in either our associated enhancers or the promoters of their target genes. Motifs for the known blood TFs KLF-1, −5, −6, −15, leukemogenesisrelated SALL4, and the MYC-interacting ZN281 were enriched in the promoters of the inclusive set of 479 paired-target genes, as compared to the promoters of all genes within 1 Mb of a tested candidate enhancer (Table S4D). Similarly, motifs for a largely distinct set of known blood TFs (TAL1, KLF-1, −3, −4, −5, −8, and GATA-1, −2, −3) and AP2C were enriched in the inclusive set of 600 paired enhancers, as compared to the overall set of 5,779 candidate enhancers tested (Table S4D).
Pairs of Transcription Factors Act Together across Enhancer-Gene Pairs
To investigate whether there was any discernible logic underlying why particular enhancers were associated with particular promoters, we next sought to identify pairs of TFs that are “co-enriched” in the inclusive set of 664 enhancer-promoter pairs(i.e., they occur across pairs at a higher frequency than expected by chance given their background frequency in each category). We identified 6 TF pairs whose sequence motifs were co-enriched in this way, suggesting potential interactions (Table S4E). For example, presence of the NR2C2 motif (implicated in regulation of the globins [Tanabe et al., 2007]) in a paired promoter was associated with presence of a KLF1 or RXRA motif in the corresponding paired enhancer. On the other hand, presence of the GATA3 motif in a paired promoter was associated with the absence of a KLF1 motif in the corresponding paired enhancer.
We also explored such pairings via ChIP-seq data. Although ChIP-seq peaks often reflect indirect binding, such secondary partners might still play a role in the restriction of enhancer-promoter interactions. We identified 24 TF pairs that are “co-enriched” in enhancer-promoter pairs (Table S4E). Unfortunately, none of the TF pairs identified in either analysis had corresponding ChIP-seq datasets or high quality consensus motifs for both TFs involved in the pair, preventing cross-confirmation between the two modalities of analysis.
Comparison of Enhancer-Gene Pairs to Hi-C-Based Measurements of Physical Proximity
We sought to evaluate whether our enhancer-gene pairs are enriched for physical proximity as measured by the global chromo-some conformation mapping technique Hi-C. To control for the dominant effects of genomic distance and TADs in Hi-C data-sets, we ranked the Hi-C contact frequencies in K562 cells (Rao et al., 2014) for the 71% of the enhancer-gene pairs that fell in the same TAD (333/470 high-confidence pairs) against all other possible interactions at similar distances within the same TAD (median 66 other genomic-loci pairs, range 6 to 260, Figures S5B and S5C). Upon plotting the fractional ranks of high-confidence pairs, we found their contact frequencies to be strongly enriched at the highest ranks (Kolmogorov-Smirnov [K-S] test against a uniform distribution p value <2e 16, Figure 6E). To ensure that this enrichment was not an artifact of paired enhancers or genes interacting more frequently with all neighboring loci (as in FIREs [Schmitt et al., 2016], we repeated this analysis twice but shuffled the genomic loci paired to either the enhancers or genes (keeping these shuffled pair sets’ overall distance distributions the same as the original enhancer-gene pair set’s distance distribution). This did not result in the same enrichment as seen in the high confidence pair distribution (K-S test of high confidence enhancer-gene pair versus enhancer-pair shuffling p value 1e–9; high confidence enhancer-gene pair versus TSS-pair shuffling p value 2e–7), consistent with more frequent looping specifically between the high confidence enhancer-gene pairs (Figure 6E). Although enriched for proximity, we note that only a minority of our hits are called as proximate to their target genes based on this analysis; as such, many enhancer-gene pairs would not have been identified if we had limited tested candidate enhancers to those physically proximate to a promoter according to Hi-C or related data.
CRISPRi Is Highly Multiplexable within Cells
To our knowledge, prior to this study, it was unknown whether extensively multiplexing gRNAs within a single cell would dilute the efficacy of CRISPRi. To evaluate this, we conducted a biological replicate of the pilot experiment, targeting the same 1,119 candidate enhancers but at a low MOI. From this experiment, we profiled the transcriptomes of 41,284 cells and identified a median of 1 ± 1.6 gRNAs per cell (Figure 7A). Each perturbation was only seen in a median of 43 ± 16 cells, as compared with 516 ± 177 cells in the high MOI pilot experiment (Figure 7B). At a 10% empirical FDR, only 316 TSSs and 69 enhancer-gene pairs were identified in the low MOI experiment, as compared with 359 TSSs and 226 enhancer-gene pairs in the high MOI pilot experiment, validating the substantial increase in power resulting from multiplexed perturbation (Figure 1B). As the same 381 TSS controls were targeted in the low MOI, pilot, and scaled experiments, we compared the degree of repression conferred by CRISPRi at increasing MOI (median 1 versus 15 versus 28 gRNAs per cell), and found them to be well-correlated (Spearman’s rho’s ranging from 0.73 to 0.87; Figure 7C). On average, the degree of repression conferred by targeting a TSS in both high MOI experiments was only ~6% less than by targeting it in the low MOI experiment (Figure 7D). Similarly, for candidate enhancers paired in the scaled experiment (10% empirical FDR) that were also targeted in the low MOI and pilot experiments, effect sizes were well correlated (Spearman’s rho’s ranging from 0.54 to 0.70; Figure 7C), and effect sizes ratios clustered around 1 (Figure 7D). Overall, these results suggest that multiplexing gRNAs within individual cells, even to MOIs of ~28, does not dilute the efficacy of CRISPRi.
Figure 7. CRISPRi Is Robust to Multiplexing within a Cell.

(A) A biological replicate of the pilot study, targeting the same 1,119 candidate enhancers and 381 TSSs, was performed at a low MOI (median 1 ± 1.6 gRNAs identified per cell).
(B) A total of 41,284 single cell transcriptional profiles were generated. Each perturbation was identified in a median of 43 ± 16 single cells.
(C) Correlation of effect sizes for TSS controls (top, purple) or enhancer-gene pairs identified in the scaled experiment (10% empirical FDR, bottom, orange) across increasing rates of gRNA per cell (left, 1 versus 15; middle, 15 versus 28; right, 1 versus 28 gRNAs/cell). Point sizes are proportional to each target gene’s expression level.
(D) The ratios of repression for each TSS control or paired candidate enhancer (as identified with a 10% empirical FDR in any experiment) in the low MOI experiment versus a high MOI experiment (top = median 1 gRNA versus 15 gRNAs; bottom = median 1 gRNA versus 28 gRNAs). The candidate enhancer outliers with stronger effect sizes in the low MOI experiment (right panel, ratios in long left tail) are likely largely due to stochastic under-sampling of lowly expressed target genes in the low MOI experiment (see also Figure S6).
DISCUSSION
Understanding the regulatory landscape of the human genome requires the validation and identification of target genes for the vast numbers of candidate enhancers that have been nominated by biochemical marks or that reside within haplotypes implicated by GWAS or eQTL studies. Our multiplexed enhancer-gene pair screening method has the potential to help address this challenge. In the scaled experiment, we evaluated 78,776 potential cis regulatory relationships involving 5,779 candidate enhancers and 10,560 expressed genes. In contrast, nine recently published CRISPR screens of noncoding sequences cumulatively studied regulatory effects on a total of 17 genes (Canver et al., 2015; Diao et al., 2016, 2017; Fulco et al., 2016; Gasperini et al., 2017; Klann et al., 2017; Korkmaz et al., 2016; Rajagopal et al., 2016; Sanjana et al., 2016). By delivering a median of 28 perturbations to each of 207,324 cells, this experiment was powered equivalently to a “one gRNA per cell” experiment profiling 5.8 million single cell transcriptomes. Of note, one recent study used scRNA-seq as a readout for the effects of CRISPR-based perturbations of 71 candidate regulatory elements on ~100 genes in seven genomic regions (Xie et al., 2017). However, its power and scope was limited by a low MOI (Figure 1B) and a gRNA barcoding strategy that suffers from a ~50% rate of template switching (Hill et al., 2018; Xie et al., 2018).
For future iterations of target prioritization for multiplexed enhancer-gene pair screening, several characteristics of our identified enhancer-gene pairs are important to keep in mind. Foremost, although a wide range of effect sizes (7.9% to97.5% for the 470 high-confidence pairs, Figure 3H) were observed on genes with a broad range of expression levels (0.0058 to 313 UMIs/cell, Figure 6C), effect sizes were correlated with expression levels (Spearman’s rho 0.53; Figure S5D). This is likely consequent to power, as small effects are more challenging to detect on lowly expressed genes. Additionally, we note that although we identified many genomic features that were significantly correlated with the likelihood of belonging to an identified pair, a pilot-trained classifier informed by biochemical marks did not appreciably increase our hit rate in the at-scale screen. Furthermore (1) 29% of enhancers did not fall within the same TAD as their target gene, (2) although enriched for proximity in 3D space as measured by Hi-C, the majority of enhancer-gene pairs are not identified as contacts in such data-sets, and (3) although enriched for sequence-level proximity, one-third of enhancer-gene pairs involved skipping of at least one closely located TSS of another K562-expressed gene. These observations underscore the difficulty of the prediction task, and we recommend that future screens do not overly bias themselves toward looking under the lamppost until additional examples accrue and the rules of mammalian gene regulation are better understood.
Although it may be surprising that cis changes in gene expression were identified for only ~10% of the candidate enhancers tested here, there are several potential caveats to bear in mind. First, previous studies have identified shadow enhancers acting to mask the effects of perturbing individual enhancers (Hong et al., 2008), although a genome-wide survey of such enhancer redundancy has yet to be conducted. To investigate such interactions more thoroughly, future iterations of our method could randomly distribute programmed pairs of multiplexed enhancer perturbations per locus. Second, other technical caveats include (1) not all enhancers may be susceptible to dCas9-KRAB perturbation, (2) gRNAs may be variably effective in targeting enhancers (Figure S2B), (3) some enhancers required for the initial establishment rather than maintenance of gene expression could be missed in a screen in a stable immortalized cell line, and (4) we did not comprehensively survey the noncoding landscape surrounding each gene, and the marks we used to define candidate enhancers may be excluding some classes of distal regulatory elements. These caveats are respectively addressable in the future by using other epigenetic modifiers or nuclease-active Cas9, by using more gRNAs per candidate enhancer, by combinatorial perturbation of selected loci (Xie et al., 2017), by using cell models of differentiation, and by densely tiling selected loci with perturbations.
Nonetheless, the fact that our paired candidate enhancers are predicted by the strength of enhancer-associated marks (e.g., H3K27ac, p300) supports the assertion that we are identifying bona fide enhancers and simultaneously weakens the case for elements that were negative. Our study provides new insights into key properties of human enhancers, e.g., the distribution of distances between at least some types of enhancers (i.e., unbuffered, upstream) and their target genes. A full understanding of the precise rules governing enhancer-promoter choice is a topic of great interest and will be facilitated by the identification of more enhancer-gene pairs.
A limitation of enhancer-gene pair screening as implemented here relates to the resolution of CRISPRi. In the future, this can potentially be improved upon by adapting enhancer-gene pair screening to use single or pairs of gRNAs with nuclease-active Cas9 to disrupt or delete candidate enhancers at the sequence level. A separate concern is whether high MOI transduction is inducing a cellular inflammatory response, and therefore biasing discovery. However, although some genes with roles in inflammation are among our paired target genes (e.g., NMU, IL6), we only observed pathway-level enrichment of one immune-system related pathway (Table S4A). Moreover, the effect sizes observed in our high MOI versus low MOI experiments were well correlated.
To date, ENCODE has cataloged over 1.3 million human candidate regulatory elements based on biochemical marks (http://screen.umassmed.edu/), while GWAS have identified over 75,000 unique haplotype-trait associations (https://www.ebi.ac.uk/gwas/). Validating candidate elements, fine-mapping of causal regulatory variants, and identifying the target genes of both enhancers and regulatory variants, represent paramount challenges for the field. Given the scale of the problem, we anticipate that the multiplex, genome-wide framework presented here for mapping gene regulation can help overcome these challenges.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jay Shendure (shendure@uw.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Culture
K562s cells are a pseudotriploid ENCODE Tier I erythroleukemia cell line derived from a female (age 53) with chronic myelogenous leukemia (Zhou et al., 2017). K562 cells expressing dCas9-BFP-KRAB (Addgene #46911, polyclonal) were a gift of the Bassik lab, grown at 37°C, and cultured in RPMI 1640 + L-Glutamine (GIBCO) supplemented with 10% fetal bovine serum (Rocky Mountain Biologicals) and 1% penicillin-streptomycin (GIBCO). K562s were authenticated by bulk/single-cell RNA-seq and visual inspection.
HEK293Ts (a human embryonic kidney female cell line) used for housemade virus production were cultured at 37°C in DMEM also supplemented with 10% fetal bovine serum and 1% penicillin-streptomycin. HEK293Ts were authenticated by visual inspection.
METHOD DETAILS
gRNA-library design
Note about terminology used below:
A gRNA-group is defined as all the gRNAs that are targeting the same candidate enhancer or positive control site. To note, all novel TSS and candidate enhancer targeting gRNA-groups are referred to as “perturbative gRNA-groups,” whereas all others are referred to as “control gRNA-groups.”
Pilot Library - 1,119 candidate enhancers as detailed in Table S1A:
Picking candidate enhancer regions:
K562 DNase-seq narrowPeaks (ENCSR000EKS) < 1 Kb away from any gene (GENCODE March 2017 v26lift37) were bedtools-intersected (Quinlan and Hall, 2010) with K562 Hi-C domains (Rao et al., 2014) that contained at least one of the top 10% most highest expressed genes in a previously generated 6,806 single-cell K562 dataset. The remaining regions were largely taken from intersections with K562 GATA1 ChIP-seq narrowPeaks (ENCSR000EFT, lifted to hg19), H3K27ac ChIP-seq narrowPeaks (ENCSR000AKP, lifted to hg19), RNA Pol II ChIP-seq narrowPeaks (ENCSR000AKY), and EP300 ChIP-seq narrow-Peaks (ENCSR000EHI) (Figure 2A). Ten further sites were handpicked and do not overlap either of these four marks.
Candidate enhancer gRNAs:
NGG-protospacers within these candidate enhancers were scored using default parameters of FlashFry (McKenna and Shendure, 2018), and the two top-quality-scoring gRNA per region were chosen as spacers to be used in the gRNA library (scores prioritized by Doench2014OnTarget > Hsu2013 > Doench2016CDFScore > otCount).
TSS positive control gRNAs:
381 genes were randomly sampled from the highly-expressed genes within the same Hi-C domains (as described above) and 2 gRNA were chosen per gene from spacers with the best empirical and predicted scores of the hCRISPRiv2 library (Horlbeck et al., 2016). To note - these spacers are designed as 19 bp, rather than the full 20 of the spacers used in the rest of our gRNAs.
NTC gRNAs:
50 scrambled-sequence spacers with no targets in the genome and 11 protospacers targeting 6 gene-devoid regions of the genome (hg19 chr4:25697737–25700237, chr5:12539119–12541619, chr6:23837183–23839683, chr8:11072736–11075236, chr8:23768553–23771053, chr9:41022164–41024664) were chosen as evaluated by Benchling’s CRISPR tool. These were randomly paired to create a gRNA group. More were chosen from 6 random regions of the hg19 genome (chr4:25697737–25700237, chr5:12539118–12541619, chr6:23837183–23839683, chr8:11072736–11075236, chr8:23768553–23771053, chr9:41022164–41024664) using FlashFry (McKenna and Shendure, 2018) to total 50 targeting these gene-devoid regions of the genome. A further 39 NTCs were sampled from those recommended by Horlbeck et al. (2016). A gRNA to the CAG promoter was additionally included as an internal control (labeled “cag_promoter” in Table S1A and Table S2A, but excluded from analysis for simplicity).
Distal enhancer positive control gRNAs:
15 gRNAs targeting the HBE1 TSS, and HS1–4 of the Globin LCR were chosen as validated from Klann et al. (2017) and Xie et al. (2017). These were manually paired based on their target sites to create gRNA-groups.
Note about Pilot Library:
Our initial FlashFry quality annotations when designing the pilot experiment did not label a small number of protospacers with perfect repeat off-targets, permitting their inclusion in our library (81 of 2,238 spacers ordered in the pilot library; only 9 gRNA-groups with both spacers affected). gRNA-groups with an impacted spacer were rare in our 145 significant enhancer-gene pairs. We also note that we still expect these guides to target their intended site, but with potentially more off-targets. This error was fixed for evaluating gRNA quality in the scaled experiment.
At-Scale Library - 5,779 candidate enhancers as detailed in Table S2A:
Choice of new and repeated sites:
A logistic regression classifier built using the 145 enhancer-gene pairs originally identified in the pilot experiment (see Aggregate analysis of enhancer-gene pairs: ChIP-seq strength quintile analysis and logistic regression classifier) was used to select the top 5,000 intergenic open chromatin regions in K562s (as defined by DNase-seq narrowPeaks (ENCSR000EKS)). Of these, 3,853 were over 1 Kb away from boundaries (GENCODE March 2017 v26lift37) of any genes expressed in the pilot 47,650 K562 single-cell dataset, were not previously included in the pilot library, and had minimum two gRNAs with high quality as again determined by FlashFry. Of the top 5,000, 120 corresponded to a candidate enhancer in one of the original 145 pilot enhancer-gene pairs, and 851 of these corresponded to candidate enhancers targeted in the pilot library but not originally identified as part of a enhancer-gene pair. We additionally included 7 more candidate enhancers not top-ranked by our model, but identified as part of the original 145 enhancer-gene pairs. The only candidate enhancer that was identified in an original 145 pilot enhancer-gene pair but not included in this library had no high quality gRNAs by this second library’s standards (see Note about Pilot Library). Only 15 sites did not overlap any of the marks shown in Figure 3A.
Two alternative gRNAs were designed for 377 of the sites repeated from the pilot library. NGG-protospacers within these candidate enhancers were again scored using default parameters of FlashFry (McKenna and Shendure, 2018), and the third and fourth top scoring spacers were chosen to be used as an alternative gRNAs.
Choice of 948 exploratory candidate enhancers:
Because the logistic regression classifier is biased toward the annotations that were used to select the initially targeted candidate enhancers (Figure 2A), we additionally used submodular subset selection to include DHSs optimized for a diversity of epigenomic features (Wei et al., 2015). We first removed from the full set of 29,833 DHSs (ENCSR000EKS) those 1,119 DHSs that were a part of the original screen. Note that we did not remove the 128 DHSs that had been selected again by the logistic regression model, because doing so would bias our remaining DHSs away from the same annotations. Then we calculated the Pearson correlation of overlapping epigenomic marks between the remaining DHSs. Lastly, we applied a facility location function (Mirchandani and Francis, 1990) to this similarity matrix and used a greedy submodular selection algorithm to identify 948 additional DHSs as exploratory candidate enhancers. The top two highest quality gRNAs (as scored by FlashFry) were included to target each candidate enhancer.
Note on choice of gRNA design for future screens of CRISPRi candidate enhancers
We used our set of enhancer-gene pairs to assess if there was a specific gRNA-target location within the candidate enhancer that increased CRISPRi efficacy. We correlated enhancer-gene pair effect size with each gRNA’s absolute distance to center of either DHS-peak or overlapping p300 ChIP-seq peak. However, neither the absolute-distance-to-center-of-DHS-peak (Pearson’s r:0.02) nor the absolute-distance-to-center-of-overlapping-p300-peak correlated with effect size (Pearson’s r: 0.07). Thus, we currently only recommend prioritizing gRNAs that fall within an open chromatin site based on quality and on-target efficiency as assessed by a gRNA quality algorithm like Flashfry (McKenna and Shendure, 2018).
gRNA-library cloning
The lentiviral CROP-seq gRNA-expression vector (Datlinger et al., 2017) was modified by Q5-Site Directed Mutagenesis (New England BioLabs, F:5-acagcatagcaagtttAAATAAGGCTAGTCCGTTATC-3 R:5-ttccagcatagctcttAAACAGAGACGTACAAAAAAG-3) to incorporate the previously described gRNA-(F+E)-combined backbone optimized for CRISPRi (Chen et al., 2013; Hill et al., 2018, Addgene #106280). Prepared vector was digested with BsmBI and alkaline phosphatase (FastDigest Esp3I and FastAP, Thermo Fisher Scientific), “filler” sequence removed by gel extraction, and cleaned (Zymo Research DNA Clean & Concentrator-5) vector without “filler” was used for all downstream cloning.
Spacer libraries were ordered as single stranded pools (CustomArray, 5-atcttgtggaaaggacgaaacaccGNNNNNNNNNNNNNNNNNNNNgtttaagagctatgctggaaacagcatagcaagt-3). 1 ng of each pool was amplified (F = 5-atcttGTGGAAAGGACGAAACA-3, R = 5-acttgctaTGCTGTTTCCAGC-3, 64C Tm, Kapa Biosystems HiFi Hotstart ReadyMix (KHF), see Special note about gRNA-library cloning below, as we now recommended a different R primer = 5-CTGTTTCCAGCATAGCTCTTAAAC-3) and purified amplicons (Zymo Research DNA Clean & Concentrator-5) were cloned into CRISPRi-optimized CROP-seq vector prepared as described above (NEBuilder® HiFi DNA Assembly Cloning Kit, NEB, 100 fmol purified vector: 200 fmol cleaned insert). 2 ul of each product was transformed into Stable Competent E. coli (NEB C3040H) in enough replicates to produce > 20 transformant clones per gRNA in the library. Plasmid DNA was purified using ZymoPURE Maxiprep kits, following by DNA Clean and Concentrator cleaning (Zymo Research).
Special note about gRNA-library cloning
In Sanger sequence of the final gRNA plasmid libraries and in the 8–15 bp immediately downstream of the spacer (7 bp of the gRNA backbone transcript captured in all single-cell RNA-sequencing datasets), we identified that ~80% of gRNAs harbored a small insertion or deletion (vast majority 1 bp deletions, Figure S7A) in between the spacer and the R primer 5-acttgctaTGCTGTTTCCAGC-3 used in the initial amplification of spacer-oligos. We inferred that this is due to slippage of the KHF polymerase as it copies the secondary structure of the first stem extension loop added as part of the more stable sgOPTI backbone. In the scRNA-seq data, ~70% of gRNA carried a 1 bp deletion, ~8% carried a 2 bp deletion, and ~2% carried a 3 bp deletion (Figure S7A).
Fortunately, 1 bp deletions did not correlate with significant disruption of CRISPRi efficacy in the scRNA-seq data. (1 bp deletion % reduction) / (full length gRNA reduction) ratio was 1.01 (high confidence enhancer-gene pair) or 0.958 (TSS control). For 2 bp deletions, this ratio was also not extreme (0.959 (high confidence pair) or 0.806 (TSS control)). However, for 3 bp deletions (very rare), the ratio was 0.908 (high confidence pair) or 0.644 (TSS control). Overall correlation of all these deletion lengths to full length efficacy was very high (Figure S7B).
Thus, the vast majority (~90%) are either wild-type or harbor 1 bp deletions that create zero-to-little effect on CRISPRi efficacy. 8% of the remaining gRNA harbor 2 bp deletions that also largely do not affect CRISPRi efficacy. However, to avoid this problem in cloning future gRNA libraries into the sgOPTI-CROP-seq plasmid, we now recommend amplifying with a reverse primer that is flush with the spacer (5-CTGTTTCCAGCATAGCTCTTAAAC-3), potentially enabling a boost in repression efficacy.
Virus production and transduction
The Fred Hutchinson Co-operative Center for Excellence in Hematology Vector Production core produced all virus for the multiplexed enhancer-gene pair screening experiments. For the singleton CRISPRi recapitulation, virus was made in-house by co-transfecting (Lipofectamine 3000, ThermoFisher, L300015) HEK293Ts with the small pools of CRISPRi-optimized CROP-seq with the ViraPower Lentiviral Packaging Mix (ThermoFisher). After 3 days, supernatant was syringe filtered with a 0.45 uM filter (cellulose acetate, VWR) to prepare virus for transduction.
Cells were transduced (8 μg/mL polybrene) with varying titers and amounts of virus to achieve differing MOI. 400,000 and ~2.5 million original cells were transduced for the pilot and at-scale experiments, respectively. At 24 hours post-transduction, cells were spun and resuspended with virus- and polybrene- free media. At a total 48 hours post-transduction, 2 μg/mL puromycin was added to the culture, and changed to 1 μg/mL puromycin at the next passage for maintenance. A total of 10 days post transduction, cells were collected for scRNA-seq or bulkRNA-seq.
Single cell transcriptome capture
~4000–8000 cells were captured per lane of a 10X Chromium device using 10X V2 Single Cell 3′ Solution reagents (10X Genomics, Inc). Six lanes were used for both the low and high MOI 1,119-pilot library experiments, and 32 lanes were used for the scaled experiment. All protocols were performed as per the Single Cell 3′ Reagent Kits v2 User Guide (Rev B), except prior to the enzymatic shearing step, 10% of full length cDNA was taken for PCR enrichment of gRNA-sequences off the CRISPRi-optimized CROP-seq transcripts as described below. After RT, the 32 lanes of the scaled experiment were split into two batches (16 lanes each) for the remainder of the prep to enable easier handling.
gRNA-transcript enrichment PCR
A three-step hemi-nested PCR reaction was performed to enrich gRNA sequences from the 3′ UTR of puromycin resistance gene transcripts produced by the CRISPRi-optimized CROP-seq integrant. PCR was monitored by qPCR to avoid overamplification, and each reaction was stopped immediately before it reached saturation.
PCR 1:
10–13 ng of full-length 10x scRNA-seq cDNA were amplified in each 50 μL KHF reaction (annealing temp 65C), spiked with SYBR Green (Invitrogen) for qPCR monitoring (10% of all unfragmented 10x cDNA).
F: U6_OUTER 5- TTTCCCATGATTCCTTCATATTTGC −3
R: R1_PCR1 5- ACACTCTTTCCCTACACGACG-3
PCR 2:
Sample replicates were pooled, cleaned with 1x Agencourt AMPure XP beads (Beckman Coulter), and 1/25th of the cleaned pooled product was amplified in a 50 μL KHF reaction spiked with SYBR Green and monitored as above (annealing temp 65C).
F: U6_INNER_with_P7_adapter 5-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGcTTGTGGAAAGGACGAAACAC −3
R: R1-P5 5-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGACG-3
PCR 3:
The PCR 2 replicate reactions were pooled and 1x AMPure cleaned. 1/25th of the cleaned pooled product was amplified in a 50 μL KHF reaction (spiked with SYBR Green and monitored as above, annealing temp 72C) and products cleaned once again via 1x Ampure.
F: 5-CAAGCAGAAGACGGCATACGAGATIIIIIIIIIIGTCTCGTGGGCTCGG-3 (standard NEXTERA P7 indexing primer) R: R1-P5 again
Sequencing of scRNA-seq libraries
Pilot library experiments
The final libraries were sequenced on a NextSeq 500 using four 75-cycle high-output kits (R1:26 I1:8, I2:0, R2:57) for each experiment (low and high MOI).
Scaled library experiments
The final library was sequenced by the Northwest Genomics Center on a NovaSeq 6000 using an S4 flow cell (R1:26, I1:8, I2:0, R2:91).
All libraries were sequenced to ~20% sequencing saturation.
Digital gene expression quantification
Sequencing data from each sample was processed using the Cell Ranger software package as provided by 10x Genomics, Inc., to generate sparse matrices of UMI counts for each gene across all cells in the experiment.
Each lane of cells was processed independently using cellranger count, aggregating data from multiple sequencing runs. The pilot library experiments were each processed with cellranger 2.0.2; the at-scale library experiment was processed with cellranger 2.1.1.
Definition of genes well-expressed or ‘detectably expressed’ in K562
Unless otherwise notes, genes were defined as well expressed or detectably expressed in K562 if they had at least one read in 0.525% of cells in their respective (pilot or at-scale screen) single cell RNA-seq datasets.
Assigning genotypes to cells
gRNAs were assigned to cells in the following method (Hill et al., 2018): Sequences corresponding to the gRNA-containing CRISPRi-optimized CROP-seq transcripts are extracted from the cellranger position sorted BAM file after running our custom indexed libraries through the cellranger pipeline to tag reads with corrected cell barcodes and UMIs. gRNA sequences are extracted and corrected to the library whitelist within an edit distance of two, and gRNA-cell pairs are tracked when a valid cell barcode and UMI are both assigned to the read. Likely chimeric reads are detected and removed to reduce noise in the assignments as previously described. We utilized thresholds to set minimum acceptable values for the total reads for a gRNA-cell pair and for the proportion of all CROP-seq transcript reads accounted for by each gRNA observed in a cell to distinguish noise from real assignments (Hill et al., 2018). Here, given the larger number of guides contained in each cell, we find that UMI counts provide a much cleaner distribution than read counts and have used UMI counts in all calculations. For the 1,119 pilot library experiments we used 0.01 read counts and 5 UMI in both our low and high MOI for each of these thresholds. For the scaled library experiment, we used 0.005 read counts and 5 UMI. Only cell barcodes that appear in the set of passing cells output by cellranger, which imposes an automated threshold on the total UMIs observed in cells, are carried forward in downstream analysis.
Differential expression tests
In our cis analyses, we tested each perturbing gRNA-group against genes within 1 Mb of the gRNA. These gRNA-gene pairs were identified by using bedtools to intersect the DHSs targeted by the gRNA library with 1 Mb windows in either direction of TSS annotations from GENCODE March 2017 v26lift37 (total of 2 Mb, centered around the TSS). In our trans analysis, all gRNA-groups were paired with all genes that were defined as expressed in K562. In both cis and trans analyses, NTCs were tested against any genes used to test perturbing-gRNAs.
For each gRNA-group we assigned a label of “1” to cells that contained a gRNA belonging to that group and a label of “0” to all other cells in the dataset. Monocle2 (Qiu et al., 2017) was used to perform a differential expression test, using the negbinomial.size family, over this categorical label to find differentially expressed genes between these two groups. Due to its support of complex model formulas, Monocle2 does not provide model coefficients as part of the differential expression results. We created a modified version of the differentialGeneTest function and associated helper functions that return both the intercept term and the coefficient of the group assignment to facilitate more robust prioritization and characterization of hits from our screen. The negative binomial family uses log as the link-function, so we can calculate the initial expression level as exp(intercept), and the fold change in expression between the two groups as exp(group_coefficient + intercept) / exp(intercept). We verified data from our power simulations that the appropriate effect sizes can be obtained with this method using the coefficients output by VGAM.
For the scaled experiment, as we collected a much larger number of lanes and observed the highest MOI, we regressed out the number of guide RNAs observed in a cell (as a proxy for the number of integrants), the percentage of total transcripts observed that are mitochondrial, and the prep batch (as following reverse transcription, the 32 lanes were prepared in two batches to make handling easier). In practice, we observe a modest boost in sensitivity when regressing out each of these factors in DE testing. This was done using the full model formula ~gRNA_group+guide_count+percent.mito+prep_batch and the reduced model ~guide_count+percent.mito+prep_batch in Monocle2.
Calling hits from differential expression test results
All differential expression test results were performed for all K562 expressed genes within 1 Mb of the target site as defined by GENCODE March 2017 v26lift37. NTCs were tested against all genes within 1 Mb of any target site.
Tests with two sources of potential false positives were excluded:
In the pilot experiment, we identified inflation of NTCs when testing them against genes highly impacted by perturbing-gRNA in our library (for example, NTCs associated with targets of our TSS and globin LCR controls). This was due to subtle yet detectable nonrandom associations of gRNA-groups with other gRNA-groups across cells, potentially due to slight bottlenecking at the transduction level (400,000 cells transduced for 1,119 pilot library versus 2.5 million transduced for 5,779 scaled library). To exclude this source of inflation in the pilot dataset, we used Fisher’s exact test to identify when an NTC was nonrandomly assorted with a perturbing-gRNA (adjusted P-value < 0.01 & odds ratio > 1). Then, any test of an NTC against a gene within 1 Mb of that gRNA’s gRNA-group was excluded from further analytical steps.
We noted Monocle was susceptible to inflating P-values when a gene was highly expressed but only in few cells. Three of our 381 TSS controls fell into this category. To avoid this problem, we excluded outlier genes that were expressed in < 20,000 cells in either the high-MOI 47,650-cell dataset and/or the scaled 207,324-cell dataset, and with log10(total UMIs / cells with a UMI) > 0.2 greater than predicted by a spline fit generated via smooth.spline() with spar = 0.85 to limit overfitting (35 genes total).
Remaining tests were filtered to those that decreased expression of the target gene.
Then, an empirical P-value was defined for each gene-gRNA-group pair test as: [(the number of NTCs with a smaller P-value than that test’s raw P-value) + 1] divided by [the total number of NTCs tests + 1].
These empirical P-values were Benjamini-Hochberg corrected, and those < 0.1 were kept for 10% empirical FDR sets.
Use of 3.5% empirical FDR to initially select enhancer-gene pairs from the pilot study
We originally used an alternative method to call the original 145 enhancer-gene pairs from the original pilot dataset (a universal cutoff of the P-value at which the proportion of passing NTC-tests/total NTC-tests was 10% of the proportion of passing candidate enhancer tests/total candidate enhancer tests). However, upon further discussion and review of the eQTL literature, we revised our method to the one defined above. This original threshold corresponded to a 3.5% empirical FDR rate, as defined above.
Inclusive versus high confidence enhancer-gene pairs as described in Table S2B
The only requirement of enhancer-gene pairs in the inclusive set was that they passed a 10% empirical FDR in the scaled experiment. To be included in the high confidence set, enhancer-gene pairs either had to be replicated at a 10% empirical FDR in the pilot dataset, or (if a candidate enhancer was unique to the scaled experiment) both gRNAs had to be individually associated with > 10% repression of the gene.
Analyses to evaluate reproducibility between gRNA
To evaluate reproducibility between gRNAs, we subset the 377 pairs (two sets of gRNA pairs targeting the same candidate enhancers in the scaled experiment) to pairs where both pairs negatively repressed at least one target gene (no significance requirement). 20 of the 377 did not meet this criteria. Then, we ranked all tested genes by average repression between the two gRNA pairs, and kept the top ranked gene for each pair. The repression levels of each type of gRNA pair on this top-ranked gene are plotted in Figure S2B, regardless of significance.
Intracellular abundance of gRNA and dCas9-KRAB transcript does not correlate with effect size
As both the dCas9-BFP-KRAB and the sgOPTI-CROP-seq construct transcripts are poly-A tagged, we are able to test if there is an association between the CRISPR components’ UMI counts and transcript abundance of a targeted gene. For the 441 candidate enhancers in a high confidence pair, we subsetted to the cells that held a guide targeting each enhancer. Within this set of cells, we tested for a significant association between the expression of the target gene and the UMI count of the dCas9-BFP-KRAB or the guides (adjusting for total cell UMI count). Of the 470 enhancer-gene pairs, only 2 and 10 had any significant (adjusted P-value <0.01; 7 and 27 for adjusted P-value < 0.05) association with dCas9-BFP-KRAB count or guide count respectively (0.4% and 2% or 1.5% and 5.7% of tests for each adjusted P-value threshold respectively). Based on this, we conclude there is not evidence for a substantial effect of dCas9-BFP-KRAB or guide counts on the observed effect size for a given enhancer-gene pair.
Quantifying gRNA abundance
In the process of assigning gRNAs to cells, we had already quantified the number of reads and UMIs associated with gRNA-cell pairs. These counts were used as is for the above analysis.
Quantifying dCas9-BFP-KRAB in cells
We constructed a bowtie2 (Langmead and Salzberg, 2012) index for a reference including both the PuroR transcript from the sgOPTI-CROP-seq vector (extending from PuroR to the 3′ LTR encoding the guide sequence as N’s) and dCas9-BFP-KRAB (including the 3′ LTR). Note that both gRNA and dCas9 transcripts were included in this analysis because several regions are identical within the 3′ UTR of the transcripts encoded by these two constructs. We then took all the unmapped reads from the unbiased (cell) libraries and converted them back into fastq format adding the final cell ID and UMI from cellranger into the read name for use downstream. We mapped these reads to the reference above using bowtie2 using the command “bowtie2 -p 8–n-ceil 20–np 0 -x <reference> -U <fastq input> -S <bam output>.” We then took only reads that map uniquely to the dCas9 contig with mapq of 30 or greater and enumerated the number of UMIs and total reads seen for each cell / barcode pair dCas9.
In each case, we tested for associations between the gRNA/dCas9 counts and the abundance of each high confidence hit in our screen, only within cells that had a guide to the target. This was done using our modified version of differentialGeneTest as described above. Note that in this case we observed that size factors typically used to account for variation in total UMI counts across cells did not appear to sufficiently correct for the strong correlation between the counts of two transcripts (the gRNA transcript / dCas9 and the target) that results from variation in total UMI counts across cells. This initially resulted in residual associations that indicated increased gRNA transcript / Cas9 resulted in higher target expression. To account for this, we added an additional term to both the full and reduced model “~ log10(total_umis)” and set all size factors to 1. This is the model from which we report the above results.
Individual replications and validations
Individual replication by CRISPRi singletons
To replicate a enhancer-gene pair’s phenotype outside of the pooled mapping format, we prepared small pools of gRNAs re-targeting 15 high-confidence candidate enhancers or the TSSs of their respective paired-target genes (Table S3A). These enhancer-gene pairs were chosen from the following requirements: candidate enhancer tested in both the pilot and at-scale study (replicated between both); target gene in upper 50% of expression of all paired genes; target gene had no strong cancer associations or growth phenotypes. Additionally, we chose 6 candidate enhancers that were not paired with any target gene using the following requirements: tested in both the pilot and at-scale screen; empirical P-values for any cis gene > 0.5 in both experiments; overlapping H3K27Ac ChIP-seq peak is in the top half of all the peaks that overlap the entire at-scale library (thus to be comparable with our paired enhancers); and within 1 Mb of a K562 expressed gene.
The two original gRNAs and two new gRNAs (making up the top 4 ranked on-target activity per candidate enhancer, filtering out those with high off-target scores using Flashfry (McKenna and Shendure, 2018); exception is candidate enhancer chr11.4680 where only 3 gRNAs passed these quality filters) were used for each respective pool, for a total of 4 gRNAs in the pool (Table S3C). The two original gRNAs were used for the TSS controls (plus two more alternative TSS gRNAs in the cases of NMU, GYPC, PTGER3, and PRKCB). These small gRNA pools were cloned into the CRISPRi-optimized-CROP-seq vector (as described above, except in the case of e-NMU targeting pool, which was cloned by ordering two reverse complement single stranded oligos and annealing them together into px459 (CRISPR-Reagent-Description_Rev20140509.pdf) (Cong et al., 2013). House-made lentiviral preps from these gRNA pools were transduced at low MOI into the K562-dCas9-BFP-KRAB line, and cultured for 10 days under puromycin selection before two technical replicates of total RNA were collected from each sample (RNeasy Mini Kit, QIAGEN).
Bulk RNA-seq libraries were prepared from each replicate via a TruSeq mRNA kit (400 ng input, Illumina, TruSeq RNA Sample Prep Kit v2 RS-122–2002 or TruSeq Stranded mRNA Library Prep 20020595), and sequenced on a NextSeq 500 (total two 150-cycle kits cycling 80/80/6 in mid output mode for e-NMU, e-PRKCB, e-GYPC, e-PTGER3; total two 75-cycle kits cycling 40/40/8 in high output for all others; aiming for 10–20 million reads/sample). Gene-level quantifications and differential expression tests were performed via kallisto (Bray et al., 2016) and sleuth (Pimentel et al., 2017). Repression percentages were calculated from the kallisto transcript per million output table (normalized by size factors): (mean between the two replicates / mean between all-non targeting samples). To note, targeting the TSS of CITED2 did not seem to successfully repress CITED2’s expression, though this is potentially due to inaccuracy of 1 of 2 technical replicates for this sample. The 3 that matched direction and magnitude of effect but were not significant in a test of differential expression potentially were not detectable due to lack of power, as we sequenced only two RNA replicates per sample.
To note: we additionally generated singleton datasets for chr6:34191315–34191338 (paired with HMGA1 in the pilot screen), but did not include this in analysis as it did not reproduce between the pilot and at-scale screen, and thus was not part of our high confidence enhancer-gene pair set.
Validation by sequence deletions
To generate monoclonal sequence lines of three candidate enhancers (Table S3B), we designed protospacers to flank the DHSs targeted in e-NMU, e-GLUL, and e-CITED2. Spacers were order as single stranded oligos (IDT, Table S3D) and then amplified (KHF, 5-GTGGAAAGGACGAAACACCg-3, 5-gctaTTTCtagctctaaaac-3, 55°C tm, 15 s extension; followed by clean-up via Zymo Research DNA Clean & Concentrator) to be made double stranded for Gibson Assembly cloning (50 ng prepared vector: 0.66 ng prepared insert) into the Cas9- and gRNA- expression vector px459 (Ran et al., 2013), expressing both the gRNA and a cassette of Cas9–2A-puromycin resistance; NEBuilder® HiFi DNA Assembly Cloning Kit). Some e-NMU targeting oligos were cloned by annealing two complementary oligos together followed by ligation into px459, in the method of CRISPR-Reagent-Description_Rev20140509 (Cong et al., 2013).
We transiently transfected the small px459 pools into the K562+dCas9-KRAB cell line using the Neon nucleofection system (500,000 cells per library, 10 uL tips, 500 ng of plasmid, pulse voltage 1450–pulse width 10–pulse number 3; ThermoFisher). Beginning 24 hours after transfection, cells were selected with 1 ug/mL puromycin for 48–72 hours, then single-cell sorted into 96 well plates using a FACSAria II (Becton Dickinson). To finally achieve clones that harbored fully homozygous deletions of e-NMU, this process was repeated on an initial set of heterozygous clones using a second round of flanking gRNAs.
After 3–4 weeks of growth, gDNA was extracted by concentrating cells into 20 uL of media, and adding 40 uL of house-made Quick Extract buffer (EB + 4 mg/mL proK + 0.45% Tween20), followed by 65°C for 6 minutes and 98°C for 2 minutes. 1 uL of this gDNA extract was used for genotyping PCRs (Kapa2G Robust PCR kit, 35 cycles 60°C-HS-3 minute extensions).
Two rounds of genotyping PCRs were performed. First, clones were screened with primers flanking the deletion to identify clones that harbored a deletion on at least one allele. Second, to confirm homozygosity, primers internal to the deleted region were used to identify candidates that still harbored wild-type alleles (Figure S4A; Table S3D). Clones that harbored full deletions with no remaining wild-type alleles were submitted to bulkRNA-sequencing (Figures 4E–4G). Two technical replicates of RNA were extracted from each monoclonal line (RNeasy Mini Kit, QIAGEN), bulkRNA-seq libraries prepared via a TruSeq mRNA kit (400 ng input, Illumina, TruSeq Stranded mRNA Library Prep 20020595), and sequenced on a NextSeq 500 (one 75-cycle kits cycling 40/40/8 in high output for monoclonal samples; aiming for 10–20 million reads/sample). Gene-level quantifications were performed as for the CRISPRi singletons, and reduction percentages calculated from kallisto transcript per million output table (normalized by size factors): (mean of all replicates per candidate enhancer) / (mean between all-non targeting samples).
Phenotyping e-NMU perturbations by flowFISH
Cells harboring e-NMU CRISPRi perturbations were generated as in Individual replication by CRISPRi singletons. A heterogeneous population of cells harboring full e-NMU deletions was generated as in Validation by sequence deletions (though without single-cell clone sorting; Table S3D). A heterogeneous population of cells harboring scanning deletions across e-NMU was generated by cloning and transfecting 19 gRNAs (Table S3D) targeted every ~100 bp across the e-NMU locus as described above in Validation: sequence deletions.
Fluorophore labeled complementary probes to NMU transcript were designed on and ordered from https://www.molecularinstruments.com/ (Table S3E). The ‘non-targeting’ probes were scrambled versions of the original NMU-targeting probes (to preserve sequence features such as GC content). RNA flowFISH was performed according to Molecular Instruments’ in situ HCR v3.0 protocol (Choi et al., 2018), which we have described again here: Cells were by resuspending in 4% formaldehyde to reach 106 cells/mL, and fixing for 1 hour. Formaldehyde was then removed, cells were washed four times in PBST (1x PBS + 0.1% Tween 20), and then resuspended in 70% ethanol. For labeling, cells were first washed twice with PBST, and then pre-hybridized by incubating at 37C for 30 minutes in 30% probe hybridization buffer (30% formamide, 4x sodium chloride sodium citrate (SSC), 9 mM citric acid, 0.1% Tween 20, 50 ug/mL heparin, 1x Denhardt’s solution, and 10% low MW dextran sulfate). Cells were then incubated overnight at 37°C in a final 4 nM probe solution (prepared by adding 2 pmol each probe (a mix of 1 uL of 2 uM stock per each probe) + 100 uL of 30% probe hybridization buffer). Cells were then repeatedly resuspended in 30% probe wash buffer and incubated for 10 minutes at 37°C, for a total of four washes. Cells were then resuspended in 5x SSCT (5x SSC + 0.1% Tween 20) and incubated at room temperature for 5 minutes before amplification.
For amplification, cells are resuspended in amplification buffer (5x SSC + 0.1% Tween 20 + 10% low MW dextran sulfate) and preamplified by incubating for 30 minutes at room temperature. 15 pmol of each fluorescently labeled hairpin was snap-cooled by heating 5 uL of 3 uM stock in hairpin storage buffer (Molecular Instruments) to 95°C and then cooling for 30 minutes to room temperature in a dark drawer. Snap-cooled hairpins were then mixed with amplification buffer, added to the sample for a final and then cooling for 30 minutes to room temperature in a dark drawer. Snap-cooled hairpins were then mixed with amplification buffer, added to the sample for a at room temperature. 15 pmol of each fluorescent0 minutes at room temperature with 0.5 uL Vybrant Dye Cycle Orange (DNA stain).
For sorting, the cells are first gated based on size and granularity using forward versus side scatter to discriminate between debris and cells. Cells in G0/G1 stage are then selected using DNA dye (Vybrant Dye Cycle Orange). Cells are then sorted into low, medium, or high bins of NMU expression using AF647 (Becton Dickinson; ~500,000 cells for the full deletion low NMU bin, ~1,000,000 cells forall other bins).
To reverse cross-link the sorted samples, cell pellets were resuspended in 500 ul of elution buffer (4 mL H2O + 500 ul 10% SDS + 500 ul NaHCO3 – 1M) + 30 ul of NaCl (5M) and incubated overnight at 65°C. 8 ul of RNase (10 mg/mL) was added to each sample, mixed by inversion, and incubated at 37°C for 2 hours. 4 ul of Proteinase K (20 mg/mL) was added, mixed by inversion, and incubated for 2 hours at 55°C. gDNA was extracted by phenol chloroform, ethanol precipitated, and resuspended in QIAGEN elution buffer.
PCR to identify e-NMU genotype enrichments in each of the NMU expression bins (Figures S4B and S4C) was performed using Kapa2G Robust (e-NMU outer PCR: F primer 5′CCAACCCCTCAACTTGTT3′ Reverse primer 5′TGCCTTCTCTGCCTTTCATT3′; anneal 60°C, extension time 1:50) on 10 ng of gDNA. PCRs were spiked with SybrGreen, and monitored on a qPCR to allow removal before overamplification to prevent excessive PCR biases. 1 uL of each PCR reaction was run on a 6% TBE polyacrylamide gel (Invitrogen) for 35 minutes at 180 V and stained with Sybr Gold for visualization. Replicate PCRs are represented by different lanes in Figures S4B and S4C.
QUANTIFICATION AND STATISTICAL ANALYSIS
Aggregate analysis of enhancer-gene pairs
The high confidence enhancer-gene pairs were used for these analyses unless otherwise noted. Details of empirical FDR and the significance thresholds used to call enhancer-gene pairs can be found above in Calling hits from differential expression test results. Singleton re-testing and validations of enhancer-gene pairs used to functionally test if the data met the assumptions of these statistical methods can be found above in Replication of enhancer-gene pairs as singletons.
Distance between perturbation and target gene
Distance was calculated between the GENCODE March 2017 v26lift37 annotated TSS of the perturbed gene and the middle of the originally targeted open chromatin region (if targeting a candidate enhancer, ENCFF001UWQ) or the GENCODE-annotated TSS of the originally targeted transcript (if targeting a TSS). To note, in Figure 6A and to calculate the median distance, we have only used enhancers that are upstream of the target gene, as the length of the gene body would confound distance-to-TSS measurements for downstream enhancer-gene pairs.
Expression distributions
Average expression of each transcript was defined as mean UMI counts per cell in the 47,650 or 207,324 cell scaled dataset. K562 expressed genes were defined as at least one read in 0.525% of cells in the same dataset.
ChIP-seq strength quintile analysis and logistic regression classifier
All candidate enhancers targeted in each library were bedtools-intersected with 170 ChIP-seq of histone-associated marks (ENCODE Project Consortium, 2012)), broken into quintiles of the 7th “signalValue” column (peak strength, usually representing overall average enrichment in the region), and the rates of enhancer-gene pairs identified in each quintile were used. In addition to average phyloP conservation score per candidate enhancer, these were used to fit both independent and multivariate logistic regression classifiers using the glm() function with binomial family. We calculated fold changes for how likely a candidate enhancers was paired by: 1 + (((odds ratio - 1) * highest quintile ChIP-seq value) - ((odds ratio - 1) * lowest quintile ChIP-seq value)).
Motif enrichment in enhancers and promoters
Using the AME tool (Analysis of Motif Enrichment) from the MEME suite (McLeay and Bailey, 2010), enhancer analysis: we compared motifs enriched in the 600 candidate enhancers in the inclusive set of 664 pairs as compared to all 5,779 in the at-scale library; promoter analysis: compared motifs enriched the 1 Kb upstream of the TSS (~promoter) of the 479 genes in the inclusive 664 pairs as compared to the ~promoters of all K562 expressed genes within 1 Mb of a tested candidate enhancer. Parameters were set to default, and Hocomoco Human v11 (core) (Kulakovskiy et al., 2013) was used as the motif library.
Motifs of TF couples across paired promoters and enhancer
To test if pairs of transcription factor (TF) motifs were enriched for co-presence across paired promoters and enhancers, we first identified 179 TFs that were expressed in K562s and had high quality motifs in Hocomoco. Using the FIMO tool (Find Individual Motif Occurrences) from the MEME suite, we annotated all 600 candidate enhancers and the promoters of all 479 genes (1 Kb upstream of the TSS) in the inclusive set of 664 pairs. Motifs in the bottom quartile of how often seen in a promoter were excluded for lack of power. Then, we looped through all possible pairs of 179 TFs in the enhancer (TFe) × 179 TFs in the promoter (TFp), and for each TFe × TFp pair, performed a Fisher’s Exact test on contingency tables designed as follows:
For the promoters of 479 paired genes: TFp in promoter or TFp not in promoter versus Promoter paired with an enhancer that contains TFe or Promoter not paired with an enhancer that contains TFe
For the 600 paired enhancers: TFe in enhancer or TFe not in enhancer versus Enhancer paired with an enhancer that contains TFp or Enhancer not paired with an enhancer that contains TFp
The six TFe × TFp co-enriched couples that had a Benjamini Hochberg corrected P-value < 0.1 for both the 479 paired promoter analysis and the 600 paired enhancers analysis were described in the main text and Table S4E.
ChIP-seq of TF couples across paired promoters and enhancer
Bedtools was used to mark when a paired enhancer or promoter in the 664 inclusive dataset overlapped a ChIP-seq peak from ENCODE generated K562 datasets were used. ChIP-seq datasets that that were in the bottom quartile of how-often-overlapping with a paired enhancer or promoter were excluded for power (leaving 168 TFe and 166 TFp). Analysis was then performed the same as in the TFe × TFp motif analysis (Fisher’s Exact Test, adjusted P-value < 0.1, pair required to be enriched when looping through both enhancers and then through promoters, TFe and TFp required to be different, Table S4E).
Functional annotation enrichment
We used the Piano package (Väremo et al., 2013) to perform functional annotation enrichment from the ‘all pathways’ Gene Ontology (http://download.baderlab.org/EM_Genesets/June_20_2014/Human/June_20_2014_versions.txt). The 10,560 K562-expressed genes within 1 Mb of a perturbing-gRNA were used as our background dataset, and randomly sampled from genes with expression greater than one standard deviation below the mean of our 353 targeted genes was used as the comparison set of “expression matched controls” (Figure 6C).
Hi-C analysis
We used the in situ Hi-C dataset for K562 cells from Rao et al. (2014), using the MAPQ 0 threshold and KR normalization, at 5 Kb resolution. We first created shuffled control loci pairs by starting with the set of enhancer-gene TSS pairs, and randomly shuffling the oriented distances between enhancer-TSS pairs, keeping either the enhancers or the TSSs intact. The rare cases where shuffling resulted in an invalid chromosomal coordinate were excluded. For each set of loci pairs, we identified the TADs (as defined in Rao et al. [2014] using Arrowhead) encompassing each loci pair. For overlapping domains, we used the farthest domain boundary on each side of the loci pair. We omitted loci pairs that were not encompassed by any TADs from further analysis. We then extracted the normalized Hi-C counts for each loci pair, along with those for all other bins representing interactions at the same genomic distance within the same TAD, and calculated its fractional rank (scaled from 0 to 1, with 1 representing the highest interaction frequency). Finally, the distributions of fractional ranks were plotted and compared. In addition to comparing interactions within TADs, we also compared loci pairs to other bins within 200 Kb or 1 Mb of each loci pair.
Analyses for multiplexability of CRISPRi within cells - low versus high MOI comparisons
In order to confirm the efficacy of repression in our high MOI experiments (pilot library MOI = ~15 and at-scale library MOI = ~28), we sought to compare the degree of repression observed in each of these experiments to that observed in our low MOI control experiment (pilot library MOI = ~1). We took all gene-target site differential expression tests passing a 10% empirical FDR in any one of the three experiments (as evaluated independently in each screen). We used this set rather than our final hit list to ensure that we were not biasing our comparison by excluding tests that would be independently called by any one screen but not the others, although we note that the results of the same set of analyses using our final set of hits are very similar.
For each of these tests, we calculated the observed fold changes of repression (where 1 is no change and 0 is complete loss of expression) for each screen and then calculated the following ratios: (pilot high MOI fold change) / (pilot low MOI fold change) and (at-scale fold change) / (pilot low MOI fold change), using a pseudocount of 0.01. As we found it potentially confusing that a higher value of these ratios represents worse efficacy of repression in the high MOI experiments, we considered making these ratios of percent repression (1 - fold change). However, as this value could be negative in some cases (where the fold change was greater than one in one of the screens), this was not compatible with display on a log scale. Therefore, in all plots showing such ratios, we are actually showing the inverse of the fold change ratios described above, which should approximately represent the ratios of percent repression without producing any negative values. Thus, in our plots and reported summary statistics, values less than one represent cases where more repression was observed in the low MOI control.
Despite the distributions of the ratios described above being centered at one, which indicates largely equivalent repression in high and low MOI experiments, there was a left tail, representing a smaller number of tests with reduced estimated efficacy in the high MOI experiments. We reasoned that this could be an artifact of these genes being more lowly expressed and/or being represented by fewer cells given the sparse sampling of the pilot library in the low MOI experiment. In either case we might tend to underestimate the amount of transcript remaining after repression or at the very least the estimates would be substantially noisier, resulting in an artifactual tail. To confirm the lower expression levels genes in the observed tail, we took all tests falling in the first quartile of each distribution and compared the expression of these genes (average expression for the pilot low MOI experiments in the group of cells without the relevant gRNA; calculated by exponentiating the intercept from the differential expression test, which in the pilot high differential test is the estimated expression in UMI counts for the group of cells without the relevant gRNA). We further scaled these values by the total number of cells observed for each gRNA group in the pilot low MOI experiment to examine the combined effect of representation and expression level, which both contribute to what we expect is simply less robust estimation of fold change. We note that this scaling does not appreciably impact the overall distributions in this case.
Power simulations
In order to predict the impact of multiplexing on the power of enhancer-gene pair screens, we developed a simulation framework. First, using single-cell RNA-seq data collected from the pilot 47,650 K562 cells, we estimated a dispersion function that relates the mean expression of a gene to its dispersion estimate (one of the two parameters required for the negative binomial distribution) calling the Monocle2 functions estimateSizeFactors and estimateDispersions. This function is typically used in differential expression testing to shrink dispersion estimates, but here we use it to estimate dispersion values for simulated transcripts. This dispersion function is then extracted from the CellDataset object output by Monocle2 and used as input to our simulations.
Next, we chose relevant ranges for each of the parameters varied in our simulation: the MOI, total cell count, effect size (fraction repressed by CRISPRi), and mean expression level of the gene being tested. By examining the range of expression values observed in our data, we chose to simulate expression data for genes having mean expression values (size parameter of the negative binomial distribution) of 0.01, 0.1, 0.32, 1.0, 3.16, and 10.0 UMIs (0.10, 0.32 and 1.00 used respectively as low, medium, and high in Figure 1B) to provide a range of representative values.
We simulated MOIs at several values from 0.3 to 50, a range which includes the MOIs estimated from our own enhancer-gene pair screens. For each MOI, we calculate the expected number of cells containing a given guide by assuming a Poisson distribution of lentiviral delivery, zero-truncating the distribution to account for drug selection for cells that contain a guide transcript, and rescaling the probability distribution of guide counts accordingly. Perfect library uniformity was assumed to obtain the expected number of cells containing a given guide and the number of cells that do not contain that guide. Effect sizes of CRISPRi repression were chosen using estimates from the literature and were simulated at several values between 10% to 90% percent repression of the average expression level of the target transcript (size parameter input to the negative binomial distribution).
Finally, we simulated several values of total cells included in the experiment ranging from 35,000 to 300,000 cells (45,000 cells shown in Figure 1B). Expression data from transcripts corresponding to 100 samplings per set of parameters were generated for the populations of cells containing the gRNA and not containing the gRNA respectively. Our expression data simulation assumed a negative binomial distribution with the appropriate size parameter for the cells with and without the gRNA, and a dispersion value estimated using the dispersion function described above given the starting mean expression level being simulated. For each set of parameters, the simulated transcripts were subjected to a differential expression test performed between cells with and without the gRNA assigned using our modified version of the Monocle2 function differentialGeneTest as described above (see Differential Expression Tests). P-values were obtained and corrected assuming an average number of 20 tests per group in the library to approximate the number of genes contained within 1 Mb on either side of each gRNA-group and the impact of multiple testing. The rate of tests falling below a adjusted P-value of 0.05 were tabulated at each set of parameters to make power curves.
Quantify errors in gRNA backbone as described in Method Details: “Special note about gRNA-library cloning,” Related to Figure S7
To quantify the rate of mismatches and indel lengths in the gRNA backbones for each library, we extracted the backbone portion of the gRNA transcript for each read in our gRNA transcript enrichment libraries and aligned it to the expected reference, (gtttAagagc taTGCTGGAAACAGCAtagcaagttTaaat), using semi-global version of the Needleman-Wunsch algorithm implemented by RecNW (Yahi et al., 2018). Mismatch and indel counts were made within the hairpin portion of the backbone (we initially screened backbone bases 8 to 31 downstream of the spacer), to restrict to bases that would be the most likely to have some if any functional impact. However, it should be noted that the overwhelming majority of all indels were small deletions observed in bases 8 to 14 or so; thus, rates provided in Figure S7A are limited to these 7 bp. For the pilot-gRNA libraries, where we had a shorter cDNA read length that does not cover the entire hairpin, so we simply quantified mismatches and indels in the 8 to 14 bp window (which again contained the overwhelming majority of all indels in our at-scale gRNA library). For each target-UMI pair in each cell, we averaged the observed mismatch and indel counts/lengths to get a consensus over all reads with a given UMI. We then averaged the statistics derived from UMIs for each target-cell assignment to get a final set of statistics for each. Each average was rounded to the nearest integer for plotting. This allowed us to quantify rates across screens and also examine how any changes in effect sizes correlated with effect sizes.
tSNE clustering of each dataset to check for biological distortions
We tested for enrichment of gRNAs in specific tSNE-based clusters of the at-scale single cell transcriptome dataset, to identify any perturbed targets that resulted in stronger changes to global expression, presumably mediated through trans effects of the target gene. For the at-scale dataset, we subsetted to genes that were expressed in at least 0.5% of cells and 50,000 cells were randomly sampled. We then processed the dataset using Seurat (Butler et al., 2018). We removed cells with greater than 10% mitochondrial transcripts, ran NormalizeData, and found the top 5,000 variable genes using FindVariableGenes. Using these top 5,000 variable genes as input we then ran ScaleData, regressing out the percent of each cell’s transcriptome accounted for by mitochondrial genes. We then computed 100 PCs using RunPCA (weighting PCs by variance explained), which were used as input to both the FI-tSNE method using RunTSNE and Louvain clustering at a resolution of 0.5 using FindClusters. Fisher’s Exact tests were performed to test for a perturbed target’s enrichment in each cluster. 8 TSS controls and 6 candidate enhancers were enriched within specific clusters (odds ratio > 5, adjusted P-value < 0.01). However, even in these cases, only 10% of cells in which the target is perturbed actually fall into the cluster in which they are found to be enriched. Thus, this is not expected to compromise the screen, as in order to be a chronic source of false positives, the gRNAs targeting these global-change genes would have to be non-randomly associated with other gRNAs in the library.
DATA AND SOFTWARE AVAILABILITY
The accession number for the sequencing data (single cell RNA-seq and bulkRNA-seq) and processed data files is GEO: GSE120861 (metadata file), and GSM3417251–GSM3417303 (actual datasets).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Stable Competent E. coli (High Efficiency) | NEB | C3040H |
| Pilot gRNA Library sgOPTI-CROP-seq Virus | Fred Hutchinson Co-operative Center for Excellence in Hematology Vector Production core | N/A |
| At-scale gRNA Library sgOPTI-CROP-seq Virus | Fred Hutchinson Co-operative Center for Excellence in Hematology Vector Production core | N/A |
| Critical Commercial Assays | ||
| Chromium Single Cell 3’ Library & Gel Bead Kit V2 | 10x Genomics | PN-120237 |
| Chromium Single Cell A Chip Kit V2 | 10x Genomics | PN-120267 |
| Chromium i7 Multiplex Kit | 10x Genomics | PN-120262 |
| TruSeq Stranded mRNA Library Prep | Illumina | 20020595 |
| TruSeq RNA UD Indexes (96 Indexes, 96 Samples) | IDT for Illumina | 20022371 |
| TruSeq RNA Sample Prep Kit v2 | Illumina | RS-122–2002 |
| NextSeq 500/550 Hi Output KT v2.5 (75 cycles) | Illumina | 20024906 |
| NextSeq 500/550 Mid Output KT v2.5 (300 cycles) | Illumina | 20024905 |
| NovaSeq 6000 S4 Reagent Kit (300 cycles) | Illumina | 20012866 |
| NEBuilder® HiFi DNA Assembly Cloning Kit | NEB | E5520S |
| DNA Clean and Concentrator | Zymo Research | D4014 |
| Agencourt AMPure XP Beads | Beckman Coulter | A63882 |
| HiFi Hotstart ReadyMix | Kapa Biosystems | KK2602 |
| Kapa2G Robust PCR kit | Kapa Biosystems | KK5702 |
| Neon Transfection System | Life Technologies | MPK1025 |
| Lipofectamine 3000 Transfection Reagent | ThermoFisher | L300015 |
| RNEasy Mini Kit | QIAGEN | 74104 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE120861 (metadata file), GSM3417251 toGSM3417303 (actual data) |
| DNase-Hypersensitivity Peaks for K562s DHS peaks | ENCODE Project Consortium, 2012 | ENCFF001UWQ |
| Human reference genome NCBI build 37, GRCh37 | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/projects/genome/assembly/grc/human/ |
| ChlP-seq Datasets, see Table S4B | ENCODE Project Consortium, 2012 | https://www.encodeproject.org/ |
| PhyloP | Pollard et al., 2010 | http://compgen.cshl.edu/phast/help-pages/phyloP.txt |
| Hocomoco Human v11 (core) | Kulakovskiy et al., 2013 | http://hocomoco11.autosome.ru/ |
| K562 Hi-C dataset | Rao et al., 2014 | GEO: GSE63525 |
| Experimental Models: Cell Lines | ||
| K562 expressing dCas9-BFP-KRAB (Addgene #46911), passage 40, re-sorted for BFP | Gift of the Bassik lab | N/A |
| HEK293Ts, passage (for making housemade lentivirus) | ATCC | CRL-3216 |
| Oligonucleotides | ||
| Array for Pilot Experiment, see Table S1 | This paper | N/A |
| Array for At-Scale Experiment, see Table S3 | This paper | N/A |
| Primers to make gRNA oligos double stranded for cloning into sgOPTI-CROP-seq - primer 1: atcttGTGGAAAGGAC GAAACA | This paper | N/A |
| Primers to make gRNA oligos double stranded for cloning into sgOPTI-CROP-seq - primer 2 (no longer recommended, please see Special note about gRNA-library cloning cloning in STAR methods): acttgctaTGCTGTTTCCAGC | This paper | N/A |
| Primers for nested PCR to enrich sgOPTI-CROP-seq from single-cell RNA-seq cDNA (round 1) - primer 1: TTTCCCATGATTCCTTCATATTTGC | This paper | N/A |
| Primers for nested PCR to enrich sgOPTI-CROP-seq from single-cell RNA-seq cDNA (round 1) - primer 2: ACACTCTTTCCCTACACGACG | This paper | N/A |
| Primers for nested PCR to enrich sgOPTI-CROP-seq from single-cell RNA-seq cDNA (round 2) - primer 1: GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGc TTGTGGAAAGGACGAAACAC | This paper | N/A |
| Primers for nested PCR to enrich sgOPTI-CROP-seq from single-cell RNA-seq cDNA (round 2) - primer 2: AATGATACGGCGACCACCGAGATCTACACTCTTTCC CTACACGACG | This paper | N/A |
| Primers for nested PCR to enrich sgOPTI-CROP-seq from single-cell RNA-seq cDNA (round 3) - primer 1: CAAGCAGAAGACGGCATACGAGATIIIIIIIIIIGTCTCGT GGGCTCGG |
This paper | N/A |
| Primers for nested PCR to enrich sgOPTI-CROP-seq from single-cell RNA-seq cDNA (round 3) - primer 2: AATGATACGGCGACCACCGAGATCTACACTCTTTC CCTACACGACG | This paper | N/A |
| Oligos and primers to engineer and genotype monoclonal sequence deletion lines, see Table S3D | This paper | N/A |
| Oligos and primers for singleton CRISPRi validations, see Table S3C | This paper | N/A |
| Probes for NMU RNA flowFISh, see Table S3E | This paper | N/A |
| Recombinant DNA | ||
| Lenti-sgOPTI-CROP-seq | Hill et al., 2018 | Addgene #106280 |
| Px459-Cas9–2A-PuroR_gRNA | Cong et al., 2013 | Addgene #62988 |
| Software and Algorithms | ||
| Bedtools Suite | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| Cell Ranger | 10x Genomics | https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger |
| Monocle2 | Qiu et al., 2017 | http://cole-trapnell-lab.github.io/monocle-release/ |
| Sleuth | Pimentel et al., 2017 | https://pachterlab.github.io/sleuth/ |
| Kallisto | Bray et al., 2016 | https://pachterlab.github.io/kallisto/ |
| FlashFry | McKenna and Shendure 2018 | https://github.com/aaronmck/FlashFry |
| Analysis of Motif Enrichment (AME - MEME Suite) | McLeay and Bailey, 2010 | http://meme-suite.org/ |
| Seurat | Butler et al., 2018 | https://satijalab.org/seurat/ |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
Highlights.
Perturbed 5,920 human candidate enhancers for impact on gene expression
Multiplexed ~28 CRISPRi perturbations per single-cell transcriptome
Adapted the eQTL analytical framework to identify 664 cis human enhancer-gene pairs
Characterized genomic features associated with these enhancer-gene pairs
ACKNOWLEDGMENTS
We thank the entire Shendure, Trapnell, and Ahituv labs, in particular D. Cusanovich, V. Agarwal, J. Tome, G. Findlay, S. Domcke, S. Regalado, J. Klein,L. Starita, D. Aghamirzaie, A. McKenna, X. Qui, F. Inoue, and O. Elor. We additionally thank J. Ousey, K. Han, M. Bassik, and M. Kircher. This work was supported by awards from the NIH (UM1HG009408 to N.A. and J. Shendure, DP1HG007811 to J. Shendure, and U24HG009446 to W.S.N.), the W.M. Keck Foundation (to C.T. and J. Shendure), and training awards from the National Science Foundation and NIH (Graduate Research Fellowship to A.J.H. and M.G. and 5T32HG000035 to M.G.). J. Shendure is an Investigator of the Howard Hughes Medical Institute.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and four tables and can be found with this article online at https://doi.org/10.1016/j.cell.2018.11.029.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adamson B, Norman TM, Jost M, Cho MY, Nuñez JK, Chen Y, Villalta JE, Gilbert LA, Horlbeck MA, Hein MY, et al. (2016). A multiplexed single-cell CRISPR screening platform enables systematic dissection of the unfolded protein response. Cell 167, 1867–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray NL, Pimentel H, Melsted P, and Pachter L (2016). Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology 34, 525–527. [DOI] [PubMed] [Google Scholar]
- Butler A, Hoffman P, Smibert P, Papalexi E, and Satija R (2018). Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol 36, 411–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver MC, Smith EC, Sher F, Pinello L, Sanjana NE, Shalem O, Chen DD, Schupp PG, Vinjamur DS, Garcia SP, et al. (2015).BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao F, Fang Y, Tan HK, Goh Y, Choy JYH, Koh BTH, Hao Tan J, Bertin N, Ramadass A, Hunter E, et al. (2017). Super-enhancers and broad H3K4me3 domains form complex gene regulatory circuits involving chromatin interactions. Sci. Rep 7, 2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Gilbert LA, Cimini BA, Schnitzbauer J, Zhang W, Li G-W, Park J, Blackburn EH, Weissman JS, Qi LS, and Huang B (2013). Dynamic imaging of genomic loci in living human cells by an optimized CRISPR/Cas system. Cell 155, 1479–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HMT, Schwarzlopf M, Fornace ME, Acharya A, Artavanis G, Stegmaier J, Cunha A, and Pierce NA (2018). Third-generation in situ hybridization chain reaction: multiplexed, quantitative, sensitive, versatile, robust. Development 145, dev165753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, and Zhang F (2013). Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datlinger P, Rendeiro AF, Schmidl C, Krausgruber T, Traxler P, Klug-hammer J, Schuster LC, Kuchler A, Alpar D, and Bock C (2017). Pooled CRISPR screening with single-cell transcriptome readout. Nat. Methods 14, 297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao Y, Li B, Meng Z, Jung I, Lee AY, Dixon J, Maliskova L, Guan K-L, Shen Y, and Ren B (2016). A new class of temporarily phenotypic enhancers identified by CRISPR/Cas9-mediated genetic screening. Genome Res. 26, 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao Y, Fang R, Li B, Meng Z, Yu J, Qiu Y, Lin KC, Huang H, Liu T, Marina RJ, et al. (2017). A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nat. Methods 14, 629–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit A, Parnas O, Li B, Chen J, Fulco CP, Jerby-Arnon L, Marjanovic ND, Dionne D, Burks T, Raychowdhury R, et al. (2016). Perturb-seq: dissecting molecular circuits with scalable single-cell RNA profiling of pooled genetic screens. Cell 167, 1853–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg E, and Levanon EY (2013). Human housekeeping genes, revisited. Trends Genet. 29, 569–574. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium (2012). An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulco CP, Munschauer M, Anyoha R, Munson G, Grossman SR, Perez EM, Kane M, Cleary B, Lander ES, and Engreitz JM (2016). Systematic mapping of functional enhancer-promoter connections with CRISPR interference. Science 354, 769–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambone JE, Dusaban SS, Loperena R, Nakata Y, and Shetzline SE (2011). The c-Myb target gene neuromedin U functions as a novel cofactor during the early stages of erythropoiesis. Blood 117, 5733–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathi M, Srivastava P, Das Sutar SK, Kumar K, Dasgupta D, Pal Singh G, Brahmachari V, and Brahmachari SK (2005). Comparative analysis of chromatin landscape in regulatory regions of human housekeeping and tissue specific genes. BMC Bioinformatics 6, 126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasperini M, Findlay GM, McKenna A, Milbank JH, Lee C, Zhang MD, Cusanovich DA, and Shendure J (2017). CRISPR/Cas9-mediated scanning for regulatory elements required for HPRT1 expression via thousands of large, programmed genomic deletions. Am. J. Hum. Genet 101, 192–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill AJ, McFaline-Figueroa JL, Starita LM, Gasperini MJ, Matreyek KA, Packer J, Jackson D, Shendure J, and Trapnell C (2018). On the design of CRISPR-based single-cell molecular screens. Nat. Methods 15, 271–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong J-W, Hendrix DA, and Levine MS (2008). Shadow enhancers as a source of evolutionary novelty. Science 321, 1314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horlbeck MA, Gilbert LA, Villalta JE, Adamson B, Pak RA, Chen Y, Fields AP, Park CY, Corn JE, Kampmann M, and Weissman JS (2016). Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5, e19760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Weiner A, Yofe I, Lara-Astiaso D, Keren-Shaul H, David E, Salame TM, Tanay A, van Oudenaarden A, and Amit I (2016). Dissecting immune circuits by linking CRISPR-pooled screens with single-cell RNA-seq. Cell 167, 1883–1896. [DOI] [PubMed] [Google Scholar]
- Klann TS, Black JB, Chellappan M, Safi A, Song L, Hilton IB, Crawford GE, Reddy TE, and Gersbach CA (2017). CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat. Biotechnol 35, 561–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korkmaz G, Lopes R, Ugalde AP, Nevedomskaya E, Han R, Myacheva K, Zwart W, Elkon R, and Agami R (2016). Functional genetic screens for enhancer elements in the human genome using CRISPR-Cas9. Nat. Biotechnol 34, 192–198. [DOI] [PubMed] [Google Scholar]
- Kulakovskiy IV, Medvedeva YA, Schaefer U, Kasianov AS, Vorontsov IE, Bajic VB, and Makeev VJ (2013). HOCOMOCO: a comprehensive collection of human transcription factor binding sites models. Nucleic Acids Res. 41, D195–D202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y, Ghazanfar S, Strbenac D, Wang A, Patrick E, Speed T, Yang J, and Yang P (2017). Housekeeping genes, revisited at the single-cell level. bioRxiv. 10.1101/229815. [DOI] [Google Scholar]
- MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, Junkins H, McMahon A, Milano A, Morales J, et al. (2017). The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res. 45 (D1), D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, and Shendure J (2018). FlashFry: a fast and flexible tool for large-scale CRISPR target design. BMC Biol. 16, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLeay RC, and Bailey TL (2010). Motif Enrichment Analysis: a unified framework and an evaluation on ChIP data. BMC Bioinformatics 11, 165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirchandani PB, and Francis RL (1990). Discrete Location Theory (Wiley). Morley M, Molony CM, Weber TM, Devlin JL, Ewens KG, Spielman RS, and Cheung VG (2004). Genetic analysis of genome-wide variation in human gene expression. Nature 430, 743–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel H, Bray NL, Puente S, Melsted P, and Pachter L (2017). Differential analysis of RNA-seq incorporating quantification uncertainty. Nat. Methods 14, 687–690. [DOI] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, and Siepel A (2010). Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20, 110–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu X, Hill A, Packer J, Lin D, Ma Y-A, and Trapnell C (2017). Single-cell mRNA quantification and differential analysis with Census. Nat. Methods 14, 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajagopal N, Srinivasan S, Kooshesh K, Guo Y, Edwards MD, Banerjee B, Syed T, Emons BJM, Gifford DK, and Sherwood RI (2016). High-throughput mapping of regulatory DNA. Nat. Biotechnol 34, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao SSP, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, and Aiden EL (2014). A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Wright J, Zheng K, Shalem O, Fontanillas P, Joung J, Cheng C, Regev A, and Zhang F (2016). High-resolution interrogation of functional elements in the noncoding genome. Science 353, 1545–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt AD, Hu M, Jung I, Xu Z, Qiu Y, Tan CL, Li Y, Lin S, Lin Y, Barr CL, and Ren B (2016). A Compendium of Chromatin Contact Maps Reveals Spatially Active Regions in the Human Genome. Cell Rep 17, 2042–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stranger BE, Montgomery SB, Dimas AS, Parts L, Stegle O, Ingle CE, Sekowska M, Smith GD, Evans D, Gutierrez-Arcelus M, et al. (2012). Patterns of cis regulatory variation in diverse human populations. PLoS Genet. 8, e1002639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe O, McPhee D, Kobayashi S, Shen Y, Brandt W, Jiang X, Campbell AD, Chen Y-T, Chang Cs., Yamamoto M, et al. (2007). Embryonic and fetal beta-globin gene repression by the orphan nuclear receptors, TR2 and TR4. EMBO J. 26, 2295–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakore PI, D’Ippolito AM, Song L, Safi A, Shivakumar NK, Kabadi AM, Reddy TE, Crawford GE, and Gersbach CA (2015). Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat. Methods 12, 1143–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Väremo L, Nielsen J, and Nookaew I (2013). Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 41, 4378–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visel A, Minovitsky S, Dubchak I, and Pennacchio LA (2007). VISTA Enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Res. 35, D88–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei K, Iyer R, and Bilmes J (2015). Submodularity in data subset selection and active learning. PMLR 37, 1954–1963. [Google Scholar]
- Xie S, Duan J, Li B, Zhou P, and Hon GC (2017). Multiplexed engineering and analysis of combinatorial enhancer activity in single cells. Mol. Cell 66, 285–299. [DOI] [PubMed] [Google Scholar]
- Xie S, Cooley A, Armendariz D, Zhou P, and Hon GC (2018). Frequent sgRNA-barcode recombination in single-cell perturbation assays. PLoS ONE 13, e0198635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahi A, Lappalainen T, Mohammadi P, and Tatonetti N (2018). RecNW: a fast pairwise aligner for targeted sequencing. bioRxiv 10.1101/371989. [DOI]
- Zhou B, Ho SS, Zhu X, Zhang X, Spies N, Byeon S, Arthur JG, Pattni R, Ben-Efraim N, Haney MS, et al. (2017). Comprehensive, integrated, and phased whole-genome analysis of the primary ENCODE cell line K562. bioRxiv. 10.1101/192344. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the sequencing data (single cell RNA-seq and bulkRNA-seq) and processed data files is GEO: GSE120861 (metadata file), and GSM3417251–GSM3417303 (actual datasets).