

ABSTRACT
Malaria is a vector-borne disease that involves multiple parasite species in a variety of ecological settings. However, the parasite species causing the disease, the prevalence of subclinical infections, the emergence of drug resistance, the scale-up of interventions, and the ecological factors affecting malaria transmission, among others, are aspects that vary across areas where malaria is endemic. Such complexities have propelled the study of parasite genetic diversity patterns in the context of epidemiologic investigations. Importantly, molecular studies indicate that the time and spatial distribution of malaria cases reflect epidemiologic processes that cannot be fully understood without characterizing the evolutionary forces shaping parasite population genetic patterns. Although broad in scope, this review in the Microbiology Spectrum Curated Collection: Advances in Molecular Epidemiology highlights the need for understanding population genetic concepts when interpreting parasite molecular data. First, we discuss malaria complexity in terms of the parasite species involved. Second, we describe how molecular data are changing our understanding of malaria incidence and infectiousness. Third, we compare different approaches to generate parasite genetic information in the context of epidemiologically relevant questions related to malaria control. Finally, we describe a few Plasmodium genomic studies as evidence of how these approaches will provide new insights into the malaria disease dynamics.
INTRODUCTION
Malaria is a vector-borne parasitic disease endemic in tropical and subtropical regions worldwide. Despite progress on reducing its burden, nearly 40% of the world’s population remains at risk of infection (1). Malaria is caused by protozoa of the genus Plasmodium (Apicomplexa: Plasmodiidae), a diverse group that infects a variety of vertebrate hosts, including primates (2–4). Such diversity has led to a disease involving multiple parasites and vector species across various ecosystems worldwide (1).
There are four species of Plasmodium that commonly infect humans: Plasmodium falciparum, Plasmodium malariae, Plasmodium ovale, and Plasmodium vivax. Of these, P. falciparum and P. vivax cause most cases of malaria morbidity, and mortality (1). These parasites emerged independently as human pathogens during the radiation of a Plasmodium clade associated with nonhuman primate and rodent hosts (3, 4) and that appears to be older than the origin of hominids (4–7) (Fig. 1). Such complex evolutionary history may explain the biological differences between parasite species causing human malaria (2–7).
FIGURE 1.
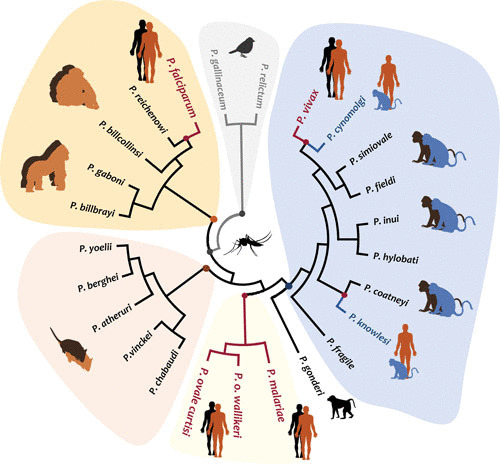
Phylogeny of Plasmodium parasites based on the mitochondrial genome. The phylogenetic tree shows all the Plasmodium species parasitic to humans, including those that cause zoonotic malarias. Although not a comprehensive phylogeny, it evidences that parasites causing human malaria are not a monophyletic group.
There are definite similarities in the life cycles among all Plasmodium species. In all species, a fraction of the circulating parasites in the blood (merozoite) differentiates into sexual stages (gametocytes) that are then taken up by the mosquito vector (Fig. 2). However, there are also marked differences between these parasite species in terms of their life histories. For example, P. falciparum infection produces gametocytes after a longer period than does P. vivax infection, but P. falciparum gametocytes are infectious longer than those in P. vivax (8). Such differences may affect their transmission and how interventions affect their fitness (8, 9).
FIGURE 2.
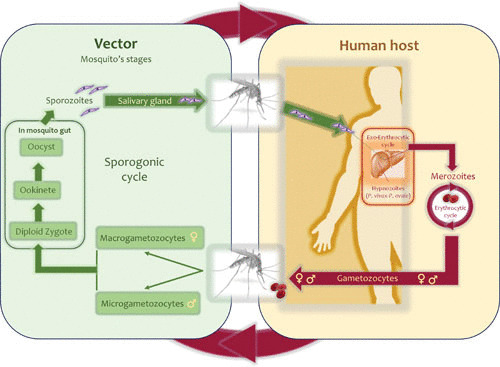
Plasmodium life cycle. The Plasmodium haplontic life cycle comprises a vertebrate host and a dipteran vector. In human malarias, an infected female Anopheles mosquito inoculates haploid sporozoites into the host. These sporozoites invade the liver cells and mature into schizonts. The schizonts rupture, releasing merozoites that infect the red blood cells. Some species develop dormant liver stages or hypnozoites that can produce merozoites at a later time (relapse). A fraction of merozoites differentiates into gametocytes (micro- and macrogametocytes). All these stages are haploid. These gametocytes are then taken up by an Anopheles mosquito, in which zygote formation takes place (diploid stage). Due to the nature of the cycle, inbreeding is common. The zygote differentiates into ookinetes and oocysts, the latter with a syncytial cell or sporoblast containing thousands of nuclei in which meiosis takes place, producing haploid sporozoites.
Plasmodium vivax and P. ovale develop a dormant liver stage (hypnozoite) that reactivates, causing relapse (infection of the red blood cells) after several weeks (to months or years) of the primary infection. Thus, a radical cure of a malaria patient with any of these two parasites requires eliminating those dormant stages not found in P. falciparum or P. malariae (1, 2). On the other hand, P. falciparum-infected erythrocytes can adhere to the endothelium of capillaries and venules, a process mediated by a gene family without clear orthologs in the other human malarias (10). This process, called sequestration, is linked to severe clinical presentations that are seldom observed in other non-falciparum malaria infections (11, 12). In contrast, P. malariae, if untreated, can produce a chronic infection that remains latent in blood for years (2).
The distributions of these malaria parasites also vary worldwide (1); there could be only one species in some areas where malaria is endemic, while all four Plasmodium species can coexist in others. Plasmodium vivax can be present in temperate zones, whereas the other human parasites are generally restricted to tropical and subtropical regions (1, 2). Plasmodium falciparum and P. vivax may have overlapping distributions in many areas of endemicity outside sub-Saharan Africa but could differ in their temporal and spatial occurrence at a local scale (13–15). In addition to these four Plasmodium species, zoonotic infections and potential animal reservoirs further complicate malaria epidemiology.
The most recognizable zoonotic malaria agent is Plasmodium knowlesi, a parasite found in nonhuman primates from Southeast Asia (Fig. 1) (4, 16, 17). Although human-to-human transmission has not been demonstrated, the incidence of this zoonosis is increasing in some areas compared to the more common human malarias (1, 18). How this zoonosis affects the goal of eliminating the disease is a matter that requires some discussion in countries such as Malaysia (1). In addition, there is growing evidence indicating that another nonhuman primate parasite from Southeast Asia, Plasmodium cynomolgi, could naturally infect humans, even as an asymptomatic infection (19, 20). The actual prevalence of human P. cynomolgi infections seems to be low, and there are no indications of human-to-human transmission (20).
The possibility of anthropozoonotic malaria cycles may also hamper elimination efforts in regions such as South America, as there are studies indicating that nonhuman primates may act as reservoirs (21, 22). Two previously accepted nonhuman primate species, Plasmodium brasilianum and Plasmodium simium, are likely synonyms of P. malariae and P. vivax, respectively, found in humans (4, 23). The interpretation of these findings, however, remains elusive. It has not been ruled out that nonhuman primates could be actively infected by human parasites without spillback to humans. This has been observed in Africa in a process in which the vectors may play a critical role (24, 25).
All these factors, together with the uneven scale-up of control interventions across regions of endemicity, drive a constantly changing malaria epidemiology. Unveiling such complex dynamics requires the use of molecular approaches and parasite population genetics.
MOLECULAR TOOLS IN MALARIA EPIDEMIOLOGY
Detecting parasite genetic material is critical when traditional epidemiologic and clinical data, on their own, cannot inform about processes leading to the observed incidence and distribution of cases. Studying parasite genetics allows us to (i) estimate the real incidence rate and prevalence by detecting subclinical infections or submicroscopic peripheral parasitemia, (ii) identify potentially infectious individuals by targeting genes expressed in gametocytes, (iii) differentiate a recrudescent case from a new infection, (iv) evaluate interventions (e.g., surveillance of mutations associated with drug resistance or alleles of an antigen targeted by a vaccine), (v) assess how spatial connectivity sustains malaria transmission by measuring the parasite gene flow-migration-colonization patterns, (vi) link parasite loci with phenotypes of interest such as drug resistance or genes that affect pathogenesis, and (vii) detect zoonotic or anthropozoonotic infections (16, 20, 22, 26–30).
Given the broad application of molecular technologies in malaria epidemiology (Fig. 3), in this review, commonly used methods are discussed intertwined with population biology and population genetics questions wherever these connections are relevant. In order to assist the reader, Fig. 3 shows a diagram linking molecular techniques in the context of problems that are commonly addressed.
FIGURE 3.
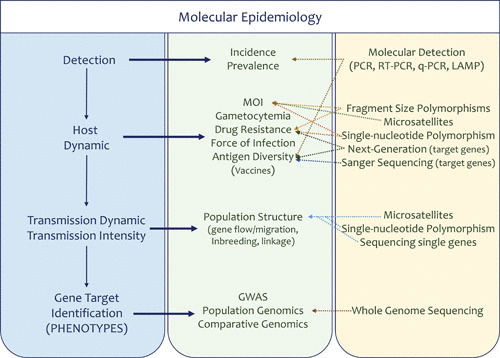
Approaches in malaria molecular epidemiology. Shown are techniques and approaches commonly used to generate molecular information in the context of epidemiologic investigations.
DETECTING THE PARASITE USING MOLECULAR METHODS
Although confirmation of a Plasmodium infection is a necessary step in managing a malaria case, scaling up the use of molecular diagnostic techniques in the context of elimination and control remains a work in progress (26, 31). A precise diagnosis (detection) allows for studying how risk factors drive incidence and the distribution of cases (27, 28). It is also essential to assess the efficacy of deployed interventions (27, 30–32). Given its importance, testing and developing molecular diagnostic methods constitute an active research area. Those readers interested in this issue could check one of the many reviews available (32–34). Here the focus is limited to some aspects of diagnostics that affect population-level investigations.
The examination of thick blood smears using light microscopy remains the standard for malaria laboratory confirmation (1, 31). Unfortunately, microscopy shows reduced sensitivity when parasitemias are low; such is the case for asymptomatic patients (26, 34–36). It also lacks the power to detect zoonotic and anthropozoonotic infections that may be important in some settings (16, 20–22). In addition, sustaining dependable microscopists is challenging in underserved areas or wherever malaria transmission is no longer perceived as a threat. These problems have led to the development of rapid diagnostic tests (RDTs) based on immunochromatographic detection by monoclonal antibodies against specific parasite proteins (32).
Current RDTs detect Plasmodium-specific antigens such as falciparum-specific histidine-rich protein 2 and histidine-rich protein 3 (HRP-2 and HRP-3, respectively) or Plasmodium-specific lactate dehydrogenase and aldolase (1, 26, 31, 32). RDT results are reproducible and rapidly available, and RDTs require minimal training and no instrumentation or dedicated facilities (31, 32). There are still technical limitations, such as reduced sensitivity with samples from malaria patients with low parasitemias (e.g., P. vivax and P. malariae), as well as handling problems that cause denaturation of antibodies. Importantly, the Pfhrp-2 and Pfhrp-3 genes are located in subtelomeric regions, areas of the Plasmodium genome that undergo frequent recombination and rearrangements. As a result, there are Pfhrp-2 gene polymorphisms (37), including different forms of Pfhrp-2 and Pfhrp-3 deletions in P. falciparum, affecting the efficacy of RDTs. Such deletions were initially discovered in Peru, but there are reports of their occurrence worldwide (34, 37–41). It has been proposed that the prevalence of these Pfhrp-2 and Pfhrp-3 deletions could be selected for by the deployment of the RDTs, as those parasites are “undetected” and not treated (28, 37, 38). A stochastic simulation model suggested that the use of Pfhrp-2-detecting RDTs was sufficient to select for Pfhrp2 and -3 double deletions in this parasite species, prompting the WHO to recommend implementation of surveillance activities in identified regions of concern (40).
In addition to RDTs, there are a variety of molecular methods, including conventional PCR, quantitative reverse transcription-PCR (qRT-PCR), RT-PCR, and loop-mediated isothermal amplification. These methods target the 18S rRNA, mitochondrial genes, and repetitive sequences (20, 26, 27, 32–34, 37, 42–44). Their commonality is that these methods target parts of the parasite genome found in multiple copies, increasing their sensitivity when parasitemias are low. However, these methods are costly, and some require dedicated infrastructure and a high level of technical training, limiting their use mostly to research projects.
Although still limited compared with the use of microscopy or RDTs (1, 26, 31, 34), the use of molecular diagnostic methods has changed our perception of malaria transmission. There is overwhelming evidence indicating that a significant proportion of the malaria incidence rate is comprised of asymptomatic infections (13, 26, 34–36, 44–46). Thus, the attention is shifting from solely focusing on passive case detection (clinical cases) to understanding how asymptomatic subclinical infections may compromise control and elimination efforts by sustaining transmission (8, 26, 30, 35, 45–47). In addition, the actual prevalence of non-falciparum malarias, such as those caused by P. malariae and P. ovale, is now considered higher than previously thought (48), particularly as part of mixed infections. Finally, the discovery of zoonoses such as those caused by P. knowlesi and P. cynomolgi has been possible because of the use of molecular methods (16–23).
INFERRING THE PREVALENCE OF INFECTIOUS PATIENTS
Understanding malaria epidemiological patterns requires deconstructing the incidence rate and clinical outcomes in terms of population-level processes. There are multispecies/multistage approaches to perform blood stage and gametocyte quantification by qPCR and qRT-PCR (8, 49, 50). Such data are helpful in parameterizing mathematical models developed to understand transmission and/or inform interventions (for examples, see references 9 and 51). As an example, many control strategies were deployed under the assumption that acquired immunity renders a person noninfectious, so asymptomatic infections in semi-immune individuals do not have a meaningful role in transmission (52, 53). Ascertaining differential patient infectiveness (e.g., those with clinical manifestations compared to asymptomatics) requires detecting and quantifying gametocytes (8, 49, 54).
Molecular assays (e.g., qRT-PCR) have been developed to detect and measure genes expressed in gametocytes, such as Pfs25 in P. falciparum and Pvs25 in P. vivax (8, 46, 49, 50, 55–61). Using such approaches, it has been shown that asymptomatic P. falciparum patients can infect mosquitoes from Africa (57). There are only a handful of studies from areas with low transmission outside Africa, or on any of the other malarial parasites. However, pioneering investigations with P. vivax have found comparable expression levels of Pvs25 in symptomatic and asymptomatic patients in South America (55, 58). There is also progress in assessing the differential contributions of lineages to gametocyte production, as well as possible within-host interactions, by targeting gametocyte-expressed polymorphic loci in P. falciparum (e.g., pfs230 and pfg377) (8, 59, 60). Analogous approaches for other malarial parasites have not been reported yet.
Nonetheless, gametocytemia and infectiousness are the results of a process that is still poorly characterized (50, 60–62); it is not a static parameter that can be estimated by sampling at a given time point and that is directly comparable across studies (50, 60, 61). Thus, developing gametocyte markers and approaches that identify groups of individuals that disproportionally sustain transmission (e.g., asymptomatic patients) is becoming a critical issue in the context of malaria elimination (8, 60–62).
STUDYING PARASITE GENETIC DIVERSITY
Beyond the detection of asexual or sexual stages (infected and infectious patients), many molecular epidemiologic investigations follow or characterize parasite genetic variants. Parasite genotyping initially focused on the prevalence of mutations conferring resistance to antimalarial drugs or on the diversity of genes encoding antigens considered vaccine candidates (27, 28). The controversies around the so-called “clonal theory of parasitic protozoa,” however, energized the incorporation of population genetics concepts into malaria epidemiology (63).
At the time that the clonal theory was proposed, many considered sexual reproduction as a simile to lack of linkage disequilibrium and high genetic diversity (63). As a result, finding population structures (nonrandom mating) (64) in Plasmodium somehow was not expected (63). The observation of linkage disequilibrium (association of alleles segregating in different loci) in P. falciparum demonstrated that population expansion of linked groups (also called clonal expansions) was a relatively common phenomenon. This early work led to a better understanding of how linkage disequilibrium in malaria parasites was driven by inbreeding or selection (27, 28, 64–66) and also brought attention to characterizing parasite genetic diversity in order to understand disease dynamics. Nowadays, measurements of parasite genetic polymorphisms are part of many epidemiologic studies covering topics ranging from assessing how transmission intensity affects the complexity of infection to identifying/following polymorphisms on genes linked to drug resistance or other forms of selection, such as antigenic variation (10, 27–29).
There are several methods available for parasite genotyping (27, 28), including single (or targeted)-gene, multilocus, and genomic approaches (Fig. 3). Researchers need to consider the advantages and limitations of each method in the context of their specific studies. An essential task in any molecular epidemiologic study, however, is to envision the expected molecular pattern under a specific epidemiologic outcome or set of assumptions.
Traditionally, the sampling in epidemiologic investigations focuses on collecting specimens in a defined population of patients in terms of time and space (27, 31). However, the time and spatial scales where the malaria cases occur will not fully account for the evolutionary time and processes behind the genetic patterns in the parasite populations. Leveraging on the differences between the evolutionary time scale in the parasite and the observed epidemiology informs about the disease dynamics in ways that cannot be achieved by using solely traditional metrics (27, 28, 65–70).
There are two sources of variation in any epidemiologic study using parasite genetics. One is the sampling of cases that is addressed by estimating the number of specimens to be collected given the question; this source of variation is considered by epidemiologists when they estimate their sample sizes. The other source of variation comes from the evolutionary processes that affected the sampled parasite loci that also need to be inferred or modeled. Those processes are genetic drift (related to historical demographic changes in transmission and random uneven reproduction of parasite lineages), type of mutations (point mutations or changes in the number of repeats), mutation rates (given by the loci functional constraints and mode of evolution), or whether such allelic variants affect the parasite fitness (genetic variants encoding phenotypes with different degrees of reproductive success or natural selection) (27–29, 64, 65, 67). These processes may also affect a set of loci differently depending on other factors, such as inbreeding (self-fertilization) and their relative locations in the chromosome (64).
As a result, a study design requires thinking about the sampling at those two levels: the number of individuals and number and type of loci. If the question involves estimating a frequency (e.g., a mutation) in a population or any other metric that consists of reporting the frequency of parasite variants at the level of infections (e.g., the multiplicity of infection; see below), the sampling should focus on individuals (cases). However, many questions require also focusing on the number of loci and whether they are linked (alleles at those loci do not segregate independently, likely due to their proximity in the chromosome) or unlinked (recombination allows alleles at those loci to segregate independently) (64). This is important when a study aims to compare whether two infections are generated by the same parasite (e.g., separating recrudescence from new infections), patterns in the parasite genetic diversity (e.g., population structure or geographic differentiation), or contrasting cases and controls given a phenotype (e.g., discovering mutations linked to a particular phenotype such as drug resistance; see below), among other investigations (27–29, 65, 71, 72). In all cases, the quality of phenotypic and epidemiologic metadata associated with the specimens will limit the information derived from the parasite genetic data. In the following sections, we describe different types of studies that interrogate parasite molecular data. Those studies will progressively show, following a historical trend, the link between epidemiology and population genetics.
Antimalarial Drug Surveillance
A method that was widely used incorporated PCR and restriction fragment length polymorphism to detect P. falciparum mutations associated with drug resistance to antimalarial drugs such as sulfadoxine-pyrimethamine and chloroquine (27, 73, 74). These methods and others follow the frequency of each mutation as an independent locus. This approach has limitations when the goal is to follow alleles with multiple mutations (e.g., double or triple mutants) that differ in their levels of resistance (69, 75). In particular, alleles sometimes cannot be adequately reconstructed from single-mutation reports because they are derived from patients infected by multiple parasite lineages that differ in their mutation profiles. A classic example is the three mutations in the Pfdhfr gene conferring resistance to pyrimethamine in P. falciparum. Those mutations could be found in a given sample; however, whether this indicates an infection with two alleles (two parasite lineages), each containing mutations with intermediate resistance (two mutations), or a single highly resistant triple mutant allele is difficult to ascertain from a table with the frequency of individual mutations (69, 75). Although the technology has changed, the approach of tracking individual mutations is still prevailing, as in the case of those reported in the Pfk13 gene associated with delayed artemisinin parasite clearance in P. falciparum (70).
Multiplicity of Infection
The first genotyping methods relied on agarose electrophoresis to detect genes encoding surface antigens with a variable number of tandem repeats (76). These methods are still in use around the world (27, 28). The loci more commonly sampled are Pfmsp2 and Pfglurp in P. falciparum (there are no orthologs of these loci in P. vivax), Pvmsp3α in P. vivax (with no ortholog in P. falciparum) (77, 78), and msp1 and csp in both species (27, 71, 76, 79). Some of these antigen genes, such as Pfmsp2 and Pfmsp1 (P. falciparum), have two or more groups of alleles that share particular motifs (24, 71). These groups are referred to as allele families. Usually, fragment size polymorphisms are studied after identifying the allele family as part of the genotyping process (71). This approach has proven useful to determine the number of different parasite genotypes coinfecting a single patient, or multiplicity of infection (MOI) (80).
MOI reflects coinfections (two or more genotypes being transmitted simultaneously by a mosquito) and superinfections (multiple but independent infections). As a result, the frequency of infections by multiple lineages is expected to decline as transmission decreases, and superinfections/coinfections become less likely (28, 65, 80–83). However, the relationship between MOI and transmission is far from linear (68, 80–84).
MOI is a summary of several processes, including the genealogy of the circulating parasites (e.g., a superinfection by identical parasites at a given locus cannot be detected) and the characteristics of the infection (e.g., superinfections may be more likely in asymptomatic patients with a long time of exposure). Studies also indicate that the prevalence of multiple infections is affected by a variety of other factors, including age and immunity (27, 28, 82). In addition, there is variance in transmission driven by spatial differences in mosquito biting rates (28, 65). Thus, large-scale geographic associations between MOI and transmission may be difficult to observe because of a combination of these factors (84, 85).
MOI can provide valuable information in the context of following changes in transmission longitudinally in a specific location (86). Indeed, cross-sectional and longitudinal studies indicate that the prevalence of P. falciparum single clonal infections increases with a reduction in malaria transmission (28, 66, 87–89). However, these trends in MOI have not been observed for P. vivax (28, 65, 84, 90, 91), for which multiclonal infections remain common even in low-transmission areas (28, 65, 84, 90). This could be the result of hypnozoites from prior infections accumulating in the liver and thus causing multiple relapses of distinct genotypes (28, 65). Unfortunately, there are no data published on P. ovale and P. malariae.
It is worth noting that MOI has also been used as a proxy for intrahost dynamics under the hypothesis that competition between parasite lineages may relate to disease severity (reviewed in reference 82). Like in the case of transmission, there is conflicting evidence on how MOI is associated with disease severity in human malarias. Part of the problem is the variety of genotyping methods used (not only fragment size polymorphism; see below) and that often studies do not include suitable controls (82, 86, 90, 91). Thus, understanding and measuring MOI require well-planned epidemiologic investigations.
Single-Gene Studies
Many studies rely on sequencing partial or complete genes from the nuclear or organelle genomes of Plasmodium spp. These investigations aim to characterize the diversity of a vaccine candidate, genes involved in pathogenesis, or genes encoding drug targets that may harbor point mutations that result in a drug-resistant phenotype (10, 27, 78, 92–95). In addition, this approach has been used to understand global patterns of diversity, including the malaria parasites’ evolutionary histories, gene flow, and/or population structure (94, 96–102).
These types of investigations have contributed to our knowledge of malaria genomic epidemiology. In the context of genes encoding antigens, single-copy genes expressed in the infective stage inoculated from the mosquito vector, the sporozoite, and the erythrocytic state (merozoite) tend to be more polymorphic than those expressed in the gametocyte or in the mosquito vector (10, 27, 94, 96). Among orthologous genes, P. falciparum and P. vivax show differences in terms of how natural selection drives the observed pattern of polymorphism (94, 103–105). Finally, patterns from the mitochondrial genome (considered a single-locus approach because of lack of recombination) and housekeeping genes indicated that P. vivax is far more diverse than P. falciparum (97–99, 101). This pattern was also evidenced whenever orthologous genes encoding antigens were studied.
The target gene approach is enriched by the use of next-generation sequencing (NGS) technologies. In particular, NGS on targeted amplification can detect the relative abundance of different alleles in a single infection by counting hundreds of overlapping reads in polymorphic regions. Thus, coupled with appropriate sampling, NGS can inform intrahost dynamics in a way that Sanger sequencing and other genotyping methods cannot. These technologies have been used in the context of evaluating vaccine efficacy, following dynamics of mutations associated with drug resistance, measuring the complexity of infections (MOI), and differentiating recrudescence from new infections (91, 106–110).
Multilocus Genotyping
Multilocus genotyping studies seek to understand processes that deviate from the expectation of random mating (population structure), as well as the dispersion of alleles linked to particular phenotypes of interest (e.g., drug resistance). The genome is sampled by genotyping several loci so linkage disequilibrium and other population-level parameters can be characterized. Linkage disequilibrium (LD), however, can emerge for a variety of reasons. First, loci could be physically close in the genome. This can be very informative if such loci are linked to mutations that confer resistance to an antimalarial drug. In such cases, characterizing multilocus haplotypes can be used to establish whether drug resistance originated one or more times. Second, linkage can emerge because of processes that affect random mating, such as inbreeding, reduction of the effective population size, epidemic expansions, and geographic isolation (28, 64, 65). The available evidence indicates that population structures are commonly observed in malarial parasite populations; the use of these patterns to deploy and evaluate the efficacy of interventions remains an area of extensive research (28, 65, 69, 83).
Two types of loci are commonly used in multilocus genotyping: microsatellites (65, 67, 68, 82, 83, 111, 112) and single nucleotide polymorphisms (SNPs) (113, 114). These loci can be used separately or in combination; they document similar processes (69, 75, 115–118). Microsatellites are abundant in Plasmodium spp., but their characteristics change across species. While the P. falciparum genome has an average of one microsatellite locus per 2 to 3 kb of sequence, the P. vivax genome has far fewer microsatellites, and they usually occur in more complex patterns (112). An advantage of microsatellites is that they have a higher mutation rate than SNPs, which allows detection of recent events with fewer loci (65, 72). A limitation, however, is that microsatellites evolve according to complex evolutionary models and are not equally suitable worldwide (67, 68, 112).
Using these loci, researchers can study different aspects of population structure, such as linkage disequilibrium and gene flow (65, 66, 72, 83). We know that P. falciparum and P. vivax have undergone clonal expansions as evidenced by extensive linkage disequilibrium (28, 65, 66, 69, 87–89, 118). Indeed, inbreeding seems to be critical to explain many of the observed patterns (65, 66, 119). Furthermore, clonal lineages tend to replace each other through time and space (65). When P. falciparum populations are compared across continents, it is found that genetic diversity and recombination rates are highest in areas of holoendemicity and lowest in regions of hypoendemicity of Central and South America (28, 65, 66, 87–89, 118). In contrast, P. vivax genetic diversity was found to vary worldwide and does not follow the same pattern (28, 65, 111, 120, 121). While transmission affects the inbreeding rate, diversity in the parasite population by itself can be affected by (among other factors) historical processes or selective sweeps (28, 64, 66, 69, 96–101). In such cases, polymorphisms physically linked to mutations that are increasing in frequency because they are being selected for (e.g., conferring drug resistance) leave a specific pattern involving a reduction of genetic diversity in the chromosome region harboring such mutations (64, 69, 116, 117, 122–125). In such investigations, microsatellites have been used to understand the multiple geographic origins and dispersion of haplotypes with mutations conferring drug resistance in P. falciparum (69, 75, 115–117, 124).
SNPs are starting to replace microsatellites in several contexts given their high reproducibility, which allows exploration of patterns over long periods and spatial scales. However, they have some limitations. Given their relatively low mutation rate compared to that of microsatellites, usually a larger number of loci is required to find the same patterns (72). In addition, ascertainment bias is a problem in some contexts. In particular, SNPs may be identified in relatively small sample sizes and may be more likely to reflect common rather than rare alleles (126), affecting assumptions in some population genetic analyses (e.g., inferences about parasite demographic history or selection). SNP data, however, are starting to yield patterns similar to those found with microsatellites, such as strong population structure explained by geographic isolation and inbreeding (91, 113, 114, 127, 128).
There are efforts directed to develop SNP typing protocols for P. falciparum and P. vivax. Their goal is to identify SNPs that can distinguish between parasites from different geographic areas (113, 114) or separate new infections from a recrudescence (91, 129). Many of the SNPs currently used in P. falciparum have been selected from different chromosomes so that demographic processes, including infection origin, can be studied (113, 114). These sets of standardized SNPs are usually referred to as a molecular barcode (113, 114). However, recombination between different parasite lineages will break such multilocus genotypes, so the term barcode used literally could be misleading. As in other approaches, the use of SNPs is challenged by multiclonal infections (28, 90, 91).
In addition to gene flow and population structure, multilocus genotyping has been used to assess changes in transmission intensity. However, the parasite genetic diversity is likely affected by events across multiple transmission events (years), whereas a change in malaria incidence may occur within weeks. Like in the case of MOI, studies seek to identify patterns of genetic diversity consistent with changes in transmission, such as a bottleneck in the parasite population as a result of scaling up interventions that should decrease the parasite population (63, 64, 127, 128). This will lead to changes in allele frequencies, with the expected outcome that genetic variation will be lost in response to the declining transmission. A first approximation is detecting changes in linkage disequilibrium. As stated earlier in the context of MOI, the proportion of infections comprised of a single genotype (monoclonal) is expected to increase when transmission declines, so the inbreeding rate is expected to rise (28, 68). Thus, the number of infections by identical nonsegregating genotypes (linkage disequilibrium or LD) is expected to increase (28, 65, 68, 113, 127, 128, 130). However, in settings where a dramatic increase in transmission occurred after a sustained decrease in malaria incidence, many infections were shown to be caused by identical or highly related parasites (a so-called clonal expansion identified by multilocus genotypes that are stable in time and space) (68, 130). Thus, the observed high LD reflects the past population reduction (clonal expansion of a few lineages) rather than the ongoing increase in transmission. Measuring LD, however, is challenging in areas where multiclonal infections are common simply because the circulating lineages may be difficult to reconstruct (28, 83, 113, 114, 130).
Alternatively, other studies have attempted to follow changes in genetic diversity (heterozygosity). However, heterozygosity may show even more complex patterns than LD since they depend on the effective population size (see below) and the mutation rate of the loci under study (28, 120). Changes in genetic diversity could be almost undetectable if there are a few genetically divergent inbred lineages coexisting in an area. The parasite heterozygosity seems to be less affected by a reduction of malaria incidence of short duration (28, 68, 83, 131).
Lastly, some studies have aimed to estimate changes in the parasite effective population size, Ne (68, 120, 131). Ne measures the uneven reproductive success of parasite lineages, so it directly measures genetic drift (64). While a reduction in the effective population size is expected if transmission is reduced (120, 131), there are many factors that could make such an outcome difficult to detect in the context of epidemiologic investigations. First, there are multiple ways to estimate Ne, each one focusing on different aspects (e.g., number of parents or differences in the number of progeny) (64, 68, 131). Second, an increase in malaria incidence could be driven by closely related parasites and/or high variance in the number of secondary infections generated by infected individuals (superspreaders) that will yield a low Ne (68). Third, Ne estimates might be inflated by migration or population substructure (131). Finally, the number of infections that sustains the parasite population between transmission seasons may have a greater impact on Ne than the total number of cases in a given year or the number of cases during the high-transmission season (131).
How to incorporate such concepts as Ne into a metric useful to those working on malaria control is a matter for discussion (68, 120, 131, 132). Nevertheless, genetic information and population genetic parameters can inform how increasing incidence may actually be driven by local parasites rather than an introduction due to migration, and these observations can be used by control programs to evaluate their efficacy in the context of elimination (28, 68, 131, 132).
Whole-Genome-Sequence-Based Methods
Population genomics is a growing field with a significant impact on malaria molecular epidemiology. However, eliminating human DNA contamination remains a challenge, especially for P. vivax given the absence of a reliable culturing system. Fortunately, there has been substantial technical progress in that regard (133, 134), leading to many genomic studies reported for malarial parasites (29, 116, 132, 135–144).
Of particular importance are genome-wide association studies (GWAS) used to determine the molecular bases of antimalarial drug resistance. The idea is to detect specific patterns in the genome indicating that a mutation has been selected for (directional selection) (122, 123). In particular, when the frequency of a mutation increases as a result of its selective advantage (e.g., it confers drug resistance), it is expected that the physically linked region of the genome will also increase in frequency via hitchhiking (122, 123), showing the same genetic background in high frequency; this process is commonly called a selective sweep. The size of the genome region affected by a selective sweep will be determined by the recombination rate, as in each generation, recombination may break up the otherwise linked sites (123). Selective sweeps also result in low genetic variation around the adaptive mutation (expected because all individuals will share the adaptive mutation and its linked genetic background) and high haplotype homozygosity. GWAS require specimens with phenotypic information so the genetic bases of such phenotypes can be characterized. Since other factors can generate LD patterns similar to what are found after a selective sweep, including epidemic/clonal expansions such as the extensive inbreeding observed in malaria (28), it is critical to consider such processes when sampling for control specimens or for specimens without the phenotype of interest (116, 135–138).
As an example, studies on the emergence of resistance against artemisinin combination therapy (ACT) in South Asia were possible because there were well-characterized P. falciparum isolates from patients showing low parasite clearance rates (135). By comparing those samples with others from sensitive parasites, GWAS approaches were able to identify genes or genomic regions associated with altered sensitivities to ACT. In particular, a region on chromosome 13 was first implicated (116, 136, 137), and then a specific locus was later identified, the kelch13 gene (138). Using a similar approach, genetic markers for dihydroartemisinin-piperaquine (an ACT) in P. falciparum have been identified (139).
Selective sweeps related to antimalarial drugs have also been found for P. vivax. A pattern consistent with a selective sweep was found in Colombia at the dhps gene, which is targeted by sulfadoxine, a drug that combined with pyrimethamine was used to treat P. falciparum but not P. vivax malaria (140, 141). These mutations have also been found in other P. vivax populations, indicating that this parasite is under drug-related selective pressure even when it is not the primary target of the drug.
One interesting finding from population genomics studies is that P. vivax populations are far more genetically diverse than P. falciparum. Even in a single population, P. vivax shows as much genetic diversity as a sample including worldwide P. falciparum isolates (140–142). This pattern was consistent with previous observations made by single-gene and multilocus approaches (90, 98–100, 103, 111). Population genomic approaches have also been used to detect patterns consistent with positive selection that may lead to the discovery of genes involved in parasite-host interaction, including novel antigens and genes involved in invasion (143, 144). These studies actually show how population genomic approaches allow modeling demographic processes (e.g., geographic structure) that may mask signatures consistent with balancing selection.
Scaling up population genomics will require, however, dealing with the high prevalence of multiple infections (145, 146). There are several promising approaches that attempt to identify chromosome fragments that are identical by descent (similar because they share a common ancestry) even as part of multiple infections (more than one lineage), and such approaches will likely improve our understanding of the role played by natural selection and demography in the variation emerging from population genomics investigations (145–147).
Genome studies of P. falciparum also include comparative approaches against P. reichenowi, a parasite from chimpanzees (5), while studies of P. vivax have been carried out using P. cynomolgi, a macaque parasite from Southeast Asia (29, 148), and a newly discovered P. vivax-like parasite in chimpanzees (6, 7). Although such studies seem distant from the questions formulated by an epidemiologist, they improve our understanding of the host-parasite relationship. In particular, these studies are revealing that the success of Plasmodium parasites in humans seems to be mediated by the evolution of gene families that are related to pathogenesis (5–7, 10). These studies have also identified genome signatures consistent with positive selection on genes that could be targeted by novel interventions. An interesting result is that positive selection appears to be more effective on single-copy genes located near telomeres (148). It has long been known that gene families, many located close to telomeres, are highly polymorphic and likely under positive selection (5). However, this process was harder to appreciate in single-copy genes. This result (148) could be explained, at least in part, by the fact that recombination is more common in these chromosome regions, making natural selection more efficient. However, such results are still preliminary.
CONCLUSION
The use of parasite genetic information empowers malaria epidemiologic investigations by unveiling factors affecting transmission and clinical outcomes in new ways. Nowadays, it is possible to study the disease dynamics with a combination of RNA- and DNA-based assays that can assess the contribution of asymptomatic or subclinical infections to malaria transmission. However, the translation of population genetic concepts into actionable and epidemiologically relevant information for malaria control is still a work in progress. Likely future work will require the integration of parasite genotyping into prospective investigations that can train/test integrated population genetics and epidemiologic models. Such models can then be used by policy makers to better deploy and evaluate interventions, particularly in the context of elimination. Nevertheless, genome approaches will likely allow the identification of novel targets for interventions or biomarkers that can be used to understand malaria transmission better in the context of control and elimination efforts.
ACKNOWLEDGMENTS
This study was supported in part by grants from the U.S. National Institutes of Health, 2U19 AI089681-08 and R01AI130473.
We declare that no conflicts of interest exist.
Footnotes
This article is part of a curated collection.
Contributor Information
Ananias A. Escalante, Department of Biology/Institute for Genomics and Evolutionary Medicine, Temple University, Philadelphia, PA 19122
M. Andreína Pacheco, Department of Biology/Institute for Genomics and Evolutionary Medicine, Temple University, Philadelphia, PA 19122.
Lee W. Riley, Divisions of Infectious Diseases and Vaccinology, School of Public Health, University of California, Berkeley, Berkeley, CA
Ronald E. Blanton, Center for Global Health & Diseases, Case Western Reserve University, Cleveland, OH
CURATED COLLECTION
REFERENCES
- 1.World Health Organization. 2018. World Malaria Report 2018. World Health Organization, Geneva, Switzerland. https://www.who.int/malaria/publications/world-malaria-report-2018/en/. [Google Scholar]
- 2.Coatney RG, Collins WE, Warren M, Contacos PG. 1971. The Primate Malaria. US Government Printing Office, Washington, DC. [Google Scholar]
- 3.Escalante AA, Cornejo OE, Freeland DE, Poe AC, Durrego E, Collins WE, Lal AA. 2005. A monkey’s tale: the origin of Plasmodium vivax as a human malaria parasite. Proc Natl Acad Sci U S A 102:1980–1985. 10.1073/pnas.0409652102. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muehlenbein MP, Pacheco MA, Taylor JE, Prall SP, Ambu L, Nathan S, Alsisto S, Ramirez D, Escalante AA. 2015. Accelerated diversification of nonhuman primate malarias in Southeast Asia: adaptive radiation or geographic speciation? Mol Biol Evol 32:422–439. 10.1093/molbev/msu310. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Otto TD, Rayner JC, Böhme U, Pain A, Spottiswoode N, Sanders M, Quail M, Ollomo B, Renaud F, Thomas AW, Prugnolle F, Conway DJ, Newbold C, Berriman M. 2014. Genome sequencing of chimpanzee malaria parasites reveals possible pathways of adaptation to human hosts. Nat Commun 5:4754. 10.1038/ncomms5754. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gilabert A, Otto TD, Rutledge GG, Franzon B, Ollomo B, Arnathau C, Durand P, Moukodoum ND, Okouga AP, Ngoubangoye B, Makanga B, Boundenga L, Paupy C, Renaud F, Prugnolle F, Rougeron V. 2018. Plasmodium vivax-like genome sequences shed new insights into Plasmodium vivax biology and evolution. PLoS Biol 16:e2006035. 10.1371/journal.pbio.2006035. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loy DE, Plenderleith LJ, Sundararaman SA, Liu W, Gruszczyk J, Chen YJ, Trimboli S, Learn GH, MacLean OA, Morgan ALK, Li Y, Avitto AN, Giles J, Calvignac-Spencer S, Sachse A, Leendertz FH, Speede S, Ayouba A, Peeters M, Rayner JC, Tham WH, Sharp PM, Hahn BH. 2018. Evolutionary history of human Plasmodium vivax revealed by genome-wide analyses of related ape parasites. Proc Natl Acad Sci U S A 115:E8450–E8459. 10.1073/pnas.1810053115. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bousema T, Drakeley C. 2011. Epidemiology and infectivity of Plasmodium falciparum and Plasmodium vivax gametocytes in relation to malaria control and elimination. Clin Microbiol Rev 24:377–410. 10.1128/CMR.00051-10. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schneider KA, Escalante AA. 2013. Fitness components and natural selection: why are there different patterns on the emergence of drug resistance in Plasmodium falciparum and Plasmodium vivax? Malar J 12:15. 10.1186/1475-2875-12-15. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wahlgren M, Goel S, Akhouri RR. 2017. Variant surface antigens of Plasmodium falciparum and their roles in severe malaria. Nat Rev Microbiol 15:479–491. 10.1038/nrmicro.2017.47. [PubMed] [DOI] [PubMed] [Google Scholar]
- 11.Wassmer SC, Taylor TE, Rathod PK, Mishra SK, Mohanty S, Arevalo-Herrera M, Duraisingh MT, Smith JD. 2015. Investigating the pathogenesis of severe malaria: a multidisciplinary and cross-geographical approach. Am J Trop Med Hyg 93(Suppl):42–56. 10.4269/ajtmh.14-0841. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mayor A, Bardají A, Macete E, Nhampossa T, Fonseca AM, González R, Maculuve S, Cisteró P, Rupérez M, Campo J, Vala A, Sigaúque B, Jiménez A, Machevo S, de la Fuente L, Nhama A, Luis L, Aponte JJ, Acácio S, Nhacolo A, Chitnis C, Dobaño C, Sevene E, Alonso PL, Menéndez C. 2015. Changing trends in P. falciparum burden, immunity, and disease in pregnancy. N Engl J Med 373:1607–1617. 10.1056/NEJMoa1406459. [PubMed] [DOI] [PubMed] [Google Scholar]
- 13.Arevalo-Herrera M, Quiñones ML, Guerra C, Céspedes N, Giron S, Ahumada M, Piñeros JG, Padilla N, Terrientes Z, Rosas A, Padilla JC, Escalante AA, Beier JC, Herrera S. 2012. Malaria in selected non-Amazonian countries of Latin America. Acta Trop 121:303–314. 10.1016/j.actatropica.2011.06.008. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hay SI, Snow RW. 2006. The malaria Atlas Project: developing global maps of malaria risk. PLoS Med 3:e473. 10.1371/journal.pmed.0030473. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chowell G, Munayco CV, Escalante AA, McKenzie FE. 2009. The spatial and temporal patterns of falciparum and vivax malaria in Perú: 1994–2006. Malar J 8:142. 10.1186/1475-2875-8-142. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Singh B, Kim Sung L, Matusop A, Radhakrishnan A, Shamsul SS, Cox-Singh J, Thomas A, Conway DJ. 2004. A large focus of naturally acquired Plasmodium knowlesi infections in human beings. Lancet 363:1017–1024. 10.1016/S0140-6736(04)15836-4. [DOI] [PubMed] [Google Scholar]
- 17.Yusof R, Ahmed MA, Jelip J, Ngian HU, Mustakim S, Hussin HM, Fong MY, Mahmud R, Sitam FA, Japning JR, Snounou G, Escalante AA, Lau YL. 2016. Phylogeographic evidence for 2 genetically distinct zoonotic Plasmodium knowlesi parasites, Malaysia. Emerg Infect Dis 22:1371–1380. 10.3201/eid2208.151885. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.William T, Jelip J, Menon J, Anderios F, Mohammad R, Awang Mohammad TA, Grigg MJ, Yeo TW, Anstey NM, Barber BE. 2014. Changing epidemiology of malaria in Sabah, Malaysia: increasing incidence of Plasmodium knowlesi. Malar J 13:390. 10.1186/1475-2875-13-390. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ta TH, Hisam S, Lanza M, Jiram AI, Ismail N, Rubio JM. 2014. First case of a naturally acquired human infection with Plasmodium cynomolgi. Malar J 13:68. 10.1186/1475-2875-13-68. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imwong M, Madmanee W, Suwannasin K, Kunasol C, Peto TJ, Tripura R, von Seidlein L, Nguon C, Davoeung C, Day NPJ, Dondorp AM, White NJ. 2019. Asymptomatic natural human infections with the simian malaria parasites Plasmodium cynomolgi and Plasmodium knowlesi. J Infect Dis 219:695–702. 10.1093/infdis/jiy519. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lalremruata A, Magris M, Vivas-Martínez S, Koehler M, Esen M, Kempaiah P, Jeyaraj S, Perkins DJ, Mordmüller B, Metzger WG. 2015. Natural infection of Plasmodium brasilianum in humans: man and monkey share quartan malaria parasites in the Venezuelan Amazon. EBioMedicine 2:1186–1192. 10.1016/j.ebiom.2015.07.033. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Brasil P, Zalis MG, de Pina-Costa A, Siqueira AM, Júnior CB, Silva S, Areas ALL, Pelajo-Machado M, de Alvarenga DAM, da Silva Santelli ACF, Albuquerque HG, Cravo P, Santos de Abreu FV, Peterka CL, Zanini GM, Suárez Mutis MC, Pissinatti A, Lourenço-de-Oliveira R, de Brito CFA, de Fátima Ferreira-da-Cruz M, Culleton R, Daniel-Ribeiro CT. 2017. Outbreak of human malaria caused by Plasmodium simium in the Atlantic Forest in Rio de Janeiro: a molecular epidemiological investigation. Lancet Glob Health 5:e1038–e1046. 10.1016/S2214-109X(17)30333-9. [DOI] [PubMed] [Google Scholar]
- 23.Escalante AA, Barrio E, Ayala FJ. 1995. Evolutionary origin of human and primate malarias: evidence from the circumsporozoite protein gene. Mol Biol Evol 12:616–626. [DOI] [PubMed] [Google Scholar]
- 24.Pacheco MA, Cranfield M, Cameron K, Escalante AA. 2013. Malarial parasite diversity in chimpanzees: the value of comparative approaches to ascertain the evolution of Plasmodium falciparum antigens. Malar J 12:328. 10.1186/1475-2875-12-328. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Makanga B, Yangari P, Rahola N, Rougeron V, Elguero E, Boundenga L, Moukodoum ND, Okouga AP, Arnathau C, Durand P, Willaume E, Ayala D, Fontenille D, Ayala FJ, Renaud F, Ollomo B, Prugnolle F, Paupy C. 2016. Ape malaria transmission and potential for ape-to-human transfers in Africa. Proc Natl Acad Sci U S A 113:5329–5334. 10.1073/pnas.1603008113. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu L, van den Hoogen LL, Slater H, Walker PG, Ghani AC, Drakeley CJ, Okell LC. 2015. Comparison of diagnostics for the detection of asymptomatic Plasmodium falciparum infections to inform control and elimination strategies. Nature 528:S86–S93. 10.1038/nature16039. [PubMed] [DOI] [PubMed] [Google Scholar]
- 27.Conway DJ. 2007. Molecular epidemiology of malaria. Clin Microbiol Rev 20:188–204. 10.1128/CMR.00021-06. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Escalante AA, Ferreira MU, Vinetz JM, Volkman SK, Cui L, Gamboa D, Krogstad DJ, Barry AE, Carlton JM, van Eijk AM, Pradhan K, Mueller I, Greenhouse B, Pacheco MA, Vallejo AF, Herrera S, Felger I. 2015. Malaria molecular epidemiology: lessons from the International Centers of Excellence for Malaria Research Network. Am J Trop Med Hyg 93(Suppl):79–86. 10.4269/ajtmh.15-0005. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Carlton JM, Das A, Escalante AA. 2013. Genomics, population genetics and evolutionary history of Plasmodium vivax. Adv Parasitol 81:203–222. 10.1016/B978-0-12-407826-0.00005-9. [PubMed] [DOI] [PubMed] [Google Scholar]
- 30.Cotter C, Sturrock HJW, Hsiang MS, Liu J, Phillips AA, Hwang J, Gueye CS, Fullman N, Gosling RD, Feachem RGJ. 2013. The changing epidemiology of malaria elimination: new strategies for new challenges. Lancet 382:900–911. 10.1016/S0140-6736(13)60310-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.World Health Organization. 2017. A Framework for Malaria Elimination. World Health Organization, Geneva, Switzerland. https://www.who.int/malaria/publications/atoz/9789241511988/en/. [Google Scholar]
- 32.Zimmerman PA, Howes RE. 2015. Malaria diagnosis for malaria elimination. Curr Opin Infect Dis 28:446–454. 10.1097/QCO.0000000000000191. [PubMed] [DOI] [PubMed] [Google Scholar]
- 33.Mukkala AN, Kwan J, Lau R, Harris D, Kain D, Boggild AK. 2018. An update on malaria rapid diagnostic tests. Curr Infect Dis Rep 20:49. 10.1007/s11908-018-0655-4. [PubMed] [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi T, Gamboa D, Ndiaye D, Cui L, Sutton PL, Vinetz JM. 2015. Malaria diagnosis across the international centers of excellence for malaria research: platforms, performance, and standardization. Am J Trop Med Hyg 93(Suppl):99–109. 10.4269/ajtmh.15-0004. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galatas B, Bassat Q, Mayor A. 2016. Malaria parasites in the asymptomatic: looking for the hay in the haystack. Trends Parasitol 32:296–308. 10.1016/j.pt.2015.11.015. [PubMed] [DOI] [PubMed] [Google Scholar]
- 36.Chen I, Clarke SE, Gosling R, Hamainza B, Killeen G, Magill A, O’Meara W, Price RN, Riley EM. 2016. “Asymptomatic” malaria: a chronic and debilitating infection that should be treated. PLoS Med 13:e1001942. 10.1371/journal.pmed.1001942. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sepúlveda N, Phelan J, Diez-Benavente E, Campino S, Clark TG, Hopkins H, Sutherland C, Drakeley CJ, Beshir KB. 2018. Global analysis of Plasmodium falciparum histidine-rich protein-2 (pfhrp2) and pfhrp3 gene deletions using whole-genome sequencing data and meta-analysis. Infect Genet Evol 62:211–219. 10.1016/j.meegid.2018.04.039. [PubMed] [DOI] [PubMed] [Google Scholar]
- 38.Gamboa D, Ho MF, Bendezu J, Torres K, Chiodini PL, Barnwell JW, Incardona S, Perkins M, Bell D, McCarthy J, Cheng Q. 2010. A large proportion of P. falciparum isolates in the Amazon region of Peru lack pfhrp2 and pfhrp3: implications for malaria rapid diagnostic tests. PLoS One 5:e8091. 10.1371/journal.pone.0008091. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Akinyi S, Hayden T, Gamboa D, Torres K, Bendezu J, Abdallah JF, Griffing SM, Quezada WM, Arrospide N, De Oliveira AM, Lucas C, Magill AJ, Bacon DJ, Barnwell JW, Udhayakumar V. 2013. Multiple genetic origins of histidine-rich protein 2 gene deletion in Plasmodium falciparum parasites from Peru. Sci Rep 3:2797. 10.1038/srep02797. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.World Health Organization. 2016. P. falciparum hrp2/3 gene deletions. Malaria Policy Advisory Committee Meeting. WHO/HTM/GMP/MPAC/2016.12. http://www.who.int/malaria/mpac/mpac-sept2016-hrp2-consultation-short-report-session7.pdf?ua=1. [Google Scholar]
- 41.Bharti PK, Chandel HS, Ahmad A, Krishna S, Udhayakumar V, Singh N. 2016. Prevalence of pfhrp2 and/or pfhrp3 gene deletion in Plasmodium falciparum population in eight highly endemic states in India. PLoS One 11:e0157949. 10.1371/journal.pone.0157949. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patel JC, Oberstaller J, Xayavong M, Narayanan J, DeBarry JD, Srinivasamoorthy G, Villegas L, Escalante AA, DaSilva A, Peterson DS, Barnwell JW, Kissinger JC, Udhayakumar V, Lucchi NW. 2013. Real-time loop-mediated isothermal amplification (RealAmp) for the species-specific identification of Plasmodium vivax. PLoS One 8:e54986. 10.1371/journal.pone.0054986. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hofmann N, Mwingira F, Shekalaghe S, Robinson LJ, Mueller I, Felger I. 2015. Ultra-sensitive detection of Plasmodium falciparum by amplification of multi-copy subtelomeric targets. PLoS Med 12:e1001788. 10.1371/journal.pmed.1001788. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vallejo AF, Martínez NL, González IJ, Arévalo-Herrera M, Herrera S. 2015. Evaluation of the loop mediated isothermal DNA amplification (LAMP) kit for malaria diagnosis in P. vivax endemic settings of Colombia. PLoS Negl Trop Dis 9:e3453. 10.1371/journal.pntd.0003453. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chaumeau V, Kajeechiwa L, Fustec B, Landier J, Naw Nyo S, Nay Hsel S, Phatharakokordbun P, Kittiphanakun P, Nosten S, Thwin MM, Win Tun S, Wiladphaingern J, Cottrell G, Parker DM, Minh MC, Kwansomboon N, Metaane S, Montazeau C, Kunjanwong K, Sawasdichai S, Andolina C, Ling C, Haohankhunnatham W, Christiensen P, Wanyatip S, Konghahong K, Cerqueira D, Imwong M, Dondorp AM, Chareonviriyaphap T, White NJ, Nosten FH, Corbel V. 2019. Contribution of asymptomatic Plasmodium infections to the transmission of malaria in Kayin State, Myanmar. J Infect Dis 219:1499–1509. 10.1093/infdis/jiy686. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sturrock HJ, Hsiang MS, Cohen JM, Smith DL, Greenhouse B, Bousema T, Gosling RD. 2013. Targeting asymptomatic malaria infections: active surveillance in control and elimination. PLoS Med 10:e1001467. 10.1371/journal.pmed.1001467. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slater HC, Ross A, Felger I, Hofmann NE, Robinson L, Cook J, Gonçalves BP, Björkman A, Ouedraogo AL, Morris U, Msellem M, Koepfli C, Mueller I, Tadesse F, Gadisa E, Das S, Domingo G, Kapulu M, Midega J, Owusu-Agyei S, Nabet C, Piarroux R, Doumbo O, Doumbo SN, Koram K, Lucchi N, Udhayakumar V, Mosha J, Tiono A, Chandramohan D, Gosling R, Mwingira F, Sauerwein R, Riley EM, White NJ, Nosten F, Imwong M, Bousema T, Drakeley C, Okell LC. 2019. The temporal dynamics and infectiousness of subpatent Plasmodium falciparum infections in relation to parasite density. Nat Commun 10:1433. 10.1038/s41467-019-09441-1. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sutherland CJ. 2016. Persistent parasitism: the adaptive biology of malariae and ovale malaria. Trends Parasitol 32:808–819. 10.1016/j.pt.2016.07.001. [PubMed] [DOI] [PubMed] [Google Scholar]
- 49.Wampfler R, Mwingira F, Javati S, Robinson L, Betuela I, Siba P, Beck HP, Mueller I, Felger I. 2013. Strategies for detection of Plasmodium species gametocytes. PLoS One 8:e76316. 10.1371/journal.pone.0076316. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koepfli C, Yan G. 2018. Plasmodium gametocytes in field studies: do we measure commitment to transmission or detectability? Trends Parasitol 34:378–387. 10.1016/j.pt.2018.02.009. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Imwong M, Stepniewska K, Tripura R, Peto TJ, Lwin KM, Vihokhern B, Wongsaen K, von Seidlein L, Dhorda M, Snounou G, Keereecharoen L, Singhasivanon P, Sirithiranont P, Chalk J, Nguon C, Day NP, Nosten F, Dondorp A, White NJ. 2016. Numerical distributions of parasite densities during asymptomatic malaria. J Infect Dis 213:1322–1329. 10.1093/infdis/jiv596. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Molineaux L, Gramiccia G. 1980. The Garki Project: Research on the Epidemiology and Control of Malaria in the Sudan Savanna of West Africa. World Health Organization, Geneva, Switzerland. [Google Scholar]
- 53.Dietz K, Molineaux L, Thomas A. 1974. A malaria model tested in the African savannah. Bull World Health Organ 50:347–357. [PMC free article] [PubMed] [Google Scholar]
- 54.Busula AO, Bousema T, Mweresa CK, Masiga D, Logan JG, Sauerwein RW, Verhulst NO, Takken W, de Boer JG. 2017. Gametocytemia and attractiveness of Plasmodium falciparum-infected Kenyan children to Anopheles gambiae mosquitoes. J Infect Dis 216:291–295. 10.1093/infdis/jix214. [PubMed] [DOI] [PubMed] [Google Scholar]
- 55.Bharti AR, Chuquiyauri R, Brouwer KC, Stancil J, Lin J, Llanos-Cuentas A, Vinetz JM. 2006. Experimental infection of the neotropical malaria vector Anopheles darlingi by human patient-derived Plasmodium vivax in the Peruvian Amazon. Am J Trop Med Hyg 75:610–616. 10.4269/ajtmh.2006.75.610. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lima NF, Bastos MS, Ferreira MU. 2012. Plasmodium vivax: reverse transcriptase real-time PCR for gametocyte detection and quantitation in clinical samples. Exp Parasitol 132:348–354. 10.1016/j.exppara.2012.08.010. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schneider P, Bousema T, Omar S, Gouagna L, Sawa P, Schallig H, Sauerwein R. 2006. (Sub)microscopic Plasmodium falciparum gametocytaemia in Kenyan children after treatment with sulphadoxine-pyrimethamine monotherapy or in combination with artesunate. Int J Parasitol 36:403–408. 10.1016/j.ijpara.2006.01.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 58.Vallejo AF, García J, Amado-Garavito AB, Arévalo-Herrera M, Herrera S. 2016. Plasmodium vivax gametocyte infectivity in sub-microscopic infections. Malar J 15:48. 10.1186/s12936-016-1104-1. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wampfler R, Timinao L, Beck HP, Soulama I, Tiono AB, Siba P, Mueller I, Felger I. 2014. Novel genotyping tools for investigating transmission dynamics of Plasmodium falciparum. J Infect Dis 210:1188–1197. 10.1093/infdis/jiu236. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bousema T, Okell L, Felger I, Drakeley C. 2014. Asymptomatic malaria infections: detectability, transmissibility and public health relevance. Nat Rev Microbiol 12:833–840. 10.1038/nrmicro3364. [PubMed] [DOI] [PubMed] [Google Scholar]
- 61.Schneider P, Greischar MA, Birget PLG, Repton C, Mideo N, Reece SE. 2018. Adaptive plasticity in the gametocyte conversion rate of malaria parasites. PLoS Pathog 14:e1007371. 10.1371/journal.ppat.1007371. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tadesse FG, Meerstein-Kessel L, Gonçalves BP, Drakeley C, Ranford-Cartwright L, Bousema T. 2019. Gametocyte sex ratio: the key to understanding Plasmodium falciparum transmission? Trends Parasitol 35:226–238. 10.1016/j.pt.2018.12.001. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tibayrenc M, Ayala FJ. 1991. Towards a population genetics of microorganisms: the clonal theory of parasitic protozoa. Parasitol Today 7:228–232. 10.1016/0169-4758(91)90234-F. [DOI] [PubMed] [Google Scholar]
- 64.Charlesworth B. 2009. Fundamental concepts in genetics: effective population size and patterns of molecular evolution and variation. Nat Rev Genet 10:195–205. 10.1038/nrg2526. [PubMed] [DOI] [PubMed] [Google Scholar]
- 65.Chenet SM, Schneider KA, Villegas L, Escalante AA. 2012. Local population structure of Plasmodium: impact on malaria control and elimination. Malar J 11:412. 10.1186/1475-2875-11-412. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Anderson TJC, Haubold B, Williams JT, Estrada-Franco JG, Richardson L, Mollinedo R, Bockarie M, Mokili J, Mharakurwa S, French N, Whitworth J, Velez ID, Brockman AH, Nosten F, Ferreira MU, Day KP. 2000. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol Biol Evol 17:1467–1482. 10.1093/oxfordjournals.molbev.a026247. [PubMed] [DOI] [PubMed] [Google Scholar]
- 67.Anderson TJ, Su XZ, Roddam A, Day KP. 2000. Complex mutations in a high proportion of microsatellite loci from the protozoan parasite Plasmodium falciparum. Mol Ecol 9:1599–1608. 10.1046/j.1365-294x.2000.01057.x. [PubMed] [DOI] [PubMed] [Google Scholar]
- 68.Chenet SM, Taylor JE, Blair S, Zuluaga L, Escalante AA. 2015. Longitudinal analysis of Plasmodium falciparum genetic variation in Turbo, Colombia: implications for malaria control and elimination. Malar J 14:363. 10.1186/s12936-015-0887-9. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Roper C, Pearce R, Nair S, Sharp B, Nosten F, Anderson T. 2004. Intercontinental spread of pyrimethamine-resistant malaria. Science 305:1124. 10.1126/science.1098876. [PubMed] [DOI] [PubMed] [Google Scholar]
- 70.Tun KM, Imwong M, Lwin KM, Win AA, Hlaing TM, Hlaing T, Lin K, Kyaw MP, Plewes K, Faiz MA, Dhorda M, Cheah PY, Pukrittayakamee S, Ashley EA, Anderson TJ, Nair S, McDew-White M, Flegg JA, Grist EP, Guerin P, Maude RJ, Smithuis F, Dondorp AM, Day NP, Nosten F, White NJ, Woodrow CJ. 2015. Spread of artemisinin-resistant Plasmodium falciparum in Myanmar: a cross-sectional survey of the K13 molecular marker. Lancet Infect Dis 15:415–421. 10.1016/S1473-3099(15)70032-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Takala SL, Escalante AA, Branch OH, Kariuki S, Biswas S, Chaiyaroj SC, Lal AA. 2006. Genetic diversity in the block 2 region of the merozoite surface protein 1 (MSP-1) of Plasmodium falciparum: additional complexity and selection and convergence in fragment size polymorphism. Infect Genet Evol 6:417–424. 10.1016/j.meegid.2006.01.009. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Haasl RJ, Payseur BA. 2011. Multi-locus inference of population structure: a comparison between single nucleotide polymorphisms and microsatellites. Heredity 106:158–171. 10.1038/hdy.2010.21. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kublin JG, Dzinjalamala FK, Kamwendo DD, Malkin EM, Cortese JF, Martino LM, Mukadam RA, Rogerson SJ, Lescano AG, Molyneux ME, Winstanley PA, Chimpeni P, Taylor TE, Plowe CV. 2002. Molecular markers for failure of sulfadoxine-pyrimethamine and chlorproguanil-dapsone treatment of Plasmodium falciparum malaria. J Infect Dis 185:380–388. 10.1086/338566. [PubMed] [DOI] [PubMed] [Google Scholar]
- 74.Djimdé A, Doumbo OK, Cortese JF, Kayentao K, Doumbo S, Diourté Y, Coulibaly D, Dicko A, Su XZ, Nomura T, Fidock DA, Wellems TE, Plowe CV. 2001. A molecular marker for chloroquine-resistant falciparum malaria. N Engl J Med 344:257–263. 10.1056/NEJM200101253440403. [PubMed] [DOI] [PubMed] [Google Scholar]
- 75.McCollum AM, Poe AC, Hamel M, Huber C, Zhou Z, Shi YP, Ouma P, Vulule J, Bloland P, Slutsker L, Barnwell JW, Udhayakumar V, Escalante AA. 2006. Antifolate resistance in Plasmodium falciparum: multiple origins and identification of novel dhfr alleles. J Infect Dis 194:189–197. 10.1086/504687. [PubMed] [DOI] [PubMed] [Google Scholar]
- 76.Snounou G, Beck HP. 1998. The use of PCR genotyping in the assessment of recrudescence or reinfection after antimalarial drug treatment. Parasitol Today 14:462–467. 10.1016/S0169-4758(98)01340-4. [DOI] [PubMed] [Google Scholar]
- 77.Rice BL, Acosta MM, Pacheco MA, Escalante AA. 2013. Merozoite surface protein-3 alpha as a genetic marker for epidemiologic studies in Plasmodium vivax: a cautionary note. Malar J 12:288. 10.1186/1475-2875-12-288. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mascorro CN, Zhao K, Khuntirat B, Sattabongkot J, Yan G, Escalante AA, Cui L. 2005. Molecular evolution and intragenic recombination of the merozoite surface protein MSP-3alpha from the malaria parasite Plasmodium vivax in Thailand. Parasitology 131:25–35. 10.1017/S0031182005007547. [PubMed] [DOI] [PubMed] [Google Scholar]
- 79.Imwong M, Pukrittayakamee S, Grüner AC, Rénia L, Letourneur F, Looareesuwan S, White NJ, Snounou G. 2005. Practical PCR genotyping protocols for Plasmodium vivax using Pvcs and Pvmsp1. Malar J 4:20. 10.1186/1475-2875-4-20. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mueller I, Schoepflin S, Smith TA, Benton KL, Bretscher MT, Lin E, Kiniboro B, Zimmerman PA, Speed TP, Siba P, Felger I. 2012. Force of infection is key to understanding the epidemiology of Plasmodium falciparum malaria in Papua New Guinean children. Proc Natl Acad Sci U S A 109:10030–10035. 10.1073/pnas.1200841109. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Koepfli C, Colborn KL, Kiniboro B, Lin E, Speed TP, Siba PM, Felger I, Mueller I. 2013. A high force of Plasmodium vivax blood-stage infection drives the rapid acquisition of immunity in Papua New Guinean children. PLoS Negl Trop Dis 7:e2403. 10.1371/journal.pntd.0002403. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Pacheco MA, Lopez-Perez M, Vallejo AF, Herrera S, Arévalo-Herrera M, Escalante AA. 2016. Multiplicity of infection and disease severity in Plasmodium vivax. PLoS Negl Trop Dis 10:e0004355. 10.1371/journal.pntd.0004355. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pacheco MA, Schneider KA, Céspedes N, Herrera S, Arévalo-Herrera M, Escalante AA. 2019. Limited differentiation among Plasmodium vivax populations from the northwest and to the south Pacific Coast of Colombia: a malaria corridor? PLoS Negl Trop Dis 13:e0007310. 10.1371/journal.pntd.0007310. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Koepfli C, Waltmann A, Ome-Kaius M, Robinson LJ, Mueller I. 2018. Multiplicity of infection is a poor predictor of village-Level Plasmodium vivax and P. falciparum population prevalence in the Southwest Pacific. Open Forum Infect Dis 5:ofy240. 10.1093/ofid/ofy240. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Karl S, White MT, Milne GJ, Gurarie D, Hay SI, Barry AE, Felger I, Mueller I. 2016. Spatial effects on the multiplicity of Plasmodium falciparum infections. PLoS One 11:e0164054. 10.1371/journal.pone.0164054. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Bei AK, Niang M, Deme AB, Daniels RF, Sarr FD, Sokhna C, Talla C, Faye J, Diagne N, Doucoure S, Mboup S, Wirth DF, Tall A, Ndiaye D, Hartl DL, Volkman SK, Toure-Balde A. 2018. Dramatic changes in malaria population genetic complexity in Dielmo and Ndiop, Senegal, revealed using genomic surveillance. J Infect Dis 217:622–627. 10.1093/infdis/jix580. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Mobegi VA, Loua KM, Ahouidi AD, Satoguina J, Nwakanma DC, Amambua-Ngwa A, Conway DJ. 2012. Population genetic structure of Plasmodium falciparum across a region of diverse endemicity in West Africa. Malar J 11:223. 10.1186/1475-2875-11-223. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Larrañaga N, Mejía RE, Hormaza JI, Montoya A, Soto A, Fontecha GA. 2013. Genetic structure of Plasmodium falciparum populations across the Honduras-Nicaragua border. Malar J 12:354. 10.1186/1475-2875-12-354. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Anthony TG, Conway DJ, Cox-Singh J, Matusop A, Ratnam S, Shamsul S, Singh B. 2005. Fragmented population structure of plasmodium falciparum in a region of declining endemicity. J Infect Dis 191:1558–1564. 10.1086/429338. [PubMed] [DOI] [PubMed] [Google Scholar]
- 90.Friedrich LR, Popovici J, Kim S, Dysoley L, Zimmerman PA, Menard D, Serre D. 2016. Complexity of infection and genetic diversity in Cambodian Plasmodium vivax. PLoS Negl Trop Dis 10:e0004526. 10.1371/journal.pntd.0004526. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhong D, Koepfli C, Cui L, Yan G. 2018. Molecular approaches to determine the multiplicity of Plasmodium infections. Malar J 17:172. 10.1186/s12936-018-2322-5. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Arnott A, Wapling J, Mueller I, Ramsland PA, Siba PM, Reeder JC, Barry AE. 2014. Distinct patterns of diversity, population structure and evolution in the AMA1 genes of sympatric Plasmodium falciparum and Plasmodium vivax populations of Papua New Guinea from an area of similarly high transmission. Malar J 13:233. 10.1186/1475-2875-13-233. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chenet SM, Branch OH, Escalante AA, Lucas CM, Bacon DJ. 2008. Genetic diversity of vaccine candidate antigens in Plasmodium falciparum isolates from the Amazon basin of Peru. Malar J 7:93. 10.1186/1475-2875-7-93. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Escalante AA, Cornejo OE, Rojas A, Udhayakumar V, Lal AA. 2004. Assessing the effect of natural selection in malaria parasites. Trends Parasitol 20:388–395. 10.1016/j.pt.2004.06.002. [PubMed] [DOI] [PubMed] [Google Scholar]
- 95.Takala SL, Coulibaly D, Thera MA, Batchelor AH, Cummings MP, Escalante AA, Ouattara A, Traoré K, Niangaly A, Djimdé AA, Doumbo OK, Plowe CV. 2009. Extreme polymorphism in a vaccine antigen and risk of clinical malaria: implications for vaccine development. Sci Transl Med 1:2ra5. 10.1126/scitranslmed.3000257. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hartl DL. 2004. The origin of malaria: mixed messages from genetic diversity. Nat Rev Microbiol 2:15–22. 10.1038/nrmicro795. [PubMed] [DOI] [PubMed] [Google Scholar]
- 97.Joy DA, Feng X, Mu J, Furuya T, Chotivanich K, Krettli AU, Ho M, Wang A, White NJ, Suh E, Beerli P, Su XZ. 2003. Early origin and recent expansion of Plasmodium falciparum. Science 300:318–321. 10.1126/science.1081449. [PubMed] [DOI] [PubMed] [Google Scholar]
- 98.Taylor JE, Pacheco MA, Bacon DJ, Beg MA, Machado RL, Fairhurst RM, Herrera S, Kim JY, Menard D, Póvoa MM, Villegas L, Mulyanto, Snounou G, Cui L, Zeyrek FY, Escalante AA. 2013. The evolutionary history of Plasmodium vivax as inferred from mitochondrial genomes: parasite genetic diversity in the Americas. Mol Biol Evol 30:2050–2064. 10.1093/molbev/mst104. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Cornejo OE, Escalante AA. 2006. The origin and age of Plasmodium vivax. Trends Parasitol 22:558–563. 10.1016/j.pt.2006.09.007. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tanabe K, Mita T, Jombart T, Eriksson A, Horibe S, Palacpac N, Ranford-Cartwright L, Sawai H, Sakihama N, Ohmae H, Nakamura M, Ferreira MU, Escalante AA, Prugnolle F, Björkman A, Färnert A, Kaneko A, Horii T, Manica A, Kishino H, Balloux F. 2010. Plasmodium falciparum accompanied the human expansion out of Africa. Curr Biol 20:1283–1289. 10.1016/j.cub.2010.05.053. [PubMed] [DOI] [PubMed] [Google Scholar]
- 101.Rodrigues PT, Valdivia HO, de Oliveira TC, Alves JMP, Duarte AMRC, Cerutti-Junior C, Buery JC, Brito CFA, de Souza JC Jr, Hirano ZMB, Bueno MG, Catão-Dias JL, Malafronte RS, Ladeia-Andrade S, Mita T, Santamaria AM, Calzada JE, Tantular IS, Kawamoto F, Raijmakers LRJ, Mueller I, Pacheco MA, Escalante AA, Felger I, Ferreira MU. 2018. Human migration and the spread of malaria parasites to the New World. Sci Rep 8:1993. 10.1038/s41598-018-19554-0. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tessema SK, Monk SL, Schultz MB, Tavul L, Reeder JC, Siba PM, Mueller I, Barry AE. 2015. Phylogeography of var gene repertoires reveals fine-scale geospatial clustering of Plasmodium falciparum populations in a highly endemic area. Mol Ecol 24:484–497. 10.1111/mec.13033. [PubMed] [DOI] [PubMed] [Google Scholar]
- 103.Cui L, Escalante AA, Imwong M, Snounou G. 2003. The genetic diversity of Plasmodium vivax populations. Trends Parasitol 19:220–226. 10.1016/S1471-4922(03)00085-0. [DOI] [PubMed] [Google Scholar]
- 104.Pacheco MA, Poe AC, Collins WE, Lal AA, Tanabe K, Kariuki SK, Udhayakumar V, Escalante AA. 2007. A comparative study of the genetic diversity of the 42kDa fragment of the merozoite surface protein 1 in Plasmodium falciparum and P. vivax. Infect Genet Evol 7:180–187. 10.1016/j.meegid.2006.08.002. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Pacheco MA, Elango AP, Rahman AA, Fisher D, Collins WE, Barnwell JW, Escalante AA. 2012. Evidence of purifying selection on merozoite surface protein 8 (MSP8) and 10 (MSP10) in Plasmodium spp. Infect Genet Evol 12:978–986. 10.1016/j.meegid.2012.02.009. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lin JT, Hathaway NJ, Saunders DL, Lon C, Balasubramanian S, Kharabora O, Gosi P, Sriwichai S, Kartchner L, Chuor CM, Satharath P, Lanteri C, Bailey JA, Juliano JJ. 2015. Using amplicon deep sequencing to detect genetic signatures of Plasmodium vivax relapse. J Infect Dis 212:999–1008. 10.1093/infdis/jiv142. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Neafsey DE, et al. 2015. Genetic diversity and protective efficacy of the RTS,S/AS01 malaria vaccine. N Engl J Med 373:2025–2037. 10.1056/NEJMoa1505819. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rao PN, Uplekar S, Kayal S, Mallick PK, Bandyopadhyay N, Kale S, Singh OP, Mohanty A, Mohanty S, Wassmer SC, Carlton JM. 2016. A method for amplicon deep sequencing of drug resistance genes in Plasmodium falciparum clinical isolates from India. J Clin Microbiol 54:1500–1511. 10.1128/JCM.00235-16. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Talundzic E, Ravishankar S, Kelley J, Patel D, Plucinski M, Schmedes S, Ljolje D, Clemons B, Madison-Antenucci S, Arguin PM, Lucchi NW, Vannberg F, Udhayakumar V. 2018. Next-generation sequencing and bioinformatics protocol for malaria drug resistance marker surveillance. Antimicrob Agents Chemother 62:e02474-17. 10.1128/AAC.02474-17. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pacheco MA, Kadakia ER, Chaudhary Z, Perkins DJ, Kelley J, Ravishankar S, Cranfield M, Talundzic E, Udhayakumar V, Escalante AA. 2019. Evolution and genetic diversity of the k13 gene associated with artemisinin delayed parasite clearance in Plasmodium falciparum. Antimicrob Agents Chemother 63:e02550-18. 10.1128/AAC.02550-18. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Orjuela-Sánchez P, Sá JM, Brandi MC, Rodrigues PT, Bastos MS, Amaratunga C, Duong S, Fairhurst RM, Ferreira MU. 2013. Higher microsatellite diversity in Plasmodium vivax than in sympatric Plasmodium falciparum populations in Pursat, Western Cambodia. Exp Parasitol 134:318–326. 10.1016/j.exppara.2013.03.029. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sutton PL. 2013. A call to arms: on refining Plasmodium vivax microsatellite marker panels for comparing global diversity. Malar J 12:447. 10.1186/1475-2875-12-447. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Daniels R, Volkman SK, Milner DA, Mahesh N, Neafsey DE, Park DJ, Rosen D, Angelino E, Sabeti PC, Wirth DF, Wiegand RC. 2008. A general SNP-based molecular barcode for Plasmodium falciparum identification and tracking. Malar J 7:223. 10.1186/1475-2875-7-223. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Baniecki ML, Faust AL, Schaffner SF, Park DJ, Galinsky K, Daniels RF, Hamilton E, Ferreira MU, Karunaweera ND, Serre D, Zimmerman PA, Sá JM, Wellems TE, Musset L, Legrand E, Melnikov A, Neafsey DE, Volkman SK, Wirth DF, Sabeti PC. 2015. Development of a single nucleotide polymorphism barcode to genotype Plasmodium vivax infections. PLoS Negl Trop Dis 9:e0003539. 10.1371/journal.pntd.0003539. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Alifrangis M, Nag S, Schousboe ML, Ishengoma D, Lusingu J, Pota H, Kavishe RA, Pearce R, Ord R, Lynch C, Dejene S, Cox J, Rwakimari J, Minja DT, Lemnge MM, Roper C. 2014. Independent origin of Plasmodium falciparum antifolate super-resistance, Uganda, Tanzania, and Ethiopia. Emerg Infect Dis 20:1280–1286. 10.3201/eid2008.131897. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cheeseman IH, Miller BA, Nair S, Nkhoma S, Tan A, Tan JC, Al Saai S, Phyo AP, Moo CL, Lwin KM, McGready R, Ashley E, Imwong M, Stepniewska K, Yi P, Dondorp AM, Mayxay M, Newton PN, White NJ, Nosten F, Ferdig MT, Anderson TJ. 2012. A major genome region underlying artemisinin resistance in malaria. Science 336:79–82. 10.1126/science.1215966. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.McCollum AM, Schneider KA, Griffing SM, Zhou Z, Kariuki S, Ter-Kuile F, Shi YP, Slutsker L, Lal AA, Udhayakumar V, Escalante AA. 2012. Differences in selective pressure on dhps and dhfr drug resistant mutations in western Kenya. Malar J 11:77. 10.1186/1475-2875-11-77. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Schultz L, Wapling J, Mueller I, Ntsuke PO, Senn N, Nale J, Kiniboro B, Buckee CO, Tavul L, Siba PM, Reeder JC, Barry AE. 2010. Multilocus haplotypes reveal variable levels of diversity and population structure of Plasmodium falciparum in Papua New Guinea, a region of intense perennial transmission. Malar J 9:336. 10.1186/1475-2875-9-336. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Waltmann A, Koepfli C, Tessier N, Karl S, Fola A, Darcy AW, Wini L, Harrison GLA, Barnadas C, Jennison C, Karunajeewa H, Boyd S, Whittaker M, Kazura J, Bahlo M, Mueller I, Barry AE. 2018. Increasingly inbred and fragmented populations of Plasmodium vivax associated with the eastward decline in malaria transmission across the Southwest Pacific. PLoS Negl Trop Dis 12:e0006146. 10.1371/journal.pntd.0006146. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gunawardena S, Ferreira MU, Kapilananda GM, Wirth DF, Karunaweera ND. 2014. The Sri Lankan paradox: high genetic diversity in Plasmodium vivax populations despite decreasing levels of malaria transmission. Parasitology 141:880–890. 10.1017/S0031182013002278. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Koepfli C, Timinao L, Antao T, Barry AE, Siba P, Mueller I, Felger I. 2013. A large Plasmodium vivax reservoir and little population structure in the South Pacific. PLoS One 8:e66041. 10.1371/journal.pone.0066041. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Smith JM, Haigh J. 1974. The hitch-hiking effect of a favourable gene. Genet Res 23:23–35. 10.1017/S0016672300014634. [DOI] [PubMed] [Google Scholar]
- 123.Kim Y, Stephan W. 2002. Detecting a local signature of genetic hitchhiking along a recombining chromosome. Genetics 160:765–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Wootton JC, Feng X, Ferdig MT, Cooper RA, Mu J, Baruch DI, Magill AJ, Su XZ. 2002. Genetic diversity and chloroquine selective sweeps in Plasmodium falciparum. Nature 418:320–323. 10.1038/nature00813. [PubMed] [DOI] [PubMed] [Google Scholar]
- 125.Nair S, Williams JT, Brockman A, Paiphun L, Mayxay M, Newton PN, Guthmann JP, Smithuis FM, Hien TT, White NJ, Nosten F, Anderson TJ. 2003. A selective sweep driven by pyrimethamine treatment in Southeast Asian malaria parasites. Mol Biol Evol 20:1526–1536. 10.1093/molbev/msg162. [PubMed] [DOI] [PubMed] [Google Scholar]
- 126.Lachance J, Tishkoff SA. 2013. SNP ascertainment bias in population genetic analyses: why it is important, and how to correct it. Bioessays 35:780–786. 10.1002/bies.201300014. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sisya TJ, Kamn’gona RM, Vareta JA, Fulakeza JM, Mukaka MF, Seydel KB, Laufer MK, Taylor TE, Nkhoma SC. 2015. Subtle changes in Plasmodium falciparum infection complexity following enhanced intervention in Malawi. Acta Trop 142:108–114. 10.1016/j.actatropica.2014.11.008. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Daniels R, Chang HH, Séne PD, Park DC, Neafsey DE, Schaffner SF, Hamilton EJ, Lukens AK, Van Tyne D, Mboup S, Sabeti PC, Ndiaye D, Wirth DF, Hartl DL, Volkman SK. 2013. Genetic surveillance detects both clonal and epidemic transmission of malaria following enhanced intervention in Senegal. PLoS One 8:e60780. 10.1371/journal.pone.0060780. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Popovici J, Pierce-Friedrich L, Kim S, Bin S, Run V, Lek D, Hee KHD, Lee Soon-U L, Cannon MV, Serre D, Menard D. 2019. Recrudescence, reinfection, or relapse? A more rigorous framework to assess chloroquine efficacy for Plasmodium vivax malaria. J Infect Dis 219:315–322. 10.1093/infdis/jiy484. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Obaldia N3rd, Baro NK, Calzada JE, Santamaria AM, Daniels R, Wong W, Chang HH, Hamilton EJ, Arevalo-Herrera M, Herrera S, Wirth DF, Hartl DL, Marti M, Volkman SK. 2015. Clonal outbreak of Plasmodium falciparum infection in eastern Panama. J Infect Dis 211:1087–1096. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Nkhoma SC, Nair S, Al-Saai S, Ashley E, McGready R, Phyo AP, Nosten F, Anderson TJ. 2013. Population genetic correlates of declining transmission in a human pathogen. Mol Ecol 22:273–285. 10.1111/mec.12099. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Auburn S, Barry AE. 2017. Dissecting malaria biology and epidemiology using population genetics and genomics. Int J Parasitol 47:77–85. 10.1016/j.ijpara.2016.08.006. [PubMed] [DOI] [PubMed] [Google Scholar]
- 133.Auburn S, Marfurt J, Maslen G, Campino S, Ruano Rubio V, Manske M, Machunter B, Kenangalem E, Noviyanti R, Trianty L, Sebayang B, Wirjanata G, Sriprawat K, Alcock D, Macinnis B, Miotto O, Clark TG, Russell B, Anstey NM, Nosten F, Kwiatkowski DP, Price RN. 2013. Effective preparation of Plasmodium vivax field isolates for high-throughput whole genome sequencing. PLoS One 8:e53160. 10.1371/journal.pone.0053160. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Oyola SO, Gu Y, Manske M, Otto TD, O’Brien J, Alcock D, Macinnis B, Berriman M, Newbold CI, Kwiatkowski DP, Swerdlow HP, Quail MA. 2013. Efficient depletion of host DNA contamination in malaria clinical sequencing. J Clin Microbiol 51:745–751. 10.1128/JCM.02507-12. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Dondorp AM, Nosten F, Yi P, Das D, Phyo AP, Tarning J, Lwin KM, Ariey F, Hanpithakpong W, Lee SJ, Ringwald P, Silamut K, Imwong M, Chotivanich K, Lim P, Herdman T, An SS, Yeung S, Singhasivanon P, Day NP, Lindegardh N, Socheat D, White NJ. 2009. Artemisinin resistance in Plasmodium falciparum malaria. N Engl J Med 361:455–467. 10.1056/NEJMoa0808859. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Takala-Harrison S, Clark TG, Jacob CG, Cummings MP, Miotto O, Dondorp AM, Fukuda MM, Nosten F, Noedl H, Imwong M, Bethell D, Se Y, Lon C, Tyner SD, Saunders DL, Socheat D, Ariey F, Phyo AP, Starzengruber P, Fuehrer HP, Swoboda P, Stepniewska K, Flegg J, Arze C, Cerqueira GC, Silva JC, Ricklefs SM, Porcella SF, Stephens RM, Adams M, Kenefic LJ, Campino S, Auburn S, MacInnis B, Kwiatkowski DP, Su XZ, White NJ, Ringwald P, Plowe CV. 2013. Genetic loci associated with delayed clearance of Plasmodium falciparum following artemisinin treatment in Southeast Asia. Proc Natl Acad Sci U S A 110:240–245. 10.1073/pnas.1211205110. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Miotto O, Amato R, Ashley EA, MacInnis B, Almagro-Garcia J, Amaratunga C, Lim P, Mead D, Oyola SO, Dhorda M, Imwong M, Woodrow C, Manske M, Stalker J, Drury E, Campino S, Amenga-Etego L, Thanh TN, Tran HT, Ringwald P, Bethell D, Nosten F, Phyo AP, Pukrittayakamee S, Chotivanich K, Chuor CM, Nguon C, Suon S, Sreng S, Newton PN, Mayxay M, Khanthavong M, Hongvanthong B, Htut Y, Han KT, Kyaw MP, Faiz MA, Fanello CI, Onyamboko M, Mokuolu OA, Jacob CG, Takala-Harrison S, Plowe CV, Day NP, Dondorp AM, Spencer CC, McVean G, Fairhurst RM, White NJ, Kwiatkowski DP. 2015. Genetic architecture of artemisinin-resistant Plasmodium falciparum. Nat Genet 47:226–234. 10.1038/ng.3189. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ariey F, Witkowski B, Amaratunga C, Beghain J, Langlois AC, Khim N, Kim S, Duru V, Bouchier C, Ma L, Lim P, Leang R, Duong S, Sreng S, Suon S, Chuor CM, Bout DM, Ménard S, Rogers WO, Genton B, Fandeur T, Miotto O, Ringwald P, Le Bras J, Berry A, Barale JC, Fairhurst RM, Benoit-Vical F, Mercereau-Puijalon O, Ménard D. 2014. A molecular marker of artemisinin-resistant Plasmodium falciparum malaria. Nature 505:50–55. 10.1038/nature12876. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Amato R, Lim P, Miotto O, Amaratunga C, Dek D, Pearson RD, Almagro-Garcia J, Neal AT, Sreng S, Suon S, Drury E, Jyothi D, Stalker J, Kwiatkowski DP, Fairhurst RM. 2017. Genetic markers associated with dihydroartemisinin-piperaquine failure in Plasmodium falciparum malaria in Cambodia: a genotype-phenotype association study. Lancet Infect Dis 17:164–173. 10.1016/S1473-3099(16)30409-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Neafsey DE, Galinsky K, Jiang RH, Young L, Sykes SM, Saif S, Gujja S, Goldberg JM, Young S, Zeng Q, Chapman SB, Dash AP, Anvikar AR, Sutton PL, Birren BW, Escalante AA, Barnwell JW, Carlton JM. 2012. The malaria parasite Plasmodium vivax exhibits greater genetic diversity than Plasmodium falciparum. Nat Genet 44:1046–1050. 10.1038/ng.2373. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Winter DJ, Pacheco MA, Vallejo AF, Schwartz RS, Arevalo-Herrera M, Herrera S, Cartwright RA, Escalante AA. 2015. Whole genome sequencing of field isolates reveals extensive genetic diversity in Plasmodium vivax from Colombia. PLoS Negl Trop Dis 9:e0004252. 10.1371/journal.pntd.0004252. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hupalo DN, Luo Z, Melnikov A, Sutton PL, Rogov P, Escalante A, Vallejo AF, Herrera S, Arévalo-Herrera M, Fan Q, Wang Y, Cui L, Lucas CM, Durand S, Sanchez JF, Baldeviano GC, Lescano AG, Laman M, Barnadas C, Barry A, Mueller I, Kazura JW, Eapen A, Kanagaraj D, Valecha N, Ferreira MU, Roobsoong W, Nguitragool W, Sattabonkot J, Gamboa D, Kosek M, Vinetz JM, González-Cerón L, Birren BW, Neafsey DE, Carlton JM. 2016. Population genomics studies identify signatures of global dispersal and drug resistance in Plasmodium vivax. Nat Genet 48:953–958. 10.1038/ng.3588. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Amambua-Ngwa A, Tetteh KK, Manske M, Gomez-Escobar N, Stewart LB, Deerhake ME, Cheeseman IH, Newbold CI, Holder AA, Knuepfer E, Janha O, Jallow M, Campino S, Macinnis B, Kwiatkowski DP, Conway DJ. 2012. Population genomic scan for candidate signatures of balancing selection to guide antigen characterization in malaria parasites. PLoS Genet 8:e1002992. 10.1371/journal.pgen.1002992. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Mobegi VA, Duffy CW, Amambua-Ngwa A, Loua KM, Laman E, Nwakanma DC, MacInnis B, Aspeling-Jones H, Murray L, Clark TG, Kwiatkowski DP, Conway DJ. 2014. Genome-wide analysis of selection on the malaria parasite Plasmodium falciparum in West African populations of differing infection endemicity. Mol Biol Evol 31:1490–1499. 10.1093/molbev/msu106. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Henden L, Lee S, Mueller I, Barry A, Bahlo M. 2018. Identity-by-descent analyses for measuring population dynamics and selection in recombining pathogens. PLoS Genet 14:e1007279. 10.1371/journal.pgen.1007279. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Schaffner SF, Taylor AR, Wong W, Wirth DF, Neafsey DE. 2018. hmmIBD: software to infer pairwise identity by descent between haploid genotypes. Malar J 17:196. 10.1186/s12936-018-2349-7. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Taylor AR, Schaffner SF, Cerqueira GC, Nkhoma SC, Anderson TJC, Sriprawat K, Pyae Phyo A, Nosten F, Neafsey DE, Buckee CO. 2017. Quantifying connectivity between local Plasmodium falciparum malaria parasite populations using identity by descent. PLoS Genet 13:e1007065. 10.1371/journal.pgen.1007065. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Cornejo OE, Fisher D, Escalante AA. 2014. Genome-wide patterns of genetic polymorphism and signatures of selection in Plasmodium vivax. Genome Biol Evol 7:106–119. 10.1093/gbe/evu267. [PubMed] [DOI] [PMC free article] [PubMed] [Google Scholar]