

Abstract
Poly(ADP-ribosyl)ation (aka PARylation) is a unique protein post-translational modification (PTM) first described over 50 years ago. PARylation regulates a number of biological processes including chromatin remodeling, the DNA damage response (DDR), transcription, apoptosis, and mitosis. The subsequent discovery of poly(ADP-ribose) polymerase-1 (PARP-1) catalyzing DNA-dependent PARylation spearheaded the field of DDR. The expanding knowledge about the poly ADP-ribose (PAR) recognition domains prompted the discovery of novel DDR factors and revealed crosstalk with other protein PTMs including phosphorylation, ubiquitination, methylation and acetylation. In this review, we highlight the current knowledge on PAR-regulated DDR, PAR recognition domain, and PARP inhibition in cancer therapy.
Introduction
Cell organisms are constantly subjected to genotoxic stress including endogenous reactive oxygen species derived from metabolism as well as exogenous ionizing radiation and ultraviolet sunlight (1, 2). It is estimated that cells can experience up to 105 DNA lesions per cell per day (2). Unrepaired DNA damage lead to aberrant chromosome rearrangement, resulting in mitotic failure and deleterious gene mutations (3). Throughout evolution, cells have developed robust DNA damage repair machineries to maintain genomic stability (4). The DNA damage response (DDR) is a concerted process involving DNA damage detection, cell cycle checkpoint regulation, DDR factor recruitment, chromatin reorganization, and DNA processing and repair (5). Poly(ADP-ribosyl)ation (PARylation) is a critical post-translational modification (PTM) that initiates and regulates DDR (6).
Poly(ADP-ribosyl)ation was first described over 50 years ago as a DNA-dependent reaction that consumes nicotinamide adenine dinucleotide (NAD+) to synthesize poly(ADP-ribose) (PAR) chains (7). ADP-ribosylation is catalyzed by poly(ADP-ribosyl)ation polymerases (PARPs) that covalently attach an ADP-ribose unit on glutamate, aspartate, arginine, lysine and serine residues on target protein using NAD+ as a substrate (8–15). The enzymes can further catalyze formation of linear PAR chains through 2′–1″-O-glycosidic bonds or branched PAR chains via α(1‴–2″)-ADP-ribose linkages (16, 17). In humans, there are 17 protein members in the PARP family proteins (18, 19). According to the structure and functional domains, PARP family proteins are categorized as DNA-dependent PARPs (PARP-1, PARP-2 and PARP-3), Tankyrases (PAR-5a and PARP-5b), CCCH (Cys-Cys-Cys-Cys-His) Zinc Finger PARPs (PARP-8, PARP-12, PARP-13), PARPs with macro domain (PARP-9, PARP-14 and PARP-15) and unclassified PARPs (PARP-4, PARP-6, PARP-8, PARP-10, PARP-11, PARP-16). Based on the ADP-ribosyltransferase activity, PARPs can be divided into three groups. PARP-1, PARP-2, PARP-5a and PARP-5b contain catalytic His-Tyr-Glu triad and catalyze PARylation (20). PARP-3, PARP-4, PARP-6, PARP-10, PARP-14, PARP-15, and PARP-16 are only capable of mono(ADP-ribosyl)ation (MARylation) (21). PARP-9 and PARP-13 are inactive proteins since they lack NAD+ binding residues (22).
In response to DNA damage, DNA-dependent PARPs, especially PARP-1, quickly recognize single strand break (SSB) and double strand break (DSB) DNA ends and start PAR synthesis. The substrates include PARPs themselves, nucleosomal and linker histones, and certain chromatin-associated proteins, which serves as a scaffold to mediate early recruitment of DDR factors and facilitates chromatin remodeling for DNA repair (23). PARylation is a reversible PTM and undergoes rapid turnover (24). The de-PARylation reaction is carried out by glycohyrdolases including PAR glycohydrolase (PARG), terminal ADP-ribose protein glycohydrolase 1 (TARG1), ADP-ribosylhydrolase 3 (ARH3), MacroD1 and MacroD2, and Nudix-Type Motif 9 and 16 (NUDT9, NUDT16) (25–29). In this review, we will summarize the role of PARylation metabolism in DDR, describe the PAR recognition domain, and review current effort of PARP inhibition in cancer therapy.
PARylation metabolism in DDR
PARP-1 and PARP-2 detect and bind DNA SSBs and DSBs using the N-terminal DNA binding domain (30). It has been shown that PARP-1 is able to recognize both SSBs and DSBs within one second of their formation in cells (31). DNA binding induces a conformation change to expose the enzymatic site of PARP1, resulting in its activation and PARylation (32). The PARylation reaction is robust and occurs immediately following DNA binding. The intranuclear PAR level can rise to 500 fold over baseline, consuming up to 90% of the cellular reservoir of NAD+ (24, 33). PARP-1 activation at the sites of DNA damage results in the synthesis of PAR chains on PARP-1 and other protein substrates, which then recruit PAR-binding DNA repair components for SSB and DSB repair (Figure 1). PAR chain at SSBs recruits XRCC1 (X-ray repair cross complementing protein 1), a pivotal scaffold protein for assembly and activation of DNA base excision repair machinery (34, 35). Recent studies from our group and others identified that the BRCT domain in XRCC1 is a strong PAR-binding domain that mediates the early recruitment of XRCC1 (36, 37).
Figure 1. The interactions between PAR recognition domains and PAR moieties.

The PBZ domain interacts with the adenines in two tandem ADP-ribose units. The WWE, FHA and OB-fold domains interact with the iso-ADP-ribose unit. The Macro domain interacts with the terminal ADP ribose unit. PARP: poly ADP-ribose polymerase; NAD+: nicotinamide adenine dinucleotide; ADPr: ADP-ribose; PBZ: PAR-binding zinc finger; FHA: forkhead-associated; OB: oligonucleotide/oligosaccharide-binding fold.
PAR at DSB lesions rapidly recruits meiotic recombination 11 (MRE11), an integral protein of the MRN (MRE11-RAD50-NBS1) complex and ataxia telangiectasia-mutated (ATM). These two key DDR factors detect DSB, halt cell cycle and activate downstream repair by homologous recombination (HR) or non-homologous end joining (NHEJ) (31, 38). HR, which mainly occurs in the S and G2 phases, requires the sister chromatid template for repair and is activated by 3′ nucleotide overhang from single-stranded DNA resection (39). Although HR is grossly normal in PARP-1-inhibited cells (40), PARylation can facilitate HR by mediating the early recruitment of breast cancer susceptibility gene 1 and 2 (BRCA1 and BRCA2) (36, 41). The BRCT domain of BRCA1-associated RING domain protein 1 (BARD1), the functional partner of BRCA1, recognizes PARylation and mediates the recruitment of the BRCA1 complex to DSBs. BRCA2 uses its oligonucleotide/oligosaccharide-binding (OB) fold domain to bind PAR, leading to the recruitment of RAD51, a crucial recombinase for strand invasion in HR (41). Additionally, PARP-1 activation at replication fork restarts the stalled fork for HR (42, 43). Furthermore, we recently demonstrated that exonuclease 1 (EXO1) is recruited by PAR through its homologues of the PilT N-terminal (PIN) domain and mediates DNA end resection in HR (44).
NHEJ rejoins the two ends of DSB without the need for a homologous template. PARP-1 binding to DNA-dependent protein kinases (DNA-PKs) has been reported to maintain genomic integrity during V(D)J recombinations carried out by NHEJ (45). Our data suggests that DNA ligase IV, a key component in NHEJ, can be recruited by PAR through interaction with its BRCT (46). Furthermore, PARP-1 recruits chromodomain helicase DNA binding protein 2 (CHD2), thereby activating chromatin expansion and depositing histone variant H3.3 at DSB sites. Subsequently, PARP-1, CHD2 and H3.3 jointly recruit the assembly of NHEJ machineries (47). In alternative NHEJ, PARP-1 directly interacts with DSB ends and activate PARylation, leading to recruitment of repair proteins of DNA ligase III/XRCC1 and poly nucleotide kinase-phosphatase (PNKP) (46, 48).
PARylation induces remodeling of chromatin into an open conformation that is amendable for DNA repair. PAR chains on chromatin-associated PARP-1 acts as an aggregate of negative charge, repelling DNA, resulting in decondensation of chromatin (49). Histone H2A, H2B, H3, and H4 can interact with PAR and cause the release of DNA from nucleosomal core particles (50, 51). Moreover, histones H1, H2A, H2B, H3 and H4 can undergo PARylation to serve as additional scaffolds for DDR factors to remodel nucleosome structures (52, 53). Furthermore, ADP-ribosylation on key lysine residues on histone tails including H3K27 and H4K16 may regulate histone methylation and acetylation (13, 54). In addition, PAR-mediated recruitment of aprataxin-poly nucleotide kinase-like factor (APLF), a histone chaperon protein, facilitates relaxation of chromatin (55).
PARylation can regulate protein ubiquitination at DNA damage sites by recruiting ubiquitin E3 ligases including checkpoint with forkhead-associated and RING finger (CHFR) and ring finger protein 146 (RNF146). CHFR recruitment by PAR catalyzes the first wave of ubiquitination at DNA damage sites, facilitating proteosomal degradation of DDR factors (56). Similarly, RNF146 recruited by PAR ubiquitinates and targets PARP-1, XRCC1, DNA ligase III, and KU70 for proteosomal degradation (57).
PAR modifications at the DNA damage sites are highly dynamic. Genotoxic stress-induced PARylation on protein targets is rapidly degraded in vivo with half-life of 40 seconds to 6 minutes (24, 58). Accumulation of undigested PAR can trigger cell death through parthanatos, a formed of programmed cell death (59). The reported PAR degradation enzymes include PARG, ARH3, TARG1, MacroD1, MacroD2, NUDT9 and NUDT16. Many of these enzymes contain the macro domain that recognizes PAR and ADP-ribose (60). The full length PARG isoform (110 kDa) localizes to the nucleus and is the major de-PARylation enzyme (61). PARG cuts the ribose-ribose bond between the ADP-ribose units and is capable of both endo-glycohydrolytic and exo-glycohydrolytic cleavage (62, 63). However, PARG is incapable of removing the terminal ADP-ribose moiety attached to amino acid residue (63). ARH3 performs the same enzymatic reaction as PARG but is reported to mainly cleave O-acetyl-ADP-ribose, a product of the NAD+-dependent Sirtuin reaction (64). ARH3 is also found to exert its activity on mitochondrial matrix-associated PAR and its PAR degradation activity in the nucleus is likely limited (26, 65). NUDT9 and NUDT16 hydrolyze the phosphodiester bond between ADP-ribose moiety and protein, resulting in a ribose-5′-phosphate (R5P) tags attached to the modified protein (66). Their contribution to PAR homeostasis is currently unclear.
The removal of terminal ADP-ribose is proposed to be the rate-limiting step in PAR degradation, evident by the significantly longer half-life of protein MARylation compared to PARylation. Recently, TARG1, MacroD1 and MacroD2 were identified to be capable of digesting MARylation by cleaving glutamate-linked ADP-ribose (27, 67). Structural analysis reveals that TARG1 catalyzes the reaction by forming a covalent lysyl-ADP-ribose intermediate, subsequently resolved by a catalytic aspartic acid (27, 68). MacroD1 and D2 perform substrate-assisted catalysis using a positioned water molecule for hydrolysis of the glutamate-ADP-ribose bond (28, 67). These enzymes have distinct subcellular localization. TARG1 predominately localizes to the nucleus (27). MacroD1 localizes to mitochondria whereas MacroD2 has both nuclear and cytoplasmic localization (69).
There is accumulating evidence supporting the important role of de-PARylation in DDR. Both PARG and TARG1 are rapidly recruited to DNA damage sites by PARylation (27, 70). Complete deletion of all isoforms of PARG in mice is embryonically lethal (71). Furthermore, PARG inhibition sensitizes cells to genotoxic stress and results in synthetic lethality with HR defect (72, 73). Failure to remove ADP-ribose from the acceptor proteins leads to a neurodegenerative disorder characterized by lysosomal accumulation of the proteins with ribose-5-phosphate attached to the glutamic acid residue (74). TARG1 truncation mutations are linked to an autosomal recessive neurodegenerative and seizure disorder (27). TARG1 knockdown also sensitizes cells to DNA toxin such as methyl methanosulphonate (27). Collectively, the data suggests that de-PARylation is an essential step in DDR while the detailed mechanism remains to be elucidated.
PAR recognition domain
PARylation mediates the fast recruitment of DDR factors to the sites of DNA damage through interactions with their PAR recognition domains. In the following, we will describe the known PAR-recognition domains, including the PAR-binding zinc finger (PBZ), the WWE domain, the BRCT domain, the FHA (forkhead-associated) domain, the OB-fold domain, the RRM (RNA recognition motif) domain, the PIN domain, and the GAR domain (Table 1). The growing numbers of the PAR recognition domains propelled the discovery in PARylation-regulated DDR, PARylation-induced PTMs and downstream signaling cascades (75). Figure 2 illustrates the interaction between various PAR recognition domains and PAR moieties. The PBZ binding-containing CHFR and the WWE domain-containing RNF146, deltex1, and TRIP12 mediate ubiquitanation of DDR factors. The Macro domain-containing MacroH2a, MacroD1, MacroD2, TARG1, and PARG mediate degradation of PAR. The FHA, BRCT, OB fold, RRM, PIN and GAR domains are crucial in mediating the interactions between scaffold proteins and DNA processing proteins in DDR.
Table 1.
List of PAR Recognition Domains
| Domain | Length (residues) | PAR interaction | Proteins | Functions in DDR | Reference |
|---|---|---|---|---|---|
| PBZ | ~ 30 | Adenines in two tandem ADP-ribose units | CHFR, APLF, CTCF | Ubiquitination, DNA processing, Maintains DDR boundaries | (56, 76–78, 80) |
| WWE | 80 – 100 | iso-ADP-ribose | RNF146, PARP-7, PARP-11, PARP-14 | Ubiquitination | (57, 81–84) |
| Macro | 130 – 190 | Terminal ADP-ribose, pyrophosphate bond | MacroH2a, ALC1, PARP-9, PARP-14, PARP-15, MacroD1, MacroD2, TARG1, PARG | PAR degradation, chromatin remodeling | (27, 28, 63, 85–88) |
| FHA | ~ 80 | Phosphate of iso-ADP- ribose | PNKP, APTX | Promotes BER and NHEJ | (46) |
| BRCT | ~ 100 | Phosphate of ADP-ribose | NBS1, BARD1, BRCA2, XRCC1, DNA Ligase IV | Promotes BER and DSB repair | (36, 46) |
| OB fold | 70 – 150 | iso-ADP- ribose | hSSB1, BRCA2 | Promotes DSB repair | (95–97) |
| RRM | ~ 90 | By structural similarity with nucleotides. | NONO, RBMX | Promotes DSB repair | (98, 99, 143) |
| PIN | 130 – 150 | By structural similarity with nucleotides. | EXO1, GEN1, SMG5 | DNA end processing | (44) |
| GAR | variable | By structural similarity with nucleotides. | FUS/TLS, EWS/EWSR1, TAF15, SAFB1, SAF-A, hnRNPUL1/2 | Chromatin remodeling, promotes DSB repair, regulates DDR factor gene expressions, R- loop removal | (100–105) |
Figure 2. PARP1 activation and recruitment of DNA damage response factors in base excision reapir (BER), nucleotide excision repair (NER) and double strand break (DSB) repair.

XRCC1: X-ray repair cross-complementing protein 1; PCNA: Proliferating cell nuclear antigen; DDB1: DNA damage-binding protein 1; DDB2: DNA damage-binding protein 2; cul4A: cullin-4A; RBX1: ring-box 1; HR: homologous recombination, a-NHEJ: alternative pathway of non-homologous end joining.
PAR-binding zinc finger (PBZ)
The PBZ domain is a small PAR-binding module found in two DDR factors, CHFR and APLF (76). The domain has a consensus sequence of [K/R]xxCx[F/Y]GxxCxbbxxxxHxxx[F/Y]xH and contains putative C2H2 zinc-finger separated by a 6–8 amino acid spacer. The domain uses a central zinc ion surrounded by two cysteine and two histidine residues that specifically recognize the adenines in tandem ADP-ribose units (77, 78). CHFR is an ubiquitin E3 ligase that regulates PARP-1 displacement from DNA damage site (56). APLF is an endonuclease that acts on auprinic-apyrimidinic site. It carries two PBZ domains, ZF1 and ZF2 with the zinc binding motif of CX5CX6HX5H (77). Simultaneous interaction of both ZF1 and ZF2 with PAR mediate the high affinity recruitment to sites of DNA damage. Recently, we discovered a novel interaction of the CCCTC-binding factor (CTCF) with PAR. CTCF has been described as a chromatin barrier that separates euchromatin and heterochromatin. (79). CTCF exhibits rapid PAR-dependent recruitment to sites of DNA damage (80). CTCF contains eleven zinc finger and deletion of zinc finger 4–6 abolishes its interaction with PAR (80). CTCF can possibly facilitate DNA repair by containing DDR to sites of DNA damage. CTCF knock-down sensitizes cells to ionizing radiation (80).
The WWE domain
The WWE domain consists of three conserved residues, tryptophan-tryptophan-glutamate that specifically recognize iso-ADP-ribose, which is the linker region between the two ADP-ribose units in PAR (81). The WWE domains are found in two categories of proteins, ubiquitin E3 ligases (RNF146, deltex1, and TRIP12) and poly-ADP ribosyltransferases (PARP-7, PARP-11, PARP-12, PARP-13, and PARP-14) (20, 82). This domain is crucial for PAR-activated downstream ubiquitination in DDR. RNF 146 (as known as Iduna) is recruited and activated by PAR (57). It subsequently ubiquitinates PARP-1, XRCC1, DNA ligase III, and KU70 and targets these factors for proteosomal degradation (57). RNF146 facilitates DNA repair and promotes cell survival after γ-irradiation (57, 83). Furthermore, RNF146 is found to promote WNT signaling by ubiquitinating Tankyrase-PARylated AXIN1/2 for proteosomal degradation (84).
The Macro domain
The Macro domain is an evolutionary conserved domain, first discovered in histone variant macroH2A (85, 86). The domain contains a 130 to 190 amino acid residues folded into a central β-sheet flanked by α-helices. It has a conserved diphosphate-binding loop that recognizes NAD metabolites including ADP-ribose and PAR (60, 69). Other macro domain-containing proteins include ALC1, PARP-9, PARP-14, PARP-15, MacroD1, MacroD2, MacroD3, TARG1 and PARG (20, 60). ALC1 is recruited by PARylation and catalyzes PARP-1-stimulated nucleosome sliding and chromatin remolding (87, 88). PARG catalyzes hydrolysis of PAR and is the major de-PARylation enzyme (63). MacroD1, MacroD2 and TARG1 share a unique enzymatic function that removes terminal ADP-ribose from ADP-ribosylated proteins (27, 28).
The BRCT domain and the FHA domain
The BRCT domain and FHA domain were first identified as motifs that recognize phosphorylated proteins. BRCT domains bind phosho-serine moiety (89–91) and FHA domains recognize phosphor-threonine moiety (92, 93). BRCT domain and FHA domain facilitate assemblies of the protein complexes that regulate cell cycle and DDR (89, 94). Recently, we discovered that some BRCT domains can serve as ADP-ribose recognition motifs and mediate PAR-dependent recruitment of DNA DSB repair factors (36). The BRCT domain of NBS1 recognizes PAR and rapidly recruits the MRN complex to DNA damage sites, leading to activation of the ATM signaling cascade in response to DNA DSBs. Moreover, the BRCA1/BARD1 complex is rapidly recruited via interaction of the BARD1 BRCT domain and PAR, promoting homologous recombination. The BRCT domain in DNA Ligase IV interacts with PAR to mediate its recruitment to sites of DNA damage and thereby facilitates NHEJ. We also discovered that the FHA domains in PNKP and APTX can recognize iso-ADP-ribose, the linker unit in PAR, with high affinity (Kd = 0.24 μM and 0.37 μM, respectively) (46). Mutations of the FHA domains abolish the rapid recruitment of PNKP and APTX to DNA damage sites. The dual phosphorylation and PARylation recognition property enables both the early recruitment and stabilization of the BRCT and FHA domain-containing DDR factors at sites of DNA damage.
The OB Fold Domain, RRM Domain, PIN domain and GAR domain
PAR and oligonucleotide are structurally similar since both contain phosphodiester backbone and ribose rings with adenine base. The OB fold domain, RRM domain, PIN domain and RGG box were discovered as RNA binding motif and were recently identified as PAR-binding modules. The OB fold domain is 70–150 amino acid domain that consists of a β barrel capped by an α helix (95, 96). The OB fold domain was first identified in bacterial and yeast as an domain that interacts with oligonucleotide and oligosaccharide (95). Recently, we discovered that the OB fold domain recognizes iso-ADP-ribose. The OB fold domain in human single strand break (hSSB1) recognizes iso-ADP-ribose and mediates the fast recruitment of hSSB1 to sites of DNA damage. Screening of other OB fold domain-containing protein reveals high affinity interaction of MEIOB (meiosis specific with OB domain), CTC1 (CST telomere replication complex component 1), hSSB2 and BRCA2 with PAR (41, 97).
The RRM domain has approximately 90 amino acid residues and consists of four anti-parallel β sheet interconnected by two α helices. The RRM domain is the most abundant RNA-binding motif in eukaryotic cells (98). The RRM domain-containing proteins regulate post-transcriptional RNA processing. Recently, the RRM domain at the N-terminus of NONO was identified to bind PAR with high affinity (99). NONO is recruited to sites of DNA damage through interaction with PAR and contributes to NHEJ.
The PIN domain has nuclease activity that cleaves specific sequence of ssDNA/ssRNA. We identified that the PIN domain of EXO1 is a PAR recognition domain (44). The PAR-mediated fast recruitment of EXO1 facilities early DNA end resection. Further screening revealed that the PIN domains of GEN1 and SMG5 could interact with PAR.
GAR (glycine-arginine-rich) domain consists of a sequence enriched in arginine and glycine. It is also termed RGG (arginine-glycine-glycine) box. This positively charged motif renders its affinity to RNA and more recently to PAR (31). The GAR domain in MRE11 mediates the rapid recruitment to sites of DNA damag. RGG region in the RNA-binding proteins including FUS/TLS, EWS/EWSR1, TAF15, SAFB1, SAF-A, and hnRNPUL1/2 were found to mediate PAR-dependent recruitment of multi-protein complex at sites of DNA damage (100–105). SAF-A is a mRNA biogenesis factor that facilitates R-loop removal from the DNA damage site (105).
PAR-binding motif (PBM)
PBM was the first PAR-interacting domain described (106). It consists of ~20 amino acids with a putative sequence of [HKR]-X-X-[AIQVY]-[KR]-[KR]-[AILV]-[FILPV] (107, 108). PBM consists of a short degenerated peptide sequence and is unlikely to form a folded structure (18). Its mode of interaction with PAR is unclear and there lacks structural data on this motif (21). There are several evidences suggesting that PBM is not a distinct PAR binding domain. First, the putative PBM in XRCC1 resides in the BRCT domain. The entire BRCT domain is required for its affinity with PAR, rather than the short PBM (46). Furthermore, many of the PBM-containing proteins, including DNA ligase3, hnRNP family proteins, and MRE11 contain the BRCT domain, RRM domain or GAR domains that are high affinity PAR-binding domains (107). Collectively, the data suggests the specific PAR interactions occur with the other PAR binding domains described above but not PBM.
PARP inhibition in cancer therapy
PARP-1 and PARP-2 are the major PARylation enzymes activated in response to genotoxic stress. PARP-1/2 were proposed to be effective targets for cancer treatment given their pivotal roles in DDR (109, 110). In 1979, it was first demonstrated that PARylation inhibition with nicotinamide analog sensitizes cancer cells to cytotoxic insults (111). Since, there are a number of PARP inhibitors developed to target the catalytic NAD+-binding pocket in PARP-1 and PARP-2 (Table 2). In 2005, Farmer et al and Bryant et al showed that PARP inhibition specifically kill BRCA1 and BRCA2 mutant cells, demonstrating synthetic lethality of PARP inhibition with HR defect (112, 113). The mechanism for PARP inhibitors-selective killing of HR defect cells was originally proposed to inhibit PARylation and cause persistent DNA SSBs, leading to formation of DSBs at replication fork and fork collapse (113). The mode of action has been revised to reflect the emerging evidence that inhibitor-inactivated PARP-1 is trapped on the DNA (114). Trapped PARP-1–DNA complexes are cytotoxic by causing replication fork collapse through direct collision and by obstructing the direct interaction of DDR factors with the broken DNA (114, 115) (Figure 3). In addition, loss of PARylation impairs early recruitment of both BRCA1 and BRCA2 to DNA lesions, suggesting that the PARylation directly contributes to HR (36, 41).
Table 2.
List of PARP inhibitors
| Compound Company | MW (g/mol) | Structure | Ki (nM) | IC50 (nM) | Status in clinical development | Reference |
|---|---|---|---|---|---|---|
| Olaparib (AZD2281) AstraZeneca |
435.08 |
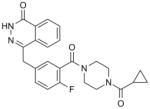
|
n/a | PARP-1: 5 PARP-2: 1 |
FDA approved for advanced ovarian cancer with gBRCAmut with ≥ 3 prior lines of chemotherapy | (116, 144) |
| Rucaparib (CO-338) Clovis |
421.36 |
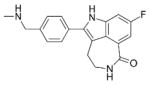
|
PARP-1: 1.4 PARP-2: n/a |
PARP-1: 0.8 PARP-2: 0.5 |
FDA approved for advanced ovarian cancer with gBRCAmut or sBRCAmut with ≥ 2 lines of chemotherapy | (145, 146) |
| Niraparib (MK-4827) Tesaro |
320.39 |
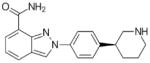
|
n/a | PARP-1: 3.8 PARP-2: 2.1 |
FDA approved for recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who have complete or partial response | (147) |
| Talazoparib (BMN-673) Pfizer |
380.35 |
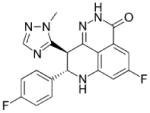
|
PARP-1: 1.2 PARP-2: 0.85 |
PARP-1: 0.57 PARP-2: n/a |
In Phase III study as to platinum monotherapy in patients chemotherapy with locally advanced or metastatic breast cancer with gBRCAmut | (148) |
| Veliparib (ABT-888) Abbvie |
244.29 |
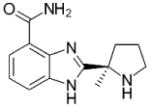
|
PARP-1: 5.2 PARP-2: 2.9 |
n/a | In Phase III studies as combination therapy with chemotherapy in patients with breast and ovarian cancer with gBRCAmut or sBRCAmut and lung cancer | (149) |
Abbreviations: MW: molecular weight, gBRCAmut: germline BRCA mutations, sBRCAmut: somatic BRCA mutations, FDA: United States Food and Drug Administration, n/a: not available
Figure 3.

Synthetic lethality of PARP inhibition in cancer cells with homologous recombination (HR) defect.
The specific activity of PARP inhibitors on cells with HR defects led to clinical trials to target cancers associated with BRCA1 and BRCA2 mutations. In 2014, the United States Food and Drug Administration (FDA) approved the first PARP inhibitor, olaparib monotherapy in patients with germline BRCA1/2 mutated advanced ovarian cancer with three more lines of chemotherapy (116). In Table 2, we list the selected PARP inhibitors that are approved or in late stage of clinical development. In addition to olaparib, there were two other PARP inhibitors approved by the FDA. Rucaparib was approved to treat patients with BRCA mutations-associated advanced ovarian cancer in December 2016. Niraparib was approved to treat patients with recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer in March 2017. Among the PARP inhibitors, talazoparib appears to be the most potent molecule to inhibit and trap PARP-1/2 (117). Active investigations are underway to test the clinical efficacy of these molecules in solid tumors including breast, castration-resistant prostate and pancreatic cancers with germline BRCA1/2 mutations (118).
Besides germline BRCA1/2 mutations, the cells with other types of HR defects confer similar sensitivity to PARP inhibitors. These include cells with somatic BRCA1/2 mutation, BRCA1/2 promoter hypermethylation or mutations in genes essential for HR including ATM, ATR, PALB2, and the FANC gene family (119, 120). Furthermore, the genomic instability including loss of heterozygosity, telomeric allelic imbalance and large-scale state transitions due to HR defect can be detected as mutational “scar” by cancer genomic analysis (121, 122). This HR defect with mutational “scar” phenotype is also termed “BRCAness” since the cells with germline BRCA1/2 mutations exhibit the same genetic aberrations (123). Using this approach, it was demonstrated that the recurrent ovarian cancer patients without germline BRCA1/2 mutations but with the biomarker of BRCAness scar have longer progression free survival when treated with niraparib (124). Moreover, the emergence of liquid biopsy to analyze the cancer genomics in blood circulating tumor cells and circulating DNA can further improve screening cancer patients for PARP inhibitor therapies (125). Besides using PARP inhibitor as monotherapy, there are several ongoing clinical trials that explore the use of PARP inhibitors in combination with platinum, taxane or alkylating chemotherapy, radiotherapy, and other inhibitors targeting cell cycle regulation and the PI3K/AKT/mTOR (phosphoinositide 3-kinase/AKT/mammalian target of rapamycin) pathway (118, 126). Taken together, these advances will expand the use of PARP inhibitors in the clinical setting.
Resistance to PARP inhibitor is an emerging clinical problem. The resistance originates from the genetic heterogeneity within a tumor, or acquired resistance following PARP inhibitor therapy. There are several reported mechanisms of drug resistance from PARP inhibitor. They include increased drug efflux, partial HR deficiency, recovery of HR defect, and activation of compensatory signaling pathways. P-glycoprotein (Pgp) is a member in the ATP binding cassette (ABC) transporter family that transports molecules (127). Overexpressoin of the Pgp transporter causes resistance to PARP inhibitor and addition of the Pgp inhibitor overcomes the PARP inhibitor resistance (128, 129). BRCA1 loss of function mutations causes HR defect and induces tumorigenesis. There are several hypomorphic mutations of BRCA1 that result in incomplete inactivation of HR (130–132). BRCA1-c61G RING-inactivating mutation induces loss of tumor suppression but retains ability to form RAD51 foci upon irradiation (130). Furthermore, BRCA1185delAG mutation, a detectable germline mutation, generates the BRCA1 mutant lacking the RING domain (131). This RING-lacking BRCA1 is not completely functional in HR but remains capable of facilitating RAD51 foci formation at the DNA damage site (131). Furthermore, Similarly, it was reported that mutation in BRCA1 exon 11 generates a splice variant lacking exon 11, BRCA1-Δ11 isoform (132). This mutant can partially compensate for full-length BRCA1. Tumor cells carrying these BRCA1 mutants are resistant to PARP inhibitors (132).
Recovery of HR defect renders cancer cells resistant PARP inhibitor. Reactivation of functional BRCA1 gene by the loss of promoter hypermethylation restores HR deficiency (133). In addition, secondary mutations in BRCA2 can cause genetic reversion of BRCA2 loss of function mutation. For example, c.6174delT in BRCA2 mutant is a truncated protein from the frameshift mutation (134). A secondary mutation in the BRCA2 mutant allele results in intragenic deletion of c.6174delT mutation, thereby restoring the open reading frame (ORF) and the HR deficiency (134). Similar ORF-restoring mutations were detected in the PARP inhibitor-resistant breast cancer cell line HCC1428 (135). 53BP1 is an important regulator of DSB repair response. Loss of 53BP1 partially recovers HR defect by promoting ATM-dependent processing of broken DNA ends to produce the recombinogenic single-stranded DNA (136). Loss of 53BP-1 causes PARP inhibitor in BRCA-1 mutated mouse mammary tumors and in ATM-deficient breast cancer cells (136, 137). The PARP inhibitor resistance in BRCA1 mutant ovarian carcinoma cells has also been observed by restoration of the HR defect by a microRNA, miR-622 targeting the Ku complex (138). Activations of signaling pathways that facilitate DDR contribute to PARP inhibitor resistance. Inhibitions of ATR, mTOR and NF-kB pathways have been shown to be effective in overcoming PARP inhibitor resistance (139–142).
Perspectives and Conclusions
Among the PARylation-regulated biology, DDR is the first one described and is one of the most studied processes. The advance in this field led to the successful development of PARP inhibitors for cancer therapy. The better characterization of the PAR recognition domains adds layers of complexities onto the DDR process. Many of the DDR factors contain multiple domains that mediate simultaneous interactions with DNA, RNA and proteins PTMs. Further research is needed to uncover the novel cross talks mediated by these interactions. Furthermore, details of ADP-ribose unit removal from the PAR chain complex remain elusive. There also lacks detailed understanding of how PAR catabolism regulates DDR. Inhibition of de-PARylation enzymes with potent, cell permeable inhibitors may present an opportunity for efficacious cancer therapy targeting PAR metabolism.
Acknowledgments
This work was supported by grants from National Institutes of Health (CA132755, CA130899 and CA187209 to X.Y.) and the Department of Defense (BA160420 to X.Y.). X.Y. is a recipient of Leukemia and Lymphoma Society Scholar Award.
Conflict of interest statement: None declared.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lindahl T, Barnes DE. Repair of endogenous DNA damage. Cold Spring Harb Symp Quant Biol. 2000;65:127–33. doi: 10.1101/sqb.2000.65.127. [DOI] [PubMed] [Google Scholar]
- 2.Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med. 2009;361(15):1475–85. doi: 10.1056/NEJMra0804615. [DOI] [PubMed] [Google Scholar]
- 3.Roos WP, Thomas AD, Kaina B. DNA damage and the balance between survival and death in cancer biology. Nat Rev Cancer. 2016;16(1):20–33. doi: 10.1038/nrc.2015.2. [DOI] [PubMed] [Google Scholar]
- 4.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40(2):179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009;461(7267):1071–8. doi: 10.1038/nature08467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.D’Amours D, Desnoyers S, D’Silva I, Poirier GG. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem J. 1999;342(Pt 2):249–68. [PMC free article] [PubMed] [Google Scholar]
- 7.Chambon P, Weill JD, Mandel P. Nicotinamide mononucleotide activation of new DNA-dependent polyadenylic acid synthesizing nuclear enzyme. Biochem Biophys Res Commun. 1963;11:39–43. doi: 10.1016/0006-291x(63)90024-x. [DOI] [PubMed] [Google Scholar]
- 8.Sekine A, Fujiwara M, Narumiya S. Asparagine residue in the rho gene product is the modification site for botulinum ADP-ribosyltransferase. J Biol Chem. 1989;264(15):8602–5. [PubMed] [Google Scholar]
- 9.Manning DR, Fraser BA, Kahn RA, Gilman AG. ADP-ribosylation of transducin by islet-activation protein. Identification of asparagine as the site of ADP-ribosylation. J Biol Chem. 1984;259(2):749–56. [PubMed] [Google Scholar]
- 10.Just I, Wollenberg P, Moss J, Aktories K. Cysteine-specific ADP-ribosylation of actin. Eur J Biochem. 1994;221(3):1047–54. doi: 10.1111/j.1432-1033.1994.tb18823.x. [DOI] [PubMed] [Google Scholar]
- 11.Ogata N, Ueda K, Kagamiyama H, Hayaishi O. ADP-ribosylation of histone H1. Identification of glutamic acid residues 2, 14, and the COOH-terminal lysine residue as modification sites. J Biol Chem. 1980;255(16):7616–20. [PubMed] [Google Scholar]
- 12.Altmeyer M, Messner S, Hassa PO, Fey M, Hottiger MO. Molecular mechanism of poly(ADP-ribosyl)ation by PARP1 and identification of lysine residues as ADP-ribose acceptor sites. Nucleic Acids Res. 2009;37(11):3723–38. doi: 10.1093/nar/gkp229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Messner S, Altmeyer M, Zhao H, Pozivil A, Roschitzki B, Gehrig P, et al. PARP1 ADP-ribosylates lysine residues of the core histone tails. Nucleic Acids Res. 2010;38(19):6350–62. doi: 10.1093/nar/gkq463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bonfiglio JJ, Fontana P, Zhang Q, Colby T, Gibbs-Seymour I, Atanassov I, et al. Serine ADP-Ribosylation Depends on HPF1. Mol Cell. 2017;65(5):932–40. e6. doi: 10.1016/j.molcel.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leidecker O, Bonfiglio JJ, Colby T, Zhang Q, Atanassov I, Zaja R, et al. Serine is a new target residue for endogenous ADP-ribosylation on histones. Nat Chem Biol. 2016;12(12):998–1000. doi: 10.1038/nchembio.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alvarez-Gonzalez R, Jacobson MK. Characterization of polymers of adenosine diphosphate ribose generated in vitro and in vivo. Biochemistry. 1987;26(11):3218–24. doi: 10.1021/bi00385a042. [DOI] [PubMed] [Google Scholar]
- 17.Miwa M, Saikawa N, Yamaizumi Z, Nishimura S, Sugimura T. Structure of poly(adenosine diphosphate ribose): identification of 2′-[1″-ribosyl-2″-(or 3″-)(1‴-ribosyl)]adenosine-5′,5″,5‴-tris(phosphate) as a branch linkage. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(2):595–9. doi: 10.1073/pnas.76.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu C, Yu X. ADP-ribosyltransferases and poly ADP-ribosylation. Curr Protein Pept Sci. 2015;16(6):491–501. doi: 10.2174/1389203716666150504122435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vyas S, Chang P. New PARP targets for cancer therapy. Nat Rev Cancer. 2014;14(7):502–9. doi: 10.1038/nrc3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibson BA, Kraus WL. New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol. 2012;13(7):411–24. doi: 10.1038/nrm3376. [DOI] [PubMed] [Google Scholar]
- 21.Gupte R, Liu Z, Kraus WL. PARPs and ADP-ribosylation: recent advances linking molecular functions to biological outcomes. Genes & development. 2017;31(2):101–26. doi: 10.1101/gad.291518.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vyas S, Matic I, Uchima L, Rood J, Zaja R, Hay RT, et al. Family-wide analysis of poly(ADP-ribose) polymerase activity. Nat Commun. 2014;5:4426. doi: 10.1038/ncomms5426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De Vos M, Schreiber V, Dantzer F. The diverse roles and clinical relevance of PARPs in DNA damage repair: current state of the art. Biochem Pharmacol. 2012;84(2):137–46. doi: 10.1016/j.bcp.2012.03.018. [DOI] [PubMed] [Google Scholar]
- 24.Alvarez-Gonzalez R, Althaus FR. Poly(ADP-ribose) catabolism in mammalian cells exposed to DNA-damaging agents. Mutat Res. 1989;218(2):67–74. doi: 10.1016/0921-8777(89)90012-8. [DOI] [PubMed] [Google Scholar]
- 25.Ueda K, Oka J, Naruniya S, Miyakawa N, Hayaishi O. Poly ADP-ribose glycohydrolase from rat liver nuclei, a novel enzyme degrading the polymer. Biochem Biophys Res Commun. 1972;46(2):516–23. doi: 10.1016/s0006-291x(72)80169-4. [DOI] [PubMed] [Google Scholar]
- 26.Niere M, Mashimo M, Agledal L, Dolle C, Kasamatsu A, Kato J, et al. ADP-ribosylhydrolase 3 (ARH3), not poly(ADP-ribose) glycohydrolase (PARG) isoforms, is responsible for degradation of mitochondrial matrix-associated poly(ADP-ribose) J Biol Chem. 2012;287(20):16088–102. doi: 10.1074/jbc.M112.349183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sharifi R, Morra R, Appel CD, Tallis M, Chioza B, Jankevicius G, et al. Deficiency of terminal ADP-ribose protein glycohydrolase TARG1/C6orf130 in neurodegenerative disease. EMBO J. 2013;32(9):1225–37. doi: 10.1038/emboj.2013.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jankevicius G, Hassler M, Golia B, Rybin V, Zacharias M, Timinszky G, et al. A family of macrodomain proteins reverses cellular mono-ADP-ribosylation. Nat Struct Mol Biol. 2013;20(4):508–14. doi: 10.1038/nsmb.2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Daniels CM, Thirawatananond P, Ong SE, Gabelli SB, Leung AK. Nudix hydrolases degrade protein-conjugated ADP-ribose. Sci Rep. 2015;5:18271. doi: 10.1038/srep18271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Langelier MF, Riccio AA, Pascal JM. PARP-2 and PARP-3 are selectively activated by 5′ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014;42(12):7762–75. doi: 10.1093/nar/gku474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ, et al. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem. 2008;283(2):1197–208. doi: 10.1074/jbc.M706734200. [DOI] [PubMed] [Google Scholar]
- 32.Langelier MF, Planck JL, Roy S, Pascal JM. Structural basis for DNA damage-dependent poly(ADP-ribosyl)ation by human PARP-1. Science. 2012;336(6082):728–32. doi: 10.1126/science.1216338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297(5579):259–63. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 34.Masson M, Niedergang C, Schreiber V, Muller S, Menissier-de Murcia J, de Murcia G. XRCC1 is specifically associated with poly(ADP-ribose) polymerase and negatively regulates its activity following DNA damage. Mol Cell Biol. 1998;18(6):3563–71. doi: 10.1128/mcb.18.6.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Okano S, Lan L, Caldecott KW, Mori T, Yasui A. Spatial and temporal cellular responses to single-strand breaks in human cells. Mol Cell Biol. 2003;23(11):3974–81. doi: 10.1128/MCB.23.11.3974-3981.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li M, Yu X. Function of BRCA1 in the DNA damage response is mediated by ADP-ribosylation. Cancer cell. 2013;23(5):693–704. doi: 10.1016/j.ccr.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Breslin C, Hornyak P, Ridley A, Rulten SL, Hanzlikova H, Oliver AW, et al. The XRCC1 phosphate-binding pocket binds poly (ADP-ribose) and is required for XRCC1 function. Nucleic Acids Res. 2015;43(14):6934–44. doi: 10.1093/nar/gkv623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aguilar-Quesada R, Munoz-Gamez JA, Martin-Oliva D, Peralta A, Valenzuela MT, Matinez-Romero R, et al. Interaction between ATM and PARP-1 in response to DNA damage and sensitization of ATM deficient cells through PARP inhibition. BMC Mol Biol. 2007;8:29. doi: 10.1186/1471-2199-8-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heyer WD, Ehmsen KT, Liu J. Regulation of homologous recombination in eukaryotes. Annu Rev Genet. 2010;44:113–39. doi: 10.1146/annurev-genet-051710-150955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schultz N, Lopez E, Saleh-Gohari N, Helleday T. Poly(ADP-ribose) polymerase (PARP-1) has a controlling role in homologous recombination. Nucleic Acids Res. 2003;31(17):4959–64. doi: 10.1093/nar/gkg703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang F, Shi J, Bian C, Yu X. Poly(ADP-Ribose) Mediates the BRCA2-Dependent Early DNA Damage Response. Cell reports. 2015;13(4):678–89. doi: 10.1016/j.celrep.2015.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, et al. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009;28(17):2601–15. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang YG, Cortes U, Patnaik S, Jasin M, Wang ZQ. Ablation of PARP-1 does not interfere with the repair of DNA double-strand breaks, but compromises the reactivation of stalled replication forks. Oncogene. 2004;23(21):3872–82. doi: 10.1038/sj.onc.1207491. [DOI] [PubMed] [Google Scholar]
- 44.Zhang F, Shi J, Chen SH, Bian C, Yu X. The PIN domain of EXO1 recognizes poly(ADP-ribose) in DNA damage response. Nucleic Acids Res. 2015;43(22):10782–94. doi: 10.1093/nar/gkv939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morrison C, Smith GC, Stingl L, Jackson SP, Wagner EF, Wang ZQ. Genetic interaction between PARP and DNA-PK in V(D)J recombination and tumorigenesis. Nat Genet. 1997;17(4):479–82. doi: 10.1038/ng1297-479. [DOI] [PubMed] [Google Scholar]
- 46.Li M, Lu LY, Yang CY, Wang S, Yu X. The FHA and BRCT domains recognize ADP-ribosylation during DNA damage response. Genes & development. 2013;27(16):1752–68. doi: 10.1101/gad.226357.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Luijsterburg MS, de Krijger I, Wiegant WW, Shah RG, Smeenk G, de Groot AJ, et al. PARP1 Links CHD2-Mediated Chromatin Expansion and H3.3 Deposition to DNA Repair by Non-homologous End-Joining. Mol Cell. 2016;61(4):547–62. doi: 10.1016/j.molcel.2016.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, et al. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34(21):6170–82. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Poirier GG, de Murcia G, Jongstra-Bilen J, Niedergang C, Mandel P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proceedings of the National Academy of Sciences of the United States of America. 1982;79(11):3423–7. doi: 10.1073/pnas.79.11.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mathis G, Althaus FR. Release of core DNA from nucleosomal core particles following (ADP-ribose)n-modification in vitro. Biochem Biophys Res Commun. 1987;143(3):1049–54. doi: 10.1016/0006-291x(87)90358-5. [DOI] [PubMed] [Google Scholar]
- 51.Realini CA, Althaus FR. Histone shuttling by poly(ADP-ribosylation) J Biol Chem. 1992;267(26):18858–65. [PubMed] [Google Scholar]
- 52.Adamietz P, Bredehorst R, Hilz H. ADP-ribosylated histone H1 from HeLa cultures. Fundamental differences to (ADP-ribose)n-histone H1 conjugates formed into vitro. Eur J Biochem. 1978;91(2):317–26. doi: 10.1111/j.1432-1033.1978.tb12682.x. [DOI] [PubMed] [Google Scholar]
- 53.Nolan NL, Butt TR, Wong M, Lambrianidou A, Smulson ME. Characterization of poly(ADP-ribose)--histone H1 complex formation in purified polynucleosomes and chromatin. Eur J Biochem. 1980;113(1):15–25. doi: 10.1111/j.1432-1033.1980.tb06133.x. [DOI] [PubMed] [Google Scholar]
- 54.Messner S, Hottiger MO. Histone ADP-ribosylation in DNA repair, replication and transcription. Trends Cell Biol. 2011;21(9):534–42. doi: 10.1016/j.tcb.2011.06.001. [DOI] [PubMed] [Google Scholar]
- 55.Mehrotra PV, Ahel D, Ryan DP, Weston R, Wiechens N, Kraehenbuehl R, et al. DNA repair factor APLF is a histone chaperone. Mol Cell. 2011;41(1):46–55. doi: 10.1016/j.molcel.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu C, Wu J, Paudyal SC, You Z, Yu X. CHFR is important for the first wave of ubiquitination at DNA damage sites. Nucleic Acids Res. 2013;41(3):1698–710. doi: 10.1093/nar/gks1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kang HC, Lee YI, Shin JH, Andrabi SA, Chi Z, Gagne JP, et al. Iduna is a poly(ADP-ribose) (PAR)-dependent E3 ubiquitin ligase that regulates DNA damage. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(34):14103–8. doi: 10.1073/pnas.1108799108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jacobson EL, Antol KM, Juarez-Salinas H, Jacobson MK. Poly(ADP-ribose) metabolism in ultraviolet irradiated human fibroblasts. J Biol Chem. 1983;258(1):103–7. [PubMed] [Google Scholar]
- 59.Conrad M, Angeli JP, Vandenabeele P, Stockwell BR. Regulated necrosis: disease relevance and therapeutic opportunities. Nat Rev Drug Discov. 2016;15(5):348–66. doi: 10.1038/nrd.2015.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feijs KL, Forst AH, Verheugd P, Luscher B. Macrodomain-containing proteins: regulating new intracellular functions of mono(ADP-ribosyl)ation. Nat Rev Mol Cell Biol. 2013;14(7):443–51. doi: 10.1038/nrm3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Meyer-Ficca ML, Meyer RG, Coyle DL, Jacobson EL, Jacobson MK. Human poly(ADP-ribose) glycohydrolase is expressed in alternative splice variants yielding isoforms that localize to different cell compartments. Exp Cell Res. 2004;297(2):521–32. doi: 10.1016/j.yexcr.2004.03.050. [DOI] [PubMed] [Google Scholar]
- 62.Hatakeyama K, Nemoto Y, Ueda K, Hayaishi O. Purification and characterization of poly(ADP-ribose) glycohydrolase. Different modes of action on large and small poly(ADP-ribose) J Biol Chem. 1986;261(32):14902–11. [PubMed] [Google Scholar]
- 63.Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, et al. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature. 2011;477(7366):616–20. doi: 10.1038/nature10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ono T, Kasamatsu A, Oka S, Moss J. The 39-kDa poly(ADP-ribose) glycohydrolase ARH3 hydrolyzes O-acetyl-ADP-ribose, a product of the Sir2 family of acetyl-histone deacetylases. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(45):16687–91. doi: 10.1073/pnas.0607911103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Niere M, Kernstock S, Koch-Nolte F, Ziegler M. Functional localization of two poly(ADP-ribose)-degrading enzymes to the mitochondrial matrix. Mol Cell Biol. 2008;28(2):814–24. doi: 10.1128/MCB.01766-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Palazzo L, Thomas B, Jemth AS, Colby T, Leidecker O, Feijs KL, et al. Processing of protein ADP-ribosylation by Nudix hydrolases. Biochem J. 2015;468(2):293–301. doi: 10.1042/BJ20141554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenthal F, Feijs KL, Frugier E, Bonalli M, Forst AH, Imhof R, et al. Macrodomain-containing proteins are new mono-ADP-ribosylhydrolases. Nat Struct Mol Biol. 2013;20(4):502–7. doi: 10.1038/nsmb.2521. [DOI] [PubMed] [Google Scholar]
- 68.Peterson FC, Chen D, Lytle BL, Rossi MN, Ahel I, Denu JM, et al. Orphan macrodomain protein (human C6orf130) is an O-acyl-ADP-ribose deacylase: solution structure and catalytic properties. J Biol Chem. 2011;286(41):35955–65. doi: 10.1074/jbc.M111.276238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Neuvonen M, Ahola T. Differential activities of cellular and viral macro domain proteins in binding of ADP-ribose metabolites. J Mol Biol. 2009;385(1):212–25. doi: 10.1016/j.jmb.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Mortusewicz O, Fouquerel E, Ame JC, Leonhardt H, Schreiber V. PARG is recruited to DNA damage sites through poly(ADP-ribose)- and PCNA-dependent mechanisms. Nucleic Acids Res. 2011;39(12):5045–56. doi: 10.1093/nar/gkr099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Koh DW, Lawler AM, Poitras MF, Sasaki M, Wattler S, Nehls MC, et al. Failure to degrade poly(ADP-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(51):17699–704. doi: 10.1073/pnas.0406182101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cortes U, Tong WM, Coyle DL, Meyer-Ficca ML, Meyer RG, Petrilli V, et al. Depletion of the 110-kilodalton isoform of poly(ADP-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol Cell Biol. 2004;24(16):7163–78. doi: 10.1128/MCB.24.16.7163-7178.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fathers C, Drayton RM, Solovieva S, Bryant HE. Inhibition of poly(ADP-ribose) glycohydrolase (PARG) specifically kills BRCA2-deficient tumor cells. Cell Cycle. 2012;11(5):990–7. doi: 10.4161/cc.11.5.19482. [DOI] [PubMed] [Google Scholar]
- 74.Williams JC, Chambers JP, Liehr JG. Glutamyl ribose 5-phosphate storage disease. A hereditary defect in the degradation of poly(ADP-ribosylated) proteins. J Biol Chem. 1984;259(2):1037–42. [PubMed] [Google Scholar]
- 75.Wei H, Yu X. Functions of PARylation in DNA Damage Repair Pathways. Genomics Proteomics Bioinformatics. 2016;14(3):131–9. doi: 10.1016/j.gpb.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ahel I, Ahel D, Matsusaka T, Clark AJ, Pines J, Boulton SJ, et al. Poly(ADP-ribose)-binding zinc finger motifs in DNA repair/checkpoint proteins. Nature. 2008;451(7174):81–5. doi: 10.1038/nature06420. [DOI] [PubMed] [Google Scholar]
- 77.Li GY, McCulloch RD, Fenton AL, Cheung M, Meng L, Ikura M, et al. Structure and identification of ADP-ribose recognition motifs of APLF and role in the DNA damage response. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(20):9129–34. doi: 10.1073/pnas.1000556107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Oberoi J, Richards MW, Crumpler S, Brown N, Blagg J, Bayliss R. Structural basis of poly(ADP-ribose) recognition by the multizinc binding domain of checkpoint with forkhead-associated and RING Domains (CHFR) J Biol Chem. 2010;285(50):39348–58. doi: 10.1074/jbc.M110.159855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ong CT, Corces VG. CTCF: an architectural protein bridging genome topology and function. Nature reviews Genetics. 2014;15(4):234–46. doi: 10.1038/nrg3663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Han D, Chen Q, Shi J, Zhang F, Yu X. CTCF participates in DNA damage response via poly(ADP-ribosyl)ation. Sci Rep. 2017;7:43530. doi: 10.1038/srep43530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Z, Michaud GA, Cheng Z, Zhang Y, Hinds TR, Fan E, et al. Recognition of the iso-ADP-ribose moiety in poly(ADP-ribose) by WWE domains suggests a general mechanism for poly(ADP-ribosyl)ation-dependent ubiquitination. Genes & development. 2012;26(3):235–40. doi: 10.1101/gad.182618.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Aravind L. The WWE domain: a common interaction module in protein ubiquitination and ADP ribosylation. Trends Biochem Sci. 2001;26(5):273–5. doi: 10.1016/s0968-0004(01)01787-x. [DOI] [PubMed] [Google Scholar]
- 83.Andrabi SA, Kang HC, Haince JF, Lee YI, Zhang J, Chi Z, et al. Iduna protects the brain from glutamate excitotoxicity and stroke by interfering with poly(ADP-ribose) polymer-induced cell death. Nat Med. 2011;17(6):692–9. doi: 10.1038/nm.2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhang Y, Liu S, Mickanin C, Feng Y, Charlat O, Michaud GA, et al. RNF146 is a poly(ADP-ribose)-directed E3 ligase that regulates axin degradation and Wnt signalling. Nat Cell Biol. 2011;13(5):623–9. doi: 10.1038/ncb2222. [DOI] [PubMed] [Google Scholar]
- 85.Kustatscher G, Hothorn M, Pugieux C, Scheffzek K, Ladurner AG. Splicing regulates NAD metabolite binding to histone macroH2A. Nat Struct Mol Biol. 2005;12(7):624–5. doi: 10.1038/nsmb956. [DOI] [PubMed] [Google Scholar]
- 86.Timinszky G, Till S, Hassa PO, Hothorn M, Kustatscher G, Nijmeijer B, et al. A macrodomain-containing histone rearranges chromatin upon sensing PARP1 activation. Nat Struct Mol Biol. 2009;16(9):923–9. doi: 10.1038/nsmb.1664. [DOI] [PubMed] [Google Scholar]
- 87.Ahel D, Horejsi Z, Wiechens N, Polo SE, Garcia-Wilson E, Ahel I, et al. Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science. 2009;325(5945):1240–3. doi: 10.1126/science.1177321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gottschalk AJ, Timinszky G, Kong SE, Jin J, Cai Y, Swanson SK, et al. Poly(ADP-ribosyl)ation directs recruitment and activation of an ATP-dependent chromatin remodeler. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(33):13770–4. doi: 10.1073/pnas.0906920106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science. 2003;302(5645):639–42. doi: 10.1126/science.1088753. [DOI] [PubMed] [Google Scholar]
- 90.Rodriguez M, Yu X, Chen J, Songyang Z. Phosphopeptide binding specificities of BRCA1 COOH-terminal (BRCT) domains. J Biol Chem. 2003;278(52):52914–8. doi: 10.1074/jbc.C300407200. [DOI] [PubMed] [Google Scholar]
- 91.Manke IA, Lowery DM, Nguyen A, Yaffe MB. BRCT repeats as phosphopeptide-binding modules involved in protein targeting. Science. 2003;302(5645):636–9. doi: 10.1126/science.1088877. [DOI] [PubMed] [Google Scholar]
- 92.Durocher D, Henckel J, Fersht AR, Jackson SP. The FHA domain is a modular phosphopeptide recognition motif. Mol Cell. 1999;4(3):387–94. doi: 10.1016/s1097-2765(00)80340-8. [DOI] [PubMed] [Google Scholar]
- 93.Mahajan A, Yuan C, Lee H, Chen ES, Wu PY, Tsai MD. Structure and function of the phosphothreonine-specific FHA domain. Sci Signal. 2008;1(51):re12. doi: 10.1126/scisignal.151re12. [DOI] [PubMed] [Google Scholar]
- 94.Li J, Williams BL, Haire LF, Goldberg M, Wilker E, Durocher D, et al. Structural and functional versatility of the FHA domain in DNA-damage signaling by the tumor suppressor kinase Chk2. Mol Cell. 2002;9(5):1045–54. doi: 10.1016/s1097-2765(02)00527-0. [DOI] [PubMed] [Google Scholar]
- 95.Murzin AG. OB(oligonucleotide/oligosaccharide binding)-fold: common structural and functional solution for non-homologous sequences. EMBO J. 1993;12(3):861–7. doi: 10.1002/j.1460-2075.1993.tb05726.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Flynn RL, Zou L. Oligonucleotide/oligosaccharide-binding fold proteins: a growing family of genome guardians. Crit Rev Biochem Mol Biol. 2010;45(4):266–75. doi: 10.3109/10409238.2010.488216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zhang F, Chen Y, Li M, Yu X. The oligonucleotide/oligosaccharide-binding fold motif is a poly(ADP-ribose)-binding domain that mediates DNA damage response. Proceedings of the National Academy of Sciences of the United States of America. 2014;111(20):7278–83. doi: 10.1073/pnas.1318367111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Maris C, Dominguez C, Allain FH. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 2005;272(9):2118–31. doi: 10.1111/j.1742-4658.2005.04653.x. [DOI] [PubMed] [Google Scholar]
- 99.Krietsch J, Caron MC, Gagne JP, Ethier C, Vignard J, Vincent M, et al. PARP activation regulates the RNA-binding protein NONO in the DNA damage response to DNA double-strand breaks. Nucleic Acids Res. 2012;40(20):10287–301. doi: 10.1093/nar/gks798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Altmeyer M, Toledo L, Gudjonsson T, Grofte M, Rask MB, Lukas C, et al. The chromatin scaffold protein SAFB1 renders chromatin permissive for DNA damage signaling. Mol Cell. 2013;52(2):206–20. doi: 10.1016/j.molcel.2013.08.025. [DOI] [PubMed] [Google Scholar]
- 101.Mastrocola AS, Kim SH, Trinh AT, Rodenkirch LA, Tibbetts RS. The RNA-binding protein fused in sarcoma (FUS) functions downstream of poly(ADP-ribose) polymerase (PARP) in response to DNA damage. J Biol Chem. 2013;288(34):24731–41. doi: 10.1074/jbc.M113.497974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rulten SL, Rotheray A, Green RL, Grundy GJ, Moore DA, Gomez-Herreros F, et al. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014;42(1):307–14. doi: 10.1093/nar/gkt835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Polo SE, Blackford AN, Chapman JR, Baskcomb L, Gravel S, Rusch A, et al. Regulation of DNA-end resection by hnRNPU-like proteins promotes DNA double-strand break signaling and repair. Mol Cell. 2012;45(4):505–16. doi: 10.1016/j.molcel.2011.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hong Z, Jiang J, Ma J, Dai S, Xu T, Li H, et al. The role of hnRPUL1 involved in DNA damage response is related to PARP1. PloS one. 2013;8(4):e60208. doi: 10.1371/journal.pone.0060208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Britton S, Dernoncourt E, Delteil C, Froment C, Schiltz O, Salles B, et al. DNA damage triggers SAF-A and RNA biogenesis factors exclusion from chromatin coupled to R-loops removal. Nucleic Acids Res. 2014;42(14):9047–62. doi: 10.1093/nar/gku601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Pleschke JM, Kleczkowska HE, Strohm M, Althaus FR. Poly(ADP-ribose) binds to specific domains in DNA damage checkpoint proteins. J Biol Chem. 2000;275(52):40974–80. doi: 10.1074/jbc.M006520200. [DOI] [PubMed] [Google Scholar]
- 107.Teloni F, Altmeyer M. Readers of poly(ADP-ribose): designed to be fit for purpose. Nucleic Acids Res. 2016;44(3):993–1006. doi: 10.1093/nar/gkv1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Krietsch J, Rouleau M, Pic E, Ethier C, Dawson TM, Dawson VL, et al. Reprogramming cellular events by poly(ADP-ribose)-binding proteins. Mol Aspects Med. 2013;34(6):1066–87. doi: 10.1016/j.mam.2012.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lindahl T, Satoh MS, Poirier GG, Klungland A. Post-translational modification of poly(ADP-ribose) polymerase induced by DNA strand breaks. Trends Biochem Sci. 1995;20(10):405–11. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- 110.de Murcia JM, Niedergang C, Trucco C, Ricoul M, Dutrillaux B, Mark M, et al. Requirement of poly(ADP-ribose) polymerase in recovery from DNA damage in mice and in cells. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(14):7303–7. doi: 10.1073/pnas.94.14.7303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Terada M, Fujiki H, Marks PA, Sugimura T. Induction of erythroid differentiation of murine erythroleukemia cells by nicotinamide and related compounds. Proceedings of the National Academy of Sciences of the United States of America. 1979;76(12):6411–4. doi: 10.1073/pnas.76.12.6411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005;434(7035):917–21. doi: 10.1038/nature03445. [DOI] [PubMed] [Google Scholar]
- 113.Bryant HE, Schultz N, Thomas HD, Parker KM, Flower D, Lopez E, et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005;434(7035):913–7. doi: 10.1038/nature03443. [DOI] [PubMed] [Google Scholar]
- 114.Murai J, Huang SY, Das BB, Renaud A, Zhang Y, Doroshow JH, et al. Trapping of PARP1 and PARP2 by Clinical PARP Inhibitors. Cancer research. 2012;72(21):5588–99. doi: 10.1158/0008-5472.CAN-12-2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Pommier Y, O’Connor MJ, de Bono J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci Transl Med. 2016;8(362):362ps17. doi: 10.1126/scitranslmed.aaf9246. [DOI] [PubMed] [Google Scholar]
- 116.Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, et al. FDA Approval Summary: Olaparib Monotherapy in Patients with Deleterious Germline BRCA-Mutated Advanced Ovarian Cancer Treated with Three or More Lines of Chemotherapy. Clin Cancer Res. 2015;21(19):4257–61. doi: 10.1158/1078-0432.CCR-15-0887. [DOI] [PubMed] [Google Scholar]
- 117.Murai J, Huang SY, Renaud A, Zhang Y, Ji J, Takeda S, et al. Stereospecific PARP trapping by BMN 673 and comparison with olaparib and rucaparib. Mol Cancer Ther. 2014;13(2):433–43. doi: 10.1158/1535-7163.MCT-13-0803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lord CJ, Ashworth A. PARP inhibitors: Synthetic lethality in the clinic. Science. 2017;355(6330):1152–8. doi: 10.1126/science.aam7344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, et al. Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP-ribose) polymerase inhibition. Cancer research. 2006;66(16):8109–15. doi: 10.1158/0008-5472.CAN-06-0140. [DOI] [PubMed] [Google Scholar]
- 120.Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer. 2016;16(2):110–20. doi: 10.1038/nrc.2015.21. [DOI] [PubMed] [Google Scholar]
- 121.Telli ML, Timms KM, Reid J, Hennessy B, Mills GB, Jensen KC, et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin Cancer Res. 2016;22(15):3764–73. doi: 10.1158/1078-0432.CCR-15-2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med. 2017 doi: 10.1038/nm.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer. 2004;4(10):814–9. doi: 10.1038/nrc1457. [DOI] [PubMed] [Google Scholar]
- 124.Mirza MR, Monk BJ, Herrstedt J, Oza AM, Mahner S, Redondo A, et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N Engl J Med. 2016;375(22):2154–64. doi: 10.1056/NEJMoa1611310. [DOI] [PubMed] [Google Scholar]
- 125.Vasant G, Dago AE, Lee J, Greene S, Lietz L, Wang Y, et al. A single cell genomic signature to detect homologous recombination deficiency (HRD) and PARP inhibitors sensitivity using patient’s circulating tumor cells (CTCs) J Clin Oncol. 2016;34(suppl) abstr e23015. [Google Scholar]
- 126.Drean A, Lord CJ, Ashworth A. PARP inhibitor combination therapy. Crit Rev Oncol Hematol. 2016;108:73–85. doi: 10.1016/j.critrevonc.2016.10.010. [DOI] [PubMed] [Google Scholar]
- 127.Verhalen B, Dastvan R, Thangapandian S, Peskova Y, Koteiche HA, Nakamoto RK, et al. Energy transduction and alternating access of the mammalian ABC transporter P-glycoprotein. Nature. 2017;543(7647):738–41. doi: 10.1038/nature21414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Rottenberg S, Jaspers JE, Kersbergen A, van der Burg E, Nygren AO, Zander SA, et al. High sensitivity of BRCA1-deficient mammary tumors to the PARP inhibitor AZD2281 alone and in combination with platinum drugs. Proceedings of the National Academy of Sciences of the United States of America. 2008;105(44):17079–84. doi: 10.1073/pnas.0806092105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Henneman L, van Miltenburg MH, Michalak EM, Braumuller TM, Jaspers JE, Drenth AP, et al. Selective resistance to the PARP inhibitor olaparib in a mouse model for BRCA1-deficient metaplastic breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2015;112(27):8409–14. doi: 10.1073/pnas.1500223112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Drost R, Bouwman P, Rottenberg S, Boon U, Schut E, Klarenbeek S, et al. BRCA1 RING function is essential for tumor suppression but dispensable for therapy resistance. Cancer cell. 2011;20(6):797–809. doi: 10.1016/j.ccr.2011.11.014. [DOI] [PubMed] [Google Scholar]
- 131.Drost R, Dhillon KK, van der Gulden H, van der Heijden I, Brandsma I, Cruz C, et al. BRCA1185delAG tumors may acquire therapy resistance through expression of RING-less BRCA1. The Journal of clinical investigation. 2016;126(8):2903–18. doi: 10.1172/JCI70196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Wang Y, Bernhardy AJ, Cruz C, Krais JJ, Nacson J, Nicolas E, et al. The BRCA1-Delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer research. 2016;76(9):2778–90. doi: 10.1158/0008-5472.CAN-16-0186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ter Brugge P, Kristel P, van der Burg E, Boon U, de Maaker M, Lips E, et al. Mechanisms of Therapy Resistance in Patient-Derived Xenograft Models of BRCA1-Deficient Breast Cancer. Journal of the National Cancer Institute. 2016;108(11) doi: 10.1093/jnci/djw148. [DOI] [PubMed] [Google Scholar]
- 134.Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature. 2008;451(7182):1111–5. doi: 10.1038/nature06548. [DOI] [PubMed] [Google Scholar]
- 135.Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature. 2008;451(7182):1116–20. doi: 10.1038/nature06633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Bunting SF, Callen E, Wong N, Chen HT, Polato F, Gunn A, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell. 2010;141(2):243–54. doi: 10.1016/j.cell.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hong R, Ma F, Zhang W, Yu X, Li Q, Luo Y, et al. 53BP1 depletion causes PARP inhibitor resistance in ATM-deficient breast cancer cells. BMC cancer. 2016;16(1):725. doi: 10.1186/s12885-016-2754-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Choi YE, Meghani K, Brault ME, Leclerc L, He YJ, Day TA, et al. Platinum and PARP Inhibitor Resistance Due to Overexpression of MicroRNA-622 in BRCA1-Mutant Ovarian Cancer. Cell reports. 2016;14(3):429–39. doi: 10.1016/j.celrep.2015.12.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, et al. ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor-resistant BRCA-deficient cancer cells. Genes & development. 2017;31(3):318–32. doi: 10.1101/gad.290957.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Xiang T, Jia Y, Sherris D, Li S, Wang H, Lu D, et al. Targeting the Akt/mTOR pathway in Brca1-deficient cancers. Oncogene. 2011;30(21):2443–50. doi: 10.1038/onc.2010.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Cardnell RJ, Feng Y, Mukherjee S, Diao L, Tong P, Stewart CA, et al. Activation of the PI3K/mTOR Pathway following PARP Inhibition in Small Cell Lung Cancer. PloS one. 2016;11(4):e0152584. doi: 10.1371/journal.pone.0152584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Nakagawa Y, Sedukhina AS, Okamoto N, Nagasawa S, Suzuki N, Ohta T, et al. NF-kappaB signaling mediates acquired resistance after PARP inhibition. Oncotarget. 2015;6(6):3825–39. doi: 10.18632/oncotarget.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Adamson B, Smogorzewska A, Sigoillot FD, King RW, Elledge SJ. A genome-wide homologous recombination screen identifies the RNA-binding protein RBMX as a component of the DNA-damage response. Nat Cell Biol. 2012;14(3):318–28. doi: 10.1038/ncb2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Menear KA, Adcock C, Boulter R, Cockcroft XL, Copsey L, Cranston A, et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2H-phthalazin- 1-one: a novel bioavailable inhibitor of poly(ADP-ribose) polymerase-1. J Med Chem. 2008;51(20):6581–91. doi: 10.1021/jm8001263. [DOI] [PubMed] [Google Scholar]
- 145.Thomas HD, Calabrese CR, Batey MA, Canan S, Hostomsky Z, Kyle S, et al. Preclinical selection of a novel poly(ADP-ribose) polymerase inhibitor for clinical trial. Mol Cancer Ther. 2007;6(3):945–56. doi: 10.1158/1535-7163.MCT-06-0552. [DOI] [PubMed] [Google Scholar]
- 146.Robillard L, Nguyen M, Harding TC, Simmons AD. In vitro and in vivo assessment of the mechanism of action of the PARP inhibitor rucaparib. AACR 2017 Proceedings: Abstracts; 2017. pp. 1–3062. abstract #2475. [Google Scholar]
- 147.Jones P, Altamura S, Boueres J, Ferrigno F, Fonsi M, Giomini C, et al. Discovery of 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole-7-carboxamide (MK-4827): a novel oral poly(ADP-ribose)polymerase (PARP) inhibitor efficacious in BRCA-1 and -2 mutant tumors. J Med Chem. 2009;52(22):7170–85. doi: 10.1021/jm901188v. [DOI] [PubMed] [Google Scholar]
- 148.Shen Y, Rehman FL, Feng Y, Boshuizen J, Bajrami I, Elliott R, et al. BMN 673, a novel and highly potent PARP1/2 inhibitor for the treatment of human cancers with DNA repair deficiency. Clin Cancer Res. 2013;19(18):5003–15. doi: 10.1158/1078-0432.CCR-13-1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, et al. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13(9):2728–37. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]