

Abstract
It is challenging to predict the docked conformations of two proteins. Current methods are susceptible to errors from treating proteins as rigid bodies and from an inability to compute relative Boltzmann populations of different docked conformations. Here, we show that by using the ClusPro server as a front end to generate possible protein–protein contacts, and using Modeling Employing Limited Data (MELD) accelerated molecular dynamics (MELD × MD) as a back end for atomistic simulations, we can find 16/20 native dimer structures of small proteins as those having the lowest free energy, starting from good–bound–backbone structures. We show that atomistic MD free energies can be used to identify native protein dimer structures.
Graphical Abstract
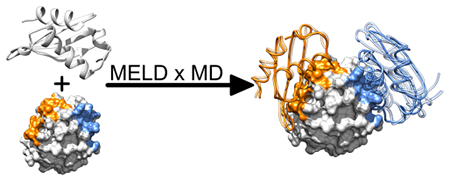
1. INTRODUCTION
We describe a way to improve computer predictions of the structures of protein–protein dimers. Much work has been done in modeling protein docking. The protein docking community has established an event, called CAPRI (Critical Assessment of PRediction of Interactions), for the blind testing of docking algorithms.1–3 This has brought the challenges into clear focus. Proteins have so many degrees of freedom of internal and relative motion that simplifications are needed to tame the computational combinatorial explosion.4–11 One main simplification is to treat each protein as a rigid body. Another simplification is to use quasi-physical “scoring functions” instead of more physically accurate free energies. Both introduce errors into structure prediction.12,13 Recently the ProPOSE approach has been developed to deal with some of these limitations allowing for flexible side chains during the docking prediction.14 Molecular dynamics (MD) simulations with atomistic force fields in principle represent an approach to such limitations, but at a extremely high computational cost. For example, using highly specialized computational resources,15 Shaw helped to refine oligomeric structures using MD16 and observed several undocking–docking events of five protein dimers starting from the bound structure.17 To reduce the computational cost of MD, Hou et al. used simplified coarse grained molecular models to quite successfully compute the binding free energy of different docking poses.18
A recently developed method called MELD (Modeling Employing Limited Data) considerably accelerates physics based simulations (like MD or Monte Carlo) using generic or vaguely directive information to restrict the search space of the problem.19,20 We call MELD × MD the application of MELD to MD simulations. MELD × MD is useful in protein structure determination,19 computing the poses and affinities of binding a peptide to a protein,21,22 and the folding of small proteins in CASP, the blind native prediction event.20,23–25 The acceleration in MELD × MD comes from a Bayesian integration of external information that can be probabilistic or combinatorial and not specific.
Here, we first use the ClusPro (CP) rigid-body docking server to estimate sets of protein–protein contacts from its 15 best docked poses.11,26–29 Those contacts are input to MELD × MD, which then explores the internal and relative degrees of freedom of the two proteins with replica-exchange sampling in a physical potential function. We find that MELD × MD adds value in predicting dimer structures by identifying the highest computed conformational population (lowest free energy). Our study here is limited to situations in which both proteins are given to have roughly the correct backbone trace, so that we can learn from our modeling the value of including the flexibility that MD provides and of computing populations (free energies) to pick out native dimer structures among options. During our MELD × MD simulation the conformations of the side chains and the backbone are allowed to fluctuate. Future work will investigate the use of this flexibility to refine the monomer backbone structure during the MELD × MD docking procedure, but this was not done here. Such refinement is particularly important when bound state monomer structures are not available.
2. EXPERIMENTAL SECTION
2.1. MELD Accelerates Conformational Exploration by Using External Information.
Because a protein has so many degrees of freedom, it is computationally expensive to sample them and find its equilibrium states by molecular dynamics simulations in a physical force field. It is even more challenging to model two proteins binding to each other. MELD is a recently developed method that can accelerate MD simulations of proteins when there is some form of target knowledge about the relevant states. Unlike traditional constraining methods, MELD does not require that the constraints be precise, accurate, or deterministic. MELD can speed up the finding and sampling of important states using external information that is even loose, partly wrong, corrupted, probabilistic, or incomplete. In the present situation, we give MELD the directive that “most of the contacts predicted from the ClusPro rigid-body server, in one of top 15 poses, are likely to be essentially right” and that “the monomers should not unfold”.
Here’s how MELD uses smart springs to direct MD simulations to focus around promising regions of conformational space. The springs are smart in two ways. (i) Not all springs are active at the same time. MELD uses Bayesian inference to parse springs into different subsets that are active at different times. The active ones are always the ones with the lowest spring energy (i.e., the ones that are least violated). Switching between different sets of springs happens without violating detailed balance. (ii) The springs energy functions are flat-bottomed. This ensures that, when they are satisfied, no energy is added to the force field’s Hamiltonian of the system. Relative populations of the different regions of the conformational space that satisfy some of the external information are consistent with the populations of unrestrained simulations and can therefore be used to compute proper free energy differences between them, based only on the force field.
The smart-springs approach focuses the computational effort around regions of the conformational space that are consistent with the data.19,20 For good sampling, MELD × MD uses the Hamiltonian and temperature replica exchange (HTRE) protocol.30,31 Higher temperatures correspond to moving up the replica ladder, which leads to weakening the springs.
2.1.1. MELD × MD Setup.
In this paper, we study protein dimers. Our conformational space is the relatively limited number of docking poses of the two monomeric proteins, in addition to some flexibility in the internal structure of each protein. We enforce these two conditions separately. (i) We limit the docking space using the intermolecular contacts proposed by CP. In the lowest energy replicas, our system is forced to explore the conformational space that is compatible with a fraction of the contacts predicted by CP. We do this by computing the intermonomer contacts of the first 15 CP poses. To avoid kinetic biases of our replica exchange protocol that would make simulation convergence harder to reach, we randomly remove contacts from poses until every pose has the same number of contacts as the pose with the least contacts. The contacts of each one of the poses are collected in different groups and our Bayesian approach allows us to have only a fraction (70%) of the springs of one of these groups active at the bottom of the replica ladder at any moment. This allows some flexibility in the final docking contacts of our poses compared to the CP contacts that we use to create the smart springs in the MELD simulations. (ii) Monomers should not unfold during the simulation. We force the monomers to maintain the native secondary structure: we weakly restrain the position of the alpha-carbons of the first monomer around their native monomer positions, and we enforce 80% of the native monomer intramolecular contacts of the second monomer to be satisfied. Our approach allows for more flexibility in the two monomers than is currently used by other docking approaches. In particular, the side chains are totally free to reorient, and the structures of the main chains are free to fluctuate around the restraints. In this work we use the same approach for all 20 pairs of dimers, but in principle it is possible to tune the flexibility of different parts of the systems according to specific needs.
The restraints that drive the docking (the one at point i) are slowly turned on while descending the replica ladder (between α = 0.83 and α = 0.33); other restraints are on at all time. The monomers are forced at all times to sit within a sphere of radius RgR + 2.5RgL + 0.5 nm, where RgR and RgL are the radius of gyration of the receptor and of the ligand, respectively. The temperature is geometrically increased climbing the replica ladder, from 300 at α = 0 to 550 at α = 0.6, and after the temperature is constant. Due to the way we enforce the MELD restraints at the highest replica, the monomers are not docked and the ligand is free to rotate. This means that, in all our simulations, every system copy experiences several undocking and docking events in different poses while climbing and descending the replica ladder.
HTRE simulations are run using 30 replicas. Replicas start equally spaced in replica index space, but their positions are adapted during the simulation to optimize exchanges. Exchange between different replicas are attempted every 50, and the simulations are 2.5 long for each replica. The initial pair of structures is the same for all replicas, and they are structures in which both monomers have their native bound structure, but the two monomers are not in contact.
2.1.2. MD Simulation Parameters.
All our MD simulations are carried out using ff14SB-side force field (FF), which uses ff99SB32 parameters for the backbone and the recent ff14SB33 parameters for the side chains. We use the GB-neck2 implicit-solvent model.34 Simulations are run with steps of 4.5 using a Langevin integrator with a friction coefficient of 1.0 ps−1. Hydrogen masses are repartitioned to allow for the long time-steps. A cutoff of 1.8 is used for all interactions. MELD is a plugin to the OpenMM35 simulation package. Non-MELD simulations used to test the dimer stability are run using AMBER.36
2.1.3. Dimer Selection and System Flexibility.
Our aim is to show how a physically accurate (but also computationally expensive) approach can accurately identify the native state of dimers when more simple docking protocols fail to do so. Of the 49 small dimers we identified from the PDB37 that are stable in our simulation condition (see SI 1 for an in depth description of the procedure used to screen PDB dimers), vanilla CP (i.e., the CP run using the standard setting of the web server) is able to correctly dock 34 of them (i.e., the CP TOP1 structure is native-like). Our test set was comprised of the 15 dimers that CP failed to correctly predict, and 5 out of the 34 it correctly predicts. For 19 of these 20 systems CP did provide at least a good structure in the first 15 CP guesses, for the twentieth dimer we provided MELD also native contacts. In this way we show how our free-energy-based prediction can add value when it is needed.
Including protein flexibilities is critical for good estimates of the free energy. We enforce weak restraints on the receptor α carbon positions, and we enforce a fraction of the native contacts within the ligand. Backbone α carbon RMSDs of the receptor and the ligand go easily up to 2.5 Å (see SI Figure 3), and the side chains are totally free to reorient. While it is true that the starting conformation of the monomers are theirs bound structures, this structure evolves quickly to a few angstroms away in the MD protocol. CP predictions are the ones that are mostly influenced for using the bound structure of the monomers, but we use bound-complex docking as a reliable source of intermolecular contacts. In principle, we can use any source of information (experimental or computational) as long as we can define the confidence associated with such information. Also we note that we use a vanilla approach to CP. A more thoughtful use of CP might yield to better results, but this is outside the scope of this paper. Future work will explore how to use less reliable sources of information compared to bound docking (like template based docking or experimental data), and the simultaneous docking and refinement of monomer structures (e.g., starting from protein structure prediction or apo structures instead of holo structures). In this initial work, we are interested in showing how well we can predict native-like dimer structures using a free energy based method. The flexibility of the proteins in the simulations presented here goes exclusively toward the evaluation of accurate free energies of the docking poses.
3. RESULTS
The quality of our predictions is discussed in the context of the metrics used by the CAPRI community; the reported values in this paper are calculated using the DockQ software.38 For heterodimers, the ligand RMSD (LRMSD) is the ligand (i.e., the shorter chain) backbone RMSD computed after super-imposing the receptor (i.e., the longer chain) backbone. For most of our structures, which are homodimers, we consider the receptor to be the first chain and the ligand to be the second one in the native dimer PDB file. For the computation of the interface RMSD (iRMSD), an interface residue is defined as any residue that has a heavy atom less than 10 Å away from an heavy atom of a residue that belongs to the other monomer chain in the native structure. We also report the percentage of true positive (fTP) and false positive (fFP) contacts, where contacts are defined with a more restrictive 5 Å heavy-atom distance cut off. We report also the CAPRI score and the DockQ score, which are a combination of the RMSD and contact distance parameters.
3.1. Using Information from CP, MELD × MD Successfully Docks 16/20 Dimers.
Our predictions are obtained by clustering the trajectories of the five lowest replicas in the HTRE simulation. The first 250 ns of each trajectory (i.e., the first 5000 exchange attempts) are considered to be equilibration period for the systems and therefore are discarded from this analysis. We cluster the frames using the DBSCAN algorithm39 implemented in sklearn.40 As the distance metric between structures, we use the LRMSD computed on the backbone alpha carbons. For a structure to be included in a cluster, it should have a maximum distance of 3 Å from core structures in the same cluster. A structure is considered a core structure of a cluster when it has at least 200 neighbors. In SI 4 we show the small effect different clustering parameters and methods have on our predictions. The centroid of each cluster is then selected as representative structure, and our predictions are ranked according to cluster population, i.e. MELD × MD TOP1 (abbreviated as MELD TOP1) structure is the centroid of the most populated cluster. No information about the native structures or the CP poses enters the clustering protocol, making this effectively a prediction.
We assess the quality of our predictions by comparing the structure of the centroid of our most populated cluster (i.e., MELD TOP1 prediction) with the crystal structure of the native protein. We use this seemingly restrictive definition of success (only TOP1) because a correct ranking of the poses is a necessary feature of free energy based methods. In section 3.1.3 we will investigate the origin of the four failed predictions and provide suggestions to fix these failures.
Figure 1 shows our results. Each box shows the native structure of the dimer in white, with the receptor depicted as surface and the ligand as a semitransparent cartoon. For each system the ligand position and structure of the MELD TOP1 prediction is represented by a blue cartoon. Boxes are colored in green if the CAPRI score is at least acceptable. The five systems on the left of the figure are the ones that CP successfully ranked (i.e., the CP TOP1 is the best pose predicted by CP according to LRMSD metric). As it is possible to notice when CP is successful in predicting a close to native pose we are also successful in predicting the docking pose. When CP fails, we are often able (11/15 cases) to correctly predict a structure close to native, making our method complementary to what is in the field. In Table 1 we report LRMSD, iRMSD, fTP, fTN, CAPRI, and DockQscores for the MELD TOP1 predictions of the 20 dimers. Out of the 16 successes according to the CAPRI score: 2 are acceptable, 13 are medium, and 1 is high quality. This suggests that, for these systems, a physically accurate model that allows for an accurate representation of the entropy and energy fluctuations of the different poses is key to obtain an accurate ranking of the structures. The relatively high LRMSD of 1Z09 is due to a rearrangement of a ligand α helix. This rearrangement happens away from the interface so it does not dramatically affect the quality of the docking prediction.
Figure 1.
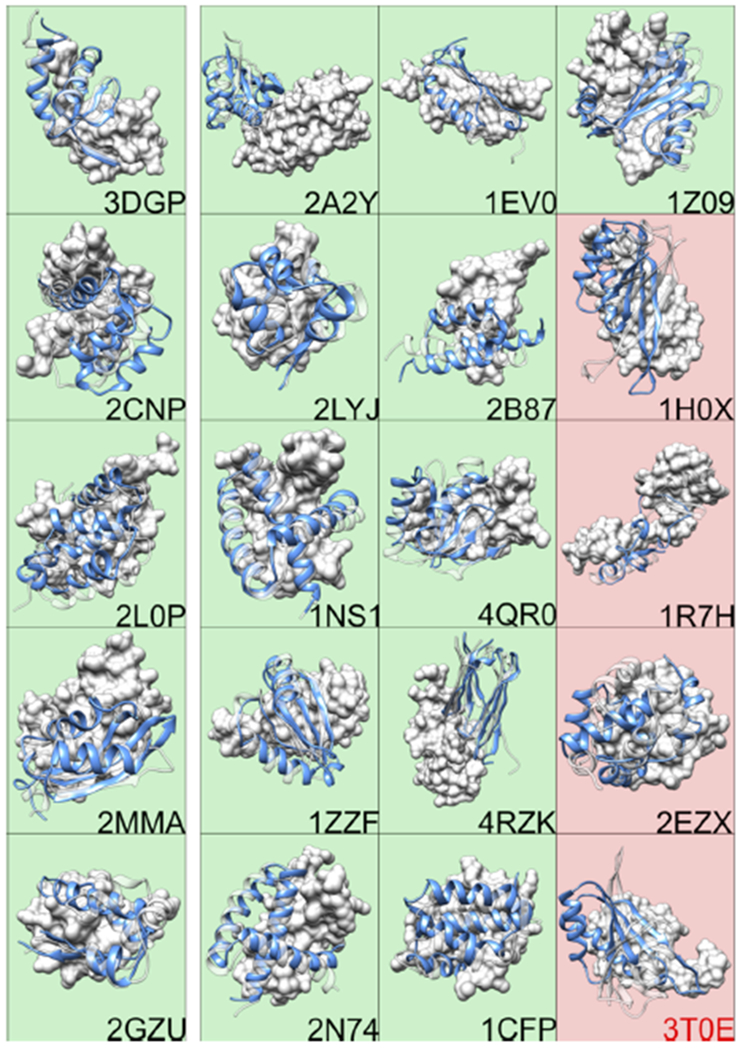
Correct predictions of the native structures of 16/20 dimers. (Green) Successful MELD TOP1 predictions. (Red) Unsuccessful. (Green, 5 left panel) Predictions are also ones that CP ranks correctly. The receptor of the native structure is depicted as white surface, and the ligand is depicted as transparent white cartoon. We only show the structure and position of the ligand of our prediction (solid blue cartoon).
Table 1.
Correct Predictions of the Native Structures of 16/20 Dimersa
| MELD TOP1 |
CP TOP15 |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| PDB id | LRMSD Å | iRMSD Å | fTP % | fFP % | CAPRI score | DockQ score | no. | LRMSD Å | iRMSD Å | fTP % | fFP % | CAPRI score | DockQ score |
| 3DGP | 1.7 | 1.5 | 73 | 18 | M | 0.73 | 0 | 6.2 | 2.1 | 74 | 32 | A | 0.58 |
| 2CNP | 2.0 | 2.1 | 73 | 28 | M | 0.67 | 0 | 4.4 | 1.9 | 70 | 22 | M | 0.63 |
| 2L0P | 2.7 | 2.7 | 81 | 44 | M | 0.65 | 0 | 3.7 | 1.4 | 80 | 28 | M | 0.73 |
| 2MMA | 4.7 | 2.4 | 71 | 56 | M | 0.59 | 0 | 6.0 | 3.4 | 25 | 82 | A | 0.36 |
| 2GZU | 5.5 | 3.4 | 78 | 47 | A | 0.55 | 0 | 2.8 | 1.6 | 71 | 10 | M | 0.69 |
| 2A2Y | 1.8 | 0.8 | 91 | 19 | H | 0.88 | 2 | 3.8 | 1.3 | 95 | 28 | M | 0.78 |
| 2LYJ | 2.0 | 1.2 | 77 | 24 | M | 0.78 | 5 | 6.0 | 3.1 | 61 | 47 | A | 0.49 |
| 1NS1 | 2.2 | 1.7 | 73 | 35 | M | 0.70 | 3 | 4.9 | 2.2 | 51 | 53 | M | 0.52 |
| 1ZZF | 2.3 | 1.4 | 87 | 21 | M | 0.78 | 6 | 3.7 | 1.4 | 90 | 28 | M | 0.76 |
| 2N74 | 2.3 | 2.6 | 72 | 42 | M | 0.64 | 1 | 4.3 | 2.5 | 57 | 59 | M | 0.54 |
| 1EV0 | 2.6 | 2.3 | 89 | 27 | M | 0.70 | 1 | 4.0 | 2.0 | 57 | 35 | M | 0.58 |
| 2B87 | 3.4 | 1.7 | 90 | 24 | M | 0.73 | 4 | 5.5 | 2.2 | 50 | 39 | A | 0.50 |
| 4QR0 | 3.9 | 2.8 | 67 | 49 | M | 0.57 | 3 | 4.8 | 2.1 | 43 | 44 | M | 0.51 |
| 4RZK | 4.8 | 1.2 | 85 | 23 | M | 0.74 | 1 | 9.0 | 1.7 | 67 | 46 | M | 0.52 |
| 1CFP | 6.8 | 4.4 | 41 | 79 | A | 0.37 | 10 | 8.0 | 4.8 | 33 | 68 | A | 0.32 |
| 1Z09 | 15.1 | 1.7 | 90 | 36 | M | 0.53 | 1 | 2.9 | 1.2 | 78 | 15 | M | 0.76 |
| 1H0X | 11.2 | 4.3 | 38 | 79 | I | 0.28 | 13 | 6.3 | 2.6 | 53 | 59 | A | 0.47 |
| 1R7H | 17.2 | 12.8 | 8 | 92 | I | 0.10 | 1 | 2.6 | 1.8 | 73 | 29 | M | 0.68 |
| 2EZX | 25.6 | 12.5 | 10 | 94 | I | 0.07 | 4 | 2.7 | 1.0 | 96 | 23 | M | 0.85 |
| 3TOE | 24.3 | 12.9 | 2 | 98 | I | 0.05 | 7 | 20.7 | 10.2 | 0 | 100 | I | 0.06 |
We give the RMSD of the ligand (LRMSD) and the interface (iRMSD) amino acids, the fraction of true (fTP) and false positive (fFP) intermolecular contacts, the CAPRI classification of the predicted model (H = high, M = medium, A = acceptable, I = incorrect), and the DockQ score. The first column of the CP TOP15 prediction (i.e., the best structure in the first 15 CP poses) reports the CP ranking of the structure with the lowest LRMSD value compared to native (ranking index is zero based). Bold PDB ID represent systems for which MELD × MD predicts the dimer structure with a higher DockQscore than the CP TOP15 prediction. 3T0E is highlighted in italic because neither MELD × MD nor CP are able to correctly predict the structure. CP is able to correctly identify the native structure of the first five dimer. The last four systems are the ones for which our protocol fails to predict the native structure. The 11 systems in between are dimers that we successfully predict.
Two factors contribute to the quality of our MELD TOP1 prediction. (i) We use the size of structure population as a proxy for a free energy evaluation. Thanks to the quality of the force field and solvent model, we are able to use/identify the most native set of CP intermolecular contacts, and to successfully dock 16/20 dimers. (ii) MELD ensures that our system sits in the vicinity of one of the first 15 CP poses (in terms of intermolecular contacts). The FF is then allowed to explore the conformational space surrounding the sets of contacts. This allows physics to identify better poses than the (vanilla) rigid docking protocol we used to predict contacts in 13/16 cases. Identifying a good set of contacts and letting physics explore the allowed conformational space to provide a good structure is a two step process in all our predictions, but we will look at them separately in sections 3.1.1 and 3.1.2. In section 3.1.3, we look at why we fail to predict the docked conformations of four systems correctly.
3.1.1. MELD “Picks” Out Which ClusPro Contacts Are Native.
MD is grounded in physics. MELD focuses MD in order to sample only certain regions of the conformational space without biasing the relative probability of the allowed conformations. It is therefore possible to use the relative population of different structural clusters in our trajectory to identify the lowest free energy cluster (i.e., the most populated). Since we allow our simulation to explore only dimer conformations that share contacts with one of the first 15 CP poses, we could say that we are able to rerank CP poses. However, in this work we allow the system some freedom to move around CP predicted contacts. So it is not straightforward to use our simulations as a pure CP reranking tool. In principle, by limiting the search space around CP docking poses it is possible to make MELD × MD an almost “pure CP poses reranking method”.
Figure 2 shows an example. For 2A2Y, the TOP1 CP structure (central column) has the dimer on the other side of the protein from the native crystal structure(left column). In the simulation, MELD × MD attempts to dock the two monomers according to the contacts from the 15 different CP poses. The fact that at the end of the simulation the centroid of the most populated cluster is extremely similar to the native structure (right column) shows how MELD was able to identify the best set of contacts to drive the monomer docking. Figure 3 shows how we consistently pick out the best pose between the 15 ones suggested by CP. It is possible to track which pose in the first 15 CP poses is favored by the FF by counting how often a pose is active in the lowest 5 replicas during our simulations. The histogram bars in Figure 3 show the relative populations of the first 15 CP poses in our MELD × MD simulations; the red lines identify the CP TOP15 (by LRMSD) for each system. It is possible to see how MELD × MD is able to consistently identify the best pose. 2LYJ, 1NS1, 4QR0, 2B87, and 1CFP have more than one good pose (LRMSD < 6.5 Å) within the first 15 predicted structures, making the populations split between those. Due to the relative relaxed way we enforce the restraint, there is some uncertainty in selecting which pose is active. First we compute the intermolecular contacts from the CP predictions. Then we force all poses to have the same number of contact as the pose with the fewest contacts by randomly removing contacts from poses that have more contacts. This is necessary to avoid kinetic bias in selecting poses along the RE ladder that could make convergence harder. Finally we enforce 70% of the contacts as active. This allows quite some freedom to explore around the original CP poses, making it relatively easy for similar poses to be selected because the FF can explore the same conformational space starting from different poses. This is not a problem for the clustering protocol we use to select the MELD TOP1 pose because it does not use any CP information, but it makes difficult to use this MELD × MD protocol as a purely CP reranking method.
Figure 2.
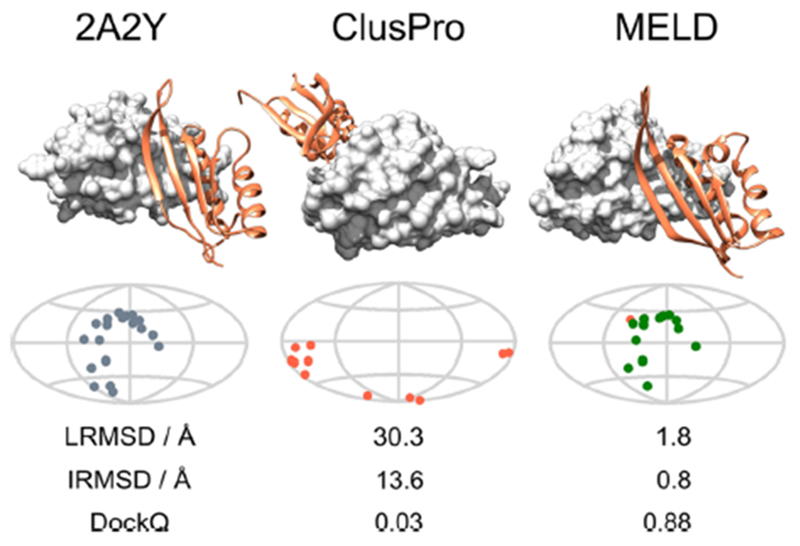
MELD can identify the correct set of contacts to use. The left column shows the native structure of the 2A2Y dimer both using a 3D view and a map of the position of native contact on the first monomer. Here the first monomer is approximated as a sphere and it is represented as a map using a Mollweide projection41 (see SI 5). In the central column we see the CP TOP1 prediction. In this case, CP places the second monomer on the wrong side of the protein. All contacts are FP and therefore are shown in red on the map representation. The wrong position of the dimer is reflected in the three metrics reported below the map. The centroid of the most populated MELD × MD cluster (right column) is strikingly similar to the native structure. The map shows that most of the contact are TP (shown in green). Comparing native and the MELD × MD contact map shows that the MELD × MD contacts are also in the right position. Metrics comparing the MELD × MD prediction with the native structure also show excellent agreement. MELD selected the best set of contacts.
Figure 3.
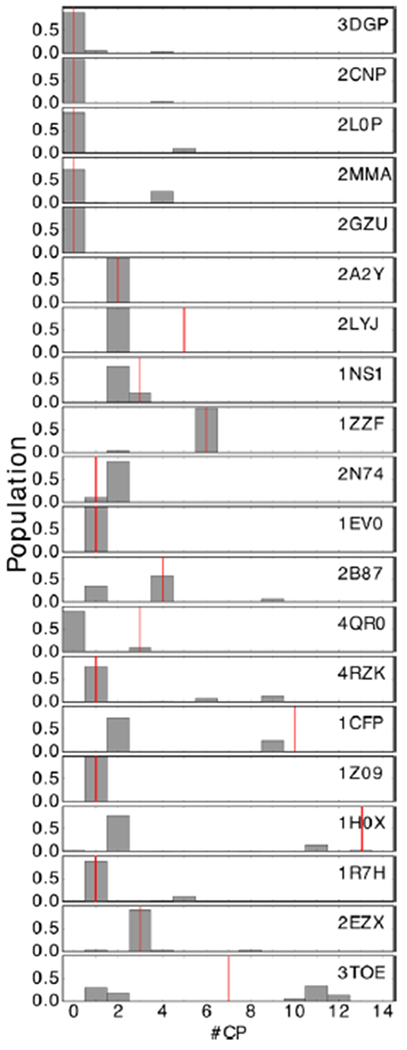
We can successfully rerank CP poses. The histograms show for each of the 20 systems how often one of the first 15 CP poses is active in the lowest 5 replicas of our MELD × MD simulations (i.e., the relative population of the first 15 CP poses). The red lines identify for each system the CP TOP15 (by LRMSD). In most cases, MELD × MD is able to consistently identify the best pose. 2LYJ, 1NS1, 4QR0, 2B87, and 1CFP have more than one good pose (LRMSD < 6.5 Å) within the first 15 predicted structures, so the MELD × MD populations tend to split between those.
3.1.2. MELD × MD Often Gives Better Structures than Vanilla CP.
As noted above, we enforce only 70% of the intermolecular contacts of any given poses, and we do not add a penalty for any additional intermolecular contact formed. This means that we allow some freedom for the monomers to explore the space around the contacts of the CP poses. An interesting example of this is shown in Figure 4. In this case, the CP TOP1 prediction is acceptable according to CAPRI metrics. MELD × MD correctly identifies that the set of contacts belonging to the CP TOP1 pose are closest to native and provides a structure that satisfies 76% of such contacts. The force field is then able to pick up and go the last mile in docking the dimer into a better structure, one that has a medium CAPRI score, a better rank than the CP pose we use to provide this set of contacts. A similar process happens in all the predictions, and only in 3 of the 16 successful predictions MELD × MD predicts slightly worse docking poses than vanilla CP does (see Table 1). This highlights also the quality of the FF for studying dimers.
Figure 4.
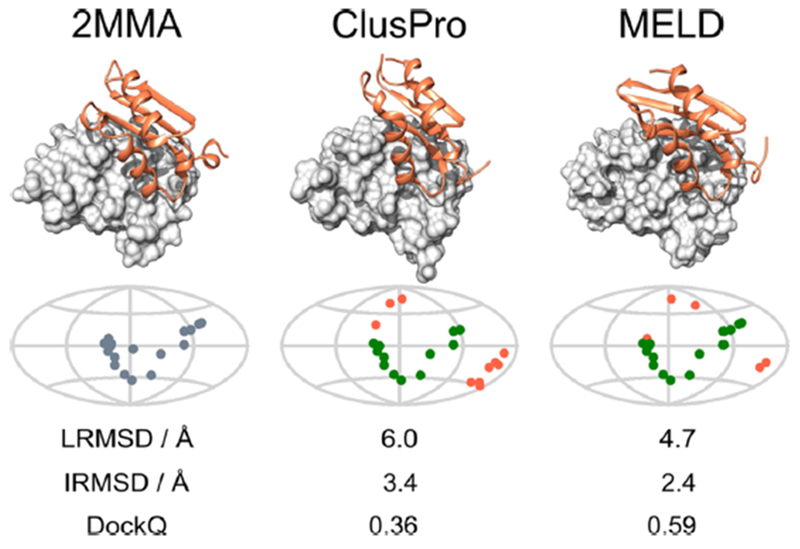
MELD × MD can provide better docking poses than CP. The left column show the native structure of the 2MMA dimer both using a 3D view and a map of the position of native contact on the first monomer. In the central column the CP TOP1 prediction is shown. In this case CP places the second monomer almost correctly; in fact from the map it is clear that this pose has 7 TP and 6 FP contacts. CAPRI ranks such pose as acceptable docking. MELD is able to use the contact of this pose for the docking process, but since we allow enough freedom to the system it predicts the final structure (right column) has 9 TP contacts and reducing the number of FP to 3 compared to native. The improvement of the CAPRI ranking from acceptable to medium reflects the benefit of this flexible approach.
3.1.3. Failures.
The four failures to predict correct dimer conformations are linked with failures of the protocol we use, or with limitations of the physical model used to describe the system. The 3T0E prediction suffered a nonconverged RE simulation. Figure 5 show that the different simulations of the RE protocol did not explore a similar set of conformations. 1R7H is also partially affected by a not totally converged RE simulation. 1R7H and 3T0E are the only two systems for which the population of the MELD TOP1 prediction is below 60% of the overall number of clustered frames. As in protein folding, low MELD cluster populations seem to flag systems for which the answer is uncertain. In 2EZX and 1R7H the ligands change conformations, making docking in our simulation time scale impossible to converge. For example, let us say that the monomers look like two hands, and the dimer native structure looks like a handshake. To achieve a correct docking the two hands must be open when the docking is attempted. It is impossible to dock correctly the two monomers if when undocked the ligand closes like a fist. This is what happens to the 2EZX and 1R7H systems. The information that is in place fails to keep the hand open, since the contacts of an open hand are a subset of the contacts of a closed one and the FF/solvent model favors a closed conformation of the ligand. A possible way to recover this is to penalize the formation of too many new intramolecular contacts, or to use a full intramolecular distances matrix instead of only a contact matrix to limit ligand conformation explorations. Interestingly, Figure 2 shows how the most often active sets of CP information for these two systems are in both cases the ones of the CP TOP15 structures. It is reasonable to assume that limiting ligand conformations for these two systems will recover the correct predictions. We briefly note here that properly tuning the flexibility of receptors and ligands has an important effect on MELD × MD ability to successfully dock dimers. Flexibility increases the size of the search space to explore. Rigidity might hinder moves that can in some case be necessary to wrap monomers around each other. In this paper we present a uniform approach for all the 20 test cases that works for most of them, but some docking attempts would benefit from individualized approaches. Finally, 1H0X prediction fails because the FF favors a different docking pose. A small bump in the third panel of the LRMSD probability highlights that the systems visits a set of native-like conformations, but this cluster appears not be the one favored by the FF. In SI 3 we show how the 16 systems that MELD × MD correctly predicts are not affected by the limitation discussed here.
Figure 5.
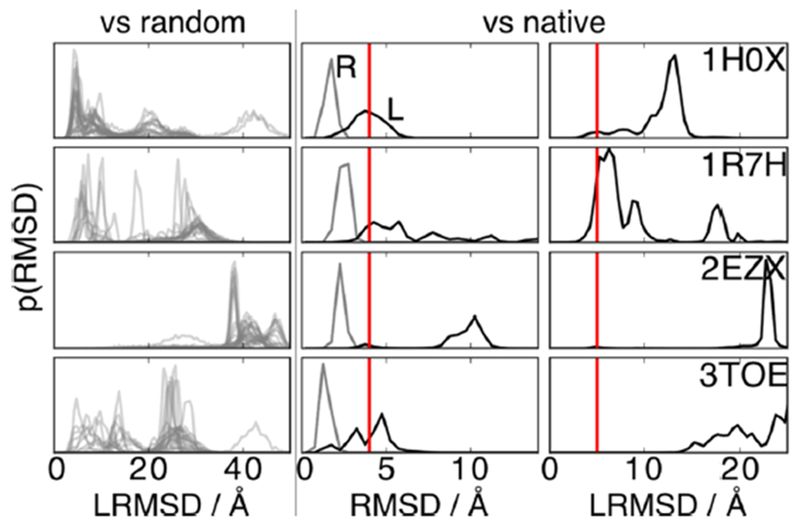
Failed predictions result from unconverged replica exchange or force field problems. We show several RMSD distributions for the four systems we fail to correctly predict (rows). The first column addresses the convergence of the RE protocol. Here are distributions of LRMSD for the 30 simulations of each RE run against a randomly selected structure from the simulation. The more similar are the traces, the more the RE simulation is converged. For 3TOE (and partially also for 1R7H) the traces vary extremely, implying a nonconverged RE simulation. The second column shows the RMSD distributions of the structure of the monomers of each dimer (receptor in gray and ligand in black) compared to their native bound structure. For 1R7H and 2EZX, the high ligand RMSD values (>4 Å to the right of the vertical red line) highlight important structural rearrangements of the second monomers during the simulations. Those rearrangements prevent the correct docking of the dimer. The last column shows the distribution of the LRMSD against native for the four systems. 1H0X visits the native conformation (<5 Å to the left of the vertical red line), but it is not the most populated state. In this case the FF/implicit solvent model combination favors another structure.
In Table 2 we look at the same quantities presented in Table 1, computed over the MELD TOP10 structure for the four systems where MELD × MD fails to predict native, and for 1Z09, for which a change in the structure of the ligand affects some of the score. All the systems affected by a change in the ligand structure (1Z09, 1R7H, and 2EZX) have small population clusters of medium quality that improve on the metrics of the TOP1 prediction. This means that when the system samples a correct ligand structure, MELD × MD is able to correctly dock the dimer. These correct ligand structures are simply sampled too rarely compared to incorrect ones, causing native docking poses not to be the most populated ones.
Table 2.
Same Quantities as in Table 1 for the MELD TOP10 Cluster for the Four Systems That MELD × MD Fails to Predict (1H0X, 1R7H, 2EZX, 3T0E) and for the 1Z09 Systema
| MELD TOP10 |
|||||||
|---|---|---|---|---|---|---|---|
| PDB id | no. | LRMSD Å | iRMSD Å | fTP % | fFP % | CAPRI score | DockQ score |
| 2EZX | 2 | 2.2 | 2.0 | 69 | 25 | M | 0.66 |
| 1R7H | 8 | 3.2 | 3.2 | 63 | 31 | M | 0.563 |
| 1Z09 | 1 | 3.4 | 1.4 | 86 | 19 | M | 0.747 |
| 1H0X | 0 | 11.2 | 4.3 | 38 | 79 | I | 0.282 |
| 3TOE | 1 | 14.9 | 6.2 | 32 | 83 | I | 0.206 |
A good structure of the ligand is crucial for correct docking predictions. Three systems (1Z09, 1R7H, 2EZX) sample native-like dimer structures often enough for them to be identified by the clustering protocol, but they are not frequent enough to be the most populated clusters of the simulations (i.e., MELD TOP1).
It is likely that limiting the flexibility of the ligand in our docking protocol would allow these three systems to be predicted correctly. It is also known that biologically relevant homodimers have a high degree of symmetry,42,43 and this implies that the structure of the two monomers needs to be similar. We can in principle use this knowledge to skim our predictions in order to eliminate homodimer predictions with significantly disparate monomer structures. We show in SI 6 that by doing so we can recover the correct predictions of these three dimers without affecting the prediction quality of the other successes.
1H0X and 3T0E are not dramatically affected by a structural rearrangement of the ligand, and they do not sample structure close to native. Failure in predicting the native state of these monomer is linked to issue of convergence of the REMD protocol and to limitations in the force field and solvent model.
3.2. We Can Apply Our Protocol to an Example CASP/CAPRI Case.
In section 3.1 we showed the success of our protocol in predicting the structure of dimers when we provide good information. Good information comes from the CP contacts prediction based on bound-complex docking and on exploring monomer conformations close to their bound structures (albeit some ligands are able to evolve to structures up to 10 Å away during the simulation). This is consistent with the aim of this paper to use flexibility to evaluate correctly the relative stability of the different binding poses. In principle, the flexibility we introduce in the system can at the same time be used to refine the monomer structures (side chains and backbone). To test to which extent our protocol is able to handle monomer structural refinement, we use the CAPRI T120 target, from the joint CAPRI round 37 - CASP 12 competition.13,44 As starting structure for the monomers we use the model # 1 submission of the Zhang-Server group (TS479),45 based on I-TASSER46–48 for the two monomers (T0921 and T0922). The monomers have a RMSD to their native structure of 3.0 and 3.4 Å, respectively. We feed these to vanilla CP and we then use the predicted contacts to limit the search space of our simulation. As the initial structure of our simulation we use the two predicted monomer structures not in contact. We run our MELD × MD simulation using the same protocol as described above. Table 3 shows a summary of the TOP1, TOP3, and TOP5MELD predictions and the TOP15 CP prediction. Our protocol is not optimized to handle the uncertainty of the monomer structures or in the intermonomer contacts. It is therefore not surprising that we fail to predict a MELD TOP1 structure close to native. However, we did identify some acceptable structures (2 and 3), and the first acceptable structure is ranked higher than the best CP structure (3). This small improvement on the CP prediction is promising. We recognize that, while this result is promising, future work (well outside the scope of this paper) is needed to fully assess the value that MELD × MD provides in more challenging conditions where monomers need to change their shapes to successfully dock. Different classes of problems fit this description: in template based docking monomers need refinement, in docking from monomer apo structure some structural rearrangement is in order, and in unstructured protein docking a folding-like process is necessary upon docking.
Table 3.
Same Quantities as in Table 1 for the CAPRI TS479 Prediction Based on Zhang-Server Monomer Structure Predictiona
| prediction | no. | LRMSD Å | iRMSD Å | fTP % | fFP % | CAPRI score | DockQ score |
|---|---|---|---|---|---|---|---|
| MELD TOP1 | 0 | 17.8 | 11.5 | 7 | 80 | I | 0.09 |
| MELD TOP3 | 2 | 8.6 | 5.5 | 13 | 82 | A | 0.23 |
| MELD TOP5 | 3 | 8.4 | 5.3 | 15 | 78 | A | 0.24 |
| CP TOP15 | 3 | 5.8 | 4.2 | 22 | 68 | A | 0.33 |
A different protocol is needed if the monomers needs refining. The protocol we used is not optimized to handle the high uncertainty of the information we input. It is therefore not surprising that we are unable to identify the most native docking pose in this case. Nonetheless the first acceptable structure is ranked 2 in MELD, while it is ranked 3 in vanilla CP. This improvement is promising.
4. CONCLUSIONS
We describe a method that gives improved protein–protein structure predictions. It starts with the 15 best structures produced by the ClusPro rigid-docking server. It then uses the interprotein contacts predicted by CP for each structure as restraints that are imposed probabalistically within the MELD Bayesian method. MELD-accelerated MD then gives improved dimer structures and produces populations (free energies) that are predictive of which dimer conformation is the best of the lot. The protocol we presents performs well if we start with the monomers having the bound backbone conformation but also demonstrates some improvement when the backbone conformation come from predictions as demonstrated in CASP/CAPRI example. In this last case, more aggressive sampling of the monomer structure would benefit the prediction. While the MELD × MD component is computationally expensive, nevertheless this approach is a promising way to compute good protein–protein binding structures.
Supplementary Material
ACKNOWLEDGMENTS
Protein images are created using the Chimera49 software. Trajectory and data are analyzed and plotted using the following libraries for python 2.7: numpy,50 SciPi,51 matplotlib,52 MDTraj,53 and Prody.54 This work has been funded by NIH grant 1R01GM125813-01 “MELD: accelerating MD modeling of proteins using Bayesian inference”, NSF DBI 1759277, and NSF AF 1816314. This research is part of the Blue Waters sustained-petascale computing project, which is supported by the National Science Foundation (Awards OCI-0725070 and ACI-1238993) and the state of Illinois. Blue Waters is a joint effort of the University of Illinois at Urbana–Champaign and its National Center for Supercomputing Applications. This work is also part of the PRAC allocation support by the NSF Awards ACI1514873 and ACI1713695. The authors thank A. Perez and O. Schueler-Furman for insightful discussions and S. Bromberg for help editing the manuscript.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jctc.8b01208.
Detailed information about the following: how we choose the 20 dimers, how to understand the metric used in the paper in the context of flexible protein docking, the convergence of the simulations, the effect of different clustering approaches on the quality of the results, how we draw the protein map, and how to use symmetry to improve predictions (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Janin J; Henrick K; Moult J; Ten Eyck L; Sternberg MJ; Vajda S; Vakser I; Wodak SJ CAPRI: a critical assessment of predicted interactions. Proteins: Struct., Funct., Genet 2003, 52, 2–9. [DOI] [PubMed] [Google Scholar]
- (2).Gray JJ; Moughon SE; Kortemme T; Schueler-Furman O; Misura KM; Morozov AV; Baker D Protein–protein docking predictions for the CAPRI experiment. Proteins: Struct., Funct., Genet 2003, 52, 118–122. [DOI] [PubMed] [Google Scholar]
- (3).Wodak SJ; Méndez R Prediction of protein–protein interactions: the CAPRI experiment, its evaluation and implications. Curr. Opin. Struct. Biol 2004, 14, 242–249. [DOI] [PubMed] [Google Scholar]
- (4).Schneidman-Duhovny D; Inbar Y; Nussinov R; Wolfson HJ PatchDock and SymmDock: servers for rigid and symmetric docking. Nucleic Acids Res 2005, 33, W363–W367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Tovchigrechko A; Vakser IA GRAMM-X public web server for protein–protein docking. Nucleic Acids Res 2006, 34, W310–W314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Torchala M; Moal IH; Chaleil RA; Fernandez-Recio J; Bates PA SwarmDock:a server for flexible protein–protein docking. Bioinformatics 2013, 29, 807–809. [DOI] [PubMed] [Google Scholar]
- (7).Shin W-H; Lee GR; Heo L; Lee H; Seok C Prediction of protein structure and interaction by GALAXY protein modeling programs. Bio Design 2014, 2, 1–11. [Google Scholar]
- (8).Esquivel-Rodriguez J; Filos-Gonzalez V; Li B; Kihara D Protein Structure Prediction; Springer, 2014; pp 209–234. [DOI] [PubMed] [Google Scholar]
- (9).Viswanath S; Ravikant D; Elber R Protein Structure Prediction; Springer, 2014; pp 199–207. [DOI] [PubMed] [Google Scholar]
- (10).Van Zundert G; Rodrigues J; Trellet M; Schmitz C; Kastritis P; Karaca E; Melquiond A; van Dijk M; De Vries S; Bonvin A The HADDOCK2. 2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol 2016, 428, 720–725. [DOI] [PubMed] [Google Scholar]
- (11).Kozakov D; Hall DR; Xia B; Porter KA; Padhorny D; Yueh C; Beglov D; Vajda S The ClusPro web server for protein–protein docking. Nat. Protoc 2017, 12, 255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Lensink MF; Velankar S; Wodak SJ Modeling protein–protein and protein–peptide complexes: CAPRI 6th edition. Proteins: Struct. Funct., Genet 2017, 85, 359–377. [DOI] [PubMed] [Google Scholar]
- (13).Lensink MF; Velankar S; Baek M; Heo L; Seok C; Wodak SJ The challenge of modeling protein assemblies: the CASP12-CAPRI experiment. Proteins: Struct. Funct., Genet 2018, 86, 257–273. [DOI] [PubMed] [Google Scholar]
- (14).Hogues H; Gaudreault F; Corbeil CR; Deprez C; Sulea T; Purisima EO Pro-POSE: Direct exhaustive protein-protein docking with side chain flexibility. J. Chem. Theory Comput 2018, 14, 4938. [DOI] [PubMed] [Google Scholar]
- (15).Shaw DE; Grossman J; Bank JA; Batson B; Butts JA; Chao JC; Deneroff MM; Dror RO; Even A; Fenton CH; et al. Anton 2: raising the bar for performance and programmability in a special-purpose molecular dynamics supercomputer. Proceedings of the international conference for high performance computing, networking, storage and analysis. 2014, 41–53. [Google Scholar]
- (16).Needham SR; Roberts SK; Arkhipov A; Mysore VP; Tynan CJ; Zanetti-Domingues LC; Kim ET; Losasso V; Korovesis D; Hirsch M; et al. EGFR oligomerization organizes kinase-active dimers into competent signalling platforms. Nat. Commun 2016, 7, 13307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Pan AC; Jacobson D; Yatsenko K; Sritharan D; Weinreich TM; Shaw DE Atomic-level characterization of protein-protein association. bioRxiv.org 2018, 303370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Hou Q; Lensink MF; Heringa J; Feenstra KA Clubmartini: selecting favourable interactions amongst available candidates, a coarse-grained simulation approach to scoring docking decoys. PLoS One 2016, 11, No. e0155251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).MacCallum JL; Perez A; Dill KA Determining protein structures by combining semireliable data with atomistic physical models by Bayesian inference. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 6985–6990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Perez A; MacCallum JL; Dill KA Accelerating molecular simulations of proteins using Bayesian inference on weak information. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 11846–11851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Morrone JA; Perez A; MacCallum J; Dill KA Computed Binding of Peptides to Proteins with MELD-Accelerated Molecular Dynamics. J. Chem. Theory Comput 2017, 13, 870–876. [DOI] [PubMed] [Google Scholar]
- (22).Morrone JA; Perez A; Deng Q; Ha SN; Holloway MK; Sawyer TK; Sherborne BS; Brown FK; Dill KA Molecular Simulations Identify Binding Poses and Approximate Affinities of Stapled α-Helical Peptides to MDM2 and MDMX. J. Chem. Theory Comput 2017, 13, 863–869. [DOI] [PubMed] [Google Scholar]
- (23).Perez A; Morrone JA; Dill KA Accelerating physical simulations of proteins by leveraging external knowledge. Wiley Interdiscip. Rev. Comput. Mol. Sci 2017, 7, No. e1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Perez A; Morrone JA; Brini E; MacCallum JL; Dill KA Blind protein structure prediction using accelerated free-energy simulations. Sci. Adv 2016, 2, No. e1601274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Pilla KB; Gaalswyk K; MacCallum JL Molecular modeling of biomolecules by paramagnetic NMR and computational hybrid methods. Biochim. Biophys. Acta, Proteins Proteomics 2017, 1865, 1654–1663. [DOI] [PubMed] [Google Scholar]
- (26).Comeau SR; Gatchell DW; Vajda S; Camacho CJ ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 2004, 20, 45–50. [DOI] [PubMed] [Google Scholar]
- (27).Comeau SR; Gatchell DW; Vajda S; Camacho CJ ClusPro: a fully automated algorithm for protein–protein docking. Nucleic Acids Res 2004, 32, W96–W99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kozakov D; Brenke R; Comeau SR; Vajda S PIPER: an FFT-based protein docking program with pairwise potentials. Proteins: Struct., Funct., Genet 2006, 65, 392–406. [DOI] [PubMed] [Google Scholar]
- (29).Kozakov D; Beglov D; Bohnuud T; Mottarella SE; Xia B; Hall DR; Vajda S How good is automated protein docking? Proteins: Struct., Funct., Genet 2013, 81, 2159–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Sugita Y; Kitao A; Okamoto Y Multidimensional replica-exchange method for freeenergy calculations. J. Chem. Phys 2000, 113, 6042–6051. [Google Scholar]
- (31).Fukunishi H; Watanabe O; Takada S On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: Application to protein structure prediction. J. Chem. Phys 2002, 116, 9058–9067. [Google Scholar]
- (32).Hornak V; Abel R; Okur A; Strockbine B; Roitberg A; Simmerling C Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins: Struct., Funct., Genet 2006, 65, 712–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Maier JA; Martinez C; Kasavajhala K; Wickstrom L; Hauser KE; Simmerling C ff14SB: improving the accuracy of protein side chain and backbone parameters from ff99SB. J. Chem. Theory Comput 2015, 11, 3696–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Nguyen H; Roe DR; Simmerling C Improved generalized born solvent model parameters for protein simulations. J. Chem. Theory Comput 2013, 9, 2020–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Eastman P; Friedrichs MS; Chodera JD; Radmer RJ; Bruns CM; Ku JP; Beauchamp KA; Lane TJ; Wang L-P; Shukla D; et al. OpenMM 4: a reusable, extensible, hardware independent library for high performance molecular simulation. J. Chem. Theory Comput 2013, 9, 461–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Case DA; Babin V; Berryman J; Betz R; Cai Q; Cerutti D; Cheatham T III; Darden T; Duke R; Gohlke H; et al. , Amber 14; 2014. [Google Scholar]
- (37).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Basu S; Wallner B DockQ: A Quality Measure for Protein-Protein Docking Models. PLoS One 2016, 11, No. e0161879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Ester M; Kriegel H-P; Sander J; Xu X A density-based algorithm for discovering clusters in large spatial databases with noise. KDD-96 Proc 1996; pp 226–231. [Google Scholar]
- (40).Pedregosa F; Varoquaux G; Gramfort A; Michel V; Thirion B; Grisel O; Blondel M; Prettenhofer P; Weiss R; Dubourg V; Vanderplas J; Passos A; Cournapeau D; Brucher M; Perrot M; Duchesnay E Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res 2011, 12, 2825–2830. [Google Scholar]
- (41).Snyder JP Map projections–A working manual; US Government Printing Office, 1987; Vol. 1395. [Google Scholar]
- (42).Lukatsky DB; Shakhnovich BE; Mintseris J; Shakhnovich EI Structural similarity enhances interaction propensity of proteins. J. Mol. Biol 2007, 365, 1596–1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).André I; Strauss CEM; Kaplan DB; Bradley P; Baker D Emergence of symmetry in homooligomeric biological assemblies. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 16148–16152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Moult J; Fidelis K; Kryshtafovych A; Schwede T; Tramontano A Critical assessment of methods of protein structure prediction (CASP)—Round XII. Proteins: Struct, Funct., Genet 2018, 86, 7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhang C; Mortuza S; He B; Wang Y; Zhang Y Template-based and free modeling of I-TASSER and QUARK pipelines using predicted contact maps in CASP12. Proteins: Struct., Funct., Genet. 2018, 86, 136–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Zhang Y I-TASSER server for protein 3D structure prediction. BMC Bioinform 2008, 9, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Roy A; Kucukural A; Zhang Y I-TASSER: a unified platform for automated protein structure and function prediction. Nat. Protoc 2010, 5, 725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Yang J; Yan R; Roy A; Xu D; Poisson J; Zhang Y The I-TASSER Suite: protein structure and function prediction. Nat. Methods 2015, 12, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- (50).Oliphant TE A guide to NumPy; Trelgol Publishing; USA, 2006; Vol. 1. [Google Scholar]
- (51).Jones E; Oliphant T; Peterson P; et al. SciPy: Open source scientific tools for Python; 2001; http://www.scipy.org/.
- (52).Hunter JD Matplotlib: A 2D graphics environment. Comput. Sci. Eng 2007, 9, 90–95. [Google Scholar]
- (53).McGibbon R T.; et al. MDTraj: A Modern Open Library for the Analysis of Molecular Dynamics Trajectories. Biophys. J 2015, 109, 1528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Bakan A; Meireles LM; Bahar I ProDy: protein dynamics inferred from theory and experiments. Bioinformatics 2011, 27, 1575–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.