

Abstract
Objective:
We tested the hypothesis that chronic fetal hypoxia, at a severity present in many types of congenital heart disease, would lead to abnormal neurodevelopment.
Methods:
Eight mid-gestation fetal sheep were cannulated onto a pumpless extracorporeal oxygenator via the umbilical vessels and supported in a fluid-filled environment for 22 ± 2 days under normoxic or hypoxic conditions. Total parenteral nutrition was provided. Control fetuses (n = 7) were harvested at gestational age 133 ± 4 days. At necropsy, brains were fixed for histopathology. Neurons were quantified in white matter tracts, and the thickness of the external granular layer of the cerebellum was measured to assess neuronal migration. Capillary density and myelination were quantified in white matter. Data were analyzed with unpaired Student t tests or 1-way analysis of variance, as appropriate.
Results:
Oxygen delivery was reduced in hypoxic fetuses (15.6 ± 1.8 mL/kg/min vs 24.3 ± 2.3 mL/kg/min, P <.01), but umbilical blood flow and caloric delivery were not different between the 2 groups. Compared with normoxic and control animals, hypoxic fetuses had reduced neuronal density and increased external granular layer thickness. Compared with normoxic and control animals, hypoxic fetuses had increased capillary density in white matter. Cortical myelin integrity score was lower in the hypoxic group compared with normoxic and control animals. There was a significant negative correlation between myelin integrity and capillary density.
Conclusions:
Chronic fetal hypoxia leads to white matter hyper-vascularity, decreased neuronal density, and impaired myelination, similar to the neuropathologic findings observed in children with congenital heart disease. These findings support the hypothesis that fetal hypoxia, even in the setting of normal caloric delivery, impairs neurodevelopment.
Keywords: congenital heart disease, neurodevelopment, fetal hypoxia
Graphical Abstract
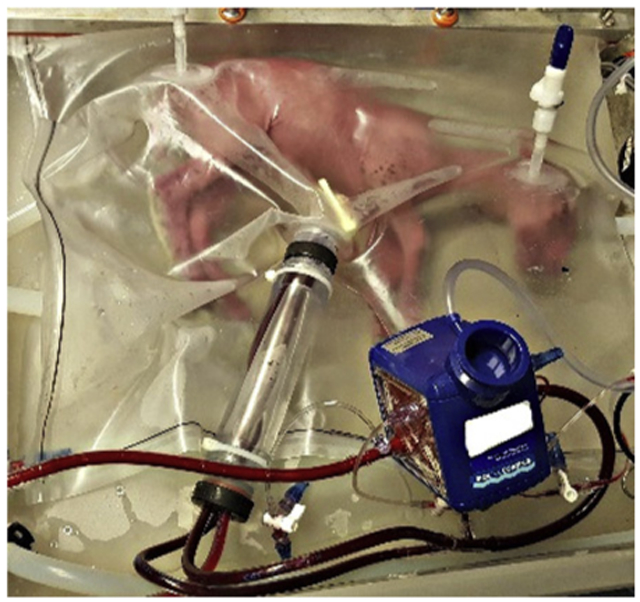
Representative hypoxemic fetal lamb supported in the extra-uterine support device.
Severe congenital heart disease (CHD) affects 6 of every 1000 live births.1 Improved surgical and perioperative care have transformed these once lethal defects into chronic conditions with greater than 90% of patients now surviving into adulthood.2 With long-term survival now the norm, there has been an increased focus on neurodevelopmental outcomes among survivors of complex CHD.3
Multiple studies have shown that children with CHD repaired in infancy lag behind peers in motor, language, and memory development, regardless of their operative approach, suggesting that neurologic injury may occur early in life.3-6 Subsequent investigations have demonstrated that anatomic and functional brain abnormalities are present in neonates before surgical repair.7-12 This relative brain immaturity increases the risk for perioperative injury and may alter subsequent brain development.12 More recently, with advances in fetal imaging, cerebral abnormalities have been identified as early as 32 weeks gestation in fetuses with CHD.13-15 This recognition of early postnatal and prenatal brain anomalies suggests that neurodevelopmental delays may be the result of in utero injuries and alterations to the normal patterns of brain development. Although considerable debate exists,16 there is speculation that inadequate cerebral oxygen delivery, a consequence of impaired cardiac output and abnormal blood streaming in the fetus with CHD, contributes to these in utero changes.13-15 Others argue that impaired glucose delivery, more than hypoxemia, contributes to in utero alterations in brain development and subsequent neurocognitive delays.16
Until now, additional insight into this pathophysiology has been extremely limited because of the challenges of studying fetuses in utero. Recently, an artificial womb was developed that mimics in utero physiology and permits the study of fetuses under both normal and pathologic intrauterine conditions.17 To gain insight into the etiology of compromised neurodevelopmental outcomes in children with CHD, we used this model to test the hypothesis that isolated chronic intrauterine hypoxia would alter neurodevelopment.
MATERIALS AND METHODS
Animal Surgery and Support
Animals were treated according to approved protocols by the institutional animal care and use committee of The Children’s Hospital of Philadelphia.
Time-dated ewes were sedated with 15 mg/kg of intramuscular ketamine and maintained under general anesthesia with inhaled isoflurane (2%-4% in O2) and propofol infusion (0.2-1.0 mg/kg/min). Eight fetuses at gestational age (GA) 109 ± 3 days (term ~145 days) were exteriorized via laparotomy and hysterotomy, cannulated via their umbilical vessels and attached to a pumpless low-resistance oxygenator circuit. They were then transitioned to a sterile fluid environment where they were supported within the Extra-uterine Environment for Neonatal Development (EXTEND) system. The surgical procedure and EXTEND support system have been described in detail by Partridge and colleagues.17
EXTEND fetuses were supported under hypoxic or normoxic conditions. Normoxia was defined as oxygen delivery 20 to 25 mL/kg/min, and hypoxia was defined as oxygen delivery 14 to 16 mL/kg/min to mimic physiologic18 and pathologic intrauterine conditions, respectively.13,15 Levels of intrauterine hypoxia reflect those observed in severe congenital cardiac lesions, particularly transposition of the great arteries.13,15 Hypoxic conditions were initiated after 1 day in the EXTEND system to permit stabilization postcannulation at GA neurologically equivalent to a 28- to 30-week preterm infant.19 Fetal oxygen delivery was continuously measured and recorded (LabChart 5, ADInstruments Inc, Colorado Springs, Colo) via measurement of weight-based umbilical blood flow (HXL Tubing Flowsensor, Transonic Systems Inc, Ithaca, NY), postmembrane saturation, and hematocrit concentration (M2-Sensor, Spectrum Medical, Gloucester, United Kingdom). When oxygen delivery fell outside target ranges, this triggered an adjustment in the oxygen tension of the sweep gas, a blended mixture of nitrogen, air, and oxygen.
All EXTEND animals received continuous total parenteral nutrition and lipids (intra-lipid 20%, 0.01-0.02 g/kg/d) to mimic sheep requirements in utero.20 Dextrose and TrophAmine (B. Braun Medical, Bethlehem, Pa) content were titrated to target a blood glucose of 20 to 30 mg/dL and blood urea nitrogen of 30 mg/dL, respectively. Unlike prior published protocols, no insulin or sedatives were used to standardize nutritional delivery and minimize experimental variables.17
EXTEND animals were humanely euthanized at GA 132 ± 3 days (pentobarbital sodium 117 mg/kg and phenytoin sodium 15 mg/kg). Brains were immediately perfusion fixed with 10% formalin at 55 cm to give a hydrostatic pressure equal to mean arterial pressure of 40 mm Hg. An additional 7 fetuses at GA of 133 ± 4 days were delivered via Caesarian section from time-dated ewes and euthanized, and underwent brain perfusion fixation in an identical manner to serve as tissue controls.
Histopathologic Analysis
After euthanasia, the cerebrum and cerebellum were removed from the skull and weighed. Coronal slices (4 mm) of the forebrain and cerebellum were cut using a brain matrix (Ted Pella Inc, Redding, Calif) and then paraffin embedded. Five-μm–thick paraffin sections were stained for hematoxylin and eosin (H&E) and reviewed by a board-certified veterinary neuro-pathologist to assess the presence, nature, and severity of histopathologic changes.
Calculation of Surface Fold Index
Gyral formation was assessed on H&E slides in comparable frontal and temporal lobe sections by calculation of the surface fold index. From the superior aspect of the lateral ventricle, perpendicular lines were drawn to the cortical surface. The area (A) and length (L) of this cortical surface were measured in triplicate (Aperio Imagescope Version 12.3, Leica Biosystems, Buffalo, Ill) and used to calculate the surface fold index (= L2/A). Surface fold index is unaffected by tissue shrinkage and used to estimate surface development relative to the volume growth.21
Neuronal Density Quantifications
Equivalent sections from the frontal and temporal lobes and cerebellum were stained with Luxol fast blue with cresyl violet counterstain to evaluate neuronal density in distal gyral projections of the corona radiata and CA1 region of the hippocampus.22 To address potential bias with this 2-dimensional approach, neuronal density was quantified in 4 high-powered fields (4 mm2at 10× objective) and averaged for each region of interest (ROI) to avoid poorly representative sampling windows.
At the level of the inferior cerebellar peduncle, the external granular layer was measured along the anterior cerebellar surface at 40 × objective. Measurements were only taken when all layers of the cerebellar cortex were identified in the field to ensure measurements occurred in the same plane. Three measurements were taken, and the mean height of the external granular cell layer was calculated for comparisons.
Immunohistochemistry
Anatomically equivalent frontal and temporal lobe sections were reacted with antibodies. Endothelial cells were visualized with rabbit CD34 antisera (1:50, 81289; ABCam, Cambridge, Mass). Myelin was visualized with rabbit myelin basic protein (1:50, MAB386; EMD Millipore Corporation. Temecula, Calif). Apoptotic cells were visualized with rabbit caspase antibody (1:100, ab4051). Sections were incubated overnight with anti-rabbit secondary antibodies (1:500, ThermoFisher, Waltham, Mass) and developed with horseradish peroxidase (SK-410; Vector Laboratories, Burlingame, Calif) for visualization of primary antibodies.
Slides were scanned at 40× magnification and evaluated using Aperio Imagescope Version 12.3 (Leica Biosystems, Buffalo Grove, Ill). Comparative morphometric analyses were performed on highly homologous and anatomically matched sections. Specific neuroanatomic landmarks, including the anterior horn of the lateral ventricle, hippocampus, and inferior cerebellar peduncle, were used to select frontal lobe, temporal lobe, and cerebellar sections, respectively (Figure 1, A-C). Periventricular white matter boundaries were defined by a horizontal line drawn tangent to the lateral ventricle at the head of the caudate nucleus, connecting to the fundi of adjacent sulci, and the white matter boundaries at those fundi.19 Grey matter was evaluated adjacent to these boundaries. Quantitative measures described below reflect these ROIs. ROIs were analyzed in 3 fields at 5 × magnification (16 mm2) for each animal, and the mean values were compared between groups.
FIGURE 1.

Brain regions of interest analyzed. Coronal, Luxol fast blue stained, hemisections of the cerebral hemisphere at the level of the frontal (A) and temporal (B) lobe and section of cerebellum (C). Squares indicate areas analyzed for histopathology. Measurements of neuronal density were made in subcortical white matter (WM) and CA1 region of the hippocampus. Myelin integrity evaluated in periventricular WM and subcortical WM. Vascular density and apoptotic bodies quantified in periventricular WM and subcortical WM and gray matter. External granular layer measured in cerebellar sections. EGL, External granular layer; GM, gray matter.
Quantification of Vascular Density
Vascular density was evaluated by counting number of vessels per high-powered field. Vessels were identified by a trained veterinary neuropathologist by the presence of a lumen lined with endothelial cells and verified with CD34 positivity.
Calculation of Myelin Integrity Score
A blinded neuropathologist evaluated the quality and quantity of myelin basic protein-positive cells at primary and tertiary projections of the corona radiata in equivalent frontal and temporal lobe sections. A validated scale of 1 to 3 was used to assess myelin quality: (1) clumpy myelin fibers and total loss of linear organization, (2) mixture of thicker and thinner processes and partial loss of linear fiber organization, and (3) fine diffuse processes and consistently linear organization of myelinated processes.2 The same blinded neuropathologist assessed myelin quantity using a scale of 1 to 4: (1) 0% to 25% myelination; (2) 26% to 50% myelination; (3) 51% to 75% myelination; and (4) 76% to 100% myelination. The sum of the myelin quality and quantity score yielded the total myelin integrity score for each ROI.
Quantification of Apoptotic Cells
For detection of apoptotic cells, caspase-3 positive cells were manually counted in ROIs. In addition to cortical ROIs, cerebellar white and grey matter were also analyzed for caspase-positive cells.
Statistical Analysis
All analyses and graphical depictions of data were performed in GraphPad Prism V6 (GraphPad Software, San Diego, Calif). Hemodynamic parameters and oxygen delivery were averaged over 12 hours and analyzed using a mixed linear effects model to compare groups over time. Normality of each dataset was assessed by Shapiro–Wilk tests. Histologic quantifications were analyzed using 1-way analysis of variance or Kruskal–Wallis tests, as appropriate based on data distribution. When significant differences existed, multiple comparisons were performed with Tukey or Dunn’s post-tests, respectively. The relationship between myelin integrity score and capillary density in matched brain sections was determined with Pearson’s correlation coefficient, and 2-tailed Student t test was used to determine the significance of the correlation. All data are presented as mean ± standard deviation.
RESULTS
Animal Groups
Four cannulated animals were maintained under hypoxic conditions and compared with 4 animals maintained under normoxic conditions. Table 1 shows the characteristics of each EXTEND group. Duration of extra-uterine support was similar between the normoxic and hypoxic groups. The hypoxic and normoxic animals also had similar umbilical blood flow and umbilical artery pressure throughout their study periods. The groups also had similar dextrose and TrophAmine supplied in their parenteral nutrition daily. Hemodynamic and caloric delivery parameters in both normoxic and hypoxic groups were similar to reported reference values for in utero sheep fetuses.20,24 The groups had similar carbon dioxide levels but significant differences in their oxygenation parameters. The hypoxic group had significantly reduced venous oxygen tension and oxygen delivery throughout the study period.
TABLE 1.
Extra-uterine environment for neonatal development groups was similar except for oxygenation parameters
| Normoxic (n = 4) | Hypoxic (n = 4) | P value | |
|---|---|---|---|
| Male sex (n) | 2 | 2 | 1.00 |
| Age at cannulation (d) | 107 ± 2 | 111 ± 3 | .07 |
| Duration of support (d) | 22.8 ± 3.4 | 22.2 ± 0.9 | .71 |
| Umbilical blood flow (mL kg−1 min−1) | 191 ± 35 | 174 ± 60 | .65 |
| Umbilical artery pressure (mm Hg) | 35 ± 3 | 34 ± 2 | .44 |
| Dextrose delivery (kcal kg−1 d−1) | 38.5 ± 4.3 | 42.2 ± 4.3 | .27 |
| TrophAmine (B. Braun Medical, Bethlehem, Pa)delivery (kcal kg−1 d−1) | 25.2 ± 4.6 | 24.4 ± 3.7 | .77 |
| Lipid delivery (kcal kg−1 d−1) | 1.9 ± 0.3 | 2.5 ± 1.0 | .29 |
| Oxygen delivery (mL kg−1min−1) | 24.3 ± 2.3 | 15.6 ± 1.8 | <.01 |
| Venous carbon dioxide tension (mm Hg) | 39.1 ± 0.2 | 39.5 ± 2.5 | .75 |
| Venous oxygen tension (mm Hg) | 40 ± 10 | 22 ± 6 | .02 |
All data shown as mean ± standard deviation.
Brain Weight and Growth
Table 2 shows brain and body weights for each group. Normoxic and hypoxic EXTEND groups had reduced body weights compared with intrauterine control animals. Cortical and cerebellar weights were also reduced in both EXTEND groups compared with control animals. When corrected for body weight, cortical and cerebellar weights were similar for all 3 groups, although they tended to be increased in the hypoxic animals.
TABLE 2.
Brain weight is preserved relative to body weight in hynoxic animals
| Intrauterine control (n = 7) | Normoxic (n = 4) | Hypoxic (n = 4) | P value | |
|---|---|---|---|---|
| GA at necropsy (d) | 133 ± 4 | 133 ± 3 | 131 ± 3 | .51 |
| Weight at necropsy (kg) | 3.82 ± 0.35 | 2.91 ± 0.43 | 2.59 ± 0.83* | <.01 |
| Cortex weight at necropsy (g) | 44.51 ± 5.18 | 37.38 ± 3.34 | 36.13 ± 0.1.50* | .01 |
| Cortex weight at necropsy (g kg−1) | 11.64 ± 0.85 | 13.11 ± 2.63 | 15.09 ± 4.68 | .18 |
| Cerebellum weight at necropsy (g) | 6.24 ± 0.74 | 5.33 ± 0.41 | 4.9 ± 0.47* | .02 |
| Cerebellum weight at necropsy (g kg−1) | 1.63 ± 0.12 | 1.88 ± 0.43 | 2.02 ± 0.54 | .23 |
| Total brain weight at necropsy (g) | 50.76 ± 5.70 | 42.7 ± 3.65 | 41.08 ± 1.98* | <.01 |
| Total brain weight at necropsy (g kg−1) | 13.28 ± 0.86 | 14.99 ± 3.05 | 17.13 ± 5.19 | .18 |
P value denotes difference among 3 groups (analysis of variance). GA, Gestational age.
Significant difference between hypoxic and control groups. Data shown as mean ± standard deviation.
Microscopic Injury Was Rare in All Groups
On H&E staining, 1 punctate area of focal necrosis was identified in the frontal lobe white matter of 1 hypoxic animal; no other necrotic or hemorrhagic lesions were identified in any animal. Median rates of apoptosis were similar in hypoxic, control, and normoxic animals’ frontal lobe (P = .87) and cerebellar white matter (P = .90) ROIs.
Hypoxic Animals Had Reduced Cortical Folding
Surface fold indices were similar in normoxic and control animals (40.5 ± 7.8 vs 41.5 ± 9.4, P = .98). However, surface fold indices were significantly reduced in hypoxic animals compared with both normoxic (24.2 ± 3.8 vs ± 7.8, P = .03) and control animals (24.2 ± 3.8 vs ± 9.4, P = .01).
Hypoxic Animals Had Reduced Neuronal Density
Hypoxic animals had reduced neuronal density in frontal lobe distal white matter tracts compared with both normoxic (4 ± 3 vs 8 ± 1 neurons/4 mm2, P =.03) and control animals (4 ± 3 vs 11 ± 4 neurons/4 mm2, P = .01) (Figure 2, A-D). In contrast, frontal lobe distal white matter neuronal density was similar when normoxic and control animals were compared (8 ± 1 vs 11 ± 4 neurons/4 mm2, P = .19). In temporal lobe sections, neuronal density was also reduced in the hippocampal CA1 region in hypoxic animals compared with both normoxic (13 ± 1 vs 22 ± 2 neurons/4 mm2, P <.01) and control animals (13 ± 1 vs 22 ± 4 neurons/4 mm2, P = .02).
FIGURE 2.

A, Neuron count was significantly reduced in the distal WM tracts of hypoxic animals compared with normoxic animals (P = .03) and control animals (P = .01) per high-powered field, which was defined at 4 mm2 (C). Neuronal density was similar between normoxic (B) and control (D) animals (P = .19). Each data point represents 1 animal. Horizontal lines represent median values. Scale bar: 100 μm and inlay, 20 μm. E, Thickness of the cerebellar external granular layer was significantly increased in hypoxic animals compared with normoxic (P < .01) and control animals (P <.01) (G). External granular layer was similar thickness in normoxic (F) and control animals (P = .70) (H). Each data point represents 1 animal. Horizontal lines represent median values. Scale bar: 100 μm and inlay, 20 μm. HPF, High-powered field.
Thickness of the external granular layer was significantly increased in hypoxic animals compared with normoxic (31 ± 7 μm vs 15 ± 6 μm, P = .01) and control animals (31 ± 7 μm vs 13 ± 3 μm, P <.01) (Figure 2, E-H). In contrast, normoxic and control animals had similar external granular layer heights (15 ± 6 μm vs 13 ± 3 μm, P = .7).
Hypoxic Animals Had Increased Vascularity in White Matter Regions of Interest
In frontal lobe white matter, capillary density was similar between normoxic and control animals (8 ± 4 vs 6 ± 3 vessels/48 mm2, P = .5), but increased in hypoxic animals compared with both normoxic (26 ± 4 vs 8 ± 4 vessels/48 mm2, P <.01) and control animals (26 ± 4 vs 6 ± 3 vessels/48 mm2, P < .01) (Figure 3, A-D). Likewise, in the temporal lobe white matter, capillary density was also increased in hypoxic animals compared with normoxic and control animals (25 ± 4 vs 13 ± 4 vs 8 ± 4 vessels/48 mm2, P < .01). In contrast, gray matter capillary density was similar among the hypoxic, control, and normoxic groups in the frontal lobe (1 ± 1 vs 2 ± 1 vs 3 ± 1 vessels/48 mm2, P = .32) and temporal lobe (3 ± 1 vs 3 ± 2 vs 3 ± 2 vessels/ 48 mm2, P = .98) ROIs.
FIGURE 3.

A, Capillary density was significantly increased in frontal lobe white matter regions of interests in hypoxic animals compared with normoxic (P <.01) and control animals (P <.01) (C). Capillary density was similar in normoxic (B) and control animals (P = .47) (D). High-powered field defined as 48 mm2. Each data point represents 1 animal. Horizontal lines represent median values. Scale bar main panels, 200 μm; inset panels 50 μm. HPF, High-powered field.
Myelination integrity scores were reduced in hypoxic animals and highly correlated with increased vascular density. In the hypoxic, normoxic, and control groups, myelin integrity scores were similar in central white matter ROIs of the frontal lobe (5.8 ± 1.0 vs 6.8 ± 0.5 vs 6.8 ± 0.5, P = .12) and temporal lobe (6.3 ± 1.0 vs 6.8 ± 0.5 vs 6.7 ± 0.8, P = .71). However, in hypoxic animals, myelin integrity was reduced in distal white matter tracts of the frontal lobe compared with normoxic (2.5 ± 1.0 vs 4.0 ± 0.8, P = .06) and control animals (2.5 ± 1.0 vs 5.0 ± 0.8, P < .01). This same pattern was observed in the temporal lobe where myelin integrity was reduced in hypoxic animals compared with normoxic (3.0 ± 0.8 vs 5.3 ± 1.7, P = .02) and control animals (3.0 ± 0.8 vs 4.9 ± 0.9, P = .03) (Figure 4, A-D). In contrast, when normoxic and control animals were compared, myelination was similar in both the frontal lobe (4 ± 0.8 vs 5 ± 0.8, P = .18) and temporal lobe (5.3 ± 1.7 vs 4.9 ± 0.9, P = .67) ROIs.
FIGURE 4.

A, Myelin integrity was significantly reduced in distal white matter tracts of hypoxic animals compared with normoxic (P = .02) and control animals (P = .03). Myelin integrity was similar in normoxic (B) and control (D) animals (P = .67). In hypoxic animals (C), myelin fibers were rare and lose their fine, linear organization. Each data point represents 1 animal. Horizontal lines represent median values. Scale bar: 50 μm.
There was a significant negative correlation between the myelin integrity score and the capillary density in the frontal lobe (Figure 5) and temporal lobe (r = −0.71, P < .01) ROIs; in hypoxic animals, reduced myelin integrity was identified in regions with increased capillary density (Figure 5).
FIGURE 5.

For all animals, there was a significant negative correlation between the myelin integrity score and the capillary density in the frontal lobe (r = −0.79, P <.01).
DISCUSSION
In this study, chronic fetal hypoxia led to reduced cortical folding, decreased neuronal density, white matter hypervascularity, and impaired myelination. Of note, there was no evidence of increased ischemic injury or cell death in the hypoxic animals, suggesting that the impact of chronic hypoxia is due to alteration of normal developmental pathways. This pattern of relative brain immaturity is similar to the findings observed in children born with CHD and may impart increased risk of perioperative brain injury.11,12 Previous magnetic resonance oximetry studies have demonstrated that fetal cerebral oxygen delivery is reduced in a variety of congenital heart lesions,13-15 but the impact of reduced cerebral oxygenation on neurodevelopment has been debated.16 By using the EXTEND device, we supported fetuses under both normoxic and hypoxic conditions while maintaining similar blood flow, umbilical pressures, and caloric delivery between groups. Normoxic and control animals had no evidence of white matter injury and similar brain maturity by histopathology. Demonstration of abnormal brain pathology under hypoxic conditions strongly supports the hypothesis that fetal hypoxia, even in the setting of normal caloric delivery, alters neurodevelopment.
Postnatal imaging studies in term infants with CHD have demonstrated asymmetric brain growth with reduced cortical in-folding, myelination, and neuronal connectivity compared with age-matched controls.11,25,26 More recently, prenatal studies have demonstrated that this relative brain immaturity even predates birth because fetuses with CHD already have reduced cortical infolding and decreased ratios of N-acetyl aspartate:choline,7 suggesting delayed dendritic development, synaptogenesis, and oligodendrocyte proliferation. We found that our hypoxic animals also tended to have asymmetric brain growth, reduced surface fold indices, a histologic measurement of cortical folding, and reduced myelin integrity compared with controls. Although we could not assess neuronal connectivity, we found that the external granular layer of the cerebellum was thickened and neuronal density was reduced in distal white matter tracts and the hippocampus. As neurons are expected to migrate outward during gestation to establish synaptic connections,21,27 these findings suggest in utero hypoxia may impair neuronal migration, especially in the absence of increased apoptosis in these areas. These histologic findings closely replicate imaging findings in fetuses and newborns with CHD, strongly supporting the hypothesis that in utero hypoxia impairs neurodevelopment in CHD.
Histopathologic correlates to these imaging findings have been less well characterized. Autopsies in newborns with CHD and fetuses with growth restriction, another condition characterized by intrauterine hypoxia, have similarly identified abnormal brain growth, impaired myelin integrity, and reduced neuronal density, but at levels more severe than we identified in our hypoxic fetuses. Given their comparably shorter duration of hypoxemia, earlier GA at evaluation, preserved cerebral blood flow, and lack of postnatal exposures, it is not surprising that morphologic and histopathologic changes are not as severe in our hypoxic animals compared with human fetuses with CHD.28,29 Animal models of growth restriction and hypoxemia have similarly demonstrated evidence of impaired neurogenesis and myelination.19,30 Of note, these studies established that alterations in brain microstructure and maturation might occur early in life, but the etiology of these brain lesions has remained elusive until now.
Recent literature suggests that chronic hypoxia impairs myelination via arrest of oligodendrocyte maturation,31,32 which is supported by our findings. Oxygen tension mediated by hypoxia inducible factor 1-alpha (HIF-1α) has been shown to be an essential regulator of myelination.31,32 Under hypoxic conditions, HIF-1α is upregulated to promote angiogenesis and increase local tissue oxygen delivery to oligodendrocytes and their precursors.33 HIF-1α activation suppresses oligodendrocyte maturation via WNT pathway activation, thereby preventing myelination.31,32 Yuen and associates31 recently demonstrated in a postnatal mouse model that constitutive HIF-1α activation under chronic hypoxic conditions suppresses oligodendrocyte maturation via sustained activation of the WNT pathway, thereby preventing oligodendrocyte maturation.31,32 By using a neonatal mouse model, Kanaan and co-workers 33 demonstrated that chronic hypoxia both induces brain angiogenesis and irreversibly reduces the extent of myelination. We speculate that this pathway may be responsible for the increased white matter vascularity and myelination deficits we identified under chronic intrauterine hypoxia. In support of this hypothesis, we have previously demonstrated significantly elevated HIF-1α serum levels in hypoxic animals supported in the EXTEND system compared with normoxic and control animals.34 We show that increased capillary density is negatively correlated to myelin integrity in white matter of hypoxic animals (Figure 5). To confirm that HIF-1α upregulation under hypoxic conditions inhibits oligodendrocyte maturation and myelination before birth, future studies are needed to quantify expression of angiogenic transcription factors and their downstream targets, including oligodendrocyte precursors, in white matter ROIs.
Clinical Significance
Studies in adolescents with CHD have shown that early changes in brain microstructure can have long-lasting implications. Relative brain immaturity at birth increases the risk of additional perioperative injury and may hinder ongoing brain development.12 Newborns with CHD have deficits in myelination and neuronal connectivity11,26 that we propose are acquired prenatally as the result of hypoxic intrauterine conditions. These alterations in brain structure persist into adolescence and are associated with worse performance on cognitive assessments.4 Improved understanding of the relationship between prenatal brain injury and postnatal neurocognitive sequelae may ultimately have implications for future fetal-based therapies, such as maternal hyper-oxygenation and normoxic extrauterine support of the hypoxic fetus. Finally, although many excellent sheep models of intrauterine hypoxemia have been described,21,23,24 establishment of this model provides a unique opportunity to study the effects of hypoxemia in isolation, investigate potential interventions, and further characterize pathways involved in brain dysmaturity.
Study Limitations
There are several limitations to this study. First, we assumed cerebral oxygen delivery was reduced in hypoxic animals based on blood flow and oxygen saturation measurements at the umbilical vessels, because we previously found invasive cerebral catheters were poorly tolerated in our model.35 Although cerebral oxygen deprivation characterizes a number of cyanotic heart lesions, there are significant differences in cerebral oxygen saturation and blood flow between lesions that may also affect neurodevelopment and thus cannot be accounted for in this study.8,15 As such, these neuropathologic findings are not universally applicable to all congenital heart lesions but do still provide important general insight into the implications of intrauterine hypoxia. Next, to demonstrate that our model recapitulated neuropathology observed in CHD, we drew comparisons between our histologic findings and lesions characterized on newborn or fetal magnetic resonance imaging studies. Ideally, we would have referenced human fetal autopsy literature or obtained live imaging studies to make more appropriate comparisons. However, given the scarcity of autopsy studies published in fetuses or newborns with CHD and prohibitive nature of the support apparatus in our study, these comparisons were limited. Finally, given the significant resources required to provide continuous, longterm support to EXTEND fetuses, sample sizes were small. Despite this limitation, histologic differences between groups were striking, so we were still able to identify statistical differences between groups.
CONCLUSIONS
In this study, we demonstrate that chronic intrauterine hypoxia leads to reduced cortical folding, reduced neuronal density, hypervascularity, and impaired myelination. These findings support the hypothesis that intrauterine hypoxia, even in the setting of preserved glucose delivery, alters neurodevelopment. We theorize that HIF-1α upregulation under hypoxic conditions contributes to this relative dysmaturity via inhibition of oligodendrocyte differentiation, but additional studies are required to better delineate these relationships. Ultimately, these data provide important insights into understanding the etiology of impaired neurodevelopment and subsequent neurocognitive disorders affecting patients with CHD and may have implications for future fetal-based therapies.
Central Message.
Chronic fetal hypoxia leads to white matter hypervascularity, decreased neuronal density, and impaired myelination, similar to the neuropathologic findings observed in children born with CHD.
Perspective.
The impact of reduced cerebral oxygenation on neurodevelopment in children with CHD has been debated. These data provide important insights into understanding the etiology of the impaired brain development and subsequent neurocognitive disorders frequently observed in children with CHD. Ultimately, these findings may have implications for future fetal-based therapies.
Acknowledgments
Supported by the Preclinical Core of the Institutional Intellectual Developmental Disabilities Research Core of the Children’s Hospital of Philadelphia (HD26979) and The Children’s Hospital of Philadelphia Departments of Surgery and Cardiothoracic Surgery, and Mortimer J. Buckley, Jr, MD, Endowed Chair.
Abbreviations and Acronyms
- CHD
congenital heart disease
- GA
gestational age
- EXTEND
Extra-Uterine Environment for Neonatal Development
- H&E
hematoxylin and eosin
- HIF-1α
hypoxia inducible factor 1-alpha
- ROI
region of interest
Footnotes
Conflict of Interest Statement
Authors have nothing to disclose with regard to commercial support.
The authors thank Rachel Rogers, MS, for assistance in statistical analyses; Grace Hwang, BA, Kathleen Young, BA, Stylianos Monos, BA, Antoneta Radu, Jerry Zsido, MS, Aaron Weilerstein, BA, and Jacqueline Amato, MS, for technical assistance; and Alistair Gunn, MBChB, PhD, Joanne Davidson, PhD, and Stephen Back, MD, PhD, for guidance with tissue preparation and staining.
References
- 1.Hoffman JI, Kaplan S. The incidence of congenital heart disease. JAm Coll Cardiol. 2002;39:1890–900. [DOI] [PubMed] [Google Scholar]
- 2.Moons P, Bovijn L, Budts W, Belmans A, Gewillig M. Temporal trends in survival to adulthood among patients born with congenital heart disease from 1970 to 1992 in Belgium. Circulation. 2010;122:2264–72. [DOI] [PubMed] [Google Scholar]
- 3.Gaynor JW, Stopp C, Wypij D, Andropoulos DB, Atallah J, Atz AM, et al. Neurodevelopmental outcomes after cardiac surgery in infancy. Pediatrics. 2015;135: 816–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bellinger DC, Wypij D, Rivkin MJ, DeMaso DR, Robertson RL Jr, Dunbar-Masterson C, et al. Adolescents with d-transposition of the great arteries corrected with the arterial switch procedure: neuropsychological assessment and structural brain imaging. Circulation. 2011;124:1361–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Neufeld RE, Clark BG, Robertson CM, Moddemann DM, Dinu IA, Joffe AR, et al. Five-year neurocognitive and health outcomes after the neonatal arterial switch operation. J Thorac Cardiovasc Surg. 2008;136:1413–21.e2. [DOI] [PubMed] [Google Scholar]
- 6.Bellinger DC, Wypij D, duPlessis AJ, Rappaport LA, Jonas RA, Wernovsky G, et al. Neurodevelopmental status at eight years in children with dextro-transposition of the great arteries: the Boston Circulatory Arrest Trial. J Thorac Cardiovasc Surg. 2003;126:1385–96. [DOI] [PubMed] [Google Scholar]
- 7.Limperopoulos C, Tworetzky W, McElhinney DB, Newburger JW, Brown DW, Robertson RL Jr, et al. Brain volume and metabolism in fetuses with congenital heart disease: evaluation with quantitative magnetic resonance imaging and spectroscopy. Circulation. 2010;121:26–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Licht DJ, Wang J, Silvestre DW, Nicolson SC, Montenegro LM, Wernovsky G, et al. Preoperative cerebral blood flow is diminished in neonates with severe congenital heart defects. J Thorac Cardiovasc Surg. 2004;128:841–9. [DOI] [PubMed] [Google Scholar]
- 9.Mahle WT, Tavani F, Zimmerman RA, Nicolson SC, Galli KK, Gaynor JW, et al. An MRI study of neurological injury before and after congenital heart surgery. Circulation. 2002;106(12 Suppl 1):I109–14 [PubMed] [Google Scholar]
- 10.Miller SP, McQuillen PS, Hamrick S, Xu D, Glidden DV, Charlton N, et al. Abnormal brain development in newborns with congenital heart disease. N Engl J Med. 2007;357:1928–38. [DOI] [PubMed] [Google Scholar]
- 11.Licht DJ, Shera DM, Clancy RR, Wernovsky G, Montenegro LM, Nicolson SC, et al. Brain maturation is delayed in infants with complex congenital heart defects. J Thorac Cardiovasc Surg. 2009;137:529–36; discussion 536-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andropoulos DB, Hunter JV, Nelson DP, Stayer SA, Stark AR, McKenzie ED, et al. Brain immaturity is associated with brain injury before and after neonatal cardiac surgery with high-flow bypass and cerebral oxygenation monitoring. J Thorac Cardiovasc Surg. 2010;139:543–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lim JM, Kingdom T, Saini B, Chau V, Post M, Blaser S, et al. Cerebral oxygen delivery is reduced in newborns with congenital heart disease. J Thorac Cardiovasc Surg. 2016;152:1095–103. [DOI] [PubMed] [Google Scholar]
- 14.Lauridsen MH, Uldbjerg N, Henriksen TB, Petersen OB, Stausbol-Gron B, Matthiesen NB, et al. Cerebral oxygenation measurements by magnetic resonance imaging in fetuses with and without heart defects. Circ Cardiovasc Imaging. 2017;10:e006459. [DOI] [PubMed] [Google Scholar]
- 15.Sun L, Macgowan CK, Sled JG, Yoo SJ, Manlhiot C, Porayette P, et al. Reduced fetal cerebral oxygen consumption is associated with smaller brain size in fetuses with congenital heart disease. Circulation. 2015;131:1313–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rudolph AM. Impaired cerebral development in fetuses with congenital cardiovascular malformations: is it the result of inadequate glucose supply? Pediatr Res. 2016;80:172–7. [DOI] [PubMed] [Google Scholar]
- 17.Partridge EA, Davey MG, Hornick MA, McGovern PE, Mejaddam AY, Vrecenak JD, et al. An extra-uterine system to physiologically support the extreme premature lamb. Nat Commun. 2017;8:15112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Itskovitz J, LaGamma EF, Rudolph AM. Effects of cord compression on fetal blood flow distribution and O2 delivery. Am J Physiol. 1987;252(1 Pt 2):H100–9. [DOI] [PubMed] [Google Scholar]
- 19.Back SA, Riddle A, Hohimer AR. Role of instrumented fetal sheep preparations in defining the pathogenesis of human periventricular white-matter injury. J Child Neurol. 2006;21:582–9. [DOI] [PubMed] [Google Scholar]
- 20.Battaglia FC, Meschia G. Principal substrates of fetal metabolism. Physiol Rev. 1978;58:499–527. [DOI] [PubMed] [Google Scholar]
- 21.Rees S, Stringer M, Just Y, Hooper SB, Harding R. The vulnerability of the fetal sheep brain to hypoxemia at mid-gestation. Brain Res Dev Brain Res. 1997;103: 103. [DOI] [PubMed] [Google Scholar]
- 22.Goni-de-Cerio F, Alvarez A, Caballero A, Mielgo VE, Alvarez FJ, Rey-Santano MC, et al. Early cell death in the brain of fetal preterm lambs after hypoxic-ischemic injury. Brain Res. 2007;1151:161–71. [DOI] [PubMed] [Google Scholar]
- 23.Davidson JO, Yuill CA, Zhang FG, Wassink G, Bennet L, Gunn AJ. Extending the duration of hypothermia does not further improve white matter protection after ischemia in term-equivalent fetal sheep. Sci Rep. 2016;6: 25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faber JJ, Green TJ. Foetal placental blood flow in the lamb. J Physiol. 1972;223: 375–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miller TA, Zak V, Shrader P, Ravishankar C, Pemberton VL, Newburger JW, et al. Growth asymmetry, head circumference, and neurodevelopmental outcomes in infants with single ventricles. J Pediatr. 2016; 168:220–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Birca A, Vakorin VA, Porayette P, Madathil S, Chau V, Seed M, et al. Interplay of brain structure and function in neonatal congenital heart disease. Ann Clin Transl Neurol. 2016;3:708–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paredes MF, James D, Gil-Perotin S, Kim H, Cotter JA, Ng C, et al. Extensive migration of young neurons into the infant human frontal lobe. Science. 2016; 354: 10.1126/science.aaf7073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kinney HC, Haynes RL, Xu G, Andiman SE, Folkerth RD, Sleeper LA, et al. Neuron deficit in the white matter and subplate in periventricular leukomalacia. Ann Neurol. 2012;71:397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Samuelsen GB, Pakkenberg B, Bogdanovic N, Gundersen HJ, Larsen JF, Graem N, et al. Severe cell reduction in the future brain cortex in human growth-restricted fetuses and infants. Am J Obstet Gynecol. 2007;197: 56.e1–7. [DOI] [PubMed] [Google Scholar]
- 30.Morton PD, Korotcova L, Lewis BK, Bhuvanendran S, Ramachandra SD, Zurakowski D, et al. Abnormal neurogenesis and cortical growth in congenital heart disease. Sci Transl Med. 2017;9:eaah7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yuen TJ, Silbereis JC, Griveau A, Chang SM, Daneman R, Fancy SP, et al. Oligodendrocyte-encoded HIF function couples postnatal myelination and white matter angiogenesis. Cell. 2014;158:383–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sim J, Johnson RS. A whiter shade of gray: HIF and coordination of angiogenesis with postnatal myelination. Dev Cell. 2014;30:116–7. [DOI] [PubMed] [Google Scholar]
- 33.Kanaan A, Farahani R, Douglas RM, Lamanna JC, Haddad GG. Effect of chronic continuous or intermittent hypoxia and reoxygenation on cerebral capillary density and myelination. Am J Physiol Regul Comp Physiol. 2006;290:R1105–14. [DOI] [PubMed] [Google Scholar]
- 34.Rossidis AC, Baumgarten H, Lawrence KM, Mejaddam A, Peranteau WH, Davey MG, et al. Chronically hypoxic fetal lambs supported by an extra-uterine device exhibit mitochondiral dysfunction and elevations of hypoxia inducible factor. Fetal Diagn Ther. 2018;1–8. [DOI] [PubMed] [Google Scholar]
- 35.Hornick MA, Davey MG, Partridge EA, Mejaddam AY, McGovern PE, Olive AM, et al. Umbilical cannulation optimizes circuit flows in premature lambs supported by the EXTra-uterine environment for Neonatal Development (EXTEND). J Physiol. 2018;596:1575–85. [DOI] [PMC free article] [PubMed] [Google Scholar]