

Abstract
We investigated the effect of bicarbonate and oxidizing agents on uranium (U) reactivity and subsequent dissolution of U(IV) and U(VI) mineral phases in the mineralized deposits from Jackpile mine, Laguna Pueblo, New Mexico, by integrating laboratory experiments with spectroscopy, microscopy and diffraction techniques. Uranium concentration in solid samples from mineralized deposit obtained for this study exceeded 7000 mg kg−1, as determined by X-ray fluorescence (XRF). Results from X-ray photoelectron spectroscopy (XPS) suggest the coexistence of U(VI) and U(IV) at a ratio of 19:1 at the near surface region of unreacted solid samples. Analyses made using X-ray diffraction (XRD) and electron microprobe detected the presence of coffinite (USiO4) and uranium-phosphorous-potassium (U-P-K) mineral phases. Imaging, mapping and spectroscopy results from scanning transmission electron microscopy (STEM) indicate that the U-P-K phases were encapsulated by carbon. Despite exposing the solid samples to strong oxidizing conditions, the highest aqueous U concentrations were measured from samples reacted with 100% air saturated 10 mM NaHCO3 solution, at pH 7.5. Analyses using X-ray absorption spectroscopy (XAS) indicate that all the U(IV) in these solid samples were oxidized to U(VI) after reaction with dissolved oxygen and hypochlorite (OCl−) in the presence of bicarbonate (HCO3−). The reaction between these organic rich deposits, and 100% air saturated bicarbonate solution (containing dissolved oxygen), can result in considerable mobilization of U in water, which has relevance to the U concentrations observed at the Rio Paguate across the Jackpile mine. Results from this investigation provide insights on the reactivity of carbon encapsulated U-phases under mild and strong oxidizing conditions that have important implication in U recovery, remediation and risk exposure assessment of sites.
Keywords: Uranium, Mineralized Deposits, Spectroscopy, Microscopy, Jackpile mine, Coffinite
Graphical.Abstract
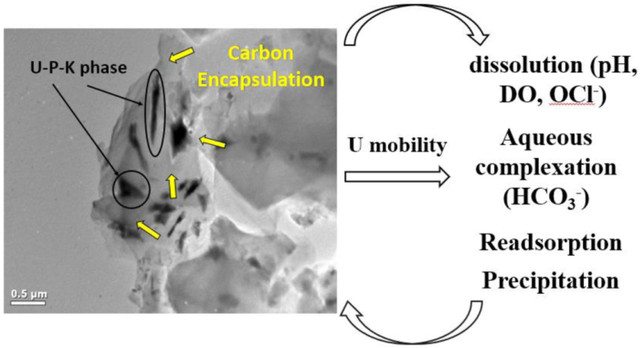
2. Introduction.
The current study is focused on mineralized deposits located at the Jackpile mine on Laguna Pueblo, in central New Mexico. In 1951, U was discovered in sandstone outcropping of the Morrison formation near the village of Paguate during an airborne radioactivity survey (Nash, 1968). Notable investigations of the Jackpile mineralized deposit have been conducted by the U.S. Geological Survey (Hilpert and Moench, 1960; Moench and Schlee, 1967), the Atomic Energy Commission (Laverty et al., 1963), and the Anaconda Company (Kittel, 1963). The deposit was mined from 1953 to 1980, affecting approximately 3,141 acres of land (Zehner, 1985). While efforts have been made to reclaim this site since 1995, U can be mobilized in surface waters of the Rio Paguate next to the Jackpile mine and can be accumulated in soils and plants (Blake et al., 2017; El Hayek et al., 2018).
The sandstone member of the Morrison formation at the Jackpile-Paguate deposit is a medium-grained, cross-stratified, 33-mile long sandstone in which the U deposits are composed of one or more semi-tabular ore layers. Un-oxidized parts of these tabular U deposits are rich in U(IV) phases including coffinite (US1O4) and lesser amounts of uraninite (UO2) (Deditius et al., 2008). Uranium mineralization is correlated with the presence of vegetative carbonaceous matter as primary minerals imparting a gray to black shade with increasing organic content (Adams et al., 1978; Beck et al., 1980; Moench and Schlee, 1967). Oxidation of these U(IV) phases resulted in the formation of secondary U(VI) minerals such as schoepite [(U02 )8 O2 (OH)1 2 .12], carnotite [K2(UO2)2(VO4)2. 3H2O], tyuyamunite [Ca(UO2)2(VO4)2.7-10H2O], meta-tyuyamunite [Ca(U02)2(V04)2. 5H2O], and uranophane [Ca(U02)2(SiO3)2(0H)2] (Charles, 1979; Hettiarachchi et al., 2018; Kelley, 1963). Additionally, other U(VI) mineral phases such as autunite [Ca(UO2)2(PO4)2.10-12H2O] and phosphuranylite [Ca(UO2)2(PO4)2(OH)4.7H2O] can occur in these deposits (Kelley, 1963). These co-existing U(IV) and U(VI) solids pose a potential source of U and other co-constituents that can be mobilized into proximate water sources (Blake et al., 2017; Blake et al., 2015).
The mobility of U from U(IV) and U(VI) phases in organic-rich U deposits can be affected by various geochemical mechanisms at the mineral-water interface. For instance, physical and chemical characteristics of the mineralized deposit (e.g., mineralogy, reaction kinetics, and crystallinity) and water chemistry (i.e., pH, Eh, and solution composition) are factors that influence U mobility (Avasarala et al., 2017; Ginder-Vogel et al., 2006; Maurice, 2009; Stumm and Morgan, 2012). Uranium transport may be enhanced when aqueous U(VI) species bind with organic matter through either ion-exchange, coordination/complexation, electron donor-receptor interactions, or Van der Waal forces at circumneutral conditions (Keiluweit and Kleber, 2009; Luo and Gu, 2008; Mibus et al., 2007; Steelink, 2002; Yang et al., 2013; Yang et al., 2012). The degree of U mobility in the presence of organic matter depends on: 1) the sorption of aqueous U(VI) onto soil minerals and solid organic matter surfaces; 2) hydrophobicity and alkyl carbon content in organic matter; and 3) the presence of U(VI)-organic colloids (Kretzschmar et al., 1999; McCarthy and Zachara, 1989; Yang et al., 2012). Uranyl ions (UO22+) form strong binary complexes with carbonate (U-CO32−) as well as ternary complexes with carbonate and alkaline earth metals such as U-Ca-CO32− (Dong and Brooks, 2006; Dong and Brooks, 2008; Langmuir, 1997). Such aqueous complexes of U(VI) adsorb onto the oppositely charged SiO2, hydrous ferric oxide, goethite, and montmorillonite surfaces (Singh et al., 2012; Troyer et al., 2016; Yang et al., 2013). Adsorption of U(IV) may also occur in organic matter aggregates [e.g. mononuclear or monomeric U(IV)] and organic-carbon coated clays (Bone et al., 2017; Wang et al., 2013). Recently, biogenic U(IV) species (amorphous) were found to be a major component of undisturbed roll front ore deposits (Bhattacharyya et al., 2017). Formation of sparingly soluble biogenic and chemogenic U(IV) through microbial reduction has been proposed as a method to immobilize aqueous U in contaminated soil and ground water (Alessi et al., 2014; Bargar et al., 2013; Lovley and Phillips, 1991; Veeramani et al., 2011). Reduction of U(VI) to U(IV) may result in the formation of crystalline nano-particulate U(IV) phases with dissolution rates similar to those of coarser U(IV) phases (Bargar et al., 2008; Nenoff et al., 2011; O’Loughlin et al., 2003; Ulrich et al., 2009). The association of U with colloids of organic matter and inorganic particles accounts for a considerable portion of U mobilized at sites affected by mining (Mkandawire and Dudel, 2008; Schindler et al., 2017). In addition to sorption, U mobility can also be affected by the solubility of U minerals. For instance, U occurs as vanadate, silicate, molybdate, phosphate, and other minerals with solubility constant values that range within several orders of magnitude (Avasarala et al., 2017; Buck et al., 1995; De Voto, 1978; Kanematsu et al., 2014; Singer et al., 2009; Singh et al., 2012; Tokunaga et al., 2009; Troyer et al., 2016). Uranium mobility from such solid U(IV) and U(VI) minerals has been widely explored in laboratory studies. However, there are very few studies on the reactivity and subsequent dissolution of U phases from natural and organic-rich U-deposits under environmentally relevant conditions.
Previous studies have investigated the mobility of U from organic-rich U-deposits at the Grants Mineral District and sediments along Rio Paguate. Deditius et al. (2008) investigated the process of formation and alteration of three different coffinites under reducing and oxidizing conditions (Deditius et al., 2008). This investigation suggested that the coffinites occur as secondary submicron-crystals within layers of organic matter that were formed as a result of alteration under oxidizing conditions (Deditius et al., 2008). Dissolution of coffinite and other U-bearing minerals in the Jackpile mine U-deposit samples was suggested as potential sources of elevated U concentrations (35.3 to 772 mg L−1) in the Rio Paguate (Blake et al., 2017). However, limited experiments have been conducted on these carbon-rich mineralized deposits to support this hypothesis.
The objective of this study was to investigate the reaction of oxidizing agents [e.g., O2 and hypochlorite (HOCl and OCl−)] and bicarbonate with U(IV) and U(VI) phases in Jackpile mine mineralized deposits using laboratory experiments, spectroscopy, and electron microscopy techniques. While there is existing knowledge about the presence of U in the mineralized deposits and waters adjacent to the Jackpile mine (Blake et al., 2017; Ruiz et al., 2016; Deditius et al., 2008; Kelley, 1963), this study identifies specific U minerals in these deposits and their reactivity under both environmentally relevant and strong oxidizing conditions. Exposure of solids from such mineralized deposits, to a range of oxidizing solutions provides new insights about the effects of natural oxidation and subsequent dissolution of U from naturally-occurring complex mineralized deposits. These mineralized deposits in the Jackpile mine have been exposed to ambient oxidizing conditions for decades since mining and reclamation have been conducted. This knowledge has relevant implications for U extraction, transport, and remediation.
3. Materials and Methods
3.1. Materials
Samples were collected in 2015 from the exposed beds in the mineralized zone of north Jackpile-Paguate mine site, Laguna Pueblo, New Mexico, USA. The Jackpile-Paguate mine site is roughly 13 miles wide, 33 miles long, and up to 65 m deep located near the village of Paguate. Its U-deposits occur in the Jurassic and cretaceous strata that were exposed by previous mining activities. These strata were located above the regional water table and thus were not saturated with ground water; however, they were exposed to the atmosphere following the mining completion. The Jackpile sandstone where all the major U-deposits occur predominantly in this strata are found in the uppermost part of the Brushy Basin Shale Member of the Jurassic Morrison formation (Blake et al., 2017; Maxwell, 1982; Moench and Schlee, 1967; Nash, 1968; Risser et al., 1984). These specific samples were selected for collection because of: 1) their dark black color and rocky texture; 2) their use in previous investigations (Blake et al., 2017; Ruiz et al., 2016; Velasco et al., 2019); and 3) their high alpha radiations (as high as 10 mR/hr) measured using our well calibrated Ludlum Model 19 MicroR Meter Geiger counter during the field trip. The intense black color of the samples could be from the high organic content measured by a 22-44% mass loss during a loss on ignition test conducted at 550°C (Ruiz et al., 2016; Velasco et al., 2019). For the current investigation, the samples were pulverized and homogenized using a Spex shatterbox and then sieved to a size of <63 μm. Only the sieved fraction was used for subsequent characterizations and batch experiments. For simplification purposes, these pulverized Laguna deposit samples are referred to as “ore” from this point on in this paper.
3.2. Procedure for Batch Experiments
Batch experiments were conducted under oxidizing conditions to investigate the effect of oxidizing conditions on the reactivity of the solid ore samples. We selected two main oxidants for these experiments: a) dissolved oxygen (natural oxidant relevant to the site) by letting solutions to achieve 100% air saturation; and b) hypochlorite, added as NaOCl at pH 7.5 as this is a strong oxidant typically used in several other studies to oxidize the organic matter fraction (Bouzidi et al., 2015; La Force and Fendorf, 2000; Love and Woronow, 1991; Regenspurg et al., 2010; Schultz et al., 1998). A 6% NaOCl concentrated solution, purchased commercially from Fisher Scientific, was selected to evaluate the leaching of U and other elements to solution caused by the oxidation of organic carbon (C) and reduced mineral phases in the ore after exposure to strong oxidizing conditions. These experiments allow evaluation of the effects of strong oxidizing conditions, like those caused by natural oxidation associated with weathering as the ore samples were exposed to ambient conditions in the Jackpile mine for decades. Considering that the pKaof hypochlorous acid (HOCl) is 7.6 (Blanco et al., 2005; Learn et al., 1987; Schultz et al., 1998), we expect that the solution at pH 7.5 will have almost equimolar concentrations of the conjugate acid (HOCl) and conjugate base (OCT−), which are both strong oxidants (Benjamin, 2014). Additionally, 7.5 was also the pH measured in the surface water collected from Rio Paguate (Blake et al., 2017).
About 2 g of the pulverized ore was reacted with 50 mL of four different 100% air saturated reagents, which include: 1) 18MΩ deionized water, pH 5.4; 2) 10 mMNaHCCL, pH 7.5; 3) 6% NaOCl, pH 7.5; and 4) 10 mM NaHC03 + 6% NaOCl, pH 7.5 at room temperature. The pH of all reagents except 18MΩ water were adjusted to 7.5 using concentrated nitric acid, 0.5N NaOH, and 0.1M HC1. Furthermore, 10 mM NaHC03 was selected to mimic the alkalinity and pH conditions measured in the Rio Paguate (Blake et al., 2017). The 18MΩ water solution was included as a well characterized baseline solution with low ionic content to approximately represent ore interactions with rainwater. All experiments were conducted in triplicate in covered flasks using air saturated reagents. The batch oxidation studies were conducted for two weeks, and aliquots were collected periodically after 0.5, 2, 4, 8, 24, 72, 144, 218, 288, and 336 hours of the experiment. In addition, total acid digestions were conducted on the unreacted and reacted ore samples by reacting 3ml of hydrochloric acid (HC1), 3 ml of nitric acid (HNO3−) and 3ml of hydrofluoric acid (HF) with 2 ± 0.002g sediments. All samples and acid extracts were filtered through 0.45μm filters and diluted as necessary for an elemental analysis using Inductively Coupled Plasma — Optical Emission Spectroscopy (ICP-OES) or Mass Spectroscopy (ICP-MS).
3.3. Aqueous analyses.
The concentrations of metals in the samples and the acid extractables were measured using a PerkinElmer Optima 5300DV ICP-OES for major cations and a PerkinElmer Nexion 300D (Dynamic Reaction Cell) ICP-MS for trace elements including As, Mo, Se, U, and V. Both ICPs were calibrated with 1) a 5-point calibration curve, and 2) QA/QC (quality analysis/quality control), by occasionally adding samples of known elemental concentrations for every 10 samples, to ensure quality results. The solution pH was measured using a Thermo Scientific Orion Versastar Advanced Electrochemistry pH meter.
3.3.1. Dissolved organic carbon (DOC) content.
After the batch experiments, dissolved organic carbon (DOC) concentrations were measured both in pre and post reaction supernatants, using the standard UV persulfate method in a Tekmar-Dohrmann Phoenix 8000 TOC analyzer. These supernatants were filtered using Millipore 0.45μm polyvinylidene fluoride filters and diluted accordingly before the analysis.
3.3.2. Acid Digestion.
Acid digestions were conducted on unreacted and reacted ore samples to assess and compare their total acid extractable trace metal concentrations. Acid digestions of unreacted ores involved the addition of 3ml of Hydrochloric acid (HC1), 3 ml of Nitric acid (HNO3−) and 3ml of Hydrofluoric acid (HF) into 50 ml Teflon digestion tubes containing 2 ± 0.002g ore samples. Similarly, for reacted ores 1 ml of HCl, HNO3− and HF was added to 50 ml Teflon digestion tubes containing 0.5 ± 0.002g of ore samples. All reagents were of high purity. The digestion tubes were then heated using a Digi prep MS SCP Science block digester at 95 °C for 2 h, followed by dilution of acid extracts from reacted and unreacted ores to 50 and 25 ml. The diluted samples are then filtered using 0.45 μm filters to remove any suspended or undissolved solids before analysis.
3.4. Solid characterization.
Characterization of ore samples was conducted using various spectroscopy, microscopy, and diffraction techniques to obtain information on occurrence of U-minerals in the ore, their crystallinity and their overall surface charge. A Rigaku ZSX Primus II wavelength dispersive X-ray fluorescence spectrometer (XRF) was used on pulverized ore samples to obtain their bulk elemental composition. A Rigaku Smart Lab x-ray diffractometer (XRD) was used on pulverized ore samples to study the minerology of the ore. We also used a JEOL JXA-8200 electron microprobe (EPMA) and synchrotron-based micro x-ray fluorescence spectroscopy (μ-SXRF) at Stanford Synchrotron Radiation Lightsource (SSRL) beam line 10.3.2 to analyze epoxy embedded solid samples for elemental mapping of minerals in the ore. A JEOL 2010F Field Emission Gun scanning transmission electron microscope (FEG/STEM) was used on pulverized ore samples to obtain structural and compositional information of the micro-sized uranium minerals. A Kratos AXIS-Ultra DLD x-ray photoelectron spectrometer (XPS) was used to collect high resolution U 4f spectra to measure oxidation states of U in the near-surface region of the ore samples following a similar procedure reported in previous studies (Blake et al., 2015; Velasco et al., 2019). All XPS spectra were processed using CASAXPS software. Synchrotron-based x-ray absorption spectrometer (XAS) ULIII measurements were conducted at SSRL beamline 4-1 to identify bulk oxidation states and molecular coordination of U following similar approaches to those described in previous studies (Blake et al., 2015; Velasco et al., 2019). The XAS spectra were analyzed and linear combination fits (LCF) were made using Athena and Artemis with standard methods (Ravel and Newville, 2005). A zeta potential on pulverized ore samples to identify their surface charge at relevant pH. Additional information on these methods is provided in the supporting information (SI).
3.5. MINEOL modelling.
The aqueous complexes and solid phases of U formed during reactions with 18MΩ water, 6% NaOCl and 10 mM NaHCO3 were estimated using MINEQL+ v4.6 (Schecher and McAvoy, 2007) to better explain the U trends observed from our experiments. Alkalinity, pH, Ca and U concentrations measured during the experiments were provided as inputs to these chemical equilibrium simulations. Simulations use these inputs to estimate the formation of Ca-U- CO32− aqueous complexes at circumneutral pH that can significantly affect the transport of U (Dong and Brooks, 2006). Concentrations of U and Ca were measured using ICP-OES. Concentrations of anions on the other hand were not measured, as U was known to predominantly form aqueous complexes and solid phases with OH− or CO32− at the experimental pH (Dong and Brooks, 2006). A MINEQL database updated with critical thermodynamic reactions such as aqueous cation-uranyl-carbonate complexes was used instead of the default MINEQL database supplied with the software (Dong and Brooks, 2006; Grenthe et al., 1992). The specific thermodynamic reactions added to the modified database are listed below.
| U(VI) Aqueous Complexes | Log K | Reference |
|---|---|---|
| UO22+ + H2O = UO2OH+ + H+ | 5.2073 | (Grenthe et al., 1992) |
| UO22+ + 2H2O = UO2(OH)2 (aq) + 2H+ | 10.3146 | (Grenthe et al., 1992) |
| UO22+ + 3H2O = UO2(OH)3− + 3H+ | 19.2218 | (Grenthe et al., 1992) |
| UO22+ + 4H2O = UO2(OH)42− + 4H+ | 33.0291 | (Grenthe et al., 1992) |
| 2UO22+ + 1H2O = (UO2)2OH3+ + H+ | 2.7072 | (Grenthe et al., 1992) |
| 2UO22+ + 2H2O = (UO2)2(OH)22+ + 2H+ | 5.6346 | (Grenthe et al., 1992) |
| 3UO22+ + 4H2O = (UO2)3(OH)42+ + 4H+ | 11.929 | (Grenthe et al., 1992) |
| 3UO22+ + 5H2O = (UO2)3(OH)5+ + 5H+ | 15.5862 | (Grenthe et al., 1992) |
| 3UO22+ + 7H2O = ( (UO2)3(OH)7− + 7H+ | 31.0508 | (Grenthe et al., 1992) |
| 4UO22+ + 7H2O = (UO2)4(OH)7+ + 7H+ | 21.9508 | (Grenthe et al., 1992) |
| UO22+ + HCO3− = UO2CO3(aq) + H+ | 0.6634 | (Grenthe et al., 1992) |
| UO22+ + 2HCO3− = UO2(CO3)22− + 2H+ | 3.7467 | (Grenthe et al., 1992) |
| UO22+ + 3HCO3− = UO2(CO3)34− + 3H+ | 9.4302 | (Grenthe et al., 1992) |
| 2UO22+ + 3H2O + HCO3− = (UO2)2CO3(OH)3− + 4H+ | 11.2229 | (Grenthe et al., 1992) |
| 2Ca2+ + UO22+ + 3CO32− = Ca2UO2(CO3)3 (aq) | −30.04 | (Dong and Brooks, 2006) |
| Ca2+ + UO22+ + 3CO32− = CaUO2(CO3)32− | −27.18 | (Dong and Brooks, 2006) |
4. Results and discussion.
4.1. Solid Analyses for unreacted ore
The co-occurrence of U(VI) and U(IV) at the near surface (top 5-10 nm) region of the unreacted Laguna ore samples (<63 μm) was identified by XPS. The XPS Survey scan of the near surface region detected the presence of 47.8% C 1s, 37.5% O 1s, 9.0 % Si 2p, 0.2% N 1s, and 0.1% U 4f (Table S2). Approximately, 95% of the U detected at the ore surface was U(VI), and the remaining 5% was U(IV) (Figure 1A, Table S3). Both measurements were based on XPS narrow scans conducted using U(IV) (uraninite [UO2]) and U(VI) (becquerelite [Ca(UO2)6O4(OH)6·8H2O]) reference materials (Figure 1E-F). Recent investigations have reported the possibility of using satellite peaks in the U4f spectra to differentiate oxidation states of U (Ilton and Bagus, 2011). However, since the samples contained nitrogen (N), magnesium (Mg), and other elements that coincide with the position of satellite peaks of U, data reported in this study are based on fits of the position of the peaks corresponding to the U4f7/2 and U4f5/2 spin-orbits. Additional microscopy, spectroscopy and diffraction analyses were conducted to obtain more specific information about these U(IV) and U(VI) mineral phases.
Figure 1.

Surface Oxidation states of U 4f in unreacted and reacted Jackpile mine ore samples A) High resolution XPS spectra of U in unreacted ore B) High resolution XPS spectra of U in ore reacted with 10 mM HCO3− C) High resolution XPS spectra of U in ore reacted with 6% NaOCl D) High resolution XPS spectra of U in ore reacted with 6% NaOCl + 10mM HCO3−. E) Reference XPS spectra for U(IV) using uraninite [UO2] F) Reference XPS spectra for U(VI) using becquerelite [Ca(UO2)6O4(OH)6·8(H2O)]
Results from electron microscopy, XRD, and μ-SXRF analyses on unreacted ores suggest that submicron U(IV) and U(VI) phases, coffinite (USiO4) and U-P-K phases, are encapsulated by carbon (Figure 2, S1). Electron microprobe analyses using back scatter electron imaging (BSE) identified submicron U-minerals (bright white spots, Figure 2A) that co-occurred with P and C (red circles, Figure 2D-G). The presence of a U-P-K phase in the samples was identified using electron microprobe, BSE imaging, and energy dispersive spectroscopy (EDS) analyses (Figure 2B, C). The co-occurrence of U, P, and Si was confirmed by micro synchrotron x-ray fluorescence (μ-SXRF) mapping analyses (Figure S2). The minerals were encapsulated by carbon-rich material as was found using back scatter electron image (Figure 2A). These results agree with previously reported observations in U-deposits at the Jackpile mine, regarding the co-occurrence of autunite, phosphuranylite, and coffinites (Blake et al., 2017; Deditius et al., 2008; Kelley, 1963). It is worth noting that another investigation had also reported a 21.8-44% mass loss during a loss on ignition test on the same organic-rich mineralized deposits used for this study, which was attributed to organic matter (Ruiz et al., 2016; Velasco et al., 2019). The co-occurrence of U, P, and Ca could correspond to uranyl phosphates (autunite [Ca(UO2)2(PO4)2.10-12H2O] and phosphuranylite [Ca(UO2)2(PO4)2(OH)4.7H2O]) that were previously identified at the Jackpile-Paguate mine (Adams et al., 1978; Moench and Schlee, 1967). The presence of U, Fe, sulfur (S), and V in addition to occurrence of U, P, Si, and C have also been recently reported (Blake et al., 2017). Electron microprobe and XRD analyses on unreacted samples suggested the co-existence of coffinite (USiO4) and U-P-K phases with other primary minerals such as kaolinite (Al2Si2O5(OH)4), quartz (SiO2), and microcline (KAlSi3O8). Additionally, based on theoretical estimations using Jade® software (Smartlab XRD), 47% of the solid material was amorphous (Figure S1), consistent with other data reported from the same site (Blake et al., 2017; Deditius et al., 2008). The presence of amorphous phases in these samples could be from the clays and other organic conglomerates that are characteristic of sandstone formations such as the Jackpile-Paguate. Furthermore, the identification of coffinite, kaolinite, quartz, and microcline in the unreacted ore samples using XRD analyses was consistent with previously reported observations (Figure S1) (Adams et al., 1978; Blake et al., 2017; Deditius et al., 2008; Moench and Schlee, 1967). These results also confirmed the observations made using μ-SXRF mapping where co-occurrence between U and Si was found (Figure S2).
Figure 2.

Electron Microprobe (EPMA) analysis of the unreacted Jackpile mine ore sediments. A) BSE image (leftmost) showing the presence of submicron U-phases encapsulated by carbon where, the mapping was performed. B) EPMA BSE image of a polished sample showing rock fragments with surface aggregates of U-P-K (red arrow). C) Energy Dispersive Spectrum (EDS) confirming the presence of a U-P-K phase showing distinct U, P and K peaks (red circle). D) Uranium map E) Phosphorous map F) pottasium map G) Carbon map. Green, yellow, pink and white represent high concentrations of elements and dark blue represents lower concentrations of elements.
The scanning transmission electron microscopy (STEM) analyses detected the presence of carbon encapsulated crystalline U-P-K phase in the solid ore samples (Figure 3). Specifically, bright field TEM imaging and EDS analyses detected a U-P-K phase that co-occurred with C and Ca (black features, Figure 3A, B) consistent with observations made by electron microprobe analysis (Figure 2). In addition, the patterned fringes (in yellow) obtained using high resolution TEM imaging suggested that these U-P-K phases were crystalline (Figure 3C). We prepared a STEM map of these U-P-K grains (Figure S3) where, the C map suggested their encapsulation by C (Figure S3B), similar to observations made in our electron microprobe mapping results. This carbon layer, composed of 33.3% C and 66.6% O (spectrum 3, Figure S4), could be from the detrital natural organic matter (Adams et al., 1978; Deditius et al., 2008; Moench and Schlee, 1967). The composition of the U-P-K phase was also estimated to be 10.7% U, 3.79% K, 9.15% P, 6.49 Si, and 69.86% O (Spectrum 1, 2 and 4, Figure S4). In another study, the sustained presence of U(IV) minerals, such as coffinites in the Jackpile-Paguate ore, was reported as a consequence of reducing conditions preserved by this vegetative organic matter (Deditius et al., 2008). However, the interactions between organic matter and U are still not well understood. Future investigations could conduct a more detailed spectroscopy, microscopy, and mass-spectrometry analyses to characterize the natural organic matter in the ore, which is beyond the scope of the present study.
Figure 3.

Scanning Transmission Electron Microscopy (STEM) imaging of U phases within unreacted ore. A) STEM image of U-P-K phases in black. B) Energy Dispersive Spectrum (EDS) on grain 1 (red circle in image 3A), confirming the presence of U-P-K phase. C) High resolution-transmission electron microscopy analysis on grain 1 (U-P-K phase) showing patterned fringes (in yellow) suggesting the phase to be crystalline. The lack of patterned fringes around the U-P-K phase (in black) suggests the presence of an amorphous phase possible organic matter in the ore sample.
The extended X-ray absorption fine edge (EXAFS) ULIII spectra suggest that the U coordination environment consists of a combination of mineralized and adsorbed U in the unreacted samples (Figure 4). For example, linear combination fitting of EXAFS spectra resulted in 45% contribution of reduced U(IV) [e.g., 40% coffinite and 5% UO2 minerals] and 55% contribution of U(VI) associated to the unreacted ore as adsorbed phases or loosely bound U(VI) (Figure 4 a-c). These results confirm the identification of both U(VI) and U(IV) with XPS. However, given that the XPS signal corresponds to the top 5-10 nm of the pulverized solids, a higher content of U(VI) was detected with XPS compared to the bulk measurement provided by EXAFS. The EXAFS analyses also confirm the presence of coffinite which was detected by XRD analyses in this and other studies (Deditius, et al 2008 and Blake et al., 2017). Additional experiments were conducted to evaluate the reactivity of these U-bearing minerals in mine ore materials.
Figure 4.

Linear X-ray absorption spectroscopy data for unreacted and reacted ore samples. a) U LiIII-edge X-ray absorption near edge spectra (XANES). b) k3-weighted extended X-ray absorption fine structure (EXAFS) spectra and U LIII-edge combination fits (dashed lines); and c) linear combination fitting results for EXAFS spectra. Among the 3 samples in each figure (A) represents the unreacted ore, (B) represents ore reacted with HCO3− under ambient oxidizing conditions, and (C) represents ore reacted with HCO3− and OCl−.
4.2. Batch experiments
4.2.1. Reaction with 18MΩ. water
Reaction of the ore with 18MΩ water containing 100% air saturated dissolved oxygen (DO) concentrations resulted in dissolution of U at concentrations as high as 200 μg L−1 (Figure 5A). The concentrations released in the first 30 minutes of the reaction (~70 μg L−1) could be attributed to release of weakly bound U. However, after eight hours of reaction, the steady increase in U concentrations up to 200 μg L−1 could be due to desorption or dissolution of readily souble U phases (Figure 5A). For example, schoepite [(UO2)8O2(OH)12·12(H2O)] or metaschoepite [UO3.2H2O] could dissolve at circumneutral pH as reported by others (Giammar and Hering, 2001; Hostetler and Garrels, 1962; Lollar, 2005; Riba et al., 2005). After about 300 hours of reaction, the U concentrations decreased to ~20 μg L−1 and a DOC concentration of 1.6 mg L−1 was measured in solution (likely released from the natural organic matter present in the ore consistent with the C detected in solid analyses presented above), after reacting the ore with 18MΩ water.
Figure 5.

Uranium trends for batch experiments on Laguna ore samples reacted with: A) 18MΩ water pH 5.4; B) 10 mM sodium bicarbonate (NaHCO3) solution pH 7.5 at ambient oxidizing conditions; C) 6% NaOCl solution, at pH 7.5; and D) 10 mM NaHCO3 + 6% NaOCl solution at pH 7.5.
The decrease in U concentrations at later times of the experiment could be due to reassociation of U onto the ore solids due to readsorption or precipitation. The surface charge of the reacted solids at an average pH of 6.5 was −25mV as shown in Figure S5A and S6A. Chemical equilibrium modelling using MINEQL was conducted to better understand this behavior and evaluate reactions that could possibly occur under these experimental conditions. The decrease in U could be attributed to the possible precipitation of schoepite (SI of Schoepite = 0, MINEQL, Table S4). Chemical equilibrium simulations also suggest that the decrease in U concentrations could be due readsorption of positively charged aqueous complexes of U formed with hydroxyl (UO2OH+, (UO2)3(OH)5+) or with organic matter (e.g., U-OM) onto the negatively charged ore surfaces (Cumberland et al., 2018; Dong and Brooks, 2006; Mikutta et al., 2016; Uyuşur et al., 2015). However, since little is known on the nature of the organic matter on these ore samples, more research is necessary to better understand the interactions between U and organic matter, and their role in U mobility from the ore samples.
After the reaction with 18MΩ water was complete, an acid digestion on the solids was performed to determine the remaining total acid extractable content of the ore samples. This analysis indicated that a 3.7% loss from solid into solution occurred on the total acid extractable U content, during reaction with 18 MΩ water (Table S6). Therefore, these results show that even a mild reaction of 18 MΩ water with ore can cause the release of U into solution. These ore samples were later subjected to complexing and oxidizing reactants such as 100% saturated with air 10 mM NaHCO3, and 6% NaOCl solutions to furrther assess the reactivity of U.
4.2.2. Reaction with 10 mM NaHCO3 solution.
The highest release of U in this study was obtained after reaction of the ore samples with a 10 mM NaHCO3 solution at, pH 7.5 under 100% air saturated DO concentrations. A maximum concentration of 25000 μg L−1 of U was released into solution during this experiment (Figure 5B). The initial release of U (~15000 μg L−1) was likely due to complexation of U with carbonate or dissolution of U-bearing minerals as reported in other studies (Giammar and Hering, 2001; Hostetler and Garrels, 1962; Lollar, 2005; Riba et al., 2005; Ulrich et al., 2009; Zhou and Gu, 2005). Additionally, the final solution contained 2.6 mg L−1 of DOC, which was higher than the DOC released after reaction with deionized water (Table S5). Thus, it is likely that U was also mobilized due to complexation with organic matter during the dissolution of U(IV) and U(VI) minerals reacting with NaHCO3 and DO. However, after eight hours of reaction, a steady increase in U concentrations was observed which could be from the dissolution of coffinite, U-P-K phase or other unidentified U minerals in the ore. Other investigations have reported dissolution of coffinite at near neutral pH (Goleva et al., 1981).
In order to futher assess U speciation for the experiments under circumneutral conditions, chemical equilibrium simulations were conducted using measured alkalinity, pH, and concentrations of Ca and U. Results from these simulations suggested that neutral or negativily-charged ternary U-Ca-CO32− aqueous complexes such as UO2(CO3)34−, CaUO2(CO3)32− and Ca2UO2(CO3)30 were predominant at pH 8 (Table S4, Figure S7B). These neutrally charged aqueous complexes would not bind as strongly as the positively charged aqueous complexes onto the negative solid surfaces (surface charge is −35mV), inhibiting readsorption, unlike observations made with 18 MΩ water (Figure S5B, Figure S6B). Other studies have reported the stability of neutrally charged ternary U-Ca-CO32− aqueous complexes in solution, and how the formation of these can inhibit adsorption of U to mineral or plant root surfaces (Blake et al., 2017; Dong and Brooks, 2006; El Hayek et al., 2018). Additionally, acid digestions of the reacted ore samples performed before and after completion of the batch experiments indicate that 17% of the total acid extractable U was extracted by 10 mM NaHCO3 solution (Table S6). This percentage loss in U was the highest among all reactivity experiments conducted in this study; and is of a great environmental relevance, considering how these conditions represent alkalinity, pH, and DO conditions observed in Rio Paguate (Blake et al., 2017).
The release of 2.6 mg L−1 DOC from ore reaction with NaHCO3 and DO requires additional investigation to better understand the role of organic matter on U speciation under conditions relevant to the Jackpile-Paguate mine site. Although information exists about the complexation of dissolved organic mater with U (Cumberland et al., 2016; Cumberland et al., 2018; Dong and Brooks, 2006; Mikutta et al., 2016; Uyuşur et al., 2015), the role of particulate and dissolved orgnic matter on U speciation under environmentally relevant conditions remains poorly understood. Subsequent experiments were perfomed to understand the reactivity of these co-occurring U(IV)-U(VI) phases under strong oxidizing conditions.
4.2.3. Reaction with 6% NaOCl solution.
The concentrations of U released during reaction with 6% NaOCl were considerably lower than those obtained from experiments with NaHCO3 solution (Figure 5C). For example, despite NaOCl being a strong oxidant, the concentrations of U (8000 μg L−1) released after reacting the ore with 6% NaOCl were less than one-third of those released after reaction with 10 mM NaHCO3 at pH 7.5 (Figure 5C). These 8000 μg L−1 U concentrations were released within the first two hours of reaction with 6% NaOCl. However, between the second and eighth hour of reaction, the U concentrations dropped to ~2000 μg L−1. The decrease in U concentrations in solution is likely due to sorption, causing the re-association of soluble U to the solids with time. In order to better interpret this result, chemical equilibrium simulations using inputs from experimentally measured pH and Ca and U concentrations were conducted (Figure S5C, S7C).
Attributing the decrease of U in solution over time to a single reaction mechanism in these experiments is not possible, given the complex organic matter chemisry and complex mineralogy of the U-ore. A variety of scenarios need to be considered, for instance, the decrease in U concentrations could be caused by precipitation of schoepite or other U—bearing minerals capable of precipitating at circumneutral pH. Oxides, vanadates and silicates such as schoepite ((UO2)8O2(OH)12·12H2O), soddiyite ((UO2)SiO4(H2O)2), haiweeite (Ca(UO2)2(Si2O5)3(H2O)5), carnotite (K2(UO2)2(VO4)2) and tyuyamunite (Ca(UO2)2(VO4)2·5-8H2O) also precipitate under circumneutral conditions (Cumberland et al., 2016; Tutu et al., 2009). Additionally, chemical equilibrium simulations indicate the prevalence of the positively charged aqueous complexes UO2OH+ at pH 6-7 (Table S4, Figure S5C). Given that the ore surfaces were negatively charged (Figure S6C), the positively charged aqueous complex UO2OH+ may readsorb onto the solids. Since U (~8000 μg L−1) and DOC (5.5 mg L−1) concentrations (Figure 5C, Table S5) were measured in solution in these experiments, it is also possible that the readsorption process was affected by U-organic matter aqueous complexes which could be stable in solution as suggested by other studies (Cumberland et al., 2016; Mikutta et al., 2016; Semião et al., 2010). However, little is known about the chemistry and characteristics of the natural organic matter from the study site. Thus, the specific U(IV)/U(VI)-aqueous complexes forming in these experiments remain unknown. Future studies are necessary to understand investigate the chemical properties and reactivity of organic matter in the ore samples to futher identify relevant interfacial inoganic and organic reaction mechanisms between U, organic matter, and other elements present in the ore samples.
After eight hours of reacting the ore samples with 6% NaOCl, an increase in U concentrations of up to ~9000 μg L−1 was observed. This increase in U concentrations could possibly be caused by the dissolution of U-bearing minerals that are soluble under acidic conditions, pH <4 (Figure 5C, S5C). Again, given the complex mineralogy of the ore, it is not possible to attribute this U release to a specific U phase such as coffinite, U-P-K, and other U-bearing minerals that were identifed in the ore based on XRD and microprobe results discussed in the previous sections. Other studies have indicated that U-bearing minerals such as uranyl sulfates, silicates -(Na, K)-boltwoodite [(Na, K) (UO2SiO3OH. H2O)], and vanadates were previously identified as products of coffinite and uraninite oxidation in solid samples from the Jackpile mine (Deditius et al., 2008; Kelley, 1963). These secondary uranyl minerals can dissolve under acidic conditions at pH <4 based on results reported in other investigations (Avasarala et al., 2017; Gorman-Lewis et al., 2009; Wang et al., 2017). Furthermore, acid digestions of the reacted ore samples indicate that about 11.7% of the total acid extractable U was extracted during batch experiments with 6% NaOCl solution (Table S6). This percentage loss in U concentrations was less than the concentrations lost during reactions with 10 mM NaHCO3. Additional experiments to investigate the combined effect of NaOCl and bicarbonate (NaHCO3) on U reactivity in the ores were conducted.
4.2.4. Reaction with 10 mM NaHCO3 + 6% NaOCl solution.
Ore samples were reacted with 6% NaOCl and 10 mM NaHCO3 to determine the combined effect of oxidation and complexation on U leachability at pH 7.5. There was no noticeable difference in the U leachability obtained from the combined reaction, compared to results from individual reactions with 10 mM NaHCO3 or 6% NaOCl (Figure 5D). A U concentration of 8000 μg L−1 was mobilized in the first two hours, which was similar to that observed during reactions with 6% NaOCl (Figure 5C). The U release was unaffected by the presence of NaHCO3, possibly because the buffering capacity of carbonate and bicarbonate was masked by the high concentration (6%) of HOC1 and OCl− in these experiments.
After two hours of the combined reaction, the U concentrations decreased in a manner similar to observations made during reaction with 6% NaOCl only. This decrease in U concentrations could not be attributed to a specific mechanism. However, based on chemical equilibrium modelling conducted using measured alkalinity, pH, and Ca and U concentrations as inputs, readsorption was unlikely due to repulsion between the neutral to negatively charged aqueous complexes (UO2CO3 (aq), UO2(CO3)22−, Ca2UO2(CO3)30 and CaUO2(CO3)32−), and the negative ore surface charge at pH 5.9 (Table S4, Figure S5D, Figure S6D, Figure S7D). As described for experiments conducted with only NaOCl, readsorption of U as U-organic matter aqueous complexes is a possible mechanism considering the presence of ~3.3 mg L−1 of DOC and ~8000 μg L−1 of U in the experimental ore extracts (Table S5, Figure 5D). The formation of stable U-organic complexes has been previously reported in other investigations (Cumberland et al., 2018; Mikutta et al., 2016). Unlike observations made during reaction with 6% NaOCl only, we found that the U concentrations during ore reaction with 10 mM NaHC03 + 6% NaOCl did not increase again after eight hours of reaction. However, acid digestions on the reacted ore samples still suggested a release of 7.2% of the total acid extractable U, after batch experiments using 6% NaOCl solution (Table S6). Although, these U concentrations are much higher than those lost during reactions with 18MΩ water, they are much lower than the concentrations lost during individual reactions with 10 mM NaHCO3 or 6% NaOCl. Additional solid analyses were conducted to characterize the reacted samples from batch experiments.
4.2.5. Solid analyses of reacted ores.
The XPS analyses on the ore samples reacted with HCO3− and OCl− under 100% air saturated dissolved oxygen concentrations, indicate that all of the U detected in the near surface region of the solids (top 5-10 nm) was oxidized U(VI). The XPS Survey scan of ore samples reacted with 10mM HCO3− detected the presence of 29.5% C 1s, 56.0% O 1s, 14.0 % Si 2p, 0.23% N 1s, and 0.02% U 4f (Table S2). Similarly, XPS survey scans of ore samples reacted with OCl− and OCl− + HCO3− were measured to be 25.6 and 31% C 1s, 60.1 and 46.3% O 1s, 13.9 and 10.85 % Si 2p, 0.23% N 1s, and 0.05 and 0.07% U 4f, respectively. As expected, the 5% U(IV) detected at the surface of unreacted samples was oxidized to U(VI) for all the reacted samples (Figure 1B-D, Table S3).
X-ray absorption spectroscopy analyses confirm that all of the U in the ore samples reacted with OCl− and HCO3− was oxidized to U(VI). Although oxidation of U was also noticeable in experiments reacted with only 100% air saturated HCO3−, some U(IV) remained in reacted ore samples. The XANES spectra indicate that the unreacted samples have similar characteristics to the reference spectra for coffinite, indicating the presence of U(IV) in these samples (Figure 4a). The samples reacted with 100% air saturated HCO3− and OCl− solutions had the characteristic uranyl (UO22+) shoulder indicating that the oxidation of this sample resulted in the increase of U(VI) (Figure 4a). Linear combination fitting (LCF) of ULIII-EXAFS spectra suggest that the bulk unreacted ore samples contained about 60% U(IV) with characteristics similar to coffinite and monomeric U(IV), as indicated in other studies (Bernier-Latmani et al., 2010) (Figure 4b). Note that U(VI) adsorbed to ferrihydrite , uranyl acetate (UO2(CH3COO)2·2H2O), autunite [(Ca(UO2)2(PO4)2(H2O)8—10)], and andersonite [Na2CaUO2(CO3)3(H2O)6] were used in the linear combination fits as references for oxidized U in the solids (Figure 4b and 4c). The LCF of ULIII-EXAFS spectra also indicate that the reaction of ores with HCO3− under 100% air saturated DO concentrations, resulted in the oxidation of coffnite and formation of U(VI) phases (Figure 4b and 4c). Additionally, U(VI) phosphate that is known for its poor solublility at circumneutral pH (Gorman-Lewis et al., 2008), dissolved during ore reaction with HCO3−. These LCF results agree with the obeservations made during batch experiments with HCO3−, that suggested dissolution of coffinite and U-P-K phases as potential contributors to the increasing U concentrations.
As expected, the LCF also suggest that all of the U(IV) present in the ore was oxidized to U(VI) by the HCO3− and OCl− solution (Figure 4b and 4c). Other studies have reported that monomeric U(IV) and UO2 are susceptible to oxidation under ambient dissolved oxygen conditions and with stronger oxidants (Cerrato et al., 2013; Ulrich et al., 2009). Additionally, the U(IV) oxidation was accompanied by simultaneous increase in uranyl acetate concentrations, which could be from U-organic matter interactions, considering the presence of carboxylic functional groups within the unreacted ore samples (Velasco et al., 2019). However, future investigations are necessary to better understand the influence of organic matter and other cooccurring elements on U redox in ores.
5. Conclusions.
The integration of experiments with microscopy, spctroscopy and diffraction analyses on ore samples collected from the Jackpile Paguate U mine, identified the co-existence of U(VI) and U(IV) as U-P-K and coffinite mineral phases encapsulated by carbon. These findings are consistent with observations made previously in ore samples from the Jackpile mine, where coffinites were associated with the organic carbon (Blake et al., 2017; Deditius et al., 2008; Kelley, 1963; Ruiz et al., 2016; Velasco et al., 2019). The association of U with different elements is indicative of the complex mineralogy of these solids. The release of U to solution was observed after reaction of the ore with NaHCO3 and strong oxidants. The highest U concentrations were mobilized after reaction with 10 mM NaHCO3 solution which, provide insights about the presence of elevated U concentrations in the Rio Paguate (35.3 to 772 mg L−1) under relevant alkalinity and pH conditions (Blake et al., 2017). Although the results from this study provide valuable initial information, the complex mineralogy of these ores keep us from attributing the release of U to solution, to specific mechanisms. Several gaps remain in our understanding, some of which are to:1) obtain more detailed solid chemistry information of the ore; 2) better understand the role of organic matter in the reactivity and mobility of U from the ore; and 3) assess the reactvity and solubility of different U minerals under relevant field conditions. More research is necessary to overcome these research gaps to better understand the mechanisms that affect the mobility of U from organic rich ores.
Supplementary Material
Highlights.
Mineralized deposits contained U(IV) and U(VI) in minerals such as coffinite (USiO4) and U-P-K phases encapsulated by carbon.
The highest U concentrations were mobilized after reaction with 100% air saturated 10 mM NaHCO3 solution.
All of the U(IV) present in the deposit samples was oxidized to U(VI) after reaction with 6% NaOCl and HCO3−.
6. Acknowledgements.
The authors would like to thank Lauren Breitner (TOC analyzer), Dr. Zachary Stoll (TOC analyzer), Emily Crowder (TOC analyzer) and Dr. Achraf Noureddine (Zeta Potential) for all their help. Special thanks to the members of the Laguna Pueblo, former Governor Richard Luarkie, current Governor Virgil Siow, and the Pueblo Council for their continued support. The authors would like to especially acknowledge the existing partnership with the Pueblo of Laguna Environment and Natural Resources Department specifically, with its members Loren Arkie, Vince Rodriguez, Sabin Chavez, Deborah Anyaibe, Dorothy Beecher, Cherylin Atcitty, Adam Ringia, and Gregory Jojola who were a great resource in the field. Part of this research was carried out at the Stanford Synchrotron Radiation Light source, a national user facility operated by Stanford University on behalf of the US DOE-OBER. Funding for this research was provided by the National Science Foundation (Grants NM EPSCoR #IIA-1301346, CREST 1345169, and CAREER 1652619) and the National Institute of Environmental Health Sciences Superfund Research Program (Award 1 P42 ES025589). Any opinions, findings, and conclusions or recommendations expressed in this publication are those of the author(s) and do not necessarily reflect the views of the National Science Foundation or the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
6. References.
- Adams S, Curtis H, Hafen P, Salek-Nejad H, 1978. Interpretation of postdepositional processes related to the formation and destruction of the Jackpile-Paguate uranium deposit, northwest New Mexico. Econ. Geol, 73(8): 1635–1654. [Google Scholar]
- Alessi DS et al. , 2014. Speciation and reactivity of uranium products formed during in situ bioremediation in a shallow alluvial aquifer. Environ. Sci. Tech, 48(21): 12842–12850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avasarala S et al. , 2017. Reactive Transport of U and V from Abandoned Uranium Mine Wastes. Environ. Sci. Tech, 51(21):12385–12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargar JR, Bernier-Latmani R, Giammar DE, Tebo BM, 2008. Biogenic uraninite nanoparticles and their importance for uranium remediation. Elements, 4(6): 407–412. [Google Scholar]
- Bargar JR et al. , 2013. Uranium redox transition pathways in acetate-amended sediments. Proc. Nat. Acad. Sci, 110(12): 4506–4511. [Google Scholar]
- Beck R, Cherrywell C, Earnest D, Feirn W, 1980. Jackpile-Paguate deposit: a review. Mem.-NMBur. Mines Miner. Resour. ;(United States), 38(CONF-7905120-). [Google Scholar]
- Benjamin MM, 2014. Water chemistry. Waveland Press. [Google Scholar]
- Bernier-Latmani R et al. , 2010. Non-uraninite products of microbial U (VI) reduction. Environ. Sci. Tech, 44(24): 9456–9462. [DOI] [PubMed] [Google Scholar]
- Bhattacharyya A et al. , 2017. Biogenic non-crystalline U (IV) revealed as major component in uranium ore deposits. Nat. Commun., 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blake J et al. , 2017. Uranium Mobility and Accumulation along the Rio Paguate, Jackpile Mine in Laguna Pueblo, New Mexico. Environ. Sci.: Processes & Impacts, 19(4): 605–621. [DOI] [PubMed] [Google Scholar]
- Blake JM et al. , 2015. Elevated concentrations of U and co-occurring metals in abandoned mine wastes in a northeastern Arizona Native American community. Environ. Sci. Tech, 49(14): 8506–8514. [DOI] [PubMed] [Google Scholar]
- Bone SE, Dynes JJ, Cliff J, Bargar JR, 2017. Uranium (IV) adsorption by natural organic matter in anoxic sediments. Proc. Nat. Acad. Sci: 201611918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouzidi A, Ararem A, Imessaoudene D, Yabrir B, 2015. Sequential extraction of Cs and Sr from Ain Oussera soils around Es-Salam research reactor facility. J. Environ, 36: 163–172. [DOI] [PubMed] [Google Scholar]
- Buck EC, Brown NR, Dietz NL, 1995. Contaminant uranium phases and leaching at the Fernald site in Ohio. Environ. Sci. Tech, 30(1): 81–88. [Google Scholar]
- Cerrato JM et al. , 2013. Relative reactivity of biogenic and chemogenic uraninite and biogenic noncrystalline U (IV). Environ. Sci. Tech, 47(17): 9756–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles A, 1979. Principal mining districts of New Mexico. New Mexico Geology: 37. [Google Scholar]
- Cumberland SA, Douglas G, Grice K, Moreau JW, 2016. Uranium mobility in organic matter-rich sediments: A review of geological and geochemical processes. Earth-Sci. Rev, 159: 160–185. [Google Scholar]
- Cumberland SA et al. , 2018. Characterization of uranium redox state in organic-rich Eocene sediments. Chemosphere, 194: 602–613. [DOI] [PubMed] [Google Scholar]
- De Voto RH, 1978. Uranium geology and exploration. Lecture notes and references. [Google Scholar]
- Deditius AP, Utsunomiya S, Ewing RC, 2008. The chemical stability of coffinite, USiO4· nH2O; 0< n< 2, associated with organic matter: A case study from Grants uranium region, New Mexico, USA. Chem. Geol, 251(1): 33–49. [Google Scholar]
- Dong W, Brooks SC, 2006. Determination of the formation constants of ternary complexes of uranyl and carbonate with alkaline earth metals (Mg2+ , Ca2+ , Sr2+ , and Ba2+ ) using anion exchange method. Environ. Sci. Tech, 40(15): 4689–4695. [DOI] [PubMed] [Google Scholar]
- Dong W, Brooks SC, 2008. Formation of aqueous MgUO2(CO3)32− complex and uranium anion exchange mechanism onto an exchange resin. E Environ. Sci. Tech, 42(6): 1979–1983. [DOI] [PubMed] [Google Scholar]
- El Hayek E et al. , 2018. Effect of Calcium on the Bioavailability of Dissolved Uranium (VI) in Plant Roots under Circumneutral pH. Environ. Sci. Tech, 52(22): 13089–13098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giammar DE, Hering JG, 2001. Time scales for sorption-desorption and surface precipitation of uranyl on goethite. E Environ. Sci. Tech, 35(16): 3332–3337. [DOI] [PubMed] [Google Scholar]
- Ginder-Vogel M, Criddle CS, Fendorf S, 2006. Thermodynamic constraints on the oxidation of biogenic UO2 by Fe(III)(hydr)oxides. Environ. Sci. Tech, 40(11): 3544–3550. [DOI] [PubMed] [Google Scholar]
- Goleva R, Dubinchuk V, Avdonin A, 1981. Relations between silicate and oxide forms of uranium mineralization in stratiform ore shows. Int. Geol. Rev, 23(8): 917–926. [Google Scholar]
- Gorman-Lewis D, Burns PC, Fein JB, 2008. Review of uranyl mineral solubility measurements. JChem Thermodyn, 40(3): 335–352. [Google Scholar]
- Gorman-Lewis D et al. , 2009. Thermodynamic properties of autunite, uranyl hydrogen phosphate, and uranyl orthophosphate from solubility and calorimetric measurements. Environ. Sci. Tech, 43(19): 7416–7422. [DOI] [PubMed] [Google Scholar]
- Grenthe I et al. , 1992. Chemical thermodynamics of uranium, 1 North-Holland Amsterdam. [Google Scholar]
- Hettiarachchi E, Paul S, Cadol D, Frey B, Rubasinghege G, 2018. Mineralogy Controlled Dissolution of Uranium from Airborne Dust in Simulated Lung Fluids (SLFs) and Possible Health Implications. Environ. Sci. Tech. Let. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilpert LS, Moench RH, 1960. Uranium deposits of the southern part of the San Juan Basin, New Mexico. Econ. Geol, 55(3): 429–464. [Google Scholar]
- Hostetler P, Garrels R, 1962. Transportation and precipitation of uranium and vanadium at low temperatures, with special reference to sandstone-type uranium deposits. Econ. Geol, 57(2): 137–167. [Google Scholar]
- Ilton ES, Bagus PS, 2011. XPS determination of uranium oxidation states. Surf. Interface Anal, 43(13): 1549–1560. [Google Scholar]
- Kanematsu M, Perdrial N, Um W, Chorover J, O’Day PA, 2014. Influence of phosphate and silica on U (VI) precipitation from acidic and neutralized wastewaters. Environ. Sci. Tech, 48(11): 6097–6106. [DOI] [PubMed] [Google Scholar]
- Keiluweit M, Kleber M, 2009. Molecular-level interactions in soils and sediments: the role of aromatic π-systems. Environ. Sci. Tech, 43(10): 3421–3429. [DOI] [PubMed] [Google Scholar]
- Kelley VC, 1963. Geology and technology of the Grants uranium region. State Bureau of Mines and Mineral Resources, Society of Economic Geologists. [Google Scholar]
- Kittel DF, 1963. Geology of the Jackpile mine area. Geology and Technology of the Grants Uranium Region, Memoir, 15. [Google Scholar]
- Kretzschmar R, Borkovec M, Grolimund D, Elimelech M, 1999. Mobile subsurface colloids and their role in contaminant transport. Adv. Agron, 66: 121–193. [Google Scholar]
- La Force MJ, Fendorf S, 2000. Solid-phase iron characterization during common selective sequential extractions. Soil Sci. Soc. Am. J, 64(5): 1608–1615. [Google Scholar]
- Langmuir D, 1997. Aqueous environmental. Prentice Hall. [Google Scholar]
- Laverty R, Ashwill W, Chenoweth W, Norton D, 1963. Ore processes. Geology and technology of the Grants uranium region: New Mexico Bureau of Mines and Mineral Resources Memoir, 15: 191–204. [Google Scholar]
- Lollar BS, 2005. Environmental geochemistry, 9 Elsevier. [Google Scholar]
- Love KM, Woronow A, 1991. Chemical changes induced in aragonite using treatments for the destruction of organic material. Chem. Geol, 93(3–4): 291–301. [Google Scholar]
- Lovley DR, Phillips EJ, 1991. Microbial reduction of uranium. Nature, 350(6317): 413. [Google Scholar]
- Luo W, Gu B, 2008. Dissolution and mobilization of uranium in a reduced sediment by natural humic substances under anaerobic conditions. Environ. Sci. Tech, 43(1): 152–156. [DOI] [PubMed] [Google Scholar]
- Maurice PA, 2009. Environmental surfaces and interfaces from the nanoscale to the global scale. Wiley. [Google Scholar]
- Maxwell CH, 1982. Mesozoic stratigraphy of the Laguna-Grants region, Albuquerque Country II. NM Geol. Soc., 33rd Annual Field Conf, pp. 261–266. [Google Scholar]
- McCarthy JF, Zachara JM, 1989. Subsurface transport of contaminants. Environ. Sci. Tech, 23(5): 496–502. [Google Scholar]
- Mibus J, Sachs S, Pfingsten W, Nebelung C, Bernhard G, 2007. Migration of uranium (IV)/(VI) in the presence of humic acids in quartz sand: A laboratory column study. J. Contam. hydrol, 89(3): 199–217. [DOI] [PubMed] [Google Scholar]
- Mikutta C, Langner P, Bargar JR, Kretzschmar R, 2016. Tetra-and hexavalent uranium forms bidentate-mononuclear complexes with particulate organic matter in a naturally uranium-enriched peatland. Environ. Sci. Tech, 50(19): 10465–10475. [DOI] [PubMed] [Google Scholar]
- Mkandawire M, Dudel EG, 2008. Natural occurring uranium nanoparticles and the implication in bioremediation of surface mine waters. Uranium, Mining and Hydrogeology: 487–496. [Google Scholar]
- Moench RH, Schlee JS, 1967. Geology and uranium deposits of the Laguna district, New Mexico: US Government Printing Office. [Google Scholar]
- Nash JT, 1968. Uranium deposits in the Jackpile Sandstone, New Mexico. Econ. Geol, 63(7): 737–750. [Google Scholar]
- Nenoff TM et al. , 2011. Synthesis and low temperature in situ sintering of uranium oxide nanoparticles. Chem. Mater, 23(23): 5185–5190. [Google Scholar]
- O’Loughlin EJ, Kelly SD, Cook RE, Csencsits R, Kemner KM, 2003. Reduction of uranium (VI) by mixed iron (II)/iron (III) hydroxide (green rust): formation of UO2 nanoparticles. Environ. Sci. Tech, 37(4): 721–727. [DOI] [PubMed] [Google Scholar]
- Ravel B, Newville M, 2005. Athena, Artemis, Hephastus: Data analysis for X-ray absorption spectroscopy using IFEFFIT. J. Synchrotron Radiat, 12(4): 537–541. [DOI] [PubMed] [Google Scholar]
- Regenspurg S et al. , 2010. Speciation of naturally-accumulated uranium in an organic-rich soil of an alpine region (Switzerland). Geochimica et Cosmochimica Acta, 74(7): 2082–2098. [Google Scholar]
- Riba O, Walker C, Ragnarsdottir KV, 2005. Kinetic studies of synthetic metaschoepite under acidic conditions in batch and flow experiments. Environ. Sci. Tech, 39(20): 7915–7920. [DOI] [PubMed] [Google Scholar]
- Risser DW, Davis PA, Baldwin JA, McAda DP, 1984. Aquifer tests at the Jackpile-Paguate uranium mine, Pueblo of Laguna, west-central New Mexico. US Geol. Surv. Water Resour. Invest. Rep, 84(4255): 26. [Google Scholar]
- Ruiz O, Thomson BM, Cerrato JM, 2016. Investigation of in situ leach (ISL) mining of uranium in New Mexico and post-mining reclamation. New Mexico Geology, 38(4). [Google Scholar]
- Schecher W, MINEQL DM, 2007. A Chemical Equilibrium Modeling System, Version 4.6, 4.5. Environmental Research Software: Hallowell, ME. [Google Scholar]
- Schindler M et al. , 2017. Mobilization and agglomeration of uraninite nanoparticles: A nano-mineralogical study of samples from the Matoush Uranium ore deposit. Am. Mineral, 102(9): 1776–1787. [Google Scholar]
- Schultz MK, Burnett WC, Inn KG, 1998. Evaluation of a sequential extraction method for determining actinide fractionation in soils and sediments. J. Environ. Radioact, 40(2): 155–174. [Google Scholar]
- Semião AJ, Rossiter HM, Schäfer A I, 2010. Impact of organic matter and speciation on the behaviour of uranium in submerged ultrafiltration. J. Memb. Sci, 348(1–2): 174–180. [Google Scholar]
- Singer DM, Zachara JM, Brown GE Jr, 2009. Uranium speciation as a function of depth in contaminated Hanford sediments-A micro-XRF, micro-XRD, and micro-and bulk-XAFS study. Environ. Sci. Tech, 43(3): 630–636. [DOI] [PubMed] [Google Scholar]
- Singh A, Catalano JG, Ulrich K-U, Giammar DE, 2012. Molecular-scale structure of uranium (VI) immobilized with goethite and phosphate. Environ. Sci. Tech., 46(12): 6594–6603. [DOI] [PubMed] [Google Scholar]
- Steelink C, 2002. Peer reviewed: investigating humic acids in soils. ACS Publications. [DOI] [PubMed] [Google Scholar]
- Stumm W, Morgan JJ, 2012. Aquatic chemistry: chemical equilibria and rates in natural waters, 126 John Wiley & Sons. [Google Scholar]
- Tokunaga TK, Kim Y, Wan J, 2009. Potential remediation approach for uranium-contaminated groundwaters through potassium uranyl vanadate precipitation. Environ. Sci. Tech, 43(14): 5467–5471. [DOI] [PubMed] [Google Scholar]
- Troyer LD et al. , 2016. Effect of phosphate on U(VI) sorption to montmorillonite: Ternary complexation and precipitation barriers. Geochim. Cosmochim. Acta, 175: 86–99. [Google Scholar]
- Tutu H, Cukrowska EM, McCarthy TS, Hart R, Chimuka L, 2009. Radioactive disequilibrium and geochemical modelling as evidence of uranium leaching from gold tailings dumps in the Witwatersrand Basin. Int. J. Environ. Anal. Chem, 89(8–12): 687–703. [Google Scholar]
- Ulrich K-U et al. , 2009. Comparative dissolution kinetics of biogenic and chemogenic uraninite under oxidizing conditions in the presence of carbonate. Geochim. Cosmochim. Acta, 73(20): 6065–6083. [Google Scholar]
- Uyuşur B, Li C, Baveye PC, Darnault CJ, 2015. pH-dependent reactive transport of uranium (VI) in unsaturated sand. J. soils and sediments, 15(3): 634–647. [Google Scholar]
- Veeramani H et al. , 2011. Products of abiotic U(VI) reduction by biogenic magnetite and vivianite. Geochim. Cosmochim. Acta, 75(9): 2512–2528. [Google Scholar]
- Velasco CA et al. , 2019. Organic Functional Group Chemistry in Mineralized Deposits Containing U (IV) and U (VI) from the Jackpile Mine in New Mexico. Environ. Sci. Tech. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G et al. , 2017. Uranium Release from Acidic Weathered Hanford Sediments: Single-Pass Flow-Through and Column Experiments. Environ. Sci. Tech.. [DOI] [PubMed] [Google Scholar]
- Wang Y et al. , 2013. Mobile uranium (IV)-bearing colloids in a mining-impacted wetland. Nat. communications, 4: 2942. [DOI] [PubMed] [Google Scholar]
- Yang Y, Saiers JE, Barnett MO, 2013. Impact of interactions between natural organic matter and metal oxides on the desorption kinetics of uranium from heterogeneous colloidal suspensions. Environ. Sci. Tech, 47(6): 2661–2669. [DOI] [PubMed] [Google Scholar]
- Yang Y et al. , 2012. Impact of natural organic matter on uranium transport through saturated geologic materials: from molecular to column scale. Environ. Sci. Tech, 46(11): 5931–5938. [DOI] [PubMed] [Google Scholar]
- Zehner HH, 1985. Hydrology and water-quality monitoring considerations, Jackpile uranium mine, northwestern New Mexico. [Google Scholar]
- Zhou P, Gu B, 2005. Extraction of oxidized and reduced forms of uranium from contaminated soils: Effects of carbonate concentration and pH. Environ. Sci. Tech, 39(12): 4435–4440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.