

SUMMARY
The NuRD complex contains both chromatin remodeling and histone deacetylase activities. Mice lacking the MTA2 subunit of NuRD show developmental defects in pro-B, pre-B, immature B, and marginal zone B cells, and abnormal germinal center B cell differentiation during immune responses. Mta2 inactivation also causes a derepression of Igll1 and VpreB1 genes in pre-B cells. Furthermore, MTA2/NuRD interacts directly with AIOLOS/IKAROS and shows a striking overlap with AIOLOS/IKAROS target genes in human pre-B cells, suggesting a functional interdependence between MTA2/NuRD and AIOLOS. Mechanistically, MTA2 deficiency in mice leads to increased H3K27 acetylation at both Igll1 and VpreB1 promoters. Gene profiling analyses also identify distinct MTA2-dependent transcription programs in pro-B and pre-B cells. In addition, we find a strong synergy between MTA2 and OCA-B in repressing Igll1 and VpreB1 at the pre-B cell stage, and in regulating both the pre-B to immature B transition and splenic B cell development.
Graphical Abstract
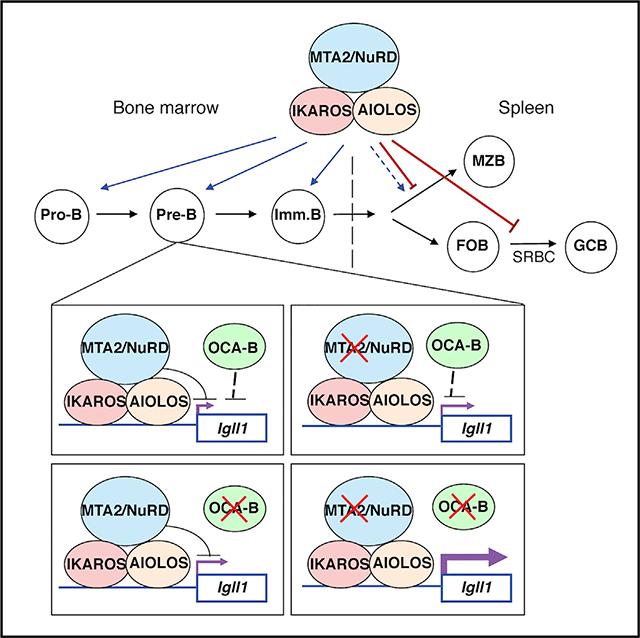
In Brief
Lu et al. examine B cell developmental defects in MTA2-deficient mice. MTA2 interacts with AIOLOS/IKAROS, represses Igll1 expression, co-binds to most AIOLOS/IKAROS target genes in pre-B cells, and cooperates with OCA-B in the pre-B to immature B transition. These data suggest that AIOLOS/IKAROS functions through MTA2/NuRD during B cell development.
INTRODUCTION
Mammalian B lymphocyte development is a tightly regulated multi-step process that proceeds from hematopoietic stem cells (HSCs) in the bone marrow through several intermediate progenitor cell stages, including multipotent progenitors (MPPs), earliest lymphocyte progenitors (ELPs), and common lymphoid progenitors (CLPs), before differentiation into B cells. Intensive studies over the past decades have implicated multiple key transcription factors (TFs) in the regulation of B cell development, including factors (e.g., PU.1, Ikaros, BCL11a, E2A, EBF, and -PAX5) that act either positively to promote B cell-specific gene expression or negatively to repress non-B lineage programs (Busslinger, 2004; Matthias and Rolink, 2005). These sequence-specific TFs achieve activation or repression of target genes through interactions both with the general transcription machinery and with chromatin regulators (e.g., histone modification enzymes and chromatin remodeling complexes), but how specific chromatin regulators contribute to B cell development remains largely unknown (Busslinger and Tarakhovsky, 2014).
Among chromatin-modifying factors, the heterogeneous NuRD (nucleosome remodeling histone deacetylase) complex is of special interest because it possesses both ATP-dependent nucleosome remodeling and histone deacetylase activities. The mammalian NuRD complexes are composed of both common factors (HDAC1/2, RbAp46/48) and variable modular factors that result in related heterogeneous complexes that likely modulate different transcriptional programs (Dege and Hagman, 2014; Feng and Zhang, 2003). Thus, beyond the common components, NuRD complexes variably contain a member (either CHD3/MI-2α or CHD4/MI-2β) of the CHD family of ATP-dependent chromatin remodeling factors, a member (MTA1, MTA2, or MTA3) of the metastasis-associated factor MTA family, a member (MBD2 or MBD3) of the methyl-CpG binding domain proteins, and either P66α or P66β (whose functions are likely to be mediated through interactions with core histones and MBD2) (Dege and Hagman, 2014; Denslow and Wade, 2007). In vivo and cell-based studies have demonstrated important and non-redundant functions of different NuRD modular components in multiple biological processes that include embryonic stem cell (ESC) maintenance, tumor progression, circadian clock regulation, synaptic differentiation, and granule neuron function in the cerebellum cortex (Dege and Hagman, 2014; Denslow and Wade, 2007; Kim et al., 2014; Sen et al., 2014; Yamada et al., 2014; Yang et al., 2016).
In relation to NuRD function in lymphogenesis, of primary interest here, previous studies have demonstrated (1) an association of MI-2β/NuRD with IKAROS and AIOLOS in T cells (Avitahl et al., 1999; Zhang et al., 2011); (2) reductions in CD4+ T cell number and Cd4 gene expression (Williams et al., 2004); (3) abnormal HSC homeostasis and defective differentiation into myeloid and lymphoid lineages (Yoshida et al., 2008), following Mi-2β inactivation; (4) an MI-2β requirement for maintaining DNA-hypermethylated chromatin at the mb1 gene promoter (Gao et al., 2009); (5) spontaneous B cell lymphomagenesis following Mta1 overexpression in vivo (Bagheri-Yarmand et al., 2007); (6) an important role for Mta3 in plasma cell differentiation (Fujita et al., 2004); and (7) MBD3/NuRD-mediated repression of the B cell transcription program in multipotent lymphoid progenitors in order to maintain a balanced differentiation of T and B lineage cells (Loughran et al., 2017). Related, our previous data showed that Mta2-deficient mice develop lupus-like autoimmune symptoms (Lu et al., 2008). However, the role of distinct NuRD complexes in normal B cell development and the transcriptional programs they regulate remain to be elucidated.
Here, we have characterized a number of conventional, conditional mutant and double-mutant mouse models in order to delineate specific phenotypes in B cell development, allowing us to link the MTA2 component of the MTA2/NuRD complex to the lymphocyte-specific transcription factor AIOLOS and the B cell-specific transcription coactivator OCA-B (Luo and Roeder, 1999; Teitell, 2003) in vivo. We also identified distinct groups of genes regulated by MTA2 in pro-B and pre-B cells.
RESULTS
B Cell Developmental Defects in Mta2 Conventional and Conditional Knockout (KO) Mice
To understand the function of MTA2/NuRD in B cell development, we first analyzed the bone marrow (BM) B cell subpopulations in 1.5- to 2.5-month-old Mta2 conventional null (Δ/Δ) (n = 5) and littermate control mice that include both wild type and Mta2 heterozygous mice (n = 11). The Mta2 null strain was derived by crossing EIIa-Cre transgenic mice with Mta2fl/fl mice (Lu et al., 2008). Mta2 null mice showed decreased frequencies of immature B (B220loIgM+), mature B (B220hiIgM+), pro-B (also called pre-BI, B220+IgM−CD43+), and pre-B (also called pre-BII, B220+IgM−CD43−) cells in BM (Figure 1A). On average, the absolute numbers of total BM B (B220+), pro-B, pre-B, and immature B cells in Mta2 Δ/Δ mice were about one-third to one-half of those in control mice (Figure 1B). Regarding the B cell repertoire in the periphery, and while a decrease of marginal zone B (MZB) cells (IgM+B220+CD21hiCD23lo) has been observed in young Mta2 Δ/Δ mice (6–8 weeks), this defect is more consistent and severe in mice older than 3 months old (Figures 1C and 1D). The number of follicular B (FOB) cells (IgM+B220+CD21−CD23hi) in adult Mta2 Δ/Δ mice remains largely normal (Figures 1C and 1D).
Figure 1. BM and Peripheral B Cell Developmental Defects in the Conventional Mta2 Null Mice.

(A) Flow cytometry data of BM cells from a 2-month-old Mta2 Δ/Δ and a littermate wild type control mouse stained for immature B (B220loIgM+), mature B (B220hiIgM+), pro-B (B220+IgM−CD43+), and pre-B (B220+IgM−CD43−) cell populations. Numbers indicate percentages of specific cell subsets in parental populations.
(B) Cell numbers of different BM B cell sub-populations (from two femurs) in 1.5- to 2.5-month-old control and Mta2 Δ/Δ mice (n = 11 for control and n = 5 for mutant mice; t test; *p < 0.05, **p <0.01, ***p < 0.001).
(C) FACS analysis of total splenic B (IgM+B220+), MZB (IgM+B220+CD21hiCD23lo), and FOB (IgM+B220+CD21−CD23hi) cells from a 4-month-old Mta2 Δ/Δ mouse and a wild-type (WT) litter-mate control mouse.
(D) Numbers of total splenic B, MZB, and FOB cells from 2- to 4-month-old wild-type and Mta2 Δ/Δ mice are plotted (n = 10 for each genotype; t test; ***p < 0.001).
To avoid the experimental obstacle caused by partial embryonic lethality of Mta2 Δ/Δ mice (Lu et al., 2008) and, more importantly, to assess the B cell-autonomous defects, we generated Cd19-Cre- and Mb1-Cre (Cd79a) Mta2 conditional KO mice. These Mta2 conditional KO mice were backcrossed with C57/BL6 mice for at least six generations. Cd19 encodes a B cell-specific cell surface signaling molecule, CD19, that is presented on all B220+CD43+HSA+ cells (a mixture of pro-B and large pre-B cells), but not on the earliest B220-expressing cells (B220+CD43+HAS−) (Krop et al., 1996). Mb1 encodes the Igα subunit of the pre-BCR (B cell receptor) complex and is expressed earlier in development than Cd19 (Hobeika et al., 2006). Surprisingly, fluorescence-activated cell sorting (FACS) analysis showed largely normal pro-B and pre-B cell compartments in Cd19-Cre Mta2fl/fl and Mb1-Cre Mta2fl/fl mice (Figures S1A–S1C). Subsequent qPCR data revealed that more than 85% of the pro-B cells from Cd19-Cre Mta2fl/fl mice and 30%of the pro-B cells from Mb1-Cre Mta2fl/fl mice still maintain the wild-type Mta2 alleles (Figure S1D), partially explaining the lack of pro-B phenotype in these mutant mice. In contrast, both pre-B and immature B cells from Cd19-Cre Mta2fl/fl mice, especially those B cell subsets from Mb1-Cre Mta2fl/fl mice, show high deletion efficiencies of the Mta2 allele (Figure S1D). Intriguingly, in relation to splenic B cell development, while Mta2 Δ/Δ mice show loss of MZB cells, both Cd19-Cre Mta2fl/fl and Mb1-Cre Mta2fl/fl show increases in MZB cell numbers and MZB frequencies in total spleen cells (Figure S2). To avoid the complexity caused by escaper pro-B cells (cells maintaining the floxed Mta2 allele) in Cd19-Cre Mta2fl/fl and Mb1-Cre Mta2fl/fl mice, we generated Vav-Cre Mta2fl/fl mice in which Cre transgene expression begins at the HSC stage (Ogilvy et al., 1999). FACS analyses revealed decreased population sizes of pro-B, pre-B, immature B cell, and recirculating B cell compartments in Vav-Cre Mta2fl/fl mice, thus recapitulating the BM B cell defects of conventional null mice (Figures 2A and 2B) and supporting the notion that Mta2 is required for normal development of pro-B cells. qPCR results also show optimal deletion efficiencies in different BM B cell subsets (Figure 2C). Within the pro-B cell population, there is more than 50 percent reduction in both Hardy Fractions B (Fr. B) and C (Fr. C) cells when MTA2 is lost (Figure 2D). qPCR analyses show that Mta2 inactivation results in abnormal rearrangement of immunoglobulin (Ig) heavy chain genes (such as VHJ558-JH3 and VHJ7183-JH3) in pro-B cells (Figure 2E). Regarding the pre-B cell population, Vav-Cre Mta2fl/fl mice contain a higher percentage of large pre-B cells and fewer small pre-B cells than control mice (Figure 2F), suggesting a cell cycle defect in these mutant pre-B cells (which might explain the decreased number of immature B cells in mutant mice). In addition, we also find abnormal Ig k chain rearrangements in Vav-Cre Mta2fl/fl small pre-B cells (Figure 2G). Furthermore, the distal recombination events seem to be more affected by Mta2 inactivation than are the proximal recombination events, suggesting a premature termination of the recombination process (Figure 2G).
Figure 2. Vav-Cre Mta2fl/fl Mice Recapitulate the BM B cell Developmental Defects of Mta2Δ/Δ Mice.

(A) BM cells from mice with indicated Mta2 status stained for immature B (B220loIgM+), mature B (B220hiIgM+), pro-B (B220+IgM−CD43+), and pre-B (B220+IgM−CD43−) cell populations. Numbers indicate percentages of specific cell subsets in parental populations.
(B) Cell numbers of different BM B cell subsets (from two femurs) in 2- to 3-month-old Vav-Cre Mta2fl/fl and littermate or age-matched control mice (n ≥ 16 for Vav-Cre Mta2fl/fl and control mice; t test; **p < 0.01, ***p < 0.001).
(C) qPCR results indicating efficiencies of Vav-Cre-mediated deletion of Mta2 allele in indicated cell populations (n = 4 for each cell subsets; t test; ***p < 0.001).
(D) Representative FACS data showing percentages of Hardy Fr. A to C within the pro-B population in Vav-Cre Mta2fl/fl and control mice. The average numbers of each fractions are plotted (n = 9 for each genotype; t test; **p < 0.01).
(E) qPCR results indicating relative abundances of different immunoglobulin heavy chain gene rearrangements in pro-B cells isolated from mice with indicated genotypes (n = 4 for each genotype).
(F) Representative FACS data showing percentages of large (L) and small (S) pre-B (B220+IgM−CD43−) cells in Vav-Cre Mta2fl/fl and control mice. The average numbers of each type of pre-B cells from mice with indicated genotypes are plotted (n ≥ 13 for Vav-Cre Mta2fl/fl and littermate control mice; t test; ***p < 0.001).
(G) qPCR results indicating relative abundances of different VJ rearrangements in small pre-B cells isolated from mice with indicated genotypes (n = 4 for each genotype; t test; *p < 0.05, **p < 0.01, ***p < 0.001).
MTA2/NuRD-AIOLOS/IKAROS Interaction and MTA2-Dependent Repression of Pre-BCR Genes
Altogether, the B cell phenotypes described above, as well as other defects of the immune system caused by Mta2 inactivation (Lu et al., 2008), were reminiscent of those found in mice lacking AIOLOS, a member of the IKAROS family and a key regulator of lymphocyte function (Wang et al., 1998). IKAROS and AIOLOS were found to be stably associated with NuRD complexes in T cells (Kim et al., 1999; Zhang et al., 2011). To assess whether MTA2/NuRD interacts with AIOLOS in B cells, nuclear extracts (NEs) from human pre-B leukemia 697 cells were subjected to immunoprecipitation with antibodies to MTA2, MI-2β, AIOLOS, and IKAROS (Figure 3A). The results show reciprocal co-immunoprecipitation between AIOLOS, IKAROS, and NuRD components MTA2 and MI-2β (Figure 3A), and were confirmed with NE from Namalwa cells (of germinal center origin) (Figure S3A). Thus, both phenotypic similarities and direct biochemical evidence suggested that Mta2-deficient mice may have other B cell developmental defects similar to those observed in Aiolos KO mice.
Figure 3. MTA2/NuRD Interacts with AIOLOS and Represses Igll1 and VpreB Genes in Pre-B Cells.

(A) Immunoprecipitation (IP)-Western blot results using 697 human pre-B leukemia cell nuclear extract (NE). Antibodies used for IPs and immunoblots are marked (AIO: AIOLOS, IKA: IKAROS).
(B) RT-qPCR results showing expression levels of Igll1 and VpreB1 genes in pre-B cells isolated from Vav-Cre Mta2fl/fl, Mx1-Cre Mta2fl/fl and their litter-mate control mice (n = 4 for Vav-Cre Mta2fl/fl and control, n = 3 for Mx1-Cre Mta2fl/fl and control; t test; *p < 0.05, **p < 0.01, ***p < 0.001).
(C) ChIP-qPCR data using human 697 cells showing enrichment of AIOLOS, MI-2β, MTA2 (left panel), and IKAROS (right panel) at the IGLL1 and VPREB1 promoter regions. The CNAP1 gene locus was used as a negative region control. Data were collected from 3 or more independent ChIP assays.
(D) ChIP-qPCR data showing enrichment of H3K27Ac and H3K4Me3 marks (normalized to H3) at promoter regions of Igll1 and VpreB1 genes in primary small pre-B cells sorted from Vav-Cre Mta2fl/fl (VavKO) and control mice (t test; *p < 0.05, **p < 0.01).
During mouse BM B cell development, immunoglobulin heavy chain (IgM) rearrangement occurs at the pro-B stage. The heavy chain then associates with the surrogate light chain (SLC), composed of LAMBDA5 (λ5) and VPREB proteins. Aiolos expression is upregulated at the pro-B to pre-B cell transition stage, and Igll1 (encoding λ5) and VpreB1 genes are downregulated at the pre-B stage. In Aiolos-null pre-B cells, both Igll1 and VpreB1 are derepressed—demonstrating a key role of Aiolos in silencing these genes at this stage (Karnowski et al., 2008; Thompson et al., 2007). In addition, Ikaros overexpression in mouse pre-B cells also results in decreased Igll1 expression (Ferreirós-Vidal et al., 2013). To test whether MTA2 is also involved in pre-BCR gene silencing, we examined Igll1 and VpreB1 expression levels in small pre-B cells (B220+IgM−CD43−) sorted from Vav-Cre Mta2fl/fl and control mice (Figure S3B). The RT-qPCR results show marked increases in expression of these genes in Mta2-deficient pre-B cells and are confirmed in Mx1-Cre Mta2fl/fl pre-B cells (Figure 3B). The increase of Igll1 and VpreB1 expression is further confirmed in Vav-Cre Mta2fl/fl pre-B cells sorted by an alternative gating scheme (B220+IgM−CD43−CD25+) (Figures S3C and S3D). To examine whether MTA2/NuRD regulates pre-BCR genes directly, chromatin immunoprecipitation (ChIP) as-says were performed using 697 human pre-B leukemia cells. The data demonstrate significant enrichments of MTA2, MI-2β, AIOLOS, and IKAROS at IGLL1 and VPREB1 promoters (Figure 3C). Thus, both genetic and biochemical experiments clearly demonstrate that MTA2/NuRD directly interacts with AIOLOS and IKAROS in developing B cells, and jointly targets pre-BCR gene loci. ChIP assays demonstrated that loss of MTA2 activity leads to increase of histone H3 lysine 27 acetylation (H3K27Ac) at both Igll1 and VpreB1 promoters (Figure 3D), indicating the mechanism underlying the increased Igll1 and VpreB1 expression in Mta2-deficient pre-B cells.
Highly Overlapping Genomic Distributions of MTA2, HDAC2, AIOLOS, and IKAROS in Human Pre-B Cells
To better understand the MTA2/NuRD-dependent gene regulation and its relationship to AIOLOS and IKAROS, we examined the genomic distributions of MTA2, HDAC2 (another subunit of NuRD complex), AIOLOS, and IKAROS in 697 cells by chromatin immunoprecipitation sequencing (ChIP-seq). These analyses identified about 28,760 MTA2 peaks, with 19% at promoter regions, 44% at intragenic regions (including introns, exons, 5′and 3′ UTR regions), and 34% at intergenic regions (including distal enhancers) (Figure 4A). HDAC2, AIOLOS, and IKAROS showed similar distribution patterns (Figure 4A). A further examination of the average distance from the binding sites of these factors to transcription start sites (TSSs) and enhancers revealed enrichment of all these factors slightly upstream of the TSS (Figure 4B). At enhancer regions, all four factors showed almost the same binding patterns (Figure 4B). Furthermore, the ChIP-seq data showed a striking overlap between target genes bound by these factors (Figure 4C). For example, about 90% of the MTA2 target genes are also AIOLOS targets, more than 90% of the HDAC2 targets are also MTA2 targets, and more than 90% of AIOLOS targets are also IKAROS targets (Figure 4C). A heatmap generated by normalized read densities from individual ChIP-seq data centered at TSSs and ranked by AIOLOS peak signals further demonstrates the high degree of AIOLOS/IKAROS and MTA2/HDAC2 colocalization at promoter regions (Figure 4D). These data strongly support a model in which AIOLOS/IKAROS recruits the MTA2/NuRD complex to repress target genes in pre-B cells.
Figure 4. Genome-Wide Mapping of MTA2, HDAC2, AIOLOS, and IKAROS in Human Pre-B Cells.

(A) Distribution of MTA2, HDAC2, AIOLOS, and IKAROS binding sites at different genomic locations in 697 human pre-B cells. Numbers indicate the percentage of total sites at each location.
(B) Meta-gene analysis of average enrichment of indicated factors around transcription start sites (TSSs) (left panel) and enhancers (right panel).
(C) Venn diagrams showing significant overlaps between AIOLOS, MTA2, and HDAC2 target genes (left panel) and between IKAROS and AIOLOS target genes (right panel).
(D) A heatmap generated by normalized read densities from individual ChIP-seq data (AIO, AIOLOS; IKA, IKAROS) centered at TSS and ranked by AIOLOS peak signals.
(E) Consensus AIOLOS-, IKAROS, MTA2, and HDAC2 recognition sequences identified by de novo-motif identification in 697 cells. The more detailed summaries of DNA motifs recognized by these factors are listed in Tables S1–S4.
(F) ChIP-seq snapshots of AIOLOS, IKAROS, HDAC2, and MTA2 binding at pre-BCR gene promoters in 697 cells. The scales of ChIP-seq signals are indicated.
Toward the identification of DNA sequence elements recognized by MTA2, HDAC2, AIOLOS, and IKAROS, respectively, motif analyses indicated that the top binding sites preferred by MTA2, HDAC2, and AIOLOS/IKAROS include motifs recognized by EBF1, RUNX1, and ETS family members (FLI1, ERG, and ETV2) (Figure 4E). The more detailed summaries of DNA motifs recognized by these factors are listed in Tables S1–S4. These data suggest a model in which AIOLOS/IKAROS/NuRD function, at least partially, by repressing target genes of EBF1, RUNX1, and ETS family transcription factors in pre-B cells. This notion is consistent with a previously proposed model that AIOLOS represses Igll1 expression at the pre-B stage by competing with EBF1 at the same DNA binding site (Thompson et al., 2007).
The ChIP-seq data also confirm that the IGLL1 and VPREB1 promoters are loaded with AIOLOS and NuRD components MTA2 and HDAC2 (Figure 4F), and that this is also the case for the promoters of the CD79A and CD79B genes (encoding the Igα and Igβ subunits of the pre-BCR/BCR complexes) (Figure 4F). For a group of genes, such as CD86 and BCL2, AIOLOS/IKAROS and NuRD do not bind to the promoter regions but instead bind to the intronic and 3′ UTR elements (Figures S4A and S4B). Interestingly, the expression levels of Cd86 and Bcl2 are elevated in Mta2-deficient murine pre-B cells (data not shown), suggesting a possible enhancer-mediated transcription repression for these genes.
MTA2-Dependent Transcriptional Program in Pro-B and Pre-B Cells
To understand the molecular basis of pro-B and pre-B defects observed in Vav-Cre Mta2fl/fl mice and to assess MTA2/NuRD-regulated transcriptional programs in these cells, we performed RNA-sequencing (RNA-seq) analyses using pro-B cells and small pre-B cells from Vav-Cre Mta2fl/fl and control mice (the cell sorting strategies are like the ones shown in Figure S3B) (Figure 5A). Consistent with the observation that MTA2 enhances HDAC activity in vitro, as well as a general role of HDACs in gene repression (Feng and Zhang, 2003), many more genes were upregulated (440 genes >1.5-fold) than downregulated (88 genes >1.5-fold) in pro-B cells upon Mta2 inactivation (Figure 5B). Among upregulated genes, about 8% (37) showed more than 3-fold increases in expression, while the rest (403) showed 1.5- to 3-fold increases in expression (Figure 5B). Among downregulated genes, about 6%(5) showed more than 3-fold reductions in expression, whereas 94% (83) showed 1.5- to 3-fold reductions (Figure 5B). A gene ontology (GO) analysis of upregulated genes in Mta2-deficient pro-B cells showed that the top enriched pathways or cellular processes include neutrophil degranulation and activation (e.g., Cd14, Cd63, Chit1, Ckap4, Lrg1, etc.); inflammatory responses (e.g., Cebpb, Csf1r, Ptgir, Cd6, Cxcr2, etc.); cell junction and extracellular matrix (ECM) relationships (e.g., Tjp1, Prtn3, Itgb4, Col5a1, etc.); macromolecule metabolism (e.g., Acvrl1, ccr1, and Cd3e, etc.); and endocytosis (e.g., Cd63 Lrp1, Hip1, Apoe, etc.) (Figure 5C). Other genes that showed more substantial (>3-fold) upregulation in Mta2-deficient pro-B cells include the following: Gpr4, Asprv1, Rcn3, Ly6a, Padi3, etc.
Figure 5. Distinct MTA2-Dependent Transcriptional Programs in Mouse Pro-B and Pre-B Cells.

(A) Heatmaps based on RNA-seq data from Vav-Cre Mta2fl/fl (VavKO) and control pro-B (left) and small pre-B (right) cells.
(B) Numbers of genes that show increased or decreased expression in pro-B and small pre-B cells upon loss of Mta2 (threshold 1.5-fold).
(C) Gene ontology (GO) analyses of MTA2-regulated pathways and processes in pro-B and small pre-B cells (black, 1.5- to 3-fold change; blue, 3–5-fold; red, >5-fold).
In the small pre-B cell compartment, there also were more genes upregulated (435 genes) than downregulated (229 genes) upon Mta2 inactivation (>1.5-fold) (Figure 5B). Among upregulated genes, about 15% (62) showed more than 3-fold increases in expression, while the rest (373) showed 1.5- to 3-fold increases in expression (Figure 5B). Among downregulated genes, only 5% (12) showed more than 3-fold reductions in expression, whereas 95% showed 1.5- to 3-fold reductions (Figure 5B). A GO analysis showed that the top enriched pathways/cellular processes in Mta2-deficient pre-B cells include the following: T cell receptor (TCR) and T cell differentiation (e.g., Lck, Thy1, Lat, Zap70, etc.), regulation of apoptosis (e.g., Bcl2, Ccnd2, Socs2, c-Kit, etc.), and inositol phosphate metabolism (e.g., Plcd3, Plcb3, Inpp4a, etc.) (Figure 5C). Intriguingly, a group of genes involved in axon guidance (e.g., Plxnd1, Plxnb2, Plxnc1, etc.) are also repressed by MTA2 in pre-B cells (Figure 5C). A comparison of our RNA-seq data with previously reported gene profiling data revealed that genes normally silenced during BM B cell differentiation from Hardy Fr C′ (cycling pre-B) to Fr D (resting pre-B) stages (Ferreirós-Vidal et al., 2013) are enriched in upregulated genes in Vav-Cre Mta2fl/fl small pre-B cells (Figure S5A), indicating an important role of MTA2/NuRD at this development stage. Other MTA2-regulated genes that showed substantial upregulation (>3.5-fold) include the following: Crhbp, Atp11a, Trim63, Ikzf2 (Helios), Gprasp2, Rcn3, H1f0, etc. Our RNA-seq data also confirmed increased expression of Igll1 and VpreB1 genes in small pre-B cells. Among MTA2-repressed genes in small pre-B cells, about 70% have MTA2 binding sites at loci of their human orthologs in 697 cells (Figure S5B), suggesting that these are potential direct MTA2 target genes in mouse pre-B cells. It is noteworthy that only 96 genes (21% of upregulated genes in Mta2-deficient pro-B cells, 22% of upregulated genes in Mta2-deficient small pre-B cells) are repressed by MTA2 in both pro-B cells and in pre-B cells (Figures 5C and S5C), suggesting that MTA2/NuRD regulates distinct transcriptional programs at different B cell developmental stages. The expression levels of a selected group of genes identified in Mta2-deficient pre-B cells by RNA-seq analyses have also been validated by RT-qPCR assays using cohorts of mice different from those used for RNA-seq analyses (Figure S5D).
Derepression of Pre-BCR Genes and a Pre-B to Immature B Transition Defect in Mice Lacking Both Mta2 and Oca-B Genes
In addition to the fact that AIOLOS is a key transcription factor in repression of lgll1 and VpreB1 genes at the pre-B cell stage (Thompson et al., 2007), we previously found that the B cell-selective transcription coactivator OCA-B also represses Igll1 and VpreB1 genes at the pre-B stage through an interaction with SYK kinase in the cytoplasm (Siegel et al., 2006). Interestingly, the joint loss of AIOLOS and OCA-B caused a greater derepression of lgll1 and VpreB1 genes in pre-B cells than was observed in either of the single-KO mice, and also caused a more severe defect in the pre-B to immature B cell transition (Karnowski et al., 2008). To determine whether MTA2 also synergizes with OCA-B in a similar fashion, we generated Vav-Cre Mta2 fl/fl;Oca-B−/− mice (DKO) and examined BM B cell development in these mice. As shown in Figures 6A and 6B, the total number of BM cells in DKO mice decreased to one-third of the number in control mice, and both immature B and mature (recirculating) B cells showed 50% reductions. However, the population sizes of immature B and mature B cells showed 10-fold or more reductions, which are much more severe than those observed in either Vav-Cre Mta2fl/fl (Figures 2A and 2B) or Oca-B−/− (Figures 6A and 6B) mice. Like Vav-Cre Mta2fl/fl mice, the DKO mice also showed an increased large pre-B to small pre-B cell ratio and abnormal Ig light chain κ rearrangements (Figures 6C and 6D).
Figure 6. Joint Loss of MTA2 and OCA-B Causes Severe BM B Cell Developmental Defects.

(A) Staining and gating of the of bone marrow B cell compartments in control, Vav-Cre Mta2fl/fl, Oca-B−/−, and DKO mice. Frequencies of indicated sub-populations are marked.
(B) Cell numbers of different B cell subsets (from two femurs) are plotted (n = 15 for control, n = 10 for Oca-B−/−, and n = 7 for DKO mice; t test; *p < 0.05, ***p < 0.001).
(C) Representative FACS data showing frequencies of large pre-B (L) and small pre-B (S) cells in mice with indicated genotypes.
(D) Real-time PCR results indicating relative abundances of different Ig light chain VJ rearrangements in small pre-B cells isolated from mice with indicated genotypes (n = 4 for each genotype; t test, *p < 0.05).
(E) RT-qPCR results showing increased Igll1 and VpreB1 expression levels in Oca-B−/− and DKO pre-B cells (n ≥ 3 for each genotype; t test; *p < 0.05, **p < 0.01).
(F) ChIP-seq snapshots of OCA-B and OCT2 binding at IGLL1 and VPREB1 promoter regions in 697 cells. The scales of ChIP-seq signals are indicated.
We further examined the expression of Igll1 and VpreB1 genes in Oca-B−/− and DKO pre-B cells. While Oca-B−/− pre-B cells exhibited an approximate 3-fold increase of Igll1 and VpreB1 expression levels, the DKO pre-B cells show a 10- to 20-fold increase of Igll1 and VpreB1 expression (Figure 6E), demonstrating a synergy between OCA-B and MTA2/NuRD in Igll1 and VpreB1 repression at the pre-B stage. To further explore the possibility that OCA-B may directly regulate transcription of Igll1 and VpreB1, ChIP and OCA-B knockdown experiments were carried out in 697 cells. As shown in Figures 6F and S6, the results not only demonstrate binding of both OCA-B and its interacting transcription factor OCT2 to IGLL1 and VPREB1 promoters but also confirm the OCA-B-mediated repression of IGLL1 and VPREB1 in pre-B cells—thus indicating a potential repression function of OCA-B.
The Role of MTA2 in the Differentiation and Function of Splenic B Cells
Unlike what is observed in Mta2 Δ/Δ mice (Figure 1C), but similar to what is observed in Cd19-Cre Mta2fl/fl or Mb1-Cre Mta2fl/fl mice (Figure S2), Vav-Cre Mta2fl/fl mice show an increased MZB population that is about 2-fold the size of that in control mice (Figures 7A and 7E). The size of the FOB population is largely unaffected in these mice (Figures 7A and 7E). To assess the effect of MTA2 inactivation on the B cell response to immunization, we immunized Vav-Cre Mta2fl/fl and control mice with SRBCs (sheep red blood cells) for 9.5 days and then examined the differentiation of germinal center B (GC B) cells (B220+CD95+CD38−CD4−) in these mice. As shown in Figures 7B and 7C, Mta2 inactivation leads to the production of more GC B cells during the immune response. In vitro Ig class switch assays also show abnormal class switching to IgG1 and to IgA in the absence of MTA2 (Figure S7), suggesting an important role of MTA2/NuRD during an immune response.
Figure 7. Abnormal Splenic B Cell Differentiation and B Cell Response in Mice Lacking MTA2 or Both MTA2 and OCA-B.

(A) Staining of total splenic B (B220+), marginal zone B (MZB) (B220+CD21hiCD23lo), and follicular B (FOB) (B220+CD21−CD23hi) cells in Vav-Cre Mta2fl/fl and control mice. Frequencies of different cell populations in parental cells are indicated.
(B) Staining of germinal center B (GCB) (B220+CD95+CD38−CD4−) cells in Vav-Cre Mta2fl/fl and control mice immunized by sheep red blood cells (SRBCs).
(C) Numbers of GCB cells and frequencies of GCB cells in splenic B cells are plotted (n = 8 for each genotype; t test; *p = 0.02).
(D) Staining of splenic B, MZB, and FOB cells in mice with indicated genotypes.
(E) Cell numbers of different B cell subsets are plotted (n = 10 for control, n = 9 for Vav-Cre Mta2fl/fl, n = 5 for Oca-B−/− and DKO mice; t test; *p < 0.05, **p < 0.01, ***p < 0.001).
Beyond a strong functional synergy between MTA2 and OCAB in the pre-B to immature B transition, as shown in Figure 6, the dramatic loss of recirculating B cells in DKO mice also suggests a severe defect in splenic B cell development in these mice. A previous study has demonstrated a greater loss of splenic B cells in Aiolos−/−, Oca-B−/− mice in comparison to either Aiolos−/− or Oca-B−/− mice (Sun et al., 2003). Similarly, while neither Vav-Cre Mta2fl/fl nor Oca-B−/− mice showed substantial loss of FOB cells, and only Oca-B−/− but not Vav-Cre Mta2fl/fl mice showed a decrease in the MZB cell population (Figures 7D and 7E), the DKO mice exhibited dramatic decreases in both FOB and MZB populations—thus demonstrating an MTA2 and/or OCA-B requirement for the development of splenic B cell compartments (Figures 7D and 7E). These data further support our hypothesis that AIOLOS functions in peripheral B cells through its interaction with MTA2/NuRD.
DISCUSSION
Despite intensive studies of key transcription factors involved in B cell lineage specification and development, less is clear about the roles of transcription cofactors such as chromatin regulators during this process. Here, through a combination of genetic, biochemical, and genomic approaches, we directly assessed the contributions of NuRD corepressor complexes to BM and splenic B cell development.
Our data demonstrate that MTA2/NuRD plays an important role at multiple stages during B cell development in BM and spleen. Initially, we found decreased BM B cell subsets (pro-B, pre-B, and immature B populations) and MZB cells in Mta2 Δ/Δ mice. Surprisingly, neither Cd19-Cre Mta2fl/fl nor Mb1-Cre Mta2fl/fl mice recapitulated the BM defects observed in Mta2 Δ/Δ mice. The incomplete deletion efficiencies observed in Cd19-Cre Mta2fl/fl and Mb1-Cre Mta2fl/fl pro-B cells are likely an important factor for causing this phenomenon. In contrast, Vav-Cre Mta2fl/fl mice exhibit optimal deletion efficiency in pro-B, pre-B, and immature B cells and recapitulate the BM B cell development defects of Mta2 Δ/Δ mice. We also show that Mta2 inactivation causes abnormal rearrangements of Ig heavy chain genes in pro-B cells. In relation to Ig light chain rearrangements in pre-B cells, our data suggest a greater effect of Mta2 inactivation on more distal events relative to proximal events, likely a result of premature termination of recombination processes. Loss of MTA2 also leads to an increased large pre-B to small pre-B ratio and a decrease in the small pre-B population, suggesting a cell cycle defect at the pre-B stage and a likely effect on the pre-B to immature B cell transition. In the spleen, where Mta2 Δ/Δ mice show an approximate 5-fold reduction of MZB cells, the Cd19-Cre Mta2fl/fl, Mb1-Cre Mta2fl/fl, and Vav-Cre Mta2fl/fl mice show, instead, an approximately 2-fold increase in MZB cells. These results suggest a potential B cell-extrinsic role for MTA2 (e.g., in stromal cells) in regulating MZB population size in Mta2 Δ/Δ mice.
At the molecular level, gene profiling analyses have identified genes suppressed by MTA2 in pro-B cells and small pre-B cells (non-dividing pre-B cells). GO analyses indicate that MTA2/NuRD regulates distinct transcriptional programs in pro-B and pre-B cells. In pro-B cells, the major pathways suppressed by MTA2/NuRD include neutrophil degranulation and activation, cell junction and ECM, inflammation response, macromolecule metabolism, and endocytosis (Figure 5C). In pre-B cells, the major pathways repressed by MTA2/NuRD include TCR signaling and T cell differentiation, regulation of apoptosis, and axon guidance (Figure 5C). It thus appears that MTA2/NuRD is involved in the repression of non-B lineage genes at both pro-B and pre-B stages. Furthermore, a gene set enrichment analysis (GSEA) indicates that genes normally silenced during Hardy Fr C′ (cycling pre-B) to Hardy Fr D (resting pre-B) differentiation are enriched in upregulated genes in Mta2-deficient small pre-B cells.
It is worth noting that, although other MTA family member genes, namely Mta3 and Mta1, are also expressed in different B subsets cells, our data demonstrate non-redundant functions of MTA2 during B cell development. Further analyses are required to elucidate the unique function of MTA1 and MTA3 and redundant function between MTA family members during B cell development.
The B cell phenotypes described above and other defects of immune system caused by loss of MTA2 function are strikingly similar to those associated with loss of AIOLOS function (Table S5; Karnowski et al., 2008; Lu et al., 2008; Sun et al., 2003; Wang et al., 1998). Consistent with previous observations of an IKAROS/AIOLOS/NuRD association in T cells (Kim et al., 1999; Zhang et al., 2011), we have shown a physical interaction between AIOLOS/IKAROS and MTA2/NuRD in B cells. This observation opened a window into the functional interplay between AIOLOS/IKAROS and MTA2/NuRD complexes. Specifically, it was already noted that Igll1 and VpreB1, two genes central to the pre-B cell differentiation, are derepressed in Aiolos−/− pre-B cells (Thompson et al., 2007). Conversely, overexpression of IKAROS in B3 mouse pre-B cells recruits MI-2β and MBD3 to its binding sites and causes rapid RNA polymerase II eviction, reduced promoter accessibility, and repression of Ikaros-bound genes that include Igll1 and VpreB1 in a MI-2β-dependent manner (Ferreirós-Vidal et al., 2013; Liang et al., 2017). In this study, our data from RT-qPCR, ChIP-qPCR, and ChIP-seq analyses all strongly support a model in which AIOLOS/IKAROS represses pre-BCR gene expression through an interaction with MTA2/NuRD.
Beyond pre-BCR gene loci, genome-wide mapping of AIOLOS, IKAROS, HDAC2, and MTA2 binding sites in human 697 cells showed a highly significant overlap between AIOLOS/IKAROS targets and NuRD targets (Figures 4C and 4D). Motif analyses showed that EBF1, RUNX1/2, and ETS family binding sites are among the top motifs enriched in DNA sequences bound by AIOLOS, IKAROS, HDAC2, and MTA2. Integration of ChIP-seq data with gene profiling data showed that the human orthologs of about 70% of the upregulated genes in Vav-Cre Mta2fl/fl pre-B cells are direct MTA2 targets in human pre-B cells. These data support the hypothesis that transcriptional repression through NuRD and transcription repressors (such as AIOLOS/IKAROS) might be achieved, at least in part, by competition with transcription activators (such as EBF1) and associated coactivators (such as p300/CBP) at promoters or enhancer elements.
At the periphery, while both Mta2 Δ/Δ and Aiolos-null mice show loss of MZB cells, the Mta2 B cell-specific null mice (Cd19-Cre-, Mb1-Cre, and Vav-Cre Mta2fl/fl) show an increase of MZB cells, implying a possible stromal cell-related MTA2 function that affects the MZB population size. It will be interesting to determine whether the B cell-specific inactivation of Aiolos leads to a similar phenotype. Consistent with the B cell activation phenotype in Aiolos-null mice, we observed increased GC B cell production in response to SRBC stimulation in Vav-Cre Mta2fl/fl mice. It is worth noting that B cell-specific Ikaros KO mice also show dramatic losses of pre-B, immature B, and recirculating B in bone marrow, and both MZB and FOB cells in spleen, but increased splenic B1-B cells (Macias-Garcia et al., 2016; Schwickert et al., 2014)—demonstrating non-redundant functions of different IKAROS family members during B cell development.
OCA-B is a transcription coactivator that plays an essential role in GC formation and transcription of secondary Ig genes, and that is also involved in B cell development in the bone marrow (Luo and Roeder, 1999; Siegel et al., 2006). Our previous study had shown that OCA-B represses pre-BCR genes Igll1 and VpreB1 in pre-B cells, likely through its interaction with SYK kinase in the cytoplasm (Siegel et al., 2006). However, the current ChIP data demonstrate a direct binding of OCA-B and its interacting transcription factor OCT2 at IGLL1 and VPREB1 promoters in 697 cells. Moreover, OCA-B knockdown in 697 cells reproduces the derepression of Igll1 and VpreB1 genes. Notably, these results indicate an important and previously unrecognized repressive function of OCA-B at these gene loci. Similar to what was observed in Aiolos−/−; Oca-B−/− mice (Karnowski et al., 2008), we observed a substantial increase in Igll1 and VpreB1 expression in Vav-Cre Mta2fl/fl; Oca-B−/− pre-B cells, along with a severe developmental block in the pre-B to immature B transition and a significant loss of MZB and FOB cells in the spleen. These data further support our model in which AIOLOS depends on MTA2/NuRD to execute its function both in BM pre-B cells and in peripheral B cells. This synergy is significant since several previous studies exploring genetic interactions of OCA-B with OCT2, NFkB, and BTK did not show severe BM B cell developmental defects in respective double-mutant mice (Kim et al., 2000; Schubart et al., 2000, 2001). The increased MZB cells in Vav-Cre Mta2fl/fl mice and the dramatic losses of MZB cells in both Oca-B−/− and DKO mice suggest that OCA-B functions downstream of MTA2/NuRD in regulating MZB cell differentiation.
In summary, our analysis of Mta2-deficient mice demonstrates an important role of the MTA2/NuRD complex in B cell development. The phenotypic similarity, physical interaction, and overlapping genome-wide binding sites between MTA2 and AIOLOS/IKAROS present a striking example in which inactivation of a transcription cofactor (MTA2) recapitulates the defects caused by inactivation of a transcription factor (AIOLOS) in vivo, strongly suggesting a mutual functional dependence between MTA2/NuRD and AIOLOS/IKAROS during B cell development. Our data have also demonstrated a strong genetic interaction between MTA2/NuRD and OCA-B in regulating Igll1 and VpreB1 expression and the pre-B to immature B cell transition. The Vav-Cre Mta2fl/fl; Oca-B−/− model could serve as a platform for future elucidation of key pathways controlling the pre-B to immature B cell checkpoint and peripheral B cell development.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Robert G. Roeder (roeder@rockefeller.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Mice were maintained in environmental control facilities at the Rockefeller University and the University of North Carolina at Chapel Hill. Mice of both sexes were 8 to 12 weeks old unless otherwise indicated. The mouse maintenance and experiments were performed following the Institutional Animal Care and Use Committee (IACUC) approved protocols of the Rockefeller University and University of North Carolina at Chapel Hill. Mice carrying Mta2 conditional alleles (Mta2fl/fl) were generated as described before (Lu et al., 2008). The Cd19-Cre mice, Mb1-Cre mice, Mx1-Cre transgenic mice, Vav1-Cre mice and Oca-B knockout (KO) mice were described previously (Hobeika et al., 2006; Kim et al., 1996; Kühn et al., 1995; Ogilvy et al., 1999; Rickert et al., 1997). The Mta2 KO mice (Δ/Δ) were derived by crossing EIIa-Cre transgenic mice with Mta2fl/fl mice (Lu et al., 2008). Mta2 Δ/Δ and littermate or age-matched control mice were of mixed B6/129 genetic background. The conditional KO mice had been backcrossed with C57/BL6 mice for at least six generations. For induction of Mx1-Cre transgene expression, Mx1-Cre Mta2flfl and littermate Mta2flfl mice received two intraperitoneal injections of poly (I:C) (250 mg) at day 28 and day 30 of age. Mouse genomic DNA was isolated using DNeasy Blood and Tissue Kit (QIAGEN) for genotyping. The primers for genotyping Mta2 alleles and Oca-B alleles can be found in Table S6. The primers for genotyping other transgenic mouse strains described above can be found at the corresponding websites of The Jackson Laboratory (https://www.jax.org/).
Cell Line
697 cells are human pre-B leukemia cells that carry the E2A-PBX1 fusion gene (Kamps et al., 1991) and were cultured with RPMI 1640, 10% fetal bovine serum (FBS). Namalwa cells are human Burkitt’s lymphoma cells and were cultured with RPMI 1640, 10% FBS.
METHOD DETAILS
Flow Cytometry
Mta2 mutant and Oca-B mutant mice, and littermate control or age-, sex-matched control mice were used for analysis. Single cell suspensions of bone marrow cells and spleen cells were stained with FITC-, PE-, PerCP/Cy5.5-, PE/Cy7-, Brilliant Violet 605-, and APC-conjugated monoclonal antibodies (described in Key Resources Table) and DAPI. Stained cells were analyzed with either BD LSR II or BD FACSCalibur (BD Biosciences). The cell staining and gating schemes were indicated in the figures, figure legends and results. The specific B cell populations were sorted with BD FACSAria (BD Biosciences). The flow cytometry data were analyzed using BD FACSDiva and FlowJo softwares. For FACS analyses of bone marrow B cell subpopulations, two femurs from each mouse were used for immunofluorescence staining and cell sorting.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PE/Cy7 anti-mouse/human CD45R/B220 | BioLegend | Cat# 103222; RRID: AB_313005 |
| PE anti-mouse IgM | BioLegend | Cat# 406508; RRID: AB_315058 |
| APC anti-mouse IgM | BioLegend | Cat# 406509; RRID: AB_315059 |
| FITC anti-mouse CD43 | BioLegend | Cat# 143204; RRID: AB_10960745 |
| FITC anti-mouse CD21/CD35 (CR2/CR1) | BioLegend | Cat# 123407; RRID: AB_940403 |
| PE anti-mouse CD23 | BioLegend | Cat# 101607; RRID: AB_312832 |
| Biotin anti-mouse Ly-51 (6C3/BP-1) | BioLegend | Cat# 108303; RRID: AB_313360 |
| PE Streptavidin | BioLegend | Cat# 405203; RRID: N/A |
| APC anti-mouse CD24 (HAS) | BioLegend | Cat# 138505; RRID: AB_2565650 |
| Brilliant Violet 605 anti-mouse IgM | BioLegend | Cat# 406523; RRID: AB_2563358 |
| PerCP/Cyanine5.5 anti-mouse CD95(Fas) | BioLegend | Cat# 152610; RRID: AB_2632905 |
| APC anti-mouse CD38 | BioLegend | Cat# 102712; RRID: AB_312933 |
| APC anti-mouse CD4 | BioLegend | Cat# 116014; RRID: AB_2563025 |
| PE anti-mouse IgG1 | BioLegend | Cat# 406607; RRID: AB_10551439 |
| Biotin anti-mouse IgG3 | BioLegend | Cat# 406803; RRID: AB_315070 |
| Biotin anti-mouse IgA | BioLegend | Cat# 407003; RRID: AB_315078 |
| Anti-mouse CD40 | BioLegend | Cat# 102802; RRID: AB_312935 |
| Anti-Mta2 | Abcam | Cat# ab8106; RRID: AB_306276 |
| Anti-CHD4 (Mi-2β) | Abcam | Cat# ab72418; RRID: AB_1268107 |
| Anti-IKZF3 (Aiolos) | Abcam | Cat# ab139408; RRID: N/A |
| Anti-IKZF1 (Ikaros) | Abcam | Cat# ab 26083; RRID: AB_881148 |
| Anti-Bob1 (Oca-B) (C-20) | Santa Cruz Biotechnology | Cat# sc-955; RRID: AB_2166917 |
| Anti-Oct-2 (C-20) | Santa Cruz Biotechnology | Cat# sc-233; RRID: AB_2167205 |
| Anti-HDAC2 | Abcam | Cat# ab7029; RRID: AB_305706 |
| Anti-Histone H3 (tri methyl K4) | Abcam | Cat# ab8580; RRID: AB_306649 |
| Anti-Histone H3 (acetyl K27) | Abcam | Cat# ab4729; RRID: AB_2118291 |
| Anti-Histone H3 | Abcam | Cat# ab1791; RRID: AB_302613 |
| Normal rabbit IgG | Santa Cruz Biotechnology | Cat# sc-2027; RRID: AB_737197 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DSG (disuccinimidyl glutarate) | Thermo Fisher Scientific | Cat# 20593 |
| Recombinant Murine IL-4 | PeproTech | Cat# 214–14 |
| Recombinant TGF-β (carrier free) | BioLegend | Cat# 763102 |
| Critical Commercial Assays | ||
| Arcturus™ PicoPure RNA isolation Kit | Applied Biosystems | Cat# KIT0204 |
| qScript cDNA SuperMix | Quanta Biosciences | Cat# 95048 |
| DNeasy Blood and Tissue Kit | QIAGEN | Cat# 69506 |
| QuantiTect SYBR Green PCR mix | QIAGEN | Cat# 204145 |
| JumpStart TaqReady Mix | Sigma-Aldrich | Cat# P2893 |
| End-It™ End-Repair Kit | Epicenter | Cat# ER81050 |
| DNA Clean & Concentrator Kit | Zymo Research | Cat# D4014 |
| NEXTflexChIP-Seq Barcodes | Perkin Elmer | Cat# NOVA-514120 |
| Dynabeads™ Protein G | Invitrogen | Cat# 10004D |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat# 20020594 |
| Mouse CD43 MicroBeads | Miltenyi Biotec | Cat# 130-049-801 |
| Deposited Data | ||
| Mta2 Control_(781t4)_pro-B | This Paper | GSM3398870 |
| Mta2 Control_(789t7)_pro-B | This Paper | GSM 3398871 |
| Vav-Cre Mta2 f/f _(781t3)_pro-B | This Paper | GSM 3398872 |
| Vav-Cre Mta2 f/f _(789t6)_pro-B | This Paper | GSM3398873 |
| Mta2 Control_(918t3)_pre-B | This Paper | GSM3398874 |
| Mta2 Control_(919t7)_pre-B | This Paper | GSM3398875 |
| Mta2 Control_(931t10)_pre-B | This Paper | GSM3398876 |
| Vav-Cre Mta2 f/f _(918t4)_pre-B | This Paper | GSM3398877 |
| Vav-Cre Mta2 f/f _(918t6)_pre-B | This Paper | GSM3398878 |
| Vav-Cre Mta2 f/f _(931t14)_pre-B | This Paper | GSM3398879 |
| INPUT_697 | This Paper | GSM2882822 |
| AIOLOS_697 | This Paper | GSM2882823 |
| IKAROS_697 | This Paper | GSM2882824 |
| MTA2_697 | This Paper | GSM2882825 |
| Mi2_697 | This Paper | GSM2882826 |
| OCAB_697 | This Paper | GSM2882827 |
| OCT2_697 | This Paper | GSM2882828 |
| HDAC2_697 | This Paper | GSM3398902 |
| Experimental Models: Cell Lines | ||
| 697 human pre-B acute lymphoblastic leukemia cell line | DSMZ | Cat# ACC42 |
| Namalwa Burkitt’s lymphoma cell line | ATCC | Cat# CRL-1432™ |
| Experimental Models: Organisms/Strains | ||
| Mta2 conventional null mice | Lu et al.,2008 | N/A |
| Cd19-Cre transgenic mice | The Jackson Lab | Stock # 004126 |
| Mb1-Cre transgenic mice | The Jackson Lab | Stock # 020505 |
| Mx-Cre transgenic mice | The Jackson Lab | Stock # 003556 |
| Vav1-Cre transgenic mice | The Jackson Lab | Stock # 008610 |
| Oca-B null mice | Kim et al., 1996 | N/A |
| Oligonucleotides | ||
| sh_hOCA-B#1 | Sigma-Aldrich | Cat# TRCN0000431764 |
| 5′-AAATG AAAG CCCATG G ACCAC-3′ | ||
| Sh_hOCA-B#2 | Sigma-Aldrich | Cat# TRCN0000423580 |
| 5′-AAATCCCTATAATTACCTCCC-3′ | ||
| Scramble shRNA | Sigma-Aldrich | Cat# SHC002 |
| 5′-CAACAAGATGAAGAGCACCAA-3′ | ||
| Oligonucleotides for qPCRs | This paper | Table S6 |
| Recombinant DNA | ||
| pLKO.1 scramble shRNA | This paper | N/A |
| pLKO.1 sh_hOCA-B#1 | This paper | N/A |
| pLKO.1 sh_hOCA-B#2 | This paper | N/A |
| Software and Algorithms | ||
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| TopHat | Kim et al., 2013 | https://github.com/DaehwanKimLab/tophat2 |
| Cufflinks | Trapnell et al., 2010 | https://github.com/cole-trapnell-lab/cufflinks |
| edgeR | Robinson et al., 2010 | http://bioconductor.org |
| Enrichr | Kuleshov et al., 2016 | http://amp.pharm.mssm.edu/Enrichr |
| Samtools | Li et al.,2009 | http://samtools.sourceforge.net/ |
| Homer | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| ngsplot | Shen et al., 2014 | https://github.com/shenlab-sinai/ngsplot |
| Integrative Genomics Viewer (IGV) | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
| GSEA | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/index.jsp |
| Other | ||
| Sheep Red Blood Cells | Cocalico Biologicals, Inc. | N/A |
| Lipopolysaccharides (LPS) | Sigma-Aldrich | Cat# L2637 |
| Poly (I:C) | Sigma-Aldrich | Cat# P9582 |
Immunoprecipitation (IP)
Dynabeads™ Protein G beads (Invitrogen) were first washed twice with PBS containing 0.5% BSA and 0.05% Triton X-100 then incubated with specific antibodies (about 2 mg antibody for each IP assay) in the same buffer for 2 to 3 h or overnight at 4°C. The conjugated antibodies then were incubated with nuclear extracts (NE) derived from 697 cells or Namalwa cells overnight at 4°C.About 200 mg of NE was used for each IP assay. After washing with buffer BC200 [20 mM Tris (pH 7.9), 0.2 mM EDTA, 1 mM DTT, 0.2 mM PMSF, 20% glycerol, and 200 mM of KCl] for three times, the immunoprecipitates were subject to immunoblot.
RNA isolation and reverse transcription (RT)
Total RNA was isolated from sorted primary B cells or cultured 697 cells using Arcturus™ PicoPure RNA isolation Kit (Applied Bio-systems). For each sample, 40 to 100 μg RNA was used for reverse transcription using qScript cDNA SuperMix (Quanta Biosciences) according to Manufacturer’s instruction.
Quantitative PCR
Quantitative PCR (qPCR) assays were performed in triplicate using QuaniTect SYBR Green PCR mix (QIAGEN) on a 7300 Real Time PCR System (Applied Biosystems). The numbers of biological samples and independent experiments used for qPCR analyses are indicated in the figure legends and text. mRNA levels were normalized to the expression level of Hprt (HPRT) or Gapdh (GAPDH) gene in mouse and human cells. For deletion efficiency and immunoglobulin gene rearrangement analyses, genomic DNA amounts were normalized to promoter regions of Cd79b or Cdk6 genes. Primer sequences for RT-qPCR and qPCR are listed in Table S6.
RNA-seq and gene ontology
For pro-B cell gene profiling, B220+IgM−CD43+ pro-B cells from 2 pairs of 10-week-old Vav-Cre Mta2fl/fl and littermate control mice were sorted for RNA isolation. For pre-B cell gene profiling, B220+IgM−CD43− small pre-B cells from 3 pairs of 10-week-old Vav-Cre Mta2fl/fl and littermate control mice were sorted for RNA isolation. The RNA library preparation was done with TruSeq Stranded mRNA Prepration Kit (Illumina). The sequencing was done by the Genomics Core Facility at The Rockefeller University using an Illumina HiSeq 2500 Sequencer. Sequencing data were aligned to mm10 reference genome on Galaxy (https://usegalaxy.org/) with default parameters using TopHat software (Kim et al., 2013). The aligned sequences were assembled using Cufflinks and then analyzed using edgeR package in R script (Robinson et al., 2010; Trapnell et al., 2010). The cut-off line for differential gene expression levels was 1.5 fold with p values smaller than 0.05. Enrichr (http://amp.pharm.mssm.edu/Enrichr/) was used for gene ontology analysis and GSEA was used for gene set enrichment analysis (Kuleshov et al., 2016; Subramanian et al., 2005).
Genomic rearrangement of the Immunoglobulin heavy chain and light chain loci
B220+IgM−CD43+ pro-B and B220+IgM−CD43− small pre-B cells from 4 pairs of 8 to 12-week-old Vav-Cre Mta2fl/fl and littermate control mice were sorted, and the genomic DNA was isolated using DNeasy Blood and Tissue Kit (QIAGEN). The VH(D)JH recombination efficiencies in pro-B cells were measured by qPCR using primers indicated in Figure 2E and described in Table S6. The VKJK recombination efficiencies in pre-B cells were measured by qPCR using primers indicated in Figure 2G and described in Table S6. A universal Vk primer was used in combination with a Jk1, Jk2, Jk4 or Jk5 primer. Promoter regions of Cd79b or Cdk6 genes were used to normalize genomic DNA amounts of different samples. The number of samples used in the assays can be found in the corresponding Figure Legends.
Chromatin immunoprecipitation (ChIP) and ChIP-sequencing
The antibodies used for ChIP are described in Key Resources Table. Normal rabbit IgG (sc-2027, Santa Cruz Biotechnology Inc.) was used as an immunoprecipitation control. In ChIP experiments, cells were incubated sequentially, at room temperature (RT), for 30 min in 2mM disuccinimidyl glutarate (DSG), for 10 min after formaldehyde addition to a final concentration of 1%, and for 5 min after addition of glycine to a final concentration of 125mM. Fixed cells were washed twice with cold PBS and resuspended in RIPA 0.3 buffer(0.1% SDS, 1% Triton X-100, 10mM Tris-HCl(pH7.4), 1mM EDTA (pH8.0), 0.1% Sodium Deoxycholate, and 0.3M NaCl). The antibodies and IgG were first incubated with Dynabeads Protein G (Invitrogen) at 4°C overnight, then washed and then incubated with sonicated cell lysate (1–2ug antibody for a ChIP-qPCR, 6–10ug antibody for ChIP-seq) at 4° overnight. For reverse crosslinking, the beads were first washed 4–8 times with high salt buffer (20mM Tris pH8, 2mM EDTA, 0.1%SDS, 1%TX100, 500mM NaCl), then washed with TE buffer (pH8.0) and then washed with elution buffer (1% SDS in TE buffer) for 15 min at RT. Eluates were then incubated at 65°C for 4 h after addition of NaCl and RNase to final concentrations of 200mM and 4ug/ml and then at 55°C for 1 h after addition of proteinase K to100ug/ml. Finally, the immunoprecipitated DNA samples were purified by QIAquick PCR Purification Kit (QIAGEN) for further analysis. About 2–5 ng of DNA precipitated by ChIP was used as the starting material for the generation of single-end sequencing libraries as described (Yu et al., 2015). Cluster generation and sequencing was performed by the Genomics Resource Center at The Rockefeller University with a HiSeq 2500 system with a read length of 50 nucleotides according to the manufacturer’s guidelines (Illumina).
Data analysis for ChIP-seq
The softwares used for ChIP-seq data analyses are listed in Key Resources Table. In brief, sequencing data were aligned using Bowtie2 (Langmead and Salzberg, 2012) to hg19 version of the reference genome on Galaxy (https://usegalaxy.org/) with default parameters. Duplicate mapped reads were removed using Samtools with the rmdup option. Bedgraph files were generated by Homer using makeUCSCfile program with default parameters (Li et al., 2009). ChIP-seq tracks generated by converting bedgraph files to tdf formats were displayed by IGV software (Robinson et al., 2011). Peak files were generated by Homer using findPeaks program with default parameters (Heinz et al., 2010). The threshold for statistical significance of peaks was set at 10−10. Annotation of filtered peaks was done by Homer with annotatePeaks program with default parameters except -style factor. ChIP-seq signals at TSSs were plotted as averaged profiles or heatmaps by ngsplot with parameter -SC global -GO max. ChIP-seq signals at enhancers were plotted as averaged profiles by ngsplot with parameter -L 10000 -F Gm12878 -SC global -GO max (Shen et al., 2014). Motif analyses were performed by Homer using findMotifsGenome program with default parameters.
shRNA knockdown
For knockdown of the OCA-B gene, 1×107 697 cells were infected with lentivirus carrying OCA-B shRNA sequences or scramble RNA sequence plus 8 mg/ml polybrene in RPMI 1640 medium with 10% FBS. After 12 h, the infected cells were changed to fresh medium with 10% FBS and cultured for 2 days. The cells were then treated with 1 mg/ml puromycin and cultured for another 2 days. Total RNAs were isolated for measuring expression levels of OCA-B by RT-qPCR.
Immunization and immunoglobulin (Ig) class switch
Sheep red blood cells (SRBC) were washed with PBS and 1:10 diluted in PBS. 500 mL diluted SRBC were intraperitoneal injected to each mouse. Splenic cells were isolated on day 9 or day 10 after immunization for FACS analysis of germinal center B cells. For Ig class switch assay, naive splenic B cells were enriched by negative selection using anti-CD43 conjugated magnetic beads (Miltenyi Biotec). Isolated cells, at a concentration of 300 to 500K cell per ml, were incubated in RPMI 1640 medium with 10% FCS. Ig class switching were induced by 5ng/ml IL-4 (Sigma) + 25ug/ml LPS (Sigma) (for IgG1), 25ug/ml LPS (for IgG3), and 5ng/ml IL-4 + 0.2mg/ml anti-CD40 antibody (BioLegend) + 0.1ng/ml TGF-β (BioLegend) (for IgA) respectively. After incubation for three days, the cells were analyzed by flow cytometry for specific Ig surface markers.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data were analyzed using Excel or GraphPad Prism softwares. t tests were used for calculation of statistical significance when comparing data from two groups. Differences were considered significant when p values were < 0.05. p values are depicted in figures using one to three asterisks (*p < 0.05, **p < 0.01, and ***p < 0.005). Data are presented as mean + SD with n indicating the number of animals or independent experiments if not stated otherwise in the figure legends or in the text.
DATA AND CODE AVAILABILITY
The RNA-seq and ChIP-seq data have been deposited in NCBI Gene Expression Omnibus. The accession number for the RNA-seq data reported in this paper is GSE107885, and the accession number for the ChIP-seq data reported in this paper is GSE107886.
Supplementary Material
Highlights.
Loss of MTA2 causes bone marrow and splenic B cell developmental defects in mice
Loss of MTA2 causes derepression of Igll1 and VpreB1 genes in pre-B cells
MTA2 binding sites highly overlap AIOLOS/IKAROS binding sites in pre-B cells
MTA2 cooperates with OCA-B in regulating pre-B to immature B transition
ACKNOWLEDGMENTS
We thank Alexander Tarakhovsky, and Joon Seok Park at The Rockefeller University, Hongsheng Wang at NIH, Ivan Maillard at University of Michigan, and Zhixin Zhang at University of Nebraska Medical Center for valuable comments and suggestions. We thank the Rockefeller University Flow Cytometry Resource Center and UNC-Chapel Hill Flow Cytometry Center for cell sorting and other technical help, and the Rockefeller University Genomics Core Facility for sequencing analysis. This work was supported by grant CA178765 to R.G.R. from NIH, United States. X.L. was supported in part by grant UL1 TR000043 from the National Center for Advancing Translational Sciences, NIH, United States and co-sponsored by the Shapiro-Silverberg Fund for the Advancement of Translational Research, United States.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.029.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Avitahl N, Winandy S, Friedrich C, Jones B, Ge Y, and Georgopoulos K (1999). Ikaros sets thresholds for T cell activation and regulates chromosome propagation. Immunity 10, 333–343. [DOI] [PubMed] [Google Scholar]
- Bagheri-Yarmand R, Balasenthil S, Gururaj AE, Talukder AH, Wang YH, Lee JH, Kim YS, Zhang X, Jones DM, Medeiros LJ, et al. (2007). Metastasis-associated protein 1 transgenic mice: a new model of spontaneous B-cell lymphomas. Cancer Res. 67, 7062–7067. [DOI] [PubMed] [Google Scholar]
- Busslinger M (2004). Transcriptional control of early B cell development. Annu. Rev. Immunol 22, 55–79. [DOI] [PubMed] [Google Scholar]
- Busslinger M, and Tarakhovsky A (2014). Epigenetic control of immunity. Cold Spring Harb. Perspect. Biol 6, a019307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dege C, and Hagman J (2014). Mi-2/NuRD chromatin remodeling complexes regulate B and T-lymphocyte development and function. Immunol. Rev 261, 126–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denslow SA, and Wade PA (2007). The human Mi-2/NuRD complex and gene regulation. Oncogene 26, 5433–5438. [DOI] [PubMed] [Google Scholar]
- Feng Q, and Zhang Y (2003). The NuRD complex: linking histone modification to nucleosome remodeling. Curr. Top. Microbiol. Immunol 274, 269–290. [DOI] [PubMed] [Google Scholar]
- Ferreirós-Vidal I, Carroll T, Taylor B, Terry A, Liang Z, Bruno L, Dharmalingam G, Khadayate S, Cobb BS, Smale ST, et al. (2013). Genome-wide identification of Ikaros targets elucidates its contribution to mouse B-cell lineage specification and pre-B-cell differentiation. Blood 121, 1769–1782. [DOI] [PubMed] [Google Scholar]
- Fujita N, Jaye DL, Geigerman C, Akyildiz A, Mooney MR, Boss JM, and Wade PA (2004). MTA3 and the Mi-2/NuRD complex regulate cell fate during B lymphocyte differentiation. Cell 119, 75–86. [DOI] [PubMed] [Google Scholar]
- Gao H, Lukin K, Ramírez J, Fields S, Lopez D, and Hagman J (2009). Opposing effects of SWI/SNF and Mi-2/NuRD chromatin remodeling complexes on epigenetic reprogramming by EBF and Pax5. Proc. Natl. Acad. Sci. USA 106, 11258–11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobeika E, Thiemann S, Storch B, Jumaa H, Nielsen PJ, Pelanda R, and Reth M (2006). Testing gene function early in the B cell lineage in mb1-cre mice. Proc. Natl. Acad. Sci. USA 103, 13789–13794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamps MP, Look AT, and Baltimore D (1991). The human t(1;19) translocation in pre-B ALL produces multiple nuclear E2A-Pbx1 fusion proteins with differing transforming potentials. Genes Dev. 5, 358–368. [DOI] [PubMed] [Google Scholar]
- Karnowski A, Cao C, Matthias G, Carotta S, Corcoran LM, Martensson IL, Skok JA, and Matthias P (2008). Silencing and nuclear repositioning of the lambda5 gene locus at the pre-B cell stage requires Aiolos and OBF-1. PLoS ONE 3, e3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim U, Qin XF, Gong S, Stevens S, Luo Y, Nussenzweig M, and Roeder RG (1996). The B-cell-specific transcription coactivator OCA-B/OBF-1/Bob-1 is essential for normal production of immunoglobulin isotypes. Nature 383, 542–547. [DOI] [PubMed] [Google Scholar]
- Kim J, Sif S, Jones B, Jackson A, Koipally J, Heller E, Winandy S, Viel A, Sawyer A, Ikeda T, et al. (1999). Ikaros DNA-binding proteins direct formation of chromatin remodeling complexes in lymphocytes. Immunity 10, 345–355. [DOI] [PubMed] [Google Scholar]
- Kim U, Gunther CS, and Roeder RG (2000). Genetic analyses of NFKB1 and OCA-B function: defects in B cells, serum IgM level, and antibody responses in Nfkb1−/−Oca-b−/− mice. J. Immunol 165, 6825–6832. [DOI] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Kwak PB, and Weitz CJ (2014). Specificity in circadian clock feedback from targeted reconstitution of the NuRD corepressor. Mol. Cell 56, 738–748. [DOI] [PubMed] [Google Scholar]
- Krop I, de Fougerolles AR, Hardy RR, Allison M, Schlissel MS, and Fearon DT (1996). Self-renewal of B-1 lymphocytes is dependent on CD19. Eur. J. Immunol 26, 238–242. [DOI] [PubMed] [Google Scholar]
- Kühn R, Schwenk F, Aguet M, and Rajewsky K (1995). Inducible gene targeting in mice. Science 269, 1427–1429. [DOI] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 44 (W1), W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Brown KE, Carroll T, Taylor B, Vidal IF, Hendrich B, Rueda D, Fisher AG, and Merkenschlager M (2017). A high-resolution map of transcriptional repression. eLife 6, e22767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loughran SJ, Comoglio F, Hamey FK, Giustacchini A, Errami Y, Earp E, Göttgens B, Jacobsen SEW, Mead AJ, Hendrich B, and Green AR (2017). Mbd3/NuRD controls lymphoid cell fate and inhibits tumorigenesis by repressing a B cell transcriptional program. J. Exp. Med 214, 3085–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Kovalev GI, Chang H, Kallin E, Knudsen G, Xia L, Mishra N, Ruiz P, Li E, Su L, and Zhang Y (2008). Inactivation of NuRD component Mta2 causes abnormal T cell activation and lupus-like autoimmune disease in mice. J. Biol. Chem 283, 13825–13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, and Roeder RG (1999). B-cell-specific coactivator OCA-B: biochemical aspects, role in B-cell development and beyond. Cold Spring Harb. Symp. Quant. Biol 64, 119–131. [DOI] [PubMed] [Google Scholar]
- Macias-Garcia A, Heizmann B, Sellars M, Marchal P, Dali H, Pasquali JL, Muller S, Kastner P, and Chan S (2016). Ikaros is a negative regulator of B1 cell development and function. J. Biol. Chem 291, 9073–9086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthias P, and Rolink AG (2005). Transcriptional networks in developing and mature B cells. Nat. Rev. Immunol 5, 497–508. [DOI] [PubMed] [Google Scholar]
- Ogilvy S, Metcalf D, Gibson L, Bath ML, Harris AW, and Adams JM (1999). Promoter elements of vav drive transgene expression in vivo throughout the hematopoietic compartment. Blood 94, 1855–1863. [PubMed] [Google Scholar]
- Rickert RC, Roes J, and Rajewsky K (1997). B lymphocyte-specific, Cre-mediated mutagenesis in mice. Nucleic Acids Res. 25, 1317–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bio-conductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubart DB, Rolink A, Schubart K, and Matthias P (2000). Cutting edge: lack of peripheral B cells and severe agammaglobulinemia in mice simultaneously lacking Bruton’s tyrosine kinase and the B cell-specific transcriptional coactivator OBF-1. J. Immunol 164, 18–22. [DOI] [PubMed] [Google Scholar]
- Schubart K, Massa S, Schubart D, Corcoran LM, Rolink AG, and Matthias P (2001). B cell development and immunoglobulin gene transcription in the absence of Oct-2 and OBF-1. Nat. Immunol 2, 69–74. [DOI] [PubMed] [Google Scholar]
- Schwickert TA, Tagoh H, Gültekin S, Dakic A, Axelsson E, Minnich M, Ebert A, Werner B, Roth M, Cimmino L, et al. (2014). Stage-specific control of early B cell development by the transcription factor Ikaros. Nat. Immunol 15, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Gui B, and Kumar R (2014). Physiological functions of MTA family of proteins. Cancer Metastasis Rev. 33, 869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L, Shao N, Liu X, and Nestler E (2014). ngs.plot: quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics 15, 284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, Kim U, Patke A, Yu X, Ren X, Tarakhovsky A, and Roeder RG (2006). Nontranscriptional regulation of SYK by the coactivator OCA-B is required at multiple stages of B cell development. Cell 125, 761–774. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun J, Matthias G, Mihatsch MJ, Georgopoulos K, and Matthias P (2003). Lack of the transcriptional coactivator OBF-1 prevents the development of systemic lupus erythematosus-like phenotypes in Aiolos mutant mice. J. Immunol 170, 1699–1706. [DOI] [PubMed] [Google Scholar]
- Teitell MA (2003). OCA-B regulation of B-cell development and function. Trends Immunol. 24, 546–553. [DOI] [PubMed] [Google Scholar]
- Thompson EC, Cobb BS, Sabbattini P, Meixlsperger S, Parelho V, Liberg D, Taylor B, Dillon N, Georgopoulos K, Jumaa H, et al. (2007). Ikaros DNA-binding proteins as integral components of B cell developmental-stage-specific regulatory circuits. Immunity 26, 335–344. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, and Pachter L (2010). Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JH, Avitahl N, Cariappa A, Friedrich C, Ikeda T, Renold A, Andrikopoulos K, Liang L, Pillai S, Morgan BA, and Georgopoulos K (1998). Aiolos regulates B cell activation and maturation to effector state. Immunity 9, 543–553. [DOI] [PubMed] [Google Scholar]
- Williams CJ, Naito T, Arco PG, Seavitt JR, Cashman SM, De Souza B, Qi X, Keables P, Von Andrian UH, and Georgopoulos K (2004). The chromatin remodeler Mi-2beta is required for CD4 expression and T cell development. Immunity 20, 719–733. [DOI] [PubMed] [Google Scholar]
- Yamada T, Yang Y, Hemberg M, Yoshida T, Cho HY, Murphy JP, Fioravante D, Regehr WG, Gygi SP, Georgopoulos K, and Bonni A (2014). Promoter decommissioning by the NuRD chromatin remodeling complex triggers synaptic connectivity in the mammalian brain. Neuron 83, 122–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Yamada T, Hill KK, Hemberg M, Reddy NC, Cho HY, Guthrie AN, Oldenborg A, Heiney SA, Ohmae S, et al. (2016). Chromatin remodeling inactivates activity genes and regulates neural coding. Science 353, 300–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T, Hazan I, Zhang J, Ng SY, Naito T, Snippert HJ, Heller EJ, Qi X, Lawton LN, Williams CJ, and Georgopoulos K (2008). The role of the chromatin remodeler Mi-2beta in hematopoietic stem cell self-renewal and multilineage differentiation. Genes Dev. 22, 1174–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Yang W, Ni T, Tang Z, Nakadai T, Zhu J, and Roeder RG (2015). RNA polymerase II-associated factor 1 regulates the release and phosphorylation of paused RNA polymerase II. Science 350, 1383–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Jackson AF, Naito T, Dose M, Seavitt J, Liu F, Heller EJ, Kashiwagi M, Yoshida T, Gounari F, et al. (2011). Harnessing of the nucleosome-remodeling-deacetylase complex controls lymphocyte development and prevents leukemogenesis. Nat. Immunol 13, 86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-seq and ChIP-seq data have been deposited in NCBI Gene Expression Omnibus. The accession number for the RNA-seq data reported in this paper is GSE107885, and the accession number for the ChIP-seq data reported in this paper is GSE107886.