

Abstract
The reversible adenine phosphoribosyltransferase enzyme (APRT) is essential for purine homeostasis in prokaryotes and eukaryotes. In humans, APRT (hAPRT) is the only enzyme known to produce AMP in cells from dietary adenine. APRT can also process adenine analogs, which are involved in plant development or neuronal homeostasis. However, the molecular mechanism underlying substrate specificity of APRT and catalysis in both directions of the reaction remains poorly understood. Here we present the crystal structures of hAPRT complexed to three cellular nucleotide analogs (hypoxanthine, IMP, and GMP) that we compare with the phosphate-bound enzyme. We established that binding to hAPRT is substrate shape–specific in the forward reaction, whereas it is base-specific in the reverse reaction. Furthermore, a quantum mechanics/molecular mechanics (QM/MM) analysis suggests that the forward reaction is mainly a nucleophilic substitution of type 2 (SN2) with a mix of SN1-type molecular mechanism. Based on our structural analysis, a magnesium-assisted SN2-type mechanism would be involved in the reverse reaction. These results provide a framework for understanding the molecular mechanism and substrate discrimination in both directions by APRTs. This knowledge can play an instrumental role in the design of inhibitors, such as antiparasitic agents, or adenine-based substrates.
Keywords: enzyme mechanism, purine, protein structure, nucleoside/nucleotide biosynthesis, quantum chemistry, adenine, hypoxanthine, QM/MM calculation, forward and reverse reactions, nucleoside monophosphate
Introduction
Purines are nitrogenous bases indispensable for living organisms. They play important roles in energy transfer, genetic information storage, and signal transduction. Purine metabolism is an essential pathway that has been conserved through evolution from prokaryotes to eukaryotes. Purines can be synthesized de novo or through salvage pathways by specific enzymes. Two such enzymes in the salvage pathway are adenine phosphoribosyltransferase (APRT, EC 2.4.2.7)4 and hypoxanthine-guanine phosphoribosyltransferase (HGPRT, EC 2.4.2.8). These are enzymes with reversible activities having an ordered sequential bi-bi reaction mechanism (1, 2). In the forward reaction, APRT synthesizes AMP from adenine (ADE) (Fig. 1), whereas HGPRT generates GMP or IMP from guanine (Gua) or hypoxanthine (Hx), respectively. Both reactions use the co-substrate α-d-5-phosphoribosyl-1-pyrophosphate (PRPP) and at least one divalent magnesium ion. In the reverse pathway, PPi and the corresponding ribonucleoside monophosphate are substrates of the reaction (8). Similarly to other phosphoribosyltransferase enzymes, both APRT and HGPRT structures are made of a Rossmann fold. They also include a PRPP-binding motif, a flexible loop, and a hood region (Fig. S1). The last seems to provide purine specificity, either ADE or Hx and Gua, in APRT or HGPRT, respectively (3, 4). Furthermore, the flexible loop is very dynamic (5), and we recently showed that a conserved tyrosine within the flexible loop of human APRT facilitates the forward reaction and is essential for cell growth (6).
Figure 1.
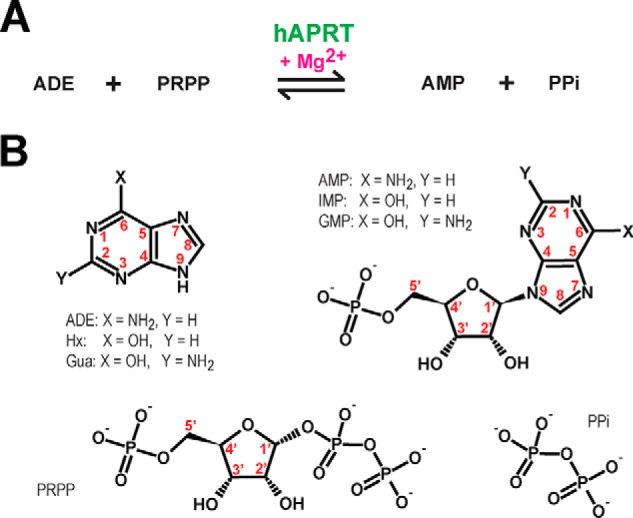
Reversible enzymatic reactions by hAPRT. A, magnesium ion is necessary for the transformation of ADE and PRPP into AMP and PPi, and vice versa. B, chemical structures of the hAPRT substrates (ADE, AMP, PRPP, PPi) and analogs (Hx, Gua, IMP, GMP) tested. Atom numbering is indicated in red.
The intracellular concentrations of adenine, hypoxanthine, and guanine are in the order of 1 μm, 10 μm, and 0.5 μm, respectively (7). For the associated mononucleotides, AMP, IMP, and GMP, the intracellular concentrations are estimated to be 200 μm, 100 μm, and 25 μm (7). For APRT, ADE and AMP concentrations in cells are therefore similar to those of other potential substrates present in the cell such as hypoxanthine, guanine, IMP, and GMP. The same is true for the HGPRT substrates. How does hAPRT thus discriminate among these molecules and achieve its specificity? For example, although Hx and ADE have the same chemical skeleton, which differs only by a hydroxyl instead of an amine at the C6 position away from the reactive N9 nitrogen, ADE seems to be the only purine metabolized by hAPRT. Furthermore, in the reverse reaction, how does hAPRT discriminate among AMP, IMP, and GMP, because these substrates have identical glycosidic bonds where the reaction takes place? In addition, the reaction mechanism in HGPRT has been well-described, and it follows an SN1-type reaction (unimolecular nucleophilic substitution reaction) which involves an oxocarbenium transition compound (9–11). However, an SN1- or SN2-type reaction mechanism is still a matter of debate for the APRT enzyme (12, 13).
Our objectives were first to define the bidirectional molecular selectivity of hAPRT with respect to the potential substrates present in cells, and second to determine the molecular mechanism of hAPRT to discriminate between SN1- and SN2-type reactions. Four crystallographic structures of human APRT in complex with either phosphate ion, Hx-PRPP-Mg2+, IMP, or GMP were obtained. Our results, compared with our recent ADE-PRPP-Mg2+-, AMP-, and AMP-PPi-Mg2+-hAPRT structures (6), show that hAPRT follows mainly an SN2-type mechanism in both directions of the reactions, with some SN1-type character, and discriminates substrates according to their molecular shapes, chemical composition, and a loop closure over the active site.
Results
Substrate-free hAPRT structure
We first used differential scanning fluorimetry (DSF) to explore the ligand-binding properties of hAPRT (Table 1). As observed previously (6), the unfolding temperature is 30 °C lower in the substrate-free hAPRT as compared with the PRPP-bound enzyme. To investigate the destabilizing effect because of the removal of PRPP from the enzyme in the reverse reaction, we solved the crystal structure of the substrate-free hAPRT to 1.70 Å resolution (Table 2). To obtain this structure, we left the very unstable PRPP molecules to naturally hydrolyze within PRPP-Mg2+-hAPRT crystal for 1 month. The structure showed a typical Rossmann fold where a central stranded twisted parallel β-sheet (β3-β4-β7-β8-β9) is sandwiched between α-helices (Fig. 2A). The conserved residues over prokaryotes and eukaryotes are all close to the active site except for the flexible loop residues (Figs. S1 and S2). The substrate-free structure showed no difference when superimposed to the PRPP-Mg2+–bound structure, with an RMSD (root mean square deviation) of 0.20 Å (Fig. 2B). However, part of the flexible loop (amino acids 103–106) was missing in both subunits. After PRPP hydrolysis, a phosphate ion, identified by a clear electron density (Fig. 2C and Note S1), remains in the active site and interacts with the conserved Ala-131–TGGT–PRPP–binding motif (Figs. 2C and S1). A network of water molecules replaced the ribose and pyrophosphate moieties and interacts with the conserved Arg-67, Asp-127, Asp-128, Ala-131 and Gly-133 residues (Fig. 2C).
Table 1.
Ligand-binding analysis by differential scanning fluorimetry
The means and the standard deviations of the measurements of the unfolding temperatures (Tm) for each protein complex are given.
| Protein complex | Unfolding temperature (Tm) in °C | References |
|---|---|---|
| hAPRT | 49.6 ± 1.2 | 6 |
| hAPRT + Hx | 47.6 ± 0.5 | This work |
| hAPRT + ADE | 50.3 ± 0.6 | 6 |
| hAPRT + PRPP | 79.6 ± 0.6 | 6 |
| hAPRT + PRPP + Hx | 79.7 ± 0.5 | This work |
| hAPRT + PRPP + ADE | 79.9 ± 0.5 | 6 |
| hAPRT + PPi | 52.1 ± 1.0 | This work |
| hAPRT + AMP | 59.0 ± 0.6 | 6 |
| hAPRT + IMP | 51.2 ± 1.1 | This work |
| hAPRT + GMP | 53.0 ± 0.7 | This work |
| hAPRT + PPi + AMP | 64.3 ± 0.7 | 6 |
| hAPRT + PPi + IMP | 53.5 ± 0.7 | This work |
| hAPRT + PPi + GMP | 53.6 ± 1.0 | This work |
Table 2.
Data collection and refinement statistics
| PO43−-hAPRT | Hx-PRPP-Mg2+-hAPRT | IMP-hAPRT | GMP-hAPRT | |
|---|---|---|---|---|
| PDB ID | 6HGP | 6HGQ | 6HGR | 6HGS |
| Data collection | ||||
| Synchrotrons | SOLEIL | ESRF | ESRF | SOLEIL |
| Beamlines | PX1 | ID14–4 | ID29 | PX1 |
| Space group | P1 | P1 | P1 | P1 |
| a, b, c (Å) | 47.4, 47.6, 47.7 | 49.1, 49.8, 71.8 | 47.6, 47.6, 47.9 | 47.4, 47.7, 47.8 |
| α, β, γ (°) | 77.1, 69.4, 61.7 | 90.1, 93.2, 102.3 | 76.4, 69.2, 61.3 | 76.7, 69.3, 61.5 |
| Resolution (Å) | 1.70 | 1.90 | 1.52 | 1.55 |
| Number of molecules in asymmetric units | 2 | 4 | 2 | 2 |
| Measured reflections | 78,393 | 112,869 | 175,078 | 98,424 |
| Redundancya | 2.2 (2.2) | 2.4 (2.4) | 3.6 (3.6) | 2.3 (2.2) |
| Rmeasb | 0.090 (0.44) | 0.092 (0.19) | 0.060 (0.79) | 0.059 (0.39) |
| 〈I/σ(I)〉 | 6.1 (1.7) | 8.6 (4.6) | 11.9 (1.7) | 9.1 (2.3) |
| Completeness (%) | 94.8 (94.2) | 90.0 (84.0) | 92.5 (89.9) | 94.6 (93.1) |
| Refinement statistics | ||||
| Unique reflections | 35,712 | 44,643 | 46,361 | 44,806 |
| Rfc | 0.182 | 0.174 | 0.170 | 0.158 |
| Rfreed | 0.205 | 0.213 | 0.196 | 0.177 |
| Number of Atoms | 2876 | 6154 | 3049 | 3034 |
| Protein | 2729 | 5681 | 2839 | 2809 |
| Ligands/ions | 10 | 132 | 46 | 54 |
| Water | 137 | 341 | 164 | 171 |
| Ramachandran plote | ||||
| Favored (%) | 100 | 100 | 100 | 100 |
| Outliers (%) | 0 | 0 | 0 | 0 |
| RMS deviations | ||||
| Bond lengths (Å) | 0.007 | 0.007 | 0.007 | 0.007 |
| Bond angles (°) | 1.1 | 1.3 | 1.3 | 1.1 |
aValues in parenthesis are for the highest shelves.
bRmeas = Σhkl(N(hkl)/(N(hkl) − 1))1/2|I − 〈I〉|/Σhkl〈I〉, where I is the intensity of the hkl reflection; 〈I〉 is the average over symmetry-related observation of the hkl reflection.
cRf = Σ|Fobs − Fcalc|/ΣFobs, where Fobs and Fcalc are the observed and the calculated structure factors, respectively.
dRfree was calculated using 5% of reflections that were selected randomly and set aside during the refinement.
e Calculated using Molprobity.
Figure 2.
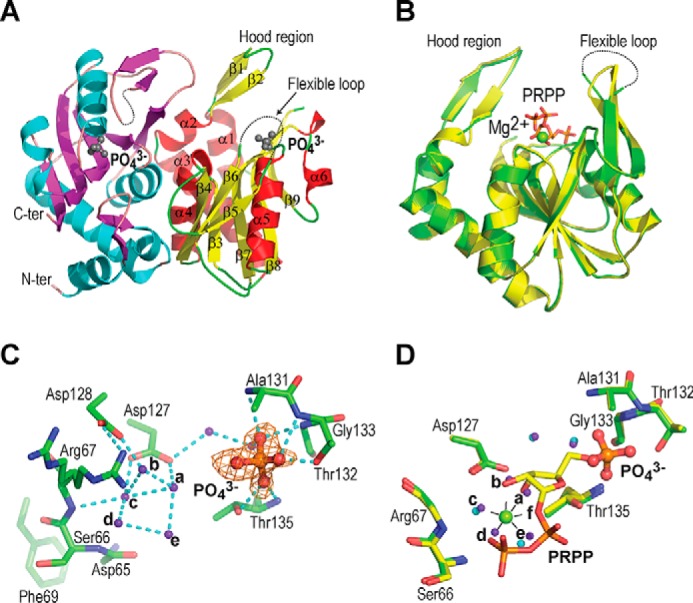
Structure of phosphate-bound hAPRT. A, structure of the hAPRT homodimer showing two phosphate molecules (gray ball-stick), the hood region, and the flexible loop. B, superimposition of the phosphate-bound (green) onto the PRPP-bound (yellow) hAPRT structure (RMSD = 0.20 Å). C, Fo – Fc omit electron density map for a phosphate ion contoured at 3σ. Without substrate bound in the active site, the conserved Arg-67 adopted two conformations. D, superimposition of the phosphate-bound (green) onto the PRPP-bound (yellow) hAPRT active site. Water molecules are shown in purple or cyan in the phosphate- or PRPP-bound structures, respectively. All the figures were generated with PyMOL. The hydrogen bonds are represented with dashed cyan lines throughout.
Five of these water molecules (called a, b, c, d, e) were located in close vicinity of the six oxygen atoms coordinating the magnesium ion in the PRPP-Mg2+-hAPRT structures (PDB IDs: 6FCH, 6FCI, and 6FD4) (Fig. 2D). In addition, the phosphate group in the substrate-free hAPRT structure interacted in a similar way compared with the 5′-phosphate group of PRPP (Fig. 2D). These observations suggest that the substrate-free enzyme is destabilized by the formation of a water molecule network in the active site and a more dynamic flexible loop.
Substrate shape recognition in the forward reaction
Next, we addressed the substrate specificity of hAPRT using other purines than adenine, which are found at similar concentrations in cells. First, we co-crystallized hAPRT with guanine, PRPP, and magnesium. Although PRPP and magnesium were perfectly identified in the complex structure, we were unable to detect guanine in the active site. Second, we used Hx, a molecule identical in shape to ADE but with a hydroxyl or a keto group in place of a primary amine (Fig. 1B). With DSF analysis, we found no difference in the unfolding temperature when adenine or hypoxanthine were added to the PRPP-Mg2+-hAPRT complex (Table 1). Using a spectroscopy assay with PRPP and Hx as substrates, we did not detect a significant amount of IMP product up to a 75 μm Hx concentration (Fig. 3A). In addition, we did not observe any inhibition by Hx while using ADE and PRPP as substrates (Fig. 3A). Nonreactivity of hAPRT toward Hx could relate to Hx chemical properties or unbinding of Hx to the active site. In both cases, Hx may not be able to induce the structural modifications of hAPRT necessary to form the active conformation, as observed in our ADE-PRPP-Mg2+-hAPRT structure where the hood and the flexible loop undergo major changes upon ADE binding (6). To understand the nonreactivity of Hx, we determined the structure of hAPRT crystallized in the presence of PRPP, magnesium ion and hypoxanthine at 1.90 Å resolution (Table 2). We observed, as for ADE, that both the hood and the flexible loop regions cap the active site where Hx is bound (Fig. 3B). The structure of the Hx-PRPP-Mg2+-hAPRT complex was identical to that of the ADE-PRPP-Mg2+-hAPRT complex (PDB ID: 6FCI) with a RMSD of 0.15 Å, with the active sites superimposing perfectly. The Fo − Fc omit density map contoured at 3σ for Hx is shown in Fig. 3C. The poorer definition of the Hx purine ring in the electron density identified in all four molecules of the asymmetric unit, as compared with the electron density of the ADE, reflects a less stable conformation or a lower affinity for the site. However, the Fo − Fc omit map densities for PRPP showed full occupancy for this substrate (Fig. 3D) (Note S1). Hx can exist in a tautomeric equilibrium with either a hydroxyl or a keto group on C6, the latter being, by far, the most stable form in solution (14). In the binding site, Hx interacts through hydrogen bonds with Arg-27, Arg-67, and Glu-104 (Fig. 3C). The interactions with Arg-27 and Arg-67 are crucial because their bonding atoms are both hydrogen donors; thus, N1 and N3 (Fig. 1) will act as hydrogen acceptors and this promotes a hydroxyl function at the C6 position. This hydroxyl group interacts on one side with the Glu-104 carboxylate side chain with an average distance of 2.9 Å over the four subunits of the asymmetric unit. On the other side, it is 3.2 Å away from the Val-25 carbonyl. This is not a favorable configuration and it may contribute to a destabilizing effect of Hx.
Figure 3.

Structure of a substrate analog of the forward reaction. A, kinetic activity of hAPRT for ADE (black), Hx (blue), and ADE in the presence of 75 μm Hx (red). B, superimposition of the Hx-PRPP-Mg2+- (orange) onto the PRPP- (yellow) hAPRT structure (RMSD = 0.62 Å). C and D, Fo – Fc omit electron density map for Hx (C) and PRPP (D) contoured at 3σ from the Hx-PRPP-Mg2+-hAPRT structure. The hydrogen bond distances are indicated in Angstroms.
Overall, the data show that, in the absence of ADE, a molecule like hypoxanthine, with an adenine shape, is able to bind to hAPRT and generate a full active site with both the hood and the flexible loop in place. However, the chemical properties of Hx appear to prevent the forward reaction to occur.
Purine base recognition in the reverse reaction
We next tried to understand why among the nucleoside monophosphates, AMP is the sole substrate metabolized by hAPRT in the reverse reaction. To this end, we measured the stability of hAPRT by DSF in the presence of PPi, AMP, IMP, and GMP. We observed that PPi increased the stability of hAPRT (the Tm increased by 2.5 °C as compared with a 9.4 °C increase with AMP) (Table 1). Combined, the two substrates (PPi and AMP) potentiated the stabilization of the enzyme (Tm of 64.3 °C with ΔTm = 14.7 °C). Stabilization of the enzyme was also observed with IMP and GMP, although to a lesser extent. The Tm increased only 2–3 °C with the addition of IMP or GMP to the enzyme as compared with substrate-free hAPRT. Moreover, addition of PPi with IMP or GMP to the enzyme did not increase the Tm values. Therefore, the complexation of IMP and GMP to hAPRT seems less favorable than with AMP, and PPi may not contribute to the binding. To probe how IMP or GMP interact with hAPRT, we determined the crystal structures of the two complexes, IMP-hAPRT and GMP-hAPRT (Table 2). We diffused the IMP and GMP molecules into substrate-free hAPRT crystals and elucidated the structures to a better than 1.6 Å resolution. The structures were then compared with the natural AMP-hAPRT complex (PDB ID: 6FCL). The electron densities in the active sites were readily attributable to IMP and GMP (Fig. 4, A and B). Both structures were similar overall and superimposable to the substrate-free, PRPP-bound hAPRT or AMP-hAPRT structures with an RMSD between 0.17 and 0.31 Å. The conformation of IMP was, however, different from that of AMP (Fig. 4A and Fig. S3). Even though the interactions with the phosphate and the sugar moieties were conserved, the base was differently positioned. Specifically, with AMP, the N1 atom faces the main chain NH group of Arg-27 and presents its lone electron pair to form a hydrogen bond of 3.0 Å (Fig. S3). In the IMP complex, the N1 atom is at a hydrogen bond distance of 2.8 Å from the carboxyl group of Arg-27 (Fig. 4A). Therefore, N1 is likely to be protonated in this configuration. This implies that the oxygen atom on C6 should be in the more stable keto tautomer configuration and not in the hydroxyl one as observed in the Hx-hAPRT complex. The consequence of these modifications is a movement of the purine core, which culminates in a 1.4 Å displacement of the N1 and C2 atoms as compared with the AMP-hAPRT structure (Fig. 4C). This movement promotes a change in the orientation of the guanidinium moiety of Arg-67. Its CZ carbon atom would now be in close contact (2.4 Å) with a pyrophosphate substrate (PPi) involved in the reverse reaction, which we modeled based on the PRPP-Mg2+-hAPRT structure (Fig. 4C). Such configuration should preclude PPi from binding and, consequently, the reverse reaction would be forbidden with IMP in the active site. In addition, if IMP were a substrate, the formation of Hx would be concomitant with a change in the hydrogen bond network. Five hydrogen bonds would need to be broken between IMP and hAPRT, and four new ones would have to be formed after the displacement of the purine moiety (Fig. 3C). This is not energetically favorable for the enzyme and, together with the Arg-67 movement, would preclude the reverse reaction to occur.
Figure 4.

Structure of two substrate analogs of the reverse reaction. A and B, Fo – Fc omit electron density maps for IMP (A) and GMP (B), contoured at 3σ from the IMP and GMP-hAPRT structures, respectively. Nonequivalent hydrogen bonds, as compared with the AMP-hAPRT structures, are indicated by their length in Angstroms. C, close-up view of the ligands after superimposition of the ADE-PRPP- (PDB ID: 6FCI), AMP- (PDB ID: 6FCL), IMP- and GMP-hAPRT structures. ADE and PRPP are in blue, AMP in magenta, IMP in green and GMP in cyan, with the corresponding amino acids in the active site colored accordingly. D, Superimposition of the AMP-, IMP-, and GMP-hAPRT structures showing the hood region and the flexible loop where the conserved Tyr-105 is located.
With GMP, we found that the GMP-hAPRT complex superimposed with that of the IMP complex with an RMSD of 0.2 Å (Fig. 4C, green and cyan). One difference comes from the extra amino group in GMP at the C2 position, which makes an additional hydrogen bond to the Arg-27 carbonyl and the Asp-128 carboxylate groups (Fig. 4B).
Hence, the hood region backbone of hAPRT is critical for substrate specificity, which allows only AMP as the sole substrate for the reverse reaction. We also noticed that in both IMP and GMP molecules, the sugar puckering adopts a 4′ endo configuration as compared with a 3′ exo configuration in the AMP structure. Finally, in all three nucleotide structures, the flexible loop is in an open conformation but not well-defined from amino acid Ser-100 to Lys-107. The nucleotides make almost no contact with the loop and therefore do not contribute to its stabilization (Fig. 4D).
Molecular basis of hAPRT catalysis in forward and reverse directions
The forward reaction catalyzed by hAPRT can occur via either an SN1 or an SN2 displacement mechanism (12, 13, 15). According to an SN1-type reaction mechanism, an oxocarbenium intermediate must exist and be located on the C1′-O4′ atoms of PRPP. It is a short-lived, solvent-sensitive intermediate that can be stabilized by an electron donor moiety (16). In the ADE-PRPP-hAPRT structure (PDB ID: 6FCI), we noticed that the hydroxyl group of Tyr-105 is the only electron donor group in close vicinity to the C1′-O4′ atoms of PRPP (6). This hydroxyl group, which is 3.4 Å and 3.8 Å away from the N9 and C1′ atoms, respectively, also contributes to their isolation from solvent molecules (Fig. 5A). Moreover, it is hydrogen-bonded to the PRPP pyrophosphate moiety (Fig. 5A). This configuration is reminiscent of the Trypanosoma cruzi trans-sialidase, in which a hydroxyl tyrosine (Tyr-342) is positioned 3.5 Å above an oxocarbenium ion and within hydrogen bond distance to a carboxylate side chain (Glu-230), which serves as a charge relay for the Tyr-342 nucleophile (17). In hAPRT, the pyrophosphate moiety could serve as a charge relay to enhance the Tyr-105 phenolate character toward the C1′ atom. We thus hypothesized that a partial electronegative charge on the Tyr-105 hydroxyl group could stabilize an oxocarbenium intermediate going through an SN1 chemical reaction. On the other hand, the C1′ atom is only 3.2 Å away from the ADE N9 atom (Fig. 5A), and the distance between the N9 and O1′ atoms from the leaving group is 4.6 Å. These short distances support an SN2-type reaction (10).
Figure 5.

Possible reaction paths for hAPRT. A, close contacts (green) and hydrogen bond (cyan) between ADE, PRPP, and Tyr-105 oxygen atom in the ADE-PRPP-Mg2+-hAPRT structure. B, proposed catalytic SN1 or SN2 pathways when hAPRT is complexed to ADE and PRPP. C, schematic representation of the reaction path for the forward reaction catalyzed by hAPRT. In green, the path found for the SN1 displacement mechanism involving ADE (only the stable intermediate was identified). The SN2 paths are shown in blue for ADE and in red for Hx. TS, transition state. D, visualization of the reduced density gradient surface. The electron density at a critical point between atoms, when the gradient of the density is null, provides information about the strength of a noncovalent interaction whereas the sign of the second derivative of the electron density distinguishes between bonded (negative value) and nonbonded (positive value) interactions. Consequently, at each critical point of a molecular system, an NCI strength coefficient is defined by multiplying the sign of the second derivative by the value of the electron density at this point. Hence, large negative values of this NCI strength coefficient are indicative of attractive interactions (dipole-dipole, hydrogen bonds, pi-pi stacking), large and positive values indicate that the interaction is nonbonding, whereas values near zero indicate weak van der Waals interactions. Critical points are indicated by yellow, pink, magenta, and cyan spheres with the associated NCI coefficient.
To discriminate between these two possible reaction paths, we applied a quantum mechanics (QM) and molecular mechanics (MM) protocol. We first placed the ADE-PRPP-Mg2+-hAPRT structure (PDB ID: 6FCI) in a minimum on its potential energy surface. To be exhaustive, we investigated four possible scenarios to identify the lowest energy path linking the reactants (ADE and PRPP) and the expected products (AMP and PPi) in the forward reaction (Fig. 5B). We first assumed that the mechanism proceeded via an SN1 pathway by dissociating the C1′-O1′ bond of PRPP to form a stable intermediate state made of three distinct species: the purine base, a phosphoribosyl oxocarbenium derivative, and a pyrophosphate. The nucleophilic attack by the purine N9 atom position on the C1′ position of the phosphoribosyl carbocation is the second step of this mechanism. We identified a stable intermediate along the path that characterizes the SN1 mechanism. We found that this intermediate relies on a minimum that is high in energy (about 100 kJ/mol) as compared not only with the energy level of the reactants but also with the transition state identified along an SN2 reaction path (Fig. 5C). We conclude that the SN1 path is theoretically possible, but its high energy level precludes the reaction from proceeding in this way.
In the case of an SN2 mechanism, the three potential pathways differ in the way the charge is transferred from Glu-104 to the pyrophosphate along the reaction path (Fig. 5B): (a) a two-step anionic SN2 pathway, (b) a one-step neutral pathway, and (c) a two-step cationic SN2 pathway (Note S2). From our QM/MM calculations, we found that only the two-step cationic SN2-(c) path was possible. Indeed, no transition state or stable intermediate corresponding to either the SN2-(a) or SN2-(b) reaction paths was found on the potential energy surface of this system, which indicates that the reaction does not proceed in either of these ways. The only transition state and stable intermediates that are unambiguously identified on the energy surface of the system correspond to a nucleophilic attack of ADE/N9 on the PRPP/C1′ position. This attack leads to the formation of an AMP-H+ derivative, which is protonated on the N7 position, and a pyrophosphate (PPi). The QM/MM energy barrier of this first step is 54.7 kJ/mol (Fig. 5C). Interestingly, we found that the positively charged intermediate is more stable than the reactants by 7.1 kJ/mol. The formation of these intermediates is, therefore, kinetically and thermodynamically favorable and, thus, drives the reaction toward the second step in the forward direction. This second step corresponds to the shift of the proton from the AMP-H+/N7 position toward the acidic side chain of Glu-104. The activation barrier for this proton shift is very low and certainly occurs quickly after the first step (6.7 kJ/mol). The calculated QM/MM relative energy of the AMP-PPi-Mg2+-hAPRT complex that is formed is −25.1 kJ/mol as compared with the reactants. The high stabilization of the products that are formed indicates that the forward reaction is thermodynamically favorable because each of the steps is reversible.
We found that the first transition state is composed of an oxocarbenium-like intermediate in which the bonds to PPi and ADE are neither totally broken nor formed, respectively. In this transition state, our calculations show the hydroxyl group of Tyr-105 to be at a suitable distance to stabilize the δ+ positively charged phosphoribosyl (Fig. 5D). The last step of this mechanism, which is of the scope of a QM/MM study, consists of the opening of the loop and the release of the PPi followed by the AMP molecule. Glu-104, in contact with the bulk water solvent, would then become deprotonated in accordance with its pKa in solution.
We applied a similar strategy to the Hx-PRPP-Mg2+-hAPRT structure to understand why Hx is not a substrate despite its ability to bind to the active site and to bring both the hood and the flexible loop in the closed conformation. The intermediate obtained after the first step is less stable than the reactants by 18.8 kJ/mol, which indicates that this reaction is neither kinetically nor thermodynamically favorable (Fig. 5C). Moreover, the proton transfer from IMP-H+/N7 to Glu-104, which occurs via a low-energy barrier, leads to the final products that are less stable than the reactants by 6.3 kJ/mol. Finally, the formation of the stable tautomer would require a large reorganization of the full complex, as observed in our IMP-hAPRT structure, which is not favorable (Fig. 4A). Taken together, the reaction path identified for the catalytic transformation of Hx into IMP by hAPRT is not favorable and this explains why Hx is not a substrate despite its chemical similarity to ADE.
In conclusion, the forward reaction in hAPRT follows a mix of SN1- and SN2-type reaction where a partial positive charge, oxocarbenium-like, is located on the C1′ atom of the transition state after the attack of the N9 nitrogen. This step is followed by the formation of a positively charged AMP intermediate prior to a proton transfer onto Glu-104 and release of the products.
Discussion
hAPRT substrate specificity in the forward and reverse reactions
The substrate-free structure corresponds to the common state just prior to the binding of PRPP or AMP to hAPRT with a cis-peptide bond between amino acids Asp-65 and Ser-66. The displacement of the water molecule network by a more stabilizing binding of PRPP should contribute to a higher unfolding temperature, in addition to a cis-trans effect (Fig. 2, C and D) and, to a lesser extent, to a stiffening of the flexible loop, as observed in the PRPP-Mg2+-hAPRT crystal structure (6). We used guanine, PRPP, and magnesium ion to test for active site specificity, but we did not visualize the purine moiety above the PRPP density. This is likely because of the extra primary amine group at C2, which would clash with the Arg-27 carbonyl in an ADE configuration. Next, we used hypoxanthine to understand why a natural analog with the same chemical shape as adenine is not a substrate. We showed that Hx was capable of binding hAPRT and, more importantly, was able to bring the hood and the flexible loop into the active conformation. Hence, hAPRT is able to recognize the shape of Hx, which selects the flexible loop and the hood conformation to generate a fully active site ready for catalysis. However, the structure was obtained with a concentration above 300 μm, which is significantly higher than the estimated concentration in the cell (10 μm), where 1 μm of adenine will compete for the same reaction site. We also showed that 75 μm Hx cannot compete with 2 μm ADE (Fig. 3A). Thus, in a cellular context, Hx is unlikely to bind to the active site and compete with ADE. In addition, QM/MM analysis as well as the IMP-hAPRT structure showed that the reaction could not occur even if Hx was bound in the active site. This means that the specificity of the enzyme in the forward reaction is not determined by the shape of the substrate but by the chemistry of the reaction where Glu-104 promotes a noneffective tautomeric form to Hx.
For the reverse reaction, hAPRT is also very specific. AMP, IMP, and GMP are chemically identical in their C1′-N9 glycosidic bonds and their intracellular concentrations are similar (7). They are therefore all potential substrates. However, IMP and GMP act as inhibitors with Ki of 350 μm and 170 μm, respectively, in the presence of 55 μm ADE (18). With 1 μm concentration of ADE in human cells, these Ki should be somewhat lower. Also, the 2-fold difference between the Kis of IMP and GMP might come from the extra amino group located in GMP at the C2 position, which makes additional contacts with the enzyme, namely Arg-27 and Asp-128 (Fig. 4B). Our structural results, which show the orientations of the nucleoside monophosphates in the active site, demonstrate the important role of the conserved Arg-67 and the proofreading capacity of the hood region in hAPRT selectivity, which both explain the aforementioned inhibition values (Fig. 4).
Proposed hAPRT molecular mechanism for the forward and reverse reactions
Our results indicate that the forward reaction proceeds through an SN2-type reaction mechanism, also known as ANDN reaction in the IUPAC nomenclature (19), and includes some SN1 character. The latter is because of the presence of a partially positively charged intermediate located on the C1′ atom and on the somewhat long bond distances (over 2 Å) between the N9, C1′, and O1′ atoms in the transition state (10). This was not expected because, for human HGPRT, an SN1-type reaction (also known as DNAN) is favored, as for purine-nucleoside phosphorylase (PNP, EC 2.4.2.1), an enzyme from the same family. In these two cases, an oxocarbenium intermediate was identified (11, 20, 21). However, this stabilized positively charged compound was undetermined in APRT enzymes. In HGPRTs and PNPs, the short-lived oxocarbenium ions are protected from the solvent by the enzymes, and it has been proposed that they are stabilized by the Ser-33 in human PNP and by the 5′OH of PRPP in human HGPRT (11, 22). In hAPRT, we identified Tyr-105 as a potential stabilizer of the reaction. Also, in the ADE-PRPP-Mg2+-hAPRT structure, the distance between the 4′ and 5′ oxygen atoms is 2.9 Å with a geometry similar to that observed in ImmGP-human HGPRT structure (PDB ID: 1BZY). However, the involvement of 5′OH in stabilization of an oxocarbenium ion is probably prevented by electrostatic interaction between the 5′OH atom and the C8 hydrogen, as predicted in QM/MM calculations (Fig. 5D). In addition, the reaction coordinate separation (the distance from the N9 to the attacking oxygen atom of PPi in the reverse reaction and vice versa) is 4.6 Å in the ADE/PRPP complex and 4.5 Å in the AMP/PPi complex. This is between 0.6 to 1Å shorter than the values for HGPRT (5.2 Å) (PDB ID: 1TC2) and for PNP (5.5 Å) in the Michaelis complex. These shorter distances are in favor of an SN2-type mechanism, even though the atomic excursion of the anomeric carbon (C1′) of the ribosyl moiety is around 2 Å from substrate to product, similar to the value for HGPRT (2.1 Å) and greater than the value for PNP (1.7 Å) (23). Together, these observations demonstrate that a pure SN1-type reaction is not possible for hAPRT and, instead, an SN2 mechanism is mainly involved.
After the binding of PRPP, the hood region and the flexible loop are preferentially in an open conformation. The interaction with an adenine molecule will select for the closed conformation of the flexible loop in which the conserved Glu-104 and Tyr-105 interact with both substrates through hydrogen bonds and van der Waals contacts. In this configuration, the C1′, O4′, and N9 atoms are isolated from the solvent. Our calculations indicate that the limiting step of the reaction is the nucleophilic attack of the ADE/N9 position onto the C1′ atom of PRPP. The high-energy level of this step is in line with the fact that this is a late transition state where the geometry of the transition step is closer to the intermediates than the reactants. To better understand how hAPRT stabilizes the transition state of the nucleophilic attack of the purine onto PRPP, we calculated and plotted the reduced density gradient surface on the geometry of the transition state (Fig. 5D). This visual method, which was proposed by Johnson et al. (24) and implemented in Maestro (25), allows analysis and visualization of a wide range of noncovalent interaction (NCI) types. Fig. 5D shows NCI with values for some critical points within the quantum region. Glu-104 appears to maintain the position of ADE (pink sphere) and also contributes to the positioning of Tyr-105. Tyr-105 is involved mainly in maintaining the position of ADE and PPi moiety (purple spheres) as well as participating weakly in the stabilization of the oxocarbenium-like moiety. As expected, two nonbonded interactions are found along the reaction path, which indicates that these bonds are not totally broken or formed (yellow spheres with high negative NCI coefficient). Specifically, the purine/N9-C1′ bond was estimated to be 2.3 Å long and the C1′-O bond 2.4 Å in length. Following this step, the N9-C1′ bond is formed concomitantly with a positively charged AMP molecule (Fig. 6A). This short-lived intermediate rapidly transfers its N7 proton to the Glu-104 carboxylic side chain. We propose that the neutralization of Glu-104 creates a nonfavorable environment in the active site and promotes the departure of the loop toward an open conformation, where the proton from Glu-104 can now leave. Altogether, these observations indicate that Glu-104 and Tyr-105 are involved in the fine stabilization of the reactants and the transition state as well as determining selectivity through the loop closer in the forward reaction. This involvement was also revealed by our kinetics analysis of a Y105F variant for which the kcat decreased by 300-fold as compared with the WT enzyme (6).
Figure 6.

Proposed molecular mechanism in both directions of the reaction for hAPRT. A, in the forward reaction, SN2-type molecular mechanism including some SN1 characteristics. B, in the reverse reaction, a proposed magnesium-assisted SN2-type mechanism. For clarity, the nucleotide is shown in a syn configuration whereas it is always found in an anti configuration in the hAPRT active site. Sugar puckering configurations are indicated in blue next to the sugar moieties.
We do not expect the reverse reaction to follow a similar path starting with loop closer, proton transfer from Glu-104 to N7 followed by rupture of the N9-C1′ bond as this would not be an energetically favorable process (Fig. 5C). Instead, in all PRPP-bound hAPRTWT structures (PDB IDs: 6FCH, 6FCI, 6FD4, 6HGQ), including the Hx-PRPP-Mg2+-hAPRTWT structure, the magnesium ion has an expected geometry (∼2.25 Å coordinating distance and ∼90° angle between atoms) except for the values of the angle between O1′-Mg2+-O2′ and O2′-Mg2+-O3′ (Table S1). A value of ∼70° was observed for these angles because of the geometry of the sugar moiety, which leads to constraints on the chelating system. However, in the ADE-PRPP-Mg2+-hAPRTY105F structures (PDB IDs: 6FD5 and 6FD6) (6), which show the transformed products AMP and PPi, the Mg2+-O1′ (from PPi) and Mg2+-O2′ (from AMP) coordinating bonds have distances of 1.8 and 2.7 Å, respectively (Fig. S4). These values are ∼0.4 Å from the ideal value. This leads to a 0.8-Å displacement of the magnesium ion toward the O2′ atom as compared with its original position, as found in the PRPP-bound hAPRTWT structures, despite an identical positioning of the three phosphate moieties from AMP, PPi, and PRPP (Fig. S5). This new geometry should move the AMP-PPi-Mg2+ complex into a catalytically competent state. This was further suggested by the comparison of the AMP-hAPRTWT and the ADE-PRPP-Mg2+-hAPRTY105F structures, in which AMP and PPi molecules had formed in crystallo (Fig. S6). In this case, despite an overall RMSD of 0.4 Å, we showed that the ribose and the base did not superimpose. This is because of a change in the ribose puckering for which the configuration changes from a C3′ exo in AMP to a C4′ endo in AMP+PPi structures. The ribose seems to have rotated around the C4′ atom, culminating to a maximum displacement of 0.7 Å for the O2′ atom (Fig. S6). This AMP movement would be because of the presence of the PPi-Mg2+ complex in the active site, which would force the O2′ and O3′ atoms of the ribose moiety to coordinate the magnesium ion. The N6 atom of AMP would then carry along the Val-25 carbonyl, which would lead to the displacement of the hood region, as observed in ADE-PRPP-Mg2+-hAPRTY105F structure (PDB ID: 6FD5) (Fig. S6). Finally, we noticed that the closest pyrophosphate oxygen atom was only 3.0 Å away from the C1′ atom in the ADE-PRPP-Mg2+-hAPRTY105F structures. This would be a favorable geometry for the attack of the C1′ atom by the pyrophosphate group.
Therefore, we propose that disruption of the conventional magnesium geometry is used in a template effect chemical reaction, to initiate the reverse enzymatic reaction (26, 27). We put forward the following mechanism for the reverse reaction (Fig. 6B). First, AMP would bind into the active site with the flexible loop in an open conformation and a sugar moiety in a C3′ exo configuration. Secondly, the PPi-Mg2+ complex would engage into the reaction site, which would rotate the ribose toward PPi and force it to adopt a C4′ endo configuration (Fig. S5). The chelate would then help to bring the C1′ atom and the pyrophosphate closer to each other to favor a metal-assisted SN2-type reaction (ANDN reaction). After the attack of the C1′ atom by PPi, a transition state would be formed with a nucleophile (PPi) and a leaving group (ADE) partially bound to the C1′ atom. The N7 atom would then be protonated through a water molecule observed in our structures (Fig. S3). Finally, the ADE and PRPP products would form (sugar moiety in a C2′ endo configuration), which would lead either to the closing of the hood and the flexible loop onto the active site or to the departure of the products. Hence, the open conformation of the loop favors the reverse reaction by keeping Glu-104 and Tyr-105 away from the substrate and allowing a new geometry of the chelate to develop. These observations are correlated with our results on the Y105F variant, which favors the reverse reaction and an open conformation for the flexible loop (6).
Conclusions
A complete understanding of both the catalytic mechanism of hAPRT and the method by which this enzyme ensures its substrate selectivity are first, but critical, steps to identify new therapeutics that could be useful as antiparasite, anticancer therapies or purine-associated diseases (28–32). In addition, we showed that the nature of the atom attached to C6 does not affect the active site formation (loop closure and hood movement), as observed in the Hx- and ADE-hAPRT structures, but a nitrogen atom is necessary to facilitate the reaction. In the future, various modifications of this chemical group could therefore be tested for their potential effects in several biological contexts, for example in neuronal diseases, or crop development (33, 34).
Materials and methods
Protein purification and crystallization
The hAPRT WT protein was purchased from Euromedex (Souffelweyersheim, France) (catalog no. ATGP0483, lot: 1046601), diluted in buffer A (20 mm Tris-HCl, pH 7.4, 5 mm MgCl2) and concentrated to 5 mg/ml (6). To prepare phosphate-bound hAPRT crystals, PRPP-Mg2+-hAPRT crystals were left 1 month at 20 °C, which led to a complete hydrolysis of PRPP and a free active site with a single phosphate ion bound. To prepare Hx-PRPP-Mg2+-hAPRT co-crystals, the protein was first buffer exchanged with a hypoxanthine-saturated buffer A (final Hx concentration above 300 μm). Then the concentrated protein was mixed with a 400 mm concentrated solution of PRPP in a 20:1 volume ratio (final PRPP concentration of 20 mm). All the solutions were prepared in buffer A and the final protein concentration of the complex was 4.5 mg/ml. Using a Mosquito robot (TTP LabTec), 400 nl of the complex was mixed with 200 nl of crystallization solutions made of 85 mm Tris-HCl, pH 8.5, 170 mm NaOAc and 19–21% PEG4000 mixed with 0–30% glycerol. Crystals appeared overnight at room temperature (20 °C). To prepare IMP- and GMP-hAPRT complexes, we transferred substrate-free crystals into crystallization solutions containing 20 mm IMP or GMP for 45 min. We repeated this transfer three times to ensure full occupancy of the hAPRT active site. The crystals were then frozen immediately in liquid nitrogen.
Data collection, processing, and structure refinement
Four diffraction data were obtained at SOLEIL (beamline PX1) and ESRF (beamlines ID14–4 and ID29) (Table 2). They were integrated with iMOSFLM (35) or XDS (36) and scaled with AIMLESS (37). The structures were solved by molecular replacement with the program PHASER using phases from the PDB ID 6FCH structure (6). Complex structures were then refined with REFMAC5 (38) using the CCP4i software suite (37) and the building program Coot (39).
DSF assays
The thermal stability of WT hAPRT protein was analyzed using a LightCycler 480 instrument (Roche) to image 384 wells simultaneously (6). Briefly, the protein was diluted to 0.2 mg/ml in 20 mm Tris-HCl, pH 7.4, 150 mm NaCl. A volume of 10 μl/well was used in plates and the protein solution was gradually increased from 30 °C to 95 °C at 0.01 °C per second with 16 acquisitions per degree. The protein stability was assessed by measuring the fluorescence of a reporter dye, SYPRO orange (Invitrogen, S6651), diluted 1000-fold. Each experiment was performed in triplicate to compute the mean value of the thermal melting profile (Tm) and its standard deviation.
In vitro hAPRT kinetic analysis
Human WT APRT activities on ADE and Hx were determined by quantifying the amount of AMP or IMP produced using a spectroscopic assay identical to the one developed in Tuttle and Krenitsky (40). Analyses were performed in 0.1 m Tris-HCl, pH 7.5, 10 mm MgCl2 (buffer D). The reaction mixture (200 μl final volume) was incubated at 23 °C. When ADE or Hx were varied, 500 μm PRPP and 200 nm WT protein were added to the mixture. The reaction started after the addition of the enzyme and was monitored at 256 nm for ADE and 244 nm for Hx, where the differential of the absorbance are maximum between AMP and ADE (ϵ = 2100 M−1/cm−1) or IMP and Hx (ϵ = 2200 M−1/cm−1) (40). The competitive assay for the ADE site was done with the above concentration of PRPP and enzyme, various amount of ADE, and in the presence of 75 μm Hx, which was the maximum limit for this spectroscopic assay. The kinetic parameters were determined with the Michaelis-Menten equation using Origin 6.1 with a hyperbolic fit. All measurements were done in triplicate.
QM/MM calculations
A computational model of the ADE-PRPP-hAPRT complex was prepared based on our X-ray structure (PDB ID: 6FCI) (6). The model of the Hx-PRPP-hAPRT complex was prepared from the model of the ADE-PRPP-hAPRT complex by mutating ADE into Hx. A model very similar to the X-ray structure of our Hx-PRPP-hAPRT complex was obtained. This was performed to place both systems initially in the same minima of the PRPP-hAPRT potential energy surface. Hence, the differences observed between the two calculated reaction paths are mainly associated with the chemical reactivity of ADE and Hx. Missing hydrogen atoms were added and optimized using the Protein Preparation Wizard in Maestro (25). For both models, the QM region included the purine base (ADE or HX), the co-substrate PRPP, the magnesium ion, and five water molecules and six side chains in close contact with these two substrates (namely, Arg-67, Lys-88, Glu-104, Tyr-105, Asp-127, Asp-128). In the end, the QM region contains 133 atoms for the Hx-PRPP-hAPRT structure and 134 atoms for the ADE-PRPP-hAPRT structure.
The optimization of the QM/MM geometry and the calculations of frequencies were performed at the B3LYP/6–31G* level of theory for all atoms except the magnesium ion (B3LYP/lacv3p**) for the QM region; for the rest of the system, the OPLS2005 molecular mechanism force field, as implemented in QSite (41), was used. Our attempt to obtain accurate energy by a single point calculation with a larger triple zeta basis set (QM region) failed because of convergence issues. For all calculations, the QM region was not constrained whereas the MM region was divided into two subregions: No constraints within a cap of 5 Å around the two substrates residues, whereas beyond this zone the residues were kept frozen. The structures corresponding to reactant and product minima are characterized by no imaginary frequency, whereas transition state structures are characterized by only one imaginary frequency that corresponds to the reaction coordinate motion.
Accession numbers
The atomic coordinates and structure factors for the phosphate-, Hx-PRPP-Mg2+–, IMP-, and GMP-bound hAPRTwt complexes have been deposited in the RCSB PDB under the accession codes 6HGP, 6HGQ, 6HGR, and 6HGS, respectively.
Author contributions
M. O., J. H., M.-C. B., F. C., J.-M. R., R. M., F. A., and P. N. data curation; M. O., J. H., M.-C. B., B. P., F. C., J.-M. R., A. R. S., R. L., G. P., C. S.-M., J.-F. G., R. M., B. D.-F., F. A., and P. N. formal analysis; M. O., J. H., A. R. S., R. L., G. P., F. A., and P. N. writing-original draft; I. C.-P., R. B., A. O.-B., F. A., and P. N. conceptualization; F. A. and P. N. resources; F. A. and P. N. software; F. A. and P. N. supervision; F. A. and P. N. funding acquisition; F. A. and P. N. validation; F. A. and P. N. investigation; F. A. and P. N. visualization; F. A. and P. N. methodology; F. A. and P. N. writing-review and editing.
Supplementary Material
Acknowledgments
We are grateful to the staffs at the X-ray crystallography beamlines of SOLEIL and ESRF. We thank Drs. Lawrence Aggerbeck, Olivia Reinaud, Mélanie Ethève-Quelquejeu, C.S. Raman, and Benoit Schneider for their very useful reading and discussion of the manuscript.
This work was supported by French National Research Agency Grant 2010-CESA-015 (to J. H. and P. N.), the ARC Foundation (to M. O.), an INSERM-University Chair (to P. N.), Paris City Hall and Ile de France Region grants (to P. N.), and Sanofi research contracts 116048 (to I. C.-P., B. P., and B. D.-F.) and EU-TL-2013-003 (to P. N.). The authors declare that they have no conflicts of interest with the contents of this article.
This article was selected as one of our Editors' Picks.
This article contains Table S1, Notes S1 and S2, and Figs. S1–S6.
The atomic coordinates and structure factors (codes 6HGP, 6HGQ, 6HGR, and 6HGS) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- APRT
- adenine phosphoribosyltransferase
- ADE
- adenine
- buffer A
- 20 mm Tris-HCl, pH 7.4, 5 mm MgCl2
- DSF
- differential scanning fluorimetry
- Gua
- guanine
- HGPRT
- hypoxanthine-guanine phosphoribosyltransferase
- Hx
- hypoxanthine
- NCI
- noncovalent interaction
- PRPP
- α-d-5-phosphoribosyl-1-pyrophosphate
- QM/MM
- quantum mechanics/molecular mechanics
- RMSD
- root mean square deviation
- SN
- nucleophilic substitution.
References
- 1. Bashor C., Denu J. M., Brennan R. G., and Ullman B. (2002) Kinetic mechanism of adenine phosphoribosyltransferase from Leishmania donovani. Biochemistry 41, 4020–4031 10.1021/bi0158730 [DOI] [PubMed] [Google Scholar]
- 2. Yuan L., Craig S. P. 3rd, McKerrow J. H., and Wang C. C. (1992) Steady-state kinetics of the schistosomal hypoxanthine-guanine phosphoribosyltransferase. Biochemistry 31, 806–810 10.1021/bi00118a024 [DOI] [PubMed] [Google Scholar]
- 3. Eads J. C., Scapin G., Xu Y., Grubmeyer C., and Sacchettini J. C. (1994) The crystal structure of human hypoxanthine-guanine phosphoribosyltransferase with bound GMP. Cell 78, 325–334 10.1016/0092-8674(94)90301-8 [DOI] [PubMed] [Google Scholar]
- 4. Shi W., Sarver A. E., Wang C. C., Tanaka K. S., Almo S. C., and Schramm V. L. (2002) Closed site complexes of adenine phosphoribosyltransferase from Giardia lamblia reveal a mechanism of ribosyl migration. J. Biol. Chem. 277, 39981–39988 10.1074/jbc.M205596200 [DOI] [PubMed] [Google Scholar]
- 5. Wang G. P., Cahill S. M., Liu X., Girvin M. E., and Grubmeyer C. (1999) Motional dynamics of the catalytic loop in OMP synthase. Biochemistry 38, 284–295 10.1021/bi982057s [DOI] [PubMed] [Google Scholar]
- 6. Huyet J., Ozeir M., Burgevin M.-C., Pinson B., Chesney F., Remy J.-M., Siddiqi A. R., Lupoli R., Pinon G., Saint-Marc C., Gibert J.-F., Morales R., Ceballos-Picot I., Barouki R., Daignan-Fornier B., et al. (2018) Structural insights into the forward and reverse enzymatic reactions in human adenine phosphoribosyltransferase. Cell Chem. Biol. 25, 666–676.e4 10.1016/j.chembiol.2018.02.011 [DOI] [PubMed] [Google Scholar]
- 7. Curto R., Voit E. O., Sorribas A., and Cascante M. (1997) Validation and steady-state analysis of a power-law model of purine metabolism in man. Biochem. J. 324, 761–775 10.1042/bj3240761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Heinonen J. K. (2001) Biological Role of Inorganic Pyrophosphate. New York, Springer US; 10.1007/978-1-4615-1433-6 [DOI] [Google Scholar]
- 9. Goitein R. K., Chelsky D., and Parsons S. M. (1978) Primary 14C and α secondary 3H substrate kinetic isotope effects for some phosphoribosyltransferases. J. Biol. Chem. 253, 2963–2971 [PubMed] [Google Scholar]
- 10. Schramm V. L., and Shi W. (2001) Atomic motion in enzymatic reaction coordinates. Curr. Opin. Struct. Biol. 11, 657–665 10.1016/S0959-440X(01)00269-X [DOI] [PubMed] [Google Scholar]
- 11. Shi W., Li C. M., Tyler P. C., Furneaux R. H., Grubmeyer C., Schramm V. L., and Almo S. C. (1999) The 2.0 Å structure of human hypoxanthine-guanine phosphoribosyltransferase in complex with a transition-state analog inhibitor. Nat. Struct. Biol. 6, 588–593 10.1038/9376 [DOI] [PubMed] [Google Scholar]
- 12. Phillips C. L., Ullman B., Brennan R. G., and Hill C. P. (1999) Crystal structures of adenine phosphoribosyltransferase from Leishmania donovani. EMBO J. 18, 3533–3545 10.1093/emboj/18.13.3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Silva M., Silva C. H., Iulek J., Oliva G., and Thiemann O. H. (2004) Crystal structure of adenine phosphoribosyltransferase from Leishmania tarentolae: Potential implications for APRT catalytic mechanism. Biochim. Biophys. Acta 1696, 31–39 10.1016/j.bbapap.2003.09.003 [DOI] [PubMed] [Google Scholar]
- 14. Costas M. E., and Acevedo-Chávez R. (1997) Density functional study of the neutral hypoxanthine tautomeric forms. J. Phys. Chem. A 101, 8309–8318 10.1021/jp970621n [DOI] [Google Scholar]
- 15. Silva C. H., Silva M., Iulek J., and Thiemann O. H. (2008) Structural complexes of human adenine phosphoribosyltransferase reveal novel features of the APRT catalytic mechanism. J. Biomol. Struct. Dyn. 25, 589–597 10.1080/07391102.2008.10507205 [DOI] [PubMed] [Google Scholar]
- 16. Chen X. Y., Link T. M., and Schramm V. L. (1998) Ricin A-chain: Kinetics, mechanism, and RNA stem-loop inhibitors. Biochemistry 37, 11605–11613 10.1021/bi980990p [DOI] [PubMed] [Google Scholar]
- 17. Amaya M. F., Watts A. G., Damager I., Wehenkel A., Nguyen T., Buschiazzo A., Paris G., Frasch A. C., Withers S. G., and Alzari P. M. (2004) Structural insights into the catalytic mechanism of Trypanosoma cruzi trans-sialidase. Structure (Lond.) 12, 775–784 10.1016/j.str.2004.02.036 [DOI] [PubMed] [Google Scholar]
- 18. Murray A. W. (1967) Studies on the nature of the regulation by purine nucleotides of adenine phosphoribosyltransferase and of hypoxanthine phosphoribosyltransferase from Ehrlich ascites-tumour cells. Biochem. J. 103, 271–279 10.1042/bj1030271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Guthrie R. D., and Jencks W. P. (1989) IUPAC recommendations for the representation of reaction-mechanisms. Acc. Chem. Res. 22, 343–349 10.1021/ar00166a001 [DOI] [Google Scholar]
- 20. Ghanem M., Murkin A. S., and Schramm V. L. (2009) Ribocation transition state capture and rebound in human purine nucleoside phosphorylase. Chem. Biol. 16, 971–979 10.1016/j.chembiol.2009.07.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stein R. L., and Cordes E. H. (1981) Kinetic α-deuterium isotope effects for Escherichia coli purine nucleoside phosphorylase-catalyzed phosphorolysis of adenosine and inosine. J. Biol. Chem. 256, 767–772 [PubMed] [Google Scholar]
- 22. Suarez J., Haapalainen A. M., Cahill S. M., Ho M. C., Yan F., Almo S. C., and Schramm V. L. (2013) Catalytic site conformations in human PNP by 19F-NMR and crystallography. Chem. Biol. 20, 212–222 10.1016/j.chembiol.2013.01.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fedorov A., Shi W., Kicska G., Fedorov E., Tyler P. C., Furneaux R. H., Hanson J. C., Gainsford G. J., Larese J. Z., Schramm V. L., and Almo S. C. (2001) Transition state structure of purine nucleoside phosphorylase and principles of atomic motion in enzymatic catalysis. Biochemistry 40, 853–860 10.1021/bi002499f [DOI] [PubMed] [Google Scholar]
- 24. Johnson E. R., Keinan S., Mori-Sánchez P., Contreras-García J., Cohen A. J., and Yang W. (2010) Revealing noncovalent interactions. J. Am. Chem. Soc. 132, 6498–6506 10.1021/ja100936w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Maestro. (2014) Schrödinger, LLC New York, NY [Google Scholar]
- 26. Otilia C., and Wolfgang L. (2004) Metal Mediated Template Synthesis of Ligands, World Scientific, Singapore [Google Scholar]
- 27. Gerbeleu N. V., Arion V. B., and Burgess J. P. (2008) Template Synthesis of Macrocyclic Compounds, John Wiley & Sons, New York, NY [Google Scholar]
- 28. Khan J. A., Tao X., and Tong L. (2006) Molecular basis for the inhibition of human NMPRTase, a novel target for anticancer agents. Nat. Struct. Mol. Biol. 13, 582–588 10.1038/nsmb1105 [DOI] [PubMed] [Google Scholar]
- 29. Nyhan W. L. (2005) Disorders of purine and pyrimidine metabolism. Mol. Genet. Metab. 86, 25–33 10.1016/j.ymgme.2005.07.027 [DOI] [PubMed] [Google Scholar]
- 30. Berens R. L., Krug E. C., and Marr J. (1995) Purine and pyrimidine metabolism. in Biochemistry and Molecular Biology of Parasites (Marr J., and Müller M., eds) pp. 89–117, Academic Press, San Diego, CA [Google Scholar]
- 31. Gutierrez J. A., Crowder T., Rinaldo-Matthis A., Ho M. C., Almo S. C., and Schramm V. L. (2009) Transition state analogs of 5′-methylthioadenosine nucleosidase disrupt quorum sensing. Nat. Chem. Biol. 5, 251–257 10.1038/nchembio.153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bollée G., Harambat J., Bensman A., Knebelmann B., Daudon M., and Ceballos-Picot I. (2012) Adenine phosphoribosyltransferase deficiency. Clin. J. Am. Soc. Nephrol. 7, 1521–1527 10.2215/CJN.02320312 [DOI] [PubMed] [Google Scholar]
- 33. Hertz N. T., Berthet A., Sos M. L., Thorn K. S., Burlingame A. L., Nakamura K., and Shokat K. M. (2013) A neo-substrate that amplifies catalytic activity of Parkinson's-disease-related kinase PINK1. Cell 154, 737–747 10.1016/j.cell.2013.07.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhang X., Chen Y., Lin X., Hong X., Zhu Y., Li W., He W., An F., and Guo H. (2013) Adenine phosphoribosyl transferase 1 is a key enzyme catalyzing cytokinin conversion from nucleobases to nucleotides in Arabidopsis. Mol. Plant. 6, 1661–1672 10.1093/mp/sst071 [DOI] [PubMed] [Google Scholar]
- 35. Leslie A. G. W., and Powell H. R. (2007) Processing diffraction data with MOSFLM. in Evolving Methods for Macromolecular Crystallography (Read R. J., and Sussman J. L., eds) Vol. 245, pp. 41–51, Berlin: 10.1007/978-1-4020-6316-9_4 [DOI] [Google Scholar]
- 36. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., Read R. J., Vagin A., and Wilson K. S. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Murshudov G. N., Vagin A. A., Dodson E. J. (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 53, 240–255 10.1107/S0907444996012255 [DOI] [PubMed] [Google Scholar]
- 39. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Tuttle J. V., and Krenitsky T. A. (1980) Purine phosphoribosyltransferases from Leishmania donovani. J. Biol. Chem. 255, 909–916 [PubMed] [Google Scholar]
- 41. QSite. (2014) Schrödinger, LLC New York, NY [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.